
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-dimensional assembled PVP-modified silver nanoprisms guided by butanol for surface-enhanced Raman scattering-based invisible printing platforms†
Kosuke
Sugawa
 *a,
Yutaro
Hayakawa
a,
Yukiko
Aida
b,
Yuto
Kajino
b and
Kaoru
Tamada
*b
*a,
Yutaro
Hayakawa
a,
Yukiko
Aida
b,
Yuto
Kajino
b and
Kaoru
Tamada
*b
aDepartment of Materials and Applied Chemistry, College of Science and Technology, Nihon University, Kanda-Surugadai, Chiyoda-ku, Tokyo 101-8308, Japan. E-mail: sugawa.kosuke@nihon-u.ac.jp
bInstitute for Materials Chemistry and Engineering (IMCE), Kyushu University, 744 Motooka, Nishiku, Fukuoka 819-0395, Japan. E-mail: tamada@ms.ifoc.kyushu-u.ac.jp
First published on 21st June 2022
Abstract
This study proposes a methodology for the fabrication of two-dimensional assembled colloidal nanocrystals based on the classical theory for the surface excess of a short-chain alcohol (butanol) in an aqueous mixture and Rayleigh–Bénard–Marangoni convection caused by temperature and/or surface tension gradients due to the volatilization of butanol at the air–water interface. When polyvinylpyrrolidone (PVP)-modified anisotropic silver nanoprisms dispersed in butanol were added into the water phase, the nanoprisms were guided to the air–water interface via adsorbed butanol together with free butanol and formed dense two-dimensional assemblies through the lateral attraction between nanoprisms as the adsorbed butanol was volatilized. The obtained dense film composed of silver nanoprisms exhibited surface-enhanced Raman scattering (SERS) activity, and in particular, the activity was largely enhanced by low-pressure plasma treatment. A SERS-based invisible printing platform that could only be recognized by x–y SERS mapping was demonstrated with the patterned nanoprism films.
The importance of metal and semiconductor nanocrystals with unique optical and electronic properties has been increasing year by year. Highly ordered assembled structures composed of these nanomaterials are currently being actively studied for a wide range of applications, including electronic devices,1 biosensing,2 and nanotherapy.3 Notably, these assembled structures have not only functions corresponding to the sum of their individual units but also unique collective properties derived from the interaction between nanomaterials as building blocks. A typical example is a dense, two-dimensional (2D) assembled structure composed of plasmonic metal nanocrystals.4,5 Within the 2D assembled structures of metal nanocrystals, the coupling of the localized surface plasmon (LSP) resonance of individual nanocrystals excites a stronger local electromagnetic field in the nanogap between adjacent particles, resulting in a significant change in the optical properties, including resonance wavelengths.5
Although various top-down approaches using beam processing technology have been proposed to fabricate nanointegrated structures, the bottom-up approach using colloidal nanocrystals synthesized by chemical synthesis still has advantages in terms of cost and convenience, especially in constructing dense 2D assemblies with sub-10 nm gap distances. To date, a sufficient number of self-assembly techniques have been established at air–liquid or liquid–liquid interfaces. These techniques mainly utilize physicochemical interactions between metal nanocrystals and/or between a metal nanocrystal and the interface induced by using surface modifiers (and promoters),6,7 adding a cosolvent,8,9 and evaporating a dispersed solvent.10,11 In these self-assembly processes, solvents are key to keeping nanocrystals monodisperse in solution or to inducing self-assembly through the volatilization of solvents in the case of the Langmuir–Blodgett (LB) technique.12 The self-assembly of symmetric particles, such as in spherical colloids, is relatively easy. However, the self-assembly process becomes drastically difficult when assembling nanocrystals of other shapes, especially asymmetric shapes with high aspect ratios, to adjust functionalities (electromagnetic field intensity, resonance wavelength, etc.). For example, triangular silver nanoplates (silver nanoprisms, AgPRs) with sharp tips are known to have excellent plasmonic properties due to the lightening rod effect, showing a significantly enhanced local electromagnetic field at controlled wavelengths. However, the fabrication of 2D assemblies of nanoprisms (side-to-side assembly) remains challenging13 because the strong attraction acting between the prism faces (i.e., face-to-face assembly) must be blocked.14
Polyvinylpyrrolidone (PVP) is known as a shape-directing polymer that leads to the growth of complex polyhedral-shaped nanocrystals in the synthetic process and plays an important role in one- to three-dimensional assemblies of metal nanocrystals.15–20 These phenomena are due to the characteristics of PVP, such as the steric barrier effect that suppresses the aggregation of metal nanocrystals even at high concentrations,16 electronic interactions with metal atomic layers,20 and the amphipathic properties of the molecular chain.16,18,19 By taking advantage of the unique physicochemical properties of PVP, the achievement of orientation-controlled, high-density 2D assemblies of asymmetric nanocrystals may be possible, even though it has never been successful.
In this study, we report a methodology to fabricate dense 2D assemblies of PVP-modified AgPRs (side-to-side assembly) at the air–water interface. The experimental procedure is extremely simple: drop an AgPR butanol dispersion solution into the water phase in a Petri dish (an open system) and wait a few hours. Here, the classical theory concerning the surface excess of a short chain alcohol in an aqueous mixture and Rayleigh–Bénard–Marangoni convection caused by temperature and/or surface tension gradients due to volatilization of a solvent are effectively utilized.
We also present the function of the obtained high-density 2D PVP-modified AgPR film as an invisible printing platform. Recently, invisible printing platforms, which refer to being invisible in normal circumstances but recognizable by specific methods, have attracted much attention for steganography and watermarking in anti-counterfeiting.21 AgPR assemblies are expected to serve as surface-enhanced Raman scattering (SERS)-based invisible printing platforms due to the properties of the film having homogeneous plasmonic nanogaps on the entire surface. SERS signals can be generated only by laser irradiation of a specific wavelength and are less susceptible to background noise due to the sharp spectral pattern peculiar to the structure of the probe molecule. In this study, the potential of using AgPR assemblies as an invisible printing platform is explored by x–y SERS mapping measurements of surfaces patterned by low-pressure plasma processing.
AgPRs were synthesized by irradiating a centre wavelength of 470 ± 5 nm light from homebuilt light-emitting diode (LED) arrays into a colloidal aqueous solution of Ag nanospheres for 24 h; during this process, the light intensity was precisely controlled to minimize nontriangular byproducts.22,23 The AgPRs synthesized by this photoinduced method were stably dispersed in an aqueous phase due to the electrostatic repulsive force of the adsorbed citrate (the detailed synthetic method is described in the ESI†). The AgPRs were sequentially modified with 16-mercaptohexadecanoic acid (MHA), polyethyleneimine (PEI, MW: ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and PVP (MW: ∼55000) and dispersed in BuOH (Fig. 1(A)). Initially, we attempted the direct modification of AgPRs with PVP, but this process resulted in the irreversible aggregation of AgPRs in solution via face-to-face stacking. Therefore, to obtain monodispersed AgPRs, MHA–PEI–PVP modification was performed in this study. The detailed surface modification procedures are described in the Experimental section, and the change in the zeta potential induced by the stepwise surface reaction is presented in Fig. S1 (ESI†). The hydrodynamic diameter of citrate-adsorbed AgPRs was 25.0 nm in the aqueous phase and that of MHA–PEI–PVP-modified AgPRs was 118.7 nm in BuOH due to swelling of PVP (and possibly PEI). The extinction spectra shown in Fig. 1(E) revealed that both citrate-adsorbed and PVP-modified AgPRs exhibit a prominent extinction peak attributed to the in-plane dipole plasmon in the longer wavelength region and a weak shoulder attributed to the in-plane quadrupole plasmon in the shorter wavelength region, which is peculiar to monodispersed AgPRs.24 The LSP resonance bands of MHA–PEI–PVP-modified AgPRs were redshifted compared with those of citrate-adsorbed AgPRs due to the increase in the local refractive index (n) of surrounding AgPRs (the refractive indices of MHA, PEI, and PVP (n = 1.48–1.53) as well as that of BuOH (n = 1.40) are larger than that of water (n = 1.33)).25
000), and PVP (MW: ∼55000) and dispersed in BuOH (Fig. 1(A)). Initially, we attempted the direct modification of AgPRs with PVP, but this process resulted in the irreversible aggregation of AgPRs in solution via face-to-face stacking. Therefore, to obtain monodispersed AgPRs, MHA–PEI–PVP modification was performed in this study. The detailed surface modification procedures are described in the Experimental section, and the change in the zeta potential induced by the stepwise surface reaction is presented in Fig. S1 (ESI†). The hydrodynamic diameter of citrate-adsorbed AgPRs was 25.0 nm in the aqueous phase and that of MHA–PEI–PVP-modified AgPRs was 118.7 nm in BuOH due to swelling of PVP (and possibly PEI). The extinction spectra shown in Fig. 1(E) revealed that both citrate-adsorbed and PVP-modified AgPRs exhibit a prominent extinction peak attributed to the in-plane dipole plasmon in the longer wavelength region and a weak shoulder attributed to the in-plane quadrupole plasmon in the shorter wavelength region, which is peculiar to monodispersed AgPRs.24 The LSP resonance bands of MHA–PEI–PVP-modified AgPRs were redshifted compared with those of citrate-adsorbed AgPRs due to the increase in the local refractive index (n) of surrounding AgPRs (the refractive indices of MHA, PEI, and PVP (n = 1.48–1.53) as well as that of BuOH (n = 1.40) are larger than that of water (n = 1.33)).25
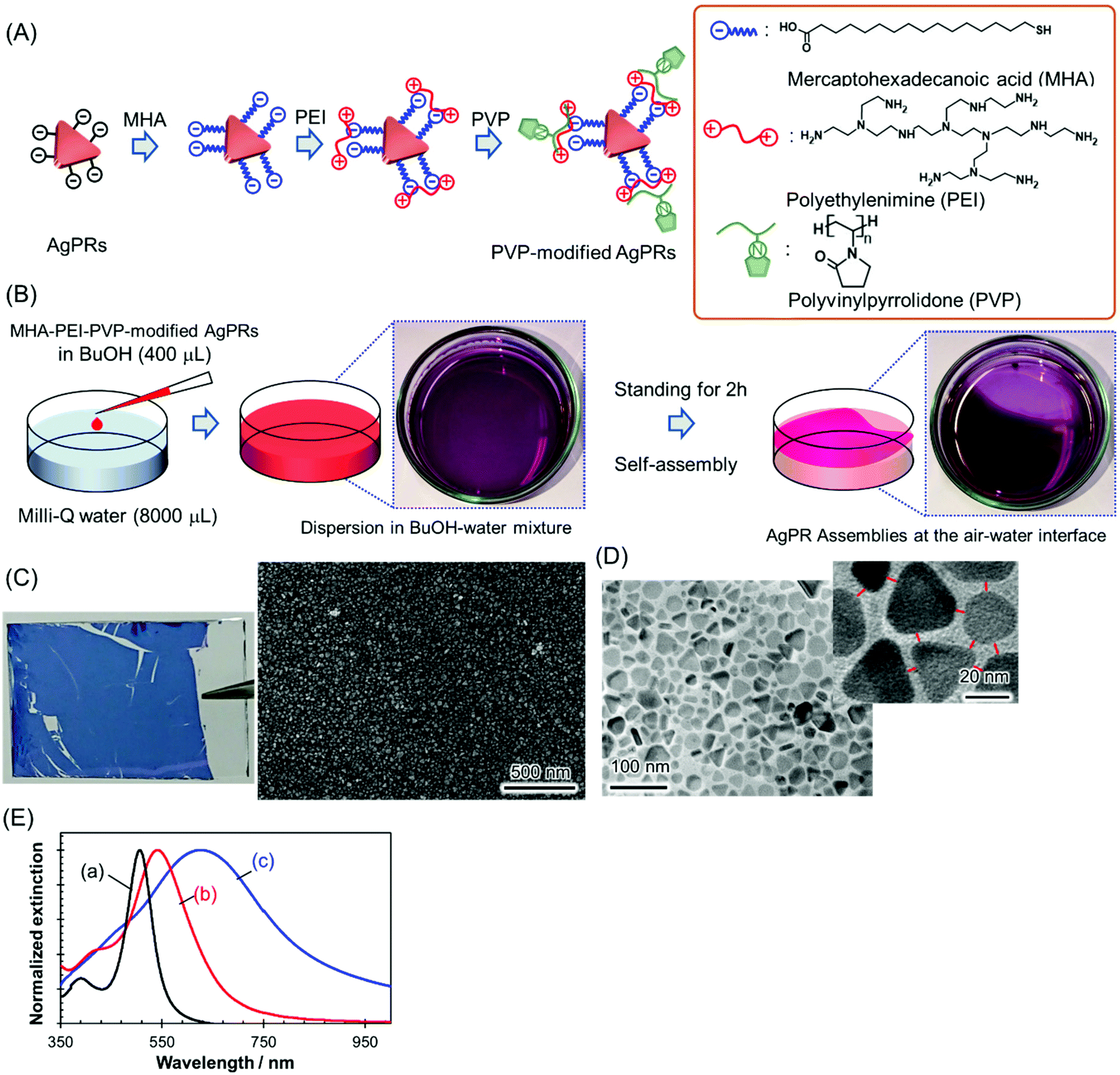 | ||
| Fig. 1 Formation processes of AgPR assemblies and their morphological and optical properties. (A) Stepwise surface modification of AgPRs with MHA, PEI, and PVP. (B) Assemblies of MHA–PEI–PVP-modified AgPRs at the air–water interface. (C) A photograph of the AgPR film transferred onto a glass substrate and SEM images of the AgPR film transferred onto a Si-wafer. (D) TEM images of the AgPR film. (E) Extinction spectra of (a) an aqueous dispersion of citrate-adsorbed AgPRs, (b) a dispersion of MHA–PEI–PVP-modified AgPRs in BuOH and (c) AgPR assemblies transferred to a glass plate (corresponding to (C)). | ||
2D assemblies of AgPRs were simply prepared through the dropwise addition of MHA–PEI–PVP-modified AgPRs (400 μL) in a BuOH dispersion to Milli-Q water (8000 μL) in a Petri dish at room temperature (Fig. 1(B)). When the BuOH dispersion was added into the water phase, most of the AgPRs were dissolved in the water phase with BuOH and formed a transparent purple solution (from this point, MHA–PEI–PVP-modified AgPRs will be simply abbreviated as “AgPRs”). However, a certain amount of AgPRs appeared at the air–water interface and spontaneously formed assemblies within a few hours. After approximately 2 h, more than 70% of the liquid surface was covered with the dark purple-coloured AgPR self-assembled film (see the video in the ESI†). The dropping of the BuOH colloidal solution of AgPRs into water at higher temperatures (e.g., 45 °C) induced faster AgPR assembly on the water surface faster than that observed at room temperature, and the AgPR assemblies became nonuniform (Fig. S2†). On the other hand, when the experiment was conducted at room temperature, uniform AgPR assemblies were formed reproducibly. The volume ratio of colloidal BuOH to water was determined by considering the number of nanoparticles necessary to fully cover the Petri dish surface (∼24 cm2) with the 2D assemblies and the critical miscibility of BuOH with water (720 μL (BuOH)/8000 μL (water)). When the volume of BuOH exceeded this critical value, BuOH and water separated into two phases.
The AgPR-assembled film transferred onto a glass plate exhibited a uniform blue colour (Fig. 1(C)). A SEM image of the AgPR film transferred onto silicon wafers confirmed the formation of uniform, dense 2D assemblies of AgPRs over a large area (Fig. S3†). The transmission electron microscopy (TEM) image (Fig. 1(D)) confirmed that the film was composed of a AgPR monolayer (side-to-side assemblies) with a gap distance of 5.0 ± 0.4 nm. The extinction spectrum of the AgPR assembled film (Fig. 1(E)) was redshifted by 84 nm with a broadened LSP resonance peak, attributed to the coupling of the in-plane dipole plasmon between AgPRs in close proximity.
The versatility of this unique nanoparticle assembly procedure was confirmed by using nanocrystals of other shapes and sizes, such as gold nanospheres (diameter of 16 ± 2 nm), large-sized AgPRs (edge length of 59 ± 7 nm), and gold nanostars (size of 93 ± 12 nm), as shown in Fig. 2(A). All these particles modified with MHA–PEI–PVP formed high-density 2D assemblies (Fig. 2(B)), could be transferred onto glass substrates (Fig. 2(C)) in a similar manner to the original AgPRs and exhibited optical properties dominated by plasmon coupling between the nanocrystals (Fig. 2(D)). It should be noted that spherical particles could form 2D self-assemblies via direct modification with PVP without MHA–PEI treatment.
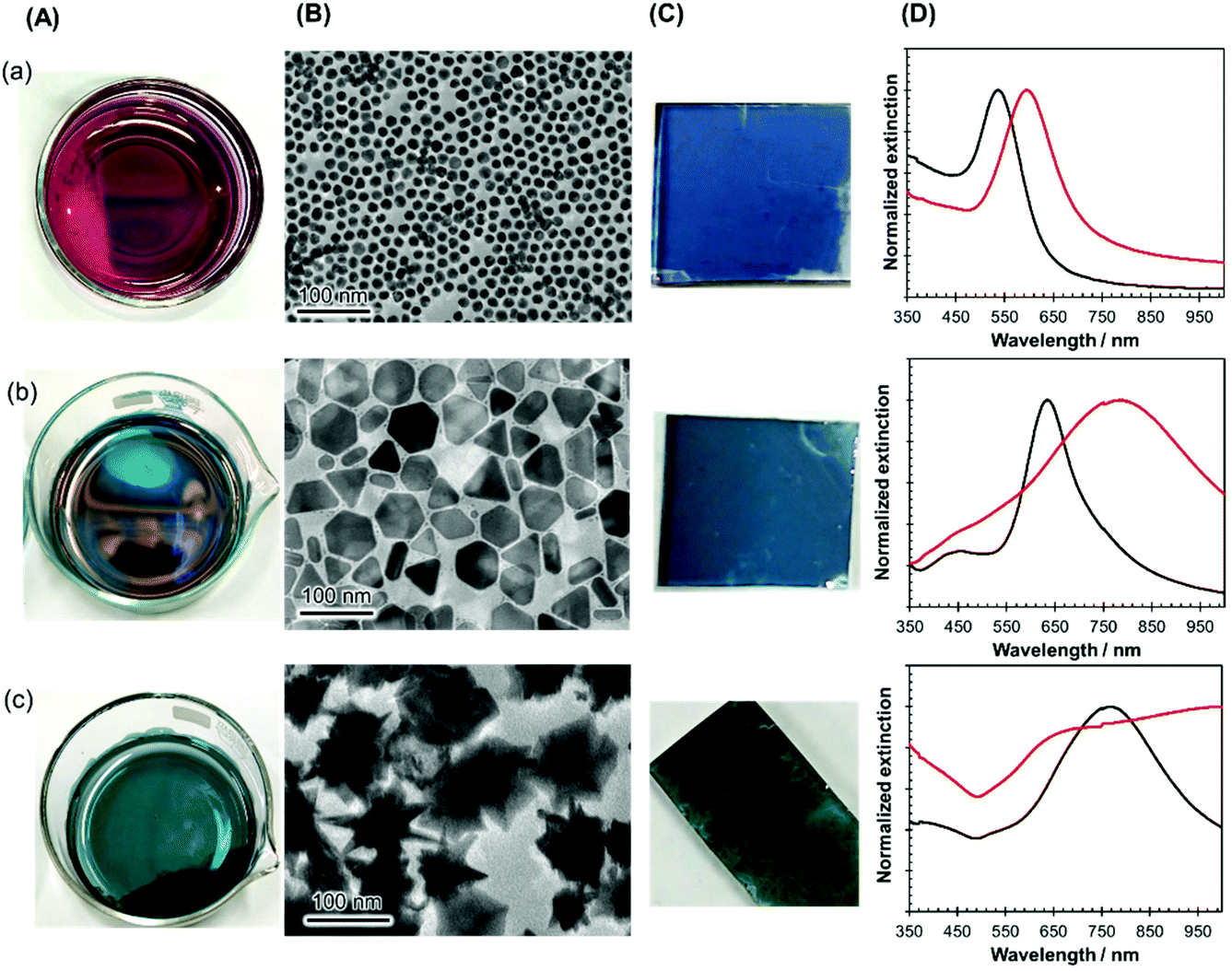 | ||
| Fig. 2 Assembly of various metal nanocrystals by using the developed methodology: (a) Au nanospheres with a diameter of 16 ± 2 nm, (b) AgPRs with an edge length of 59 ± 7 nm, and (c) Au nanostars with a size of 93 ± 12 nm. These assembled films were prepared at the air–water interface by dropping a BuOH dispersion (400 μL) into the water phase (8000 μL) and leaving it for 2 h. (A) Photographs of the films at the air–water interface, (B) TEM images, (C) photographs of films transferred onto glass substrates, and (D) extinction spectra of the BuOH dispersion (black lines) and the transferred films (red lines). | ||
Compared with other self-assembly techniques involving metal nanocrystals at the air–water (or liquid/liquid) interface using PVP modification,17–19,26 our proposed method has a unique characteristic of self-assembly due to its combination of (i) spontaneous ascent and self-assembly from a dispersed system, (ii) the necessity of BuOH as a solvent mixed with the water phase, and (iii) the necessity of an “open” system. The extinction spectrum of the AgPRs in the BuOH–Milli-Q water mixture (after the dropwise addition of BuOH dispersion into Milli-Q water) maintained the in-plane dipole mode at 532 nm and the in-plane quadrupole at 420 nm, as in the BuOH dispersion, and these LSP resonance bands gradually attenuated with the formation of assembled films at the air–water interface, as shown in Fig. S4.† This result indicates that the AgPRs spontaneously ascended to the air–water interface and assembled while maintaining a monodispersed state in the solution (no particle aggregation in the solution).
The necessity of BuOH was confirmed by performing the same experiments with other solvents, such as ethanol, acetone, methanol and ethylene glycol (Fig. 3 and S5†). In the series of experiments, magnetic stir-bar mixing was used to mix the solution for 10 s in consideration of the different miscibilities of the solvents with water (Fig. 3(A)). The result for the BuOH system (Fig. 3(B)(a)) was the same as that in Fig. 1: close-packed monolayer formation with a constant gap distance between AgPRs was clearly visualized even though some nontriangular Ag nanoplates were incorporated. In contrast, only a slight film formation was observed with ethanol and acetone (Fig. 3(B)(c) and (e)), and no signs of film formation were observed with methanol and ethylene glycol (Fig. S5†). The degree of assembly with different solvents was estimated not only by the macroscopic surface coverage at the air–water interface (Fig. 3(B)(a), (c) and (e)) but also from the density of particles obtained from the TEM images (Fig. 3(B) (b), (d) and (f)), as summarized in Fig. 3(C). The AgPRs modified with MHA–PEI–PVP could be dispersed in Milli-Q water without organic solvents, but no clear AgPR assemblies were detected in the pure water system. Furthermore, when the BuOH–Milli-Q water mixed solution was kept in a Petri dish covered with a lid, no AgPR assemblies were found over 40 h (Fig. S6†). The necessity of an open system is key to understanding the self-assembly mechanism.
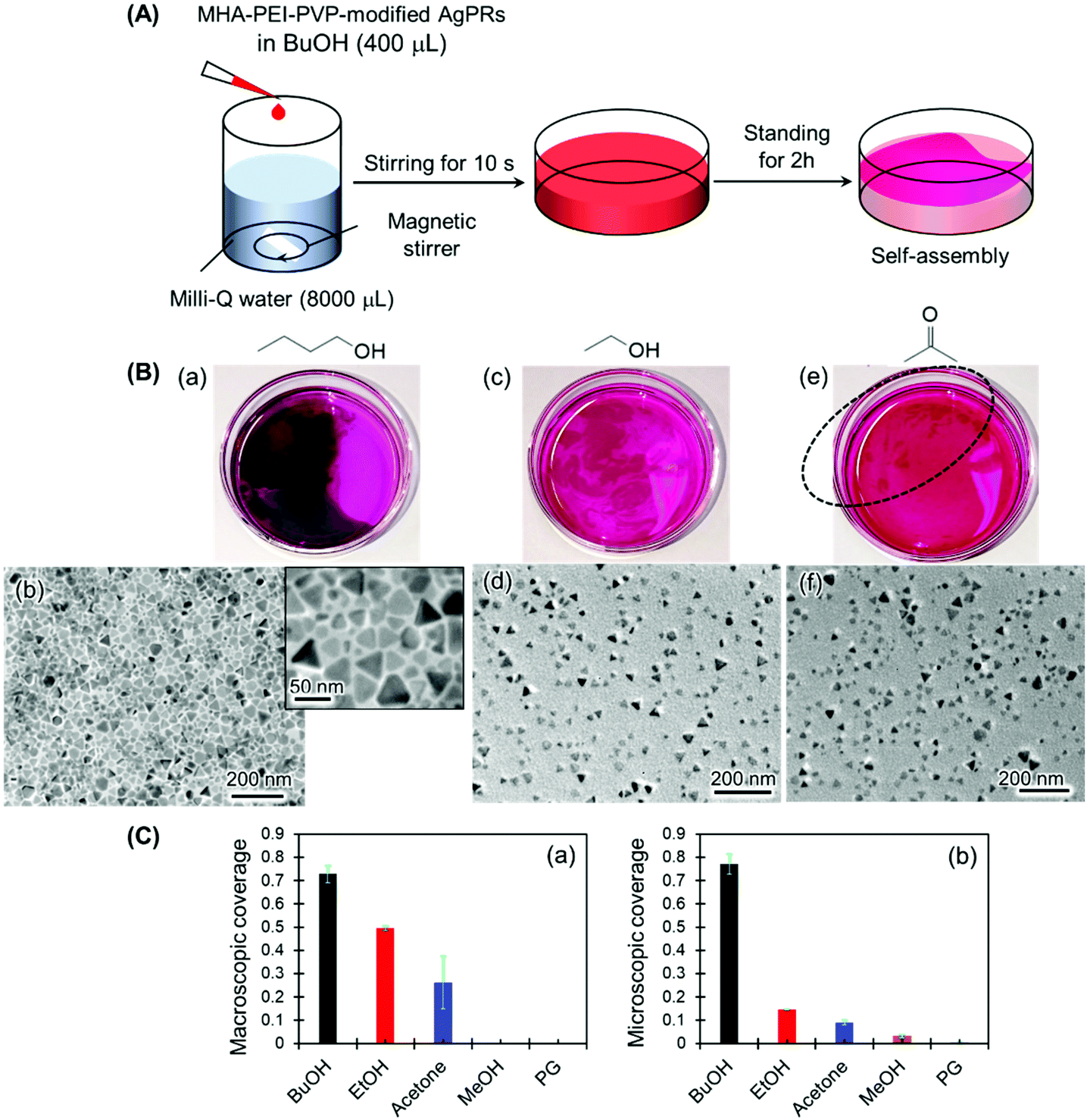 | ||
| Fig. 3 Influence of mixing solvents on AgPR assembly: (a) BuOH, (c) EtOH, and (e) acetone. (A) Schematic of the experimental procedure. A AgPR dispersion was added dropwise into Milli-Q water and mixed with a magnetic stirrer for 10 s. The mixed solution was left for 2 h in a Petri dish. (B) Photographs of the air–water interface after 2 h, and TEM images of the transferred films. (C) Macroscopic surface coverage obtained from the photographs of the air–water interface (a), and microscopic surface coverage obtained from the TEM images (b). Both values were estimated by using ImageJ software. No film formation was observed with methanol and ethylene glycol (data not shown). | ||
The solvent-selective AgPR assembly mechanism seems to be composed of the following two processes: (1) transportation of the nanocrystals from the bulk phase to the air–water interface and (2) self-assembly of the nanocrystals at the air–water interface. We assume that the strong interaction between BuOH and PVP adsorbed on the outermost AgPRs plays an important role in these processes (Scheme 1).
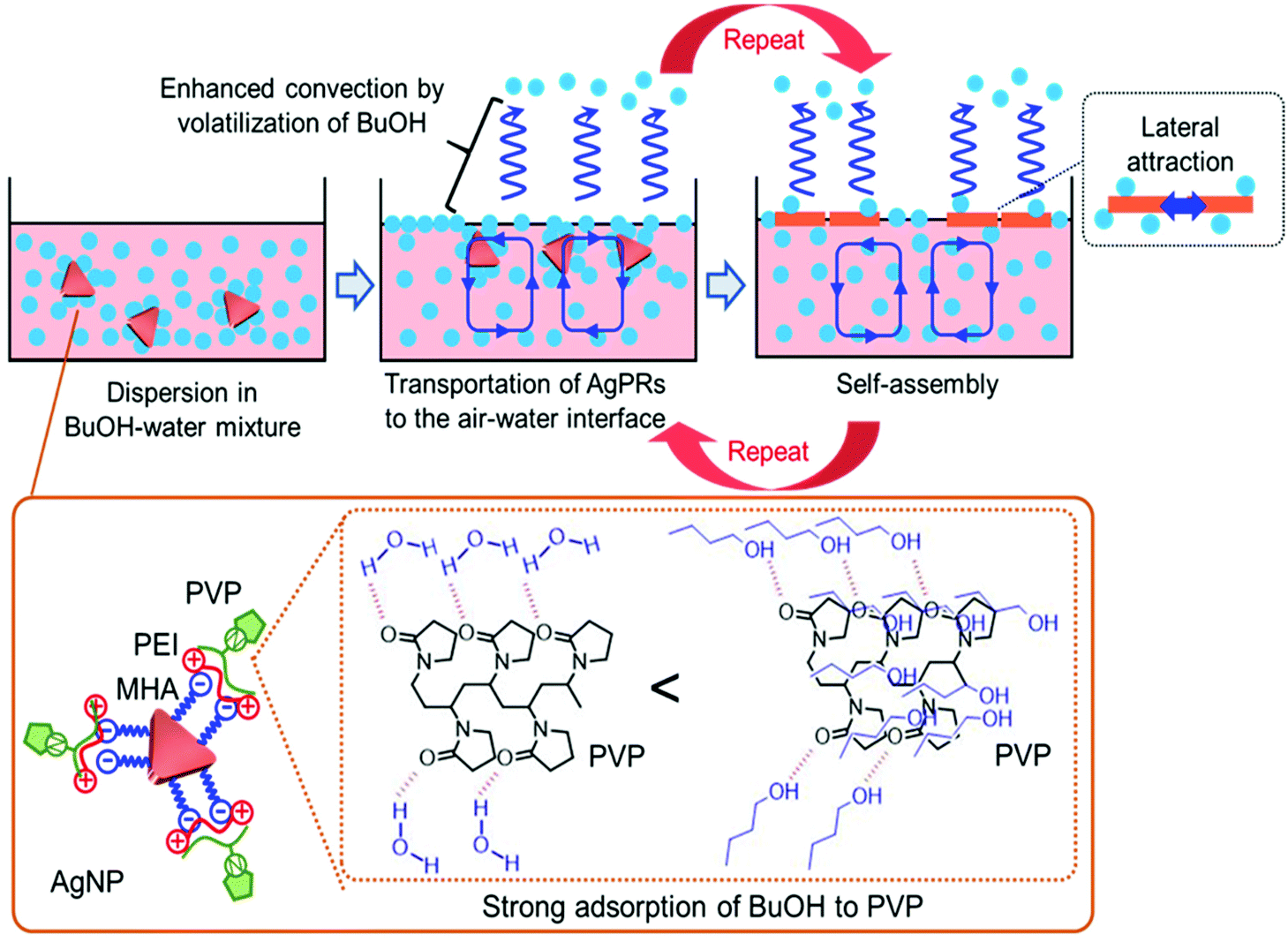 | ||
| Scheme 1 Suggested mechanism for AgPR assembly at the air–liquid interface. | ||
PVP adsorbed on AgPRs is an amphipathic polymer. Amide groups in the pyrrolidone rings of PVP can be hydrated in an aqueous solution, whereas neutral BuOH interacts with PVP more strongly than water via an additional hydrophobic interaction between the butyl group and the hydrocarbon backbone of PVP.27 The extinction spectra and zeta potential data shown in Fig. S7 and S8† confirmed that BuOH is retained on the AgPR surface in the BuOH–Milli-Q water mixed solution. A short-chain alcohol is known to adsorb strongly onto the air–water interface in an aqueous mixture and to form a stable monolayer at maximum surface excess, where hydrophobic hydration is no longer retained.28–31 The adsorption of butanol could be experimentally confirmed by surface tension measurements and analysis based on the Gibbs adsorption equation (see Fig. S9†). This phenomenon occurs even in our three-component system composed of water, BuOH, and BuOH-adsorbed AgPRs. Furthermore, the surface excess BuOH at the air–water interface preferentially volatizes in an open system,32 as evidenced by an increase in the surface tension over time (see Fig. S9†). Consequently, microscopic temperature and surface tension gradients due to the volatilization of BuOH cause Rayleigh–Bénard–Marangoni convection33 and transport the BuOH-adsorbed AgPRs to the air–water interface. The BuOH-adsorbed AgPRs, which are floating at the air–water interface, are spontaneously assembled by the lateral force along the interface induced by the convection flow and lateral attraction (probably both hydrophobic and van der Waals interactions) between AgPRs when surface excess BuOH is volatilized. The contact angle of the AgPR surface was ∼50 degrees soon after the film formation due to the adsorbed BuOH but reduced with time.
The solvent selectivity for AgPR assembly shown in Fig. 3 proves the rationality of our description. Ethanol and acetone produced incomplete, small amounts of AgPR assemblies at the interface, while methanol and ethylene glycol hardly produced any assemblies. When the hydrocarbon chains of alcohols become too short, preferential adsorption of alcohols on AgPRs with PVP and surface excess at the air–water interface (surface tension gradients between the bulk and interface) are significantly reduced (see Fig. S9†). As a result, Rayleigh–Bénard–Marangoni convection is not sufficiently induced, and the AgPRs are not transported to the interface for self-assembly. Ethylene glycol strongly adsorbs on PVP via two hydroxyl groups compared with ethanol, but its surface excess is smaller than that of ethanol (see Fig. S9†),34 and AgPR assemblies cannot be achieved. The fact that BuOH with a saturated vapour pressure lower than that of EtOH and acetone was the most effective solvent for assembly refutes the possibility that the assembly is caused by temperature gradients due to volatilization of the solvent.
Although a previous study used colloidal BuOH for self-assembly at the air–water interface,18 the assembly mechanism is quite different. In the previous study, BuOH was used as a spreading solvent in a conventional Langmuir film formation. In contrast, in our study, BuOH was used as a material that drives Rayleigh–Bénard–Marangoni convection via surface excess and volatilization. In our method, individual AgPRs ascend at the air–water interface from the bulk aqueous phase and form highly oriented side-to-side assemblies after floating at the interface; i.e., face-to-face nonuniform assemblies are unlikely to form even for anisotropic nanoparticles with high aspect ratios.
This assembly methodology of nanocrystals at the air–water interface from a dispersion solution using the surface excess in an aqueous mixture is considered to be highly versatile and effective because the reaction is thermodynamically controlled and quite reproducible and the solvent involved is eventually removed by volatilization during the assembly process.
In this study, the AgPRs, which exhibit a strong electromagnetic field through excitation of the in-plane dipole mode,35 formed dense 2D assemblies with parallel arrangements. Here, the plasmon coupling between the in-plane dipole modes generated a stronger electromagnetic field in the nanogaps between AgPRs. The AgPRs were sufficiently protected with MHA, PEI, and PVP. By taking advantage of these characteristics, in this study, we demonstrated an invisible printing platform application based on the SERS phenomenon, which can be realized on AgPR assemblies. As shown in Fig. 4(A), a glass plate mask (thickness: 1.5 mm) hollowed out in an “S” shape (S-shape size: 6.0 × 4.5 mm2) was placed on the AgPR assemblies on a glass substrate and crimped. The sample plate was treated with low-pressure plasma (intensity: 10; time: 60 s), the mask was removed, and the plate was immersed in a 1 mM MeOH solution of mercaptobenzoic acid (MBA) for 1 h to immobilize the MBA on the AgPR assemblies. After washing the plate with MeOH, an x–y SERS mapping measurement was performed on the assemblies (the detailed measurement conditions are described in the Experimental section in the ESI†). Fig. 4(B)(a) shows a photograph of AgPR assemblies modified with MBA after low-pressure plasma treatment. The AgPR assemblies were observed as a uniform blue film. Under normal circumstances, the masked and unmasked areas were indistinguishable, a so-called “invisible state”. In contrast, as shown in Fig. 4(B)(b), the SERS mapping image of the same region clearly visualized the masked and unmasked regions based on the signal intensity derived from the SERS spectrum of MBA (Fig. 4(C)). The signal intensity in the unmasked area (Fig. 4(C)(c)) was 2.5-fold higher than that in the masked area (Fig. 4(C)(b)). The SERS spectrum of AgPR assemblies before the treatment did not show a SERS peak at approximately 1587 cm−1 (Fig. 4(C)(a)), indicating that the SERS signals were derived from immobilized MBA. The assignment of SERS signals is shown in Fig. S10.†36,37 The amplification of the SERS signal in the patterned region is due to the increase in the amount of adsorbed MBA induced by decomposition of the surface modifiers and to the plasmon-derived signal enhancement induced by the direct adsorption of MBA on the bare AgPRs. Consequently, the SERS signal of the AgPR assemblies was selectively amplified in the area hollowed out in the S-shape, and the S-shape was visually recognized by 1587 cm−1 SERS signal mapping as a “visible state”.
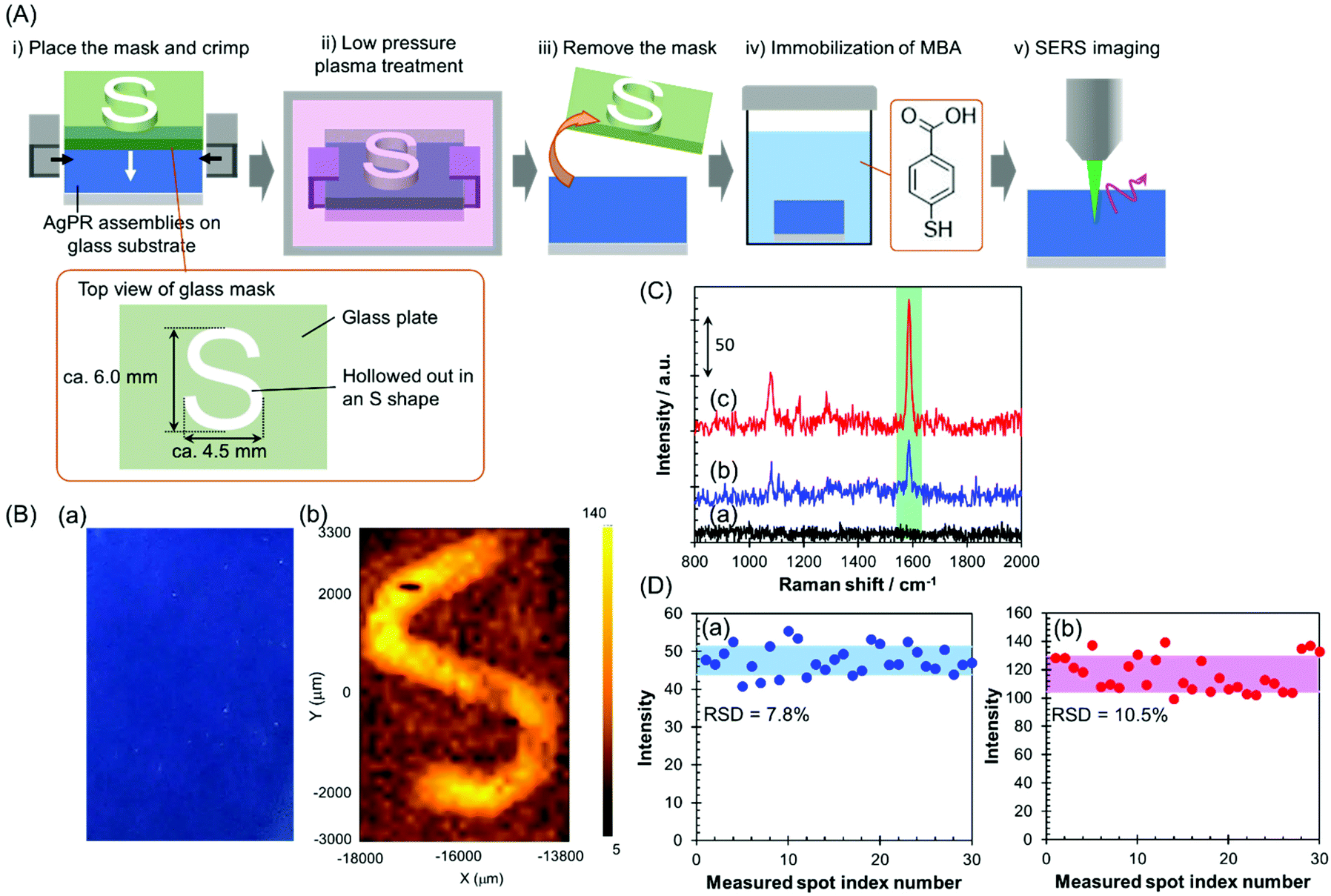 | ||
| Fig. 4 Application of AgPR assemblies to invisible information recording based on SERS mapping. (A) Process of recording invisible information on AgPR assemblies. (B) A photograph of AgPR assemblies after low-pressure plasma treatment (a), and a SERS mapping image of the same surface (b). (C) SERS spectra of the AgPR assemblies as-prepared (a) and in the masked (b) and unmasked (S-shaped) (c) regions after low-pressure plasma treatment and immobilization of MBA. (D) Relative standard deviation (RSD) of the SERS signal intensities at 1587 cm−1 obtained at 30 random points in the masked (a) and unmasked (b) regions. | ||
These results demonstrate that the AgPR assemblies function as a SERS-based invisible printing platform that can only be recognized by x–y SERS signal mapping. Fig. 4(D) shows the 1587 cm−1 SERS intensity obtained by measuring 30 random points within the masked and unmasked areas. The relative standard deviations of the SERS intensities in the masked and unmasked regions were 7.8% and 10.5%, respectively. This result guarantees the high resolution of information visible by SERS mapping measurements.
Conclusions
In summary, we succeeded in forming a high-density 2D assembled film composed of parallelly arranged anisotropic AgPRs by spontaneous adsorption of AgPRs from a BuOH aqueous mixture solution onto the air–water interface, in which surface excess butanol and Rayleigh–Bénard–Marangoni convection due to volatilization of butanol were key. This technique is applicable to forming assemblies of plasmonic nanocrystals with various sizes, shapes, and metallic species. This assembly methodology for nanocrystals is highly versatile and effective for forming unique higher-order structures because the thermodynamically controlled reaction is quite reproducible and the solvent involved is eventually removed by volatilization during the assembly process. The AgPR assemblies function as a SERS-based invisible printing platform only recognizable by x–y SERS mapping. This developed technology has potential applications for steganography and watermarking in banknote anti-counterfeiting, for example. Although x–y SERS mapping measurements are not currently a high-throughput process, it is expected that the applications of plasmonic invisible printing platforms will be enhanced by improvements in SERS measurement technologies.Author contributions
KS and KT initiated and supervised the study. YH, YA, YK and KS carried out the experiments and data analyses. KS and KT cowrote the paper. All the authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by financial aid from JSPS KAKENHI (grant numbers 20H02850 and 19H05627).Notes and references
- D. M. Balazs and M. A. Loi, Adv. Mater., 2018, 30, 1800082 CrossRef.
- L. Scarabelli, D. Vila-Liarte, A. Mihi and L. M. Liz-Marzan, Acc. Mater. Res., 2021, 2, 816–827 CrossRef CAS.
- C. Yi, S. Zhang, K. T. Webb and Z. Nie, Acc. Chem. Res., 2017, 50, 12–21 CrossRef CAS PubMed.
- S. Shi and T. P. Russell, Adv. Mater., 2018, 30, 1800714 CrossRef PubMed.
- Z. Ye, C. Li, Q. Chen, Y. Xu and S. E. J. Bell, Nanoscale, 2021, 13, 5937–5953 RSC.
- Y. Xu, M. P. Konrad, W. W. Y. Lee, Z. Ye, S. Bell and E. J. Steven, Nano Lett., 2016, 16, 5255–5260 CrossRef CAS PubMed.
- H. Duan, D. Wang, D. G. Kurth and H. Moehwald, Angew. Chem., Int. Ed., 2004, 43, 5639–5642 CrossRef CAS PubMed.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS PubMed.
- J.-W. Liu, S.-Y. Zhang, H. Qi, W.-C. Wen and S.-H. Yu, Small, 2012, 8, 2412–2420 CrossRef CAS PubMed.
- H. Wang, C. J. Levin and N. J. Halas, J. Am. Chem. Soc., 2005, 127, 14992–14993 CrossRef CAS PubMed.
- T. P. Bigioni, X.-M. Lin, T. T. Nguyen, E. I. Corwin, T. A. Witten and H. M. Jaeger, Nat. Mater., 2006, 5, 265–270 CrossRef CAS PubMed.
- A. Yoshida, K. Imazu, X. Li, K. Okamoto and K. Tamada, Langmuir, 2012, 28, 17153–17158 CrossRef CAS PubMed.
- S. Xu, H. Li, M. Guo, L. Wang, X. Li and Q. Xue, Analyst, 2021, 146, 4858–4864 RSC.
- B. Luo, J. W. Smith, Z. Wu, J. Kim, Z. Ou and Q. Chen, ACS Nano, 2017, 11, 7626–7633 CrossRef CAS PubMed.
- J. Henzie, M. Gruenwald, A. Widmer-Cooper, P. L. Geissler and P. Yang, Nat. Mater., 2012, 11, 131–137 CrossRef CAS PubMed.
- A. Tao, P. Sinsermsuksakul and P. Yang, Nat. Nanotechnol., 2007, 2, 435–440 CrossRef CAS PubMed.
- Q. Guo, M. Xu, Y. Yuan, R. Gu and J. Yao, Langmuir, 2016, 32, 4530–4537 CrossRef CAS PubMed.
- G. D. Moon, T. I. Lee, B. Kim, G. S. Chae, J. Kim, S. H. Kim, J.-M. Myoung and U. Jeong, ACS Nano, 2011, 5, 8600–8612 CrossRef CAS PubMed.
- Y. H. Lee, W. Shi, H. K. Lee, R. Jiang, I. Y. Phang, Y. Cui, L. Isa, Y. Yang, J. Wang, S. Li and X. Y. Ling, Nat. Commun., 2015, 6, 6990 CrossRef CAS PubMed.
- M.-R. Gao, S.-R. Zhang, Y.-F. Xu, Y.-R. Zheng, J. Jiang and S.-H. Yu, Adv. Funct. Mater., 2014, 24, 916–924 CrossRef CAS.
- S. Ye, Q. Fu and J. Ge, Adv. Funct. Mater., 2014, 24, 6430–6438 CrossRef CAS.
- N. Takeshima, K. Sugawa, M. Noguchi, H. Tahara, S. Jin, K. Takase, J. Otsuki and K. Tamada, Chem. Lett., 2020, 49, 240–243 CrossRef CAS.
- C. Xue and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 2036–2038 CrossRef CAS PubMed.
- R. Jin, Y. C. Cao, E. Hao, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS PubMed.
- D. E. Charles, D. Aherne, M. Gara, D. M. Ledwith, Y. K. Gun'ko, J. M. Kelly, W. J. Blau and M. E. Brennan-Fournet, ACS Nano, 2010, 4, 55–64 CrossRef CAS PubMed.
- X. Lin, G. Fang, Y. Liu, Y. He, L. Wang and B. Dong, J. Phys. Chem. Lett., 2020, 11, 3573–3581 CrossRef CAS PubMed.
- N. Gacem and P. Diao, Colloids Surf., A, 2013, 417, 32–38 CrossRef CAS.
- J. Glinski, G. Chavepeyer, J.-K. Platten and P. Smet, J. Chem. Phys., 1998, 109, 5050–5053 CrossRef CAS.
- R. Strey, Y. Viisanen, M. Aratono, J. P. Kratohvil, Q. Yin and S. E. Friberg, J. Phys. Chem. B, 1999, 103, 9112–9116 CrossRef CAS.
- K. Habrdova, S. Hovorka and L. Bartovska, J. Chem. Eng. Data, 2004, 49, 1003–1007 CrossRef CAS.
- Y. F. Yano, J. Colloid Interface Sci., 2005, 284, 255–259 CrossRef CAS PubMed.
- M.-M. Walz, C. Caleman, J. Werner, V. Ekholm, D. Lundberg, N. L. Prisle, G. Oehrwall and O. Bjoerneholm, Phys. Chem. Chem. Phys., 2015, 17, 14036–14044 RSC.
- F. Doumenc, T. Boeck, B. Guerrier and M. Rossi, J. Fluid Mech., 2010, 648, 521–539 CrossRef CAS.
- A. Horibe, S. Fukusako and M. Yamada, Int. J. Thermophys., 1996, 17, 483–493 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- W. K. H. Ho, Z. Y. Bao, X. Gan, K.-Y. Wong, J. Dai and D. Lei, J. Phys. Chem. Lett., 2019, 10, 4692–4698 CrossRef CAS PubMed.
- Y. Wang, W. Ji, H. Sui, Y. Kitahama, W. Ruan, Y. Ozaki and B. Zhao, J. Phys. Chem. C, 2014, 118, 10191–10197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01725c |
| This journal is © The Royal Society of Chemistry 2022 |