
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates
Liping
Guo†
 *ab,
Katie J.
Lamb†
*a and
Michael
North
a
*ab,
Katie J.
Lamb†
*a and
Michael
North
a
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: michael.north@york.ac.uk
bCollege of Biological, Chemical Science and Engineering, Jiaxing University, Jiaxing 314001, China
First published on 7th December 2020
Abstract
This review is focused on the rapidly developing area of organocatalysed cyclic carbonate synthesis. It starts with an introduction to carbon capture and utilisation in general, which introduces the importance of cyclic carbonates. The various mechanisms for cyclic carbonate synthesis are next discussed with a focus on the relative and absolute stereochemical consequences of each mechanism and how this combined with other physical organic chemistry techniques can be used to distinguish between three main mechanistic classes: epoxide activation; carbon dioxide activation and dual activation. Section 2 of the review then discusses recently reported homogeneous organocatalysed systems whilst section 3 surveys recently reported heterogeneous catalyst systems. For each catalyst system, the mechanism of action is discussed with reference to the mechanisms introduced at the start of the review and key parameters such as the optimal temperature, pressure and catalyst loading are given combined with information on substrate scope and yields. This allows the various catalysts to be critically compared on the basis of whichever parameter(s) are of most significance to the reader. A Final Thoughts/Perspective section focusses on the limitations of the use of turnover frequency and turnover number to compare catalysts in this reaction and makes the case for authors to use of a wider range of green metrics when reporting new catalysts. Finally, the Conclusions section focuses on future challenges such as the use of biomass derived epoxides, use of impure carbon dioxide and the need to develop new and existing highly active catalysts out of the laboratory and into real world applications.
 Liping Guo | Liping Guo obtained her Ph.D. degree in 2010 from Zhejiang University, P. R. China. Currently, she is a Lecturer in Chemistry at the College of Biological, Chemical Science and Engineering at Jiaxing University. Since 2020, she has been working as a visiting scholar for a period of one year in the group of Professor Michael North in the Green Chemistry Centre of Excellence at the University of York. Her current research interests lie in the development of the novel and sustainable catalytic systems for carbon dioxide utilization. |
 Katie J. Lamb | Katie Lamb obtained her MChem degree from the University of York in 2013. In 2017, she obtained her Ph.D. in Green Chemistry from the same university under the guidance of Professor Michael North on the development of novel and sustainable carbon dioxide utilization and carbon capture and storage methodologies. From 2018 to 2020, Katie was a Postdoctoral Research Associate at the Green Chemistry Centre of Excellence of the University of York, researching the development of sustainable catalysts for cyclic carbonate synthesis. She is now working at the Central Teaching Laboratories at the University of Liverpool. |
 Michael North | Michael North is professor of Green Chemistry at the University of York. His research focusses on developing organic chemistry for a circular economy. Specific interests include: carbon dioxide capture and utilisation; sustainable solvent development; synthesis of biomass derived polymers and catalysis with Earth crust abundant elements. He has published over 260 articles; is named on 6 patents; edited a four volume book set on Sustainable Catalysis and co-edited a two volume book set on Carbon Dioxide Utilisation. He was awarded the 2001 Descartes Prize by the European Commission and received the 2014 Green chemistry award from the RSC in 2014. |
1. Introduction
1.1. Carbon dioxide generation and utilisation
If there is one chemical reaction that best represents the current state of human civilisation, it is surely the reaction of low oxidation state carbon containing species with molecular oxygen to form carbon dioxide. This is the basis of the generation of energy by the combustion of solid, liquid and gaseous fossil fuels (Fig. 1). Fossil fuel combustion has been the basis of almost all electricity generation for the last 140 years; is still responsible for almost all land, sea and air transportation; provides the fuel needed to heat buildings and provides the thermal energy needed to drive other commercially important chemical reactions. | ||
| Fig. 1 Combustion reactions of fossil fuels. Liquid fuels are a complex mixture represented here by octane as a typical component. | ||
The carbon dioxide produced by the processes shown in Fig. 1 is currently vented to the atmosphere and as a result, atmospheric carbon dioxide levels have risen from about 280 ppm at the start of the industrial revolution1 (ca. 1750) to 316 ppm in 1958 when the Moana Loa carbon dioxide observatory opened, to 417 ppm in May 2020.2 This is an increase of 49% from the pre-industrial revolution value and 33% of all carbon dioxide currently in the Earth's atmosphere now comes from anthropogenic activities.
This substantial increase in atmospheric carbon dioxide levels is now accepted as being the main cause of climate changes such as sea level rise; melting of glaciers and ice shelves; desertification and increasingly frequent severe weather events.3 As a result, it is now imperative that we find something to do with waste carbon dioxide other than dumping it into the atmosphere. Two solutions are available: carbon capture and storage (CCS)4 and carbon capture and utilisation (CCU).5–7 CCS is the engineering solution and involves capturing and purifying waste carbon dioxide (from point sources or from the atmosphere), then pressurising the purified gas before transporting it to a location where it can be stored underground or in depleted oil fields. A feature of CCS is that the carbon dioxide is treated as a waste of no value, so all the costs of capture, purification, transport and storage have to be borne by the carbon dioxide producer and ultimately by the consumer of the principle product of the point source. This makes CCS an inherently expensive process. It is also a continuation of the linear economy approach to energy generation and as such is unsustainable in the medium term. A variation on CCS is enhanced oil recovery (EOR) in which the carbon dioxide is stored not in a depleted oil field, but in one that is only partly depleted, allowing more crude oil to be extracted.8 This gives the carbon dioxide some value, but the crude oil that is extracted will, when used as fuel, generate even more carbon dioxide, which then needs to be captured and stored. A recent study has shown that in every reasonable scenario, more carbon dioxide will be generated by EOR than is stored.9
In contrast, in CCU the carbon dioxide is treated not as waste, but as a valuable resource for the chemical or biological synthesis of commercially important chemicals and fuels. This gives the carbon dioxide value as a chemical feedstock and represents a switch from a linear to a circular economy approach to energy and chemicals production. As such it is a sustainable process. There are only three important reactions of carbon dioxide that lead to fuels (Fig. 2). These are the hydrogenation reactions to methane and methanol and the reduction of carbon dioxide to carbon monoxide. Production of methane (the Sabatier reaction10) directly produces the main component of natural gas. Methanol has a low cetane number which limits its use as a fuel, but it can be dehydrated to dimethyl ether which has a higher cetane number and which is a potential replacement for diesel oil.11 Of these two carbon dioxide hydrogenation reactions, methanol production is more commercially attractive as it uses only three moles of hydrogen per mole of carbon dioxide reduced and two thirds of the hydrogen is incorporated into the methanol. In contrast, for methane synthesis, four molar equivalents of hydrogen are required and only half of this is incorporated into the methane. Thus, methanol production has been commercialised by Carbon Recycling International with the first plant being built in Iceland.12 Reduction of carbon dioxide to carbon monoxide can be achieved by a variety of methods including electrochemically,13 photochemically,14 plasmatically15 and thermally in processes such as the dry reforming of methane.16 The carbon monoxide can then be combined with hydrogen in a Fischer–Tropsch reaction17 to produce liquid hydrocarbons suitable for use as liquid transport fuels. This is a well-established commercial process.
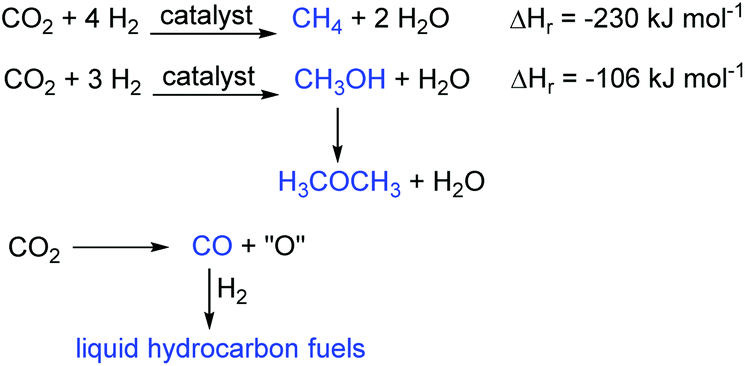 | ||
| Fig. 2 Conversion of CO2 to fuels. | ||
Conversion of carbon dioxide to chemicals is a far more varied process with many different products being available.18,19 Four which have already been commercialised are shown in Fig. 3. The reaction between carbon dioxide and ammonia to produce urea was first commercialised in 1921 and is now the largest scale commercial chemical synthesis; being used to prepare around 150 million tonnes of urea annually.20 The synthesis of salicylic acid from phenol and carbon dioxide under basic conditions is the first step in the industrial synthesis of aspirin and is an even older process, dating back to 1890.21 The syntheses of urea and salicylic acid occur spontaneously in the absence of a catalyst. In contrast, the reaction between carbon dioxide and epoxides occurs only in the presence of a catalyst and can be controlled to give either a cyclic carbonate22 or a polycarbonate.23 Cyclic carbonate synthesis has been commercialised since 1958 and cyclic carbonates have numerous commercial applications including as components of the electrolytes in lithium ion batteries, as polar aprotic solvents and in gas separations.24 Pure polycarbonates derived from carbon dioxide and epoxides are not yet commercial products, but the same concept is being used by Covestro in their ‘Dream’ process to produce random copolymers of polyether and polycarbonate for use as the polyol in polyurethane foam.25
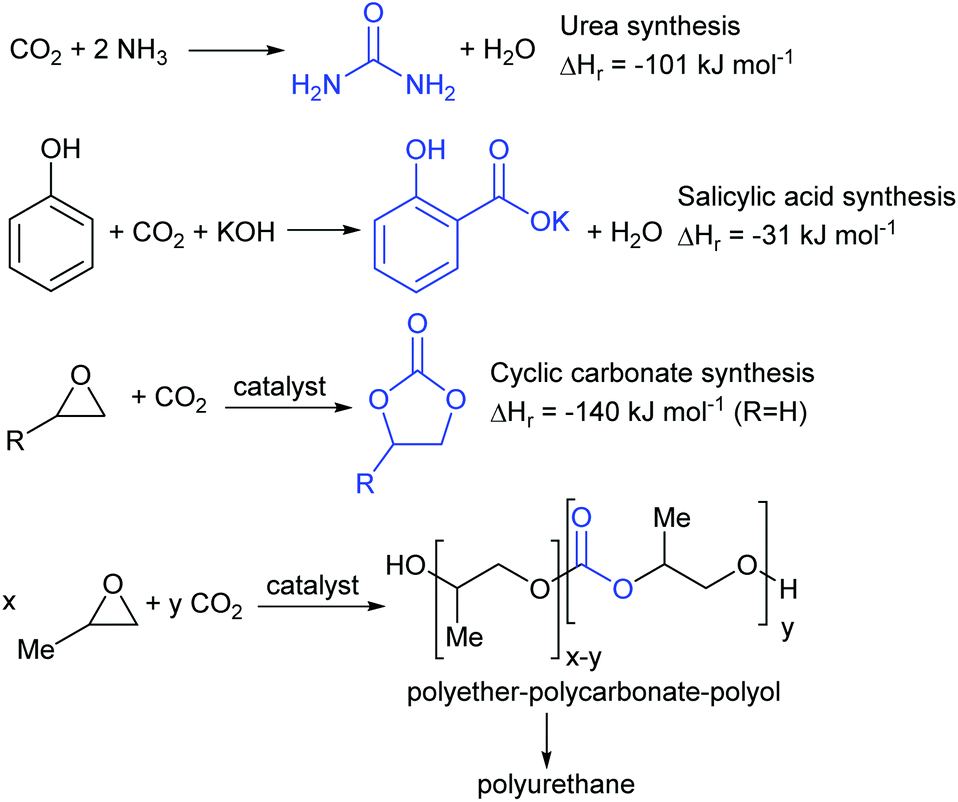 | ||
| Fig. 3 Commercialised examples of chemicals produced from CO2. | ||
The syntheses of fuels and chemicals from carbon dioxide are in some ways complementary processes. Fuel is a relatively low value product, but with a very large market and so with the capability to utilise globally significant amounts of carbon dioxide. In contrast, chemicals production is on a much smaller scale and so can make only a limited contribution to the utilisation of global waste carbon dioxide. However, chemicals are a much higher value product. This leads to the concept of a carbon dioxide refinery26 in which both fuel and chemicals are prepared from carbon dioxide (Fig. 4). Fuel production is responsible for most of the carbon dioxide utilisation whilst chemicals production is responsible for most of the profit of the refinery. Only by producing fuel and chemicals together can such a carbon dioxide refinery be both environmentally and commercially sustainable. A recent analysis27 showed that just two reactions of carbon dioxide: dry reforming of methane (fuel production) and cyclic carbonate synthesis (chemical production) could consume up to 25% of the waste carbon dioxide produced annually.
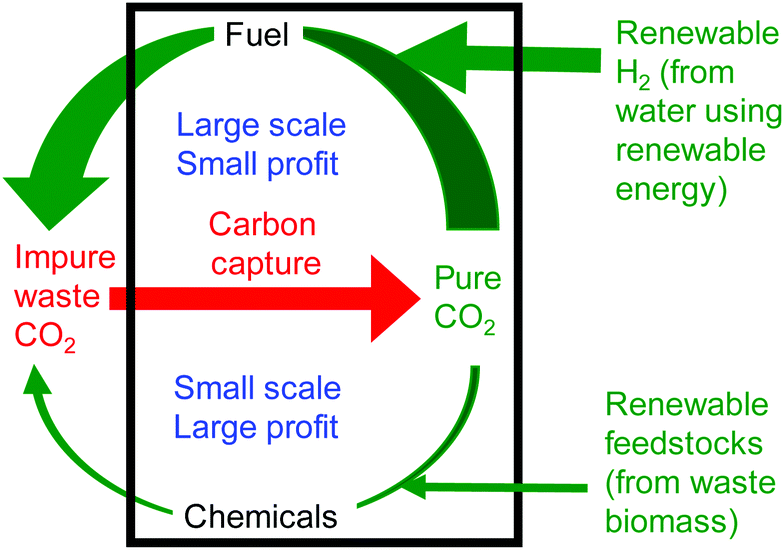 | ||
| Fig. 4 The carbon dioxide refinery concept. | ||
The development of catalysts for cyclic carbonate synthesis is a topic of enormous current activity. The vast majority of this work involves the use of metal-based catalysts. However, there is growing interest in the development of non-metal based catalysts and this review provides a survey of this important aspect of cyclic carbonate synthesis, focussing on the main developments since the last review of organocatalysed cyclic carbonate synthesis in 2017.28 The following section provides a mechanistic overview of cyclic carbonate synthesis and the remaining sections survey the various classes of organocatalysts used for cyclic carbonate synthesis.
1.2. Mechanisms of cyclic carbonate synthesis
Most papers reporting a new catalyst for cyclic carbonate synthesis include a mechanism or catalytic cycle.29 In the best cases these are based on detailed physical organic chemistry studies including: study of reaction kinetics;30 determination of reaction stereochemistry;31 isotopic labelling;32,33 detection of reaction intermediates32 and DFT calculations.32 In other cases however, they are little more than speculation on the part of the authors. The proposed mechanisms for cyclic carbonate synthesis can be grouped into three classes: epoxide activation; carbon dioxide activation and activation of both the epoxide and carbon dioxide. Each of these is discussed below; then referred too throughout subsequent sections of this review.Epoxide activation is by far the most commonly proposed mechanism and requires the catalyst to be able to act as a Lewis or Brønsted acid. A catalytic cycle corresponding to this mechanism is given in Scheme 1. The Lewis or Brønsted acid catalyst first coordinates to the epoxide. Metal-based catalysts act as Lewis acids,34 whilst organocatalysts are most commonly Brønsted acids, though Lewis acidic examples are also known. This coordination activates the epoxide towards ring-opening by a nucleophile (step a). The nucleophile (which must also be capable of acting as a leaving group) is almost always a halide and may be part of the Lewis or Brønsted acid catalyst or added separately, most commonly as a tetraalkylammonium salt. The resulting alcohol or alkoxide intermediate then reacts with carbon dioxide (step b) to form a carbonate intermediate which can undergo a 5-exo-tet cyclisation to form the cyclic carbonate and regenerate the nucleophile and acid catalyst (step c).
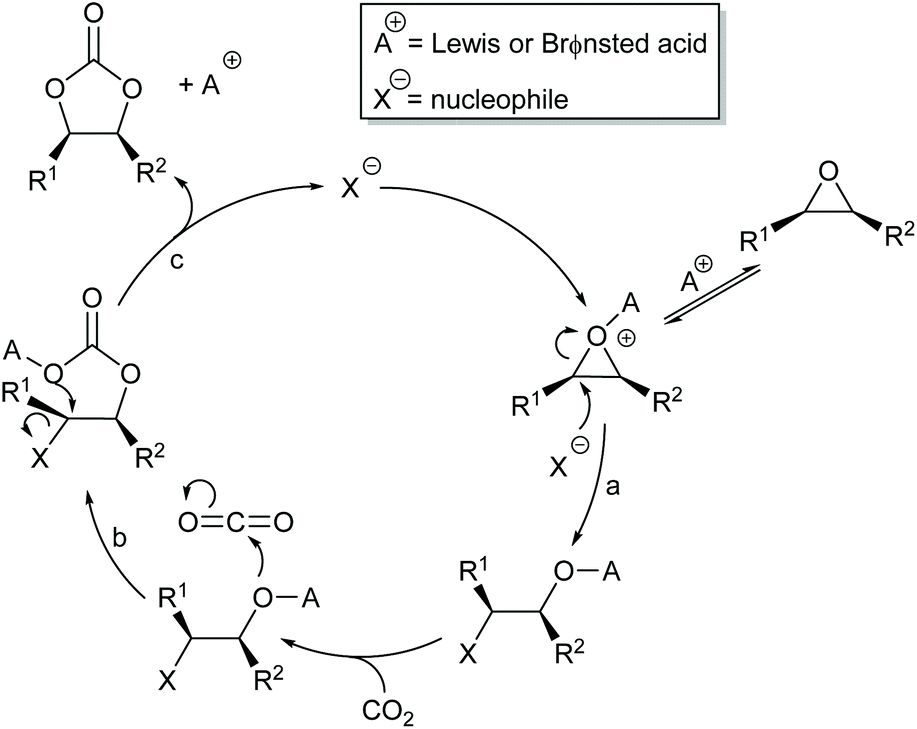 | ||
| Scheme 1 Epoxide activation mechanism for cyclic carbonate synthesis. | ||
The key feature of this mechanism is that it involves two substitution reactions at the same carbon atom (steps a and c). In most cases, the first of these will be an SN2 reaction (and hence will take place at the less hindered end of the epoxide) and the second is an intramolecular substitution. Both these reactions occur with inversion of stereochemistry, so overall retention of the epoxide stereochemistry is observed. This is easily detected using a 1,2-disubstituted epoxide as shown in Scheme 2. For those catalysts that will accept 1,2-disubstituted epoxides as substrates, cyclohexene oxide 1 provides a commercially available and convenient test substrate. For epoxide 1, only the cis-isomer is possible, but the derived cyclic carbonate 2 exists as both cis- and trans-isomers and these are readily distinguished by both IR and NMR spectroscopy.35 The epoxide activation mechanism should give cis-2 as the only product. Unfortunately, many catalyst systems either give no reaction with cyclohexene oxide or convert it to polycarbonate. In these cases, the readily prepared 1-deuterated decene oxides cis-3 and trans-3 provide an alternative.31 The stereochemistry of the resulting cyclic carbonate 4 can be determined by 1H NMR analysis. DFT calculations commonly indicate that either step a or step c of the catalytic cycle shown in Scheme 1 can be rate determining, depending on the catalyst, nucleophile and substrate used. Analysis of the reaction kinetics can also be used to probe this as carbon dioxide is only involved in the catalytic cycle from step b onwards.
 | ||
| Scheme 2 Stereochemical tests for the epoxide activation mechanism. | ||
There are however two classes of substrates which give different regio- and stereo-chemical outcomes under epoxide activation conditions. Styrene oxide 5 (and other electron-rich aromatic epoxides) often show evidence of reaction occurring at the more substituted end of the epoxide. This is apparent by the partial or complete racemisation observed during the reaction, when enantiomerically pure epoxides are used, and is due to the substitution reactions in Scheme 1 steps a and c having appreciable SN1 character, due to stabilisation of the carbenium ion by the electron-rich aromatic ring (Scheme 3).
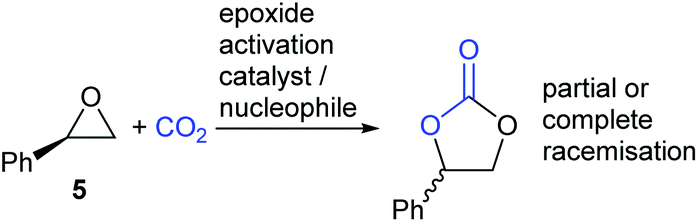 | ||
| Scheme 3 Epoxide activation mechanism with (R)-styrene oxide 5. | ||
Finally, glycidol 6 (and other hydroxymethyl epoxides) can react by a modified epoxide activation mechanism (Scheme 4) in which the hydroxyl group is involved in neighbouring group participation. In this case, the reaction involves a single substitution reaction, though still gives retention of configuration due to the concomitant change in regiochemistry. The occurrence of this mechanism is apparent from the regiochemistry of the product if the glycidol is isotopically labelled or bears substituents at C1 and/or C3.
 | ||
| Scheme 4 Epoxide activation with glycidol 6. | ||
Notably, the mechanism shown in Scheme 4 needs no nucleophilic co-catalyst, the carbon dioxide derived carbonic acid or carbonate fulfils that role. This leads to the second general mechanism for cyclic carbonate synthesis: carbon dioxide activation in which the carbon dioxide is reversibly converted into a nucleophile. In this mechanism (Scheme 5), the catalyst reacts with carbon dioxide to form a carboxylate or carbonic acid (step a). This species acts as a nucleophile to ring-open the epoxide (step b) and subsequent ring-closure to form the cyclic carbonate occurs onto the carbonyl group with elimination of the catalyst (step c).
 | ||
| Scheme 5 Carbon dioxide activation mechanism for cyclic carbonate synthesis. | ||
The carbon dioxide activation mechanism requires a catalyst which is nucleophilic towards carbon dioxide, but not towards epoxides. Catalysts which have been proposed to act in this way include carbenes and nitrogen based nucleophiles such as DMAP, tertiary amines, guanidines and amidines. In some cases the carbon dioxide adduct of the nucleophile is an isolatable species.36,37 The most readily recognised difference between the epoxide activation and carbon dioxide activation mechanisms is in the stereochemistry of the cyclic carbonate product. Retention of the stereochemistry of an appropriately substituted epoxide (Scheme 2) is normally observed with epoxide activation (Scheme 1), whilst inversion of the epoxide stereochemistry is observed for carbon dioxide activation proceeding as shown in Scheme 5. Thus, by controlling the reaction mechanism, it is possible to convert a single stereoisomer of an epoxide into either the cis- or trans-isomer of a cyclic carbonate. This is can be achieved by choice of an appropriate catalyst and sections 2 and 3 of this review will highlight many organocatalysts which function by each of the epoxide and carbon dioxide activation mechanisms.
Cyclic carbonates are often formed as a by-product during polycarbonate synthesis,34 arising by back-biting depolymerisation of a growing polycarbonate chain as illustrated in Scheme 6. Polycarbonate synthesis occurs with a single inversion of the epoxide stereochemistry and the subsequent depolymerisation occurs by reaction of the terminal alkoxide with an internal carbonate group and so does not involve any further stereochemistry changing reactions. Thus, depolymerisation of polycarbonates can be considered to involve a carbon dioxide activation mechanism with the internal carbonate unit playing the role of an activated carbon dioxide unit. In 2019, Pescarmona et al. published an excellent review comparing the ability of various classes of catalysts (metal and metal-free) to form cyclic versus polymeric carbonates.38
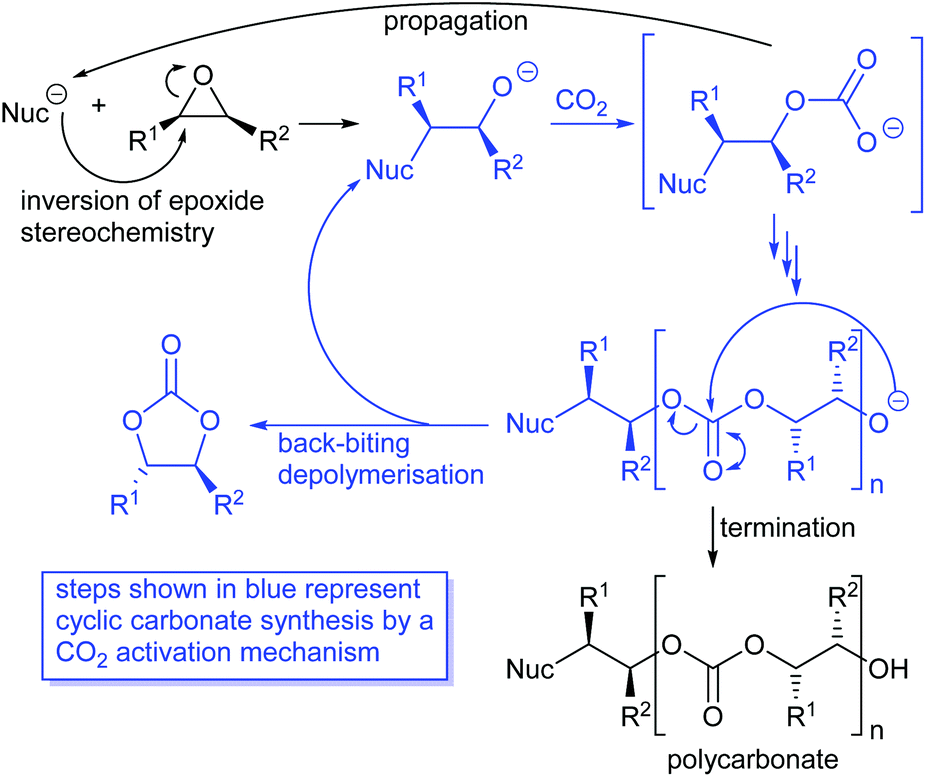 | ||
| Scheme 6 Synthesis and depolymerisation of polycarbonates. | ||
The two mechanisms discussed above, both activate one of the two components (carbon dioxide or epoxide) of the reaction and can achieve significant rate enhancements compared to uncatalysed cyclic carbonate synthesis. As a result, many researchers have developed catalysts to simultaneously activate both the carbon dioxide and epoxide, aiming to achieve even greater rate enhancements. Whilst there is a limit to how fast it is desirable for a reaction to occur, increasing the rate constant for a reaction allows it to be carried out at a lower temperature and thus reduces the thermal demand and hence the carbon dioxide generated and emitted during the reaction.
Scheme 7 illustrates two possible catalytic cycles for dual activation mechanisms, though this is a very diverse area. The nucleophilic and acidic catalyst components may be distinct species, or they can be parts of the same catalyst. The latter has the entropic advantage of bringing all the reaction components together in a single self-assembled species prior to reaction. The upper catalytic cycle of Scheme 7 (steps a–c) is essentially the carbon dioxide activated mechanism, modified by also having the epoxide activated towards ring-opening. As such, this catalytic cycle results in inversion of the epoxide stereochemistry. However, the lower catalytic cycle of Scheme 7 (steps d and e) shows that by inserting a second carbon dioxide into the ring-opened epoxide, a second reaction pathway becomes possible which results in a second inversion of stereochemistry at the carbon attached to R1 and hence to overall retention of epoxide stereochemistry. Thus, cyclic carbonate synthesis proceeding via activation of both substrates can lead to either stereochemical outcome. Indeed it was recently shown that when using a bimetallic aluminium(salen) complex as catalyst with epoxide 3 as substrate, both pathways shown in Scheme 7 can operate simultaneously, resulting in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of cyclic carbonates 4 arising from retention and inversion of epoxide stereochemistry.32 As a result, stereochemical control of cyclic carbonate synthesis can be achieved not only by choice of the catalyst, but also, for some catalyst systems, by optimising the reaction conditions; especially the carbon dioxide pressure and concentration of any halide co-catalyst used.
:1 ratio of cyclic carbonates 4 arising from retention and inversion of epoxide stereochemistry.32 As a result, stereochemical control of cyclic carbonate synthesis can be achieved not only by choice of the catalyst, but also, for some catalyst systems, by optimising the reaction conditions; especially the carbon dioxide pressure and concentration of any halide co-catalyst used.
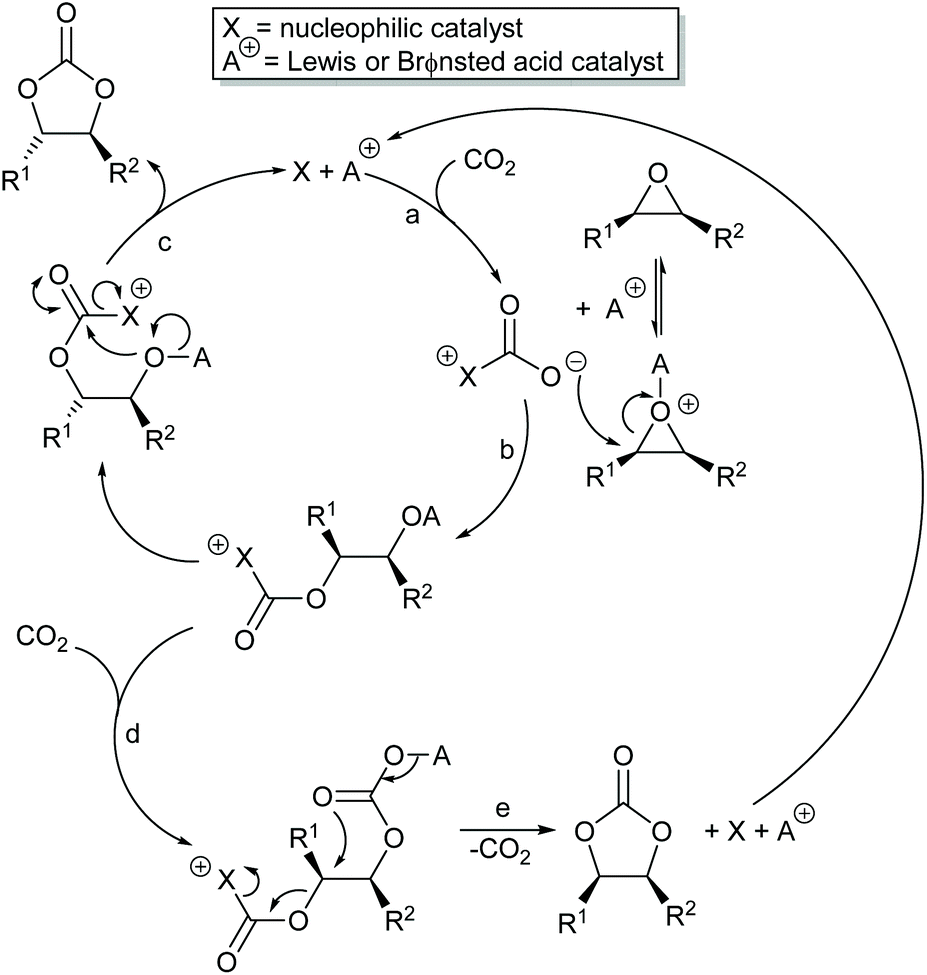 | ||
| Scheme 7 Dual activation mechanisms for cyclic carbonate synthesis. | ||
The best evidence to support a dual activation mechanism, will usually come from a comparative study of the reaction kinetics of the proposed dual activating system and closely related systems, which can activate only one component of the reaction. When the dual activation is achieved by two separate catalysts (A+ and X in Scheme 7), this can be simply achieved by comparing the rates of reactions catalysed by both catalyst components with those in which only one component (A+ or X in Scheme 7) is present. However, when the two catalyst components are part of the same molecule (i.e. A+ and X are covalently linked together), then additional synthetic work is required to produce minimally modified catalysts in which one of the catalytic sites (A+ or X in Scheme 7) is deleted or blocked. Ideally, the dual activating catalyst should not only give a faster rate of reaction than either A+ or X alone, it should have a totally different kinetic profile so that rate changes resulting from inevitable changes to the catalyst structure can be ruled out.
In summary, the synthesis of cyclic carbonates from epoxides and carbon dioxide is a mechanistically diverse area with multiple possible catalytic cycles. This enables catalysts with very different structures and functionalities to be developed to catalyse this reaction. The majority of these catalysts are metal-based, but there is rapidly growing interest in the development of metal-free organocatalysts. The following sections of this review survey the various classes of organocatalysts that have been shown to be effective for cyclic carbonate synthesis.
2. Homogeneous catalysts
Homogeneous catalysts are easier to study mechanistically and therefore to optimise compared to heterogeneous ones. However, they are more difficult to separate from the cyclic carbonate product and thus reuse than heterogeneous catalysts. Optimised homogeneous catalysts can however subsequently be immobilised to convert them into heterogeneous catalysts.39 Thus, the development of new homogeneous catalysts for cyclic carbonate synthesis remains an important and widely studied area.Recent efforts to develop improved homogeneous catalysts for cyclic carbonate synthesis have focused on three main approaches. The first of these is development of functionalised organic halide salts to provide new, highly active, one-component catalysts. The functional groups present in these catalysts can be epoxide activating groups such as hydrogen bond donors, and/or carbon dioxide activating groups such as organic bases; both of which could enhance the reaction of epoxides with carbon dioxide as discussed in section 1.2. The second approach is to develop additives which possess epoxide activating and/or carbon dioxide activating functional groups for use as co-catalysts with conventional organic salts. Such a catalytic system consists of at least two components. Finally, catalytic systems with both highly active epoxide activating groups and carbon dioxide activating groups have been developed. In these systems, halide ions are not necessary, thus permitting the development of halide-free catalytic systems.40
2.1. One-component systems
 | ||
| Fig. 5 Imidazolium based catalysts 7–8. | ||
In 2019, Gao et al. developed a one-pot reaction catalysed by 1-butyl-3-[(3-hydroxyphenyl)methyl]imidazolium bromide, 8, capable of reacting epibromohydrin with carbon dioxide and acids including phenols, thiophenols and aromatic carboxylic acids, to form cyclic carbonates (Scheme 8).42 Under the optimised reaction conditions (60–70 °C, 10 bar of carbon dioxide for 6 h, using 5.0 mol% of catalyst 8) 27 different combinations of acids and epoxides (in 5:1 ratios) were converted into ether, thioether and ester functionalised aromatic cyclic carbonates, in 41–93% isolated yields. This work is interesting as the acids were varied and non-conventional epoxides were screened to determine the substrate scope. The range of substrates screened for this reaction is impressive and it would be interesting to see if this system functions with other haloalkyl epoxides. Fairly toxic reagents, including 1-butylimidazole, are however required to prepare catalyst 8. It was proposed that a key part of the reaction mechanism was proton exchange between the phenol of catalyst 8 and ring-opened epibromohydrin derivatives.
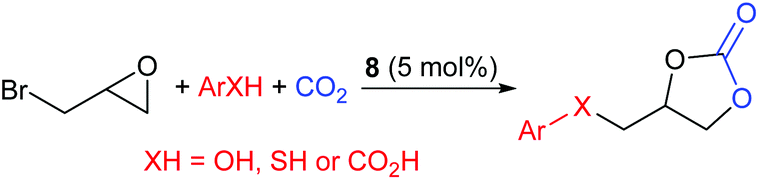 | ||
| Scheme 8 Synthesis of cyclic carbonates from epibromohydrin, CO2 and acids. | ||
In 2017, Zhang et al. performed a DFT study into how amino-functionalised imidazolium ionic liquids catalyse the formation of propylene carbonate from propylene oxide.43 By considering how changes in the concentration of the ionic liquid [APmim]Cl can affect the reaction pathway, a total of nine potential reaction pathways were identified and analysed. Out of these nine pathways, Zhang et al. proposed that unimolecular catalysis will form propylene carbonate via the traditional epoxide activation route (Scheme 1). In the case of bimolecular catalysis by [APmim]Cl, it was proposed that a carbamic-acid functionalised imidazolium ionic liquid ([CAPmim]Cl) would form first, before then inducing an epoxide activation mechanism. This route was predicted to be more favourable due to the stronger acidity of the carbamic acid group, which is key to promoting ring-opening of the epoxide. However, neither of these routes were determined to be the most favourable mechanism, as it was calculated that a binary mixture of [APmim]Cl and [CAPmim]Cl led to the most favourable pathway (Scheme 9). Ultimately, Zhang highlighted the importance of hydrogen bond and dispersive interactions in imidazolium ionic liquid catalysed reactions. Zhang's work also highlights the importance of not just assuming that a standard reaction mechanism occurs and that further analysis such as kinetic studies combined with DFT calculations should be considered.
 | ||
| Scheme 9 Reaction mechanisms determined by Zhang et al. via the [CAPmim]Cl intermediate. | ||
Also in 2017, Zhang et al. studied the ability of eight variations of a benzyl substituted imidazolium ionic liquid (3-benzyl-1-methylimidazolium chloride) to form cyclic carbonates.44 Under the optimum reaction conditions of 0.25 mol% of catalyst, 130 °C and 20 bar of carbon dioxide for 4 h, isolated yields of 89–96% were obtained for four terminal epoxides and cyclohexene carbonate was obtained in 52% yield. DFT calculations were performed and predicted that epoxide activation was the first step of the reaction mechanism (Scheme 1). The substrate scope however was low and the synthesis of the ionic liquids required light-free conditions, which may not be ideal for industrial applications.
In 2018, Li et al. studied the ability of four amino functionalised imidazolium ionic liquids; [APbim]Br, [APmim]Br, [APeim]Br and [AEeim]Br (where AP and AE stand for 3-amimnopropyl and 2-aminoethyl respectively), to convert five terminal epoxides into cyclic carbonates.45 Under the optimum reaction conditions of 120 °C and 10 bar of carbon dioxide, with 0.6 mol% of [APbim]Br for 2 h, 80–99% yields with 99% selectivity were reported, but no internal epoxides were tested. Unusually, the recyclability of these homogeneous catalysts was tested, and they could be recycled up to eight times, without any drop in epoxide conversions. A thorough kinetic study was also performed into order to justify the proposed dual activation reaction mechanism (Scheme 7).
Also in 2018, Dyson et al. performed a catalyst optimisation study of imidazolium ionic liquids to investigate how using different halide counteranions, and changing the presence and location of acidic protons in the ionic liquid, affected their ability to catalyse the conversion of epichlorohydrin into 3-chloropropylene carbonate.46 By studying the ability of four imidazolium ionic liquids in combination with chloride, bromide or iodide anions to catalyse cyclic carbonate formation, Dyson hypothesised that the presence of acidic protons to facilitate ring-opening of the epoxide (in a similar manner to Scheme 1) is not necessarily important for the reaction mechanism. The interaction between the ionic liquid cation and halide anion, and thus the overall nucleophilicity of the halide anion, also affects the reaction mechanism. This can be influenced by hydrogen bonding occurring between the epoxide and ionic liquid, during epoxide ring-opening. Achieving catalytically active imidazolium ionic liquids is thus a delicate balancing act between the nucleophilicity of the halide and proton acidity. In screening the different ionic liquids used in this study, 3-chloropropylene carbonate yields of 36–62% were obtained using 5 mol% of catalyst, at 50 °C and 1 bar of carbon dioxide for 3 h, under solvent free conditions. Testing this catalyst system against a wider substrate scope, to see if the product yields follow the same trend reported with epichlorohydrin, would be interesting. In general, performing more studies into optimising and understanding how catalytic systems truly work, rather than simply aiming for high conversions (and yields) for a few terminal epoxides, would be extremely beneficial.
In a slightly different approach, Tejeda et al. studied the activity of phenol containing imidazole-based organocatalysts, rather than ionic liquid based systems.47 Neutral phenolic imidazoles 9a,b were both found to be extremely active organocatalysts, though 9b had better solubility than 9a. Imidazoles 9a,b were then converted into bifunctional imidazolium salts 10a–c (Scheme 10) which possessed both a phenolic group to activate the epoxide via hydrogen bonding and a halide anion to act as a nucleophile. Compounds 10a–c were found to be even more active catalysts.
 | ||
| Scheme 10 Synthesis of imidazolium salts 10a–c. | ||
The most active phenolic imidazolium catalyst was determined to be 10c, which gave excellent conversions (61–100%) and yields (49–99%) for both internal and bio-based furan and diacid epoxides under very mild conditions (Scheme 11). The catalyst could also be recycled five times without any changes in catalytic activity. On the basis of NMR experiments, an epoxide activation reaction mechanism was proposed for catalyst 10c (Scheme 1) in which the phenol activates the epoxide. This approach of screening novel catalysts against numerous epoxides in order to test the full capability of the catalyst and to guide catalyst structure development when designing more active catalysts, should be adopted more often in future studies. The majority of studies only screen a catalyst against one epoxide. As a result, key information such as solubility, functional group tolerance and reactivity of the catalyst can easily be missed. The screening of catalyst 10c against internal epoxides and more complex bio-based epoxides is also highly noteworthy.
 | ||
| Scheme 11 Conversions and yields reported for catalyst 10c under optimal reaction conditions. | ||
In 2019, Kühn et al. discovered that bridge-functionalised bisimidazolium bromides could catalyse the transformation of epoxides into cyclic carbonates, obtaining 89–93% yields for six terminal epoxides using the most active catalyst 11 (Fig. 6) with 5 mol% of catalyst and 4 bar of carbon dioxide at 70 °C for 16 h.48 This catalyst could be used up to six times without any drop in conversion but was completely ineffective in ring-opening cyclohexene oxide 1. Some of these catalysts were also hydroscopic, which would hinder their ability to be used under ‘flue gas’ type conditions. Fairly mild conditions were used though compared to other organocatalysts.
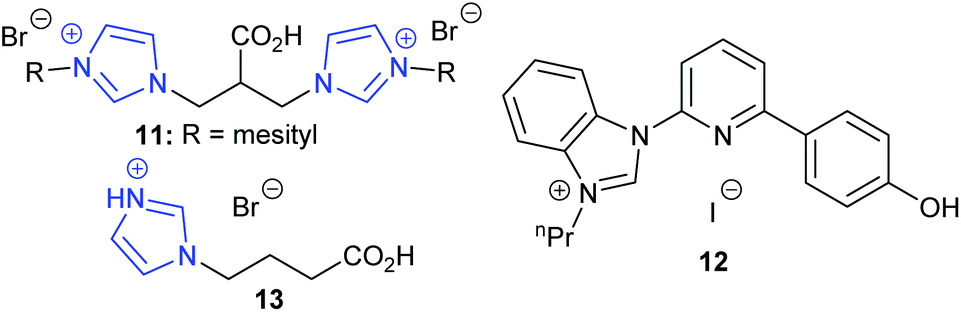 | ||
| Fig. 6 Imidazolium based catalysts 11–13. | ||
Also in 2019, Xia et al. performed DFT calculations to investigate how amine-functionalised ionic liquids, in conjunction with halide anions, could ring-open epibromohydrin.49 Following protonation of the ionic liquid in solution, the catalyst was predicted to form cyclic carbonates by acting as a hydrogen bond donor to activate the epoxide (Scheme 1), followed by ring-opening of the epoxide by the halide anion. The ring-opened intermediate was then predicted to interact with carbon dioxide, rather than the ionic liquid catalyst. The amine-functionalised ionic liquids were computationally compared to similar catalysts in the literature, but no laboratory results were obtained to confirm the conclusions.
In the same year, Liu et al. studied hydroxyl- and carboxyl-functionalised imidazolium halides,50 which were designed to activate epoxides via hydrogen bond formation followed by epoxide ring-opening by a halide, in a similar manner to Xia's DFT work.48 The synthesis of these catalysts required palladium(II) chloride and other metal complexes, which may impact on the sustainability of this system. The most active organocatalyst 12 out of the ten bifunctional catalysts synthesised was tested against sixteen epoxides, five of which were internal epoxides, giving 87–97% isolated yields for cyclic carbonates obtained from terminal epoxides and 11–87% yields for cyclic carbonates obtained from internal epoxides. These yields are impressive, especially for the internal epoxides, considering the fairly mild reaction conditions and low catalytic loadings that were required (2 mol% of catalyst at 5 bar of carbon dioxide and 60 °C or 120 °C for 24 h). Control experiments indicated that the mechanism was epoxide activation (Scheme 1) based on hydrogen bond formation between the phenol group of the catalyst and the epoxide, followed by ring-opening of the activated epoxide by a halide. Not only did this research provide an active catalyst, but it also added support to Xia's proposed mechanism. The recyclability of these catalysts needs improving however, as catalyst leaching was reported over just five reactions.
Zhang et al. studied the ability of nine carboxylic acid containing protic imidazolium ionic liquids to form propylene carbonate, using 5 mol% of catalyst at 15 bar of carbon dioxide and 120–130 °C for 2 or 12 h.51 Isolated yields of 82–95% for terminal cyclic carbonates and 53% for cyclohexene carbonate were reported for the most active catalyst 13. Similar results were obtained over five runs and varying the chain length of the carboxyalkyl chain was found to affect the reaction mechanism, which involved dual activation (Scheme 7).
An interesting piece of work was published by Kleij et al. in 2020, reporting the ability to convert five-membered cyclic carbonates into six-membered cyclic carbonates.52 By synthesising alcohol-functionalised five-membered cyclic carbonates, it was discovered that these compounds could undergo an organocatalysed cascade reaction in the presence of the guanidine-superbase 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1-acetylimidazole (AcIm). By first converting homoallylic alcohols into epoxides, followed by carbon dioxide insertion to form five-membered cyclic carbonates, the desired compounds could be isolated via an induced isomerisation and selective acylation route (Scheme 12). This route was capable of forming fifteen functionalised six-membered cyclic carbonates in 65–96% yields under mild reaction conditions, using 30 mol% of TBD and 1.2 equivalents of AcIm in acetonitrile under a nitrogen atmosphere at 25 °C for 2 h. This reaction could also be performed on a gram scale. The choice of base was critical for this reaction process, with N-heterocyclic bases giving the highest yields and a cooperative reaction mechanism occurring with TBD and AcIm. Control experiments, along with DFT calculations, indicated the crucial role of a free alcohol group in the carbonate, prior to equilibration of the five-membered cyclic carbonate into a six-membered cyclic carbonate. It was also determined that the reaction occurs via a two-step consecutive process primarily dictated by a kinetically controlled acylation step. Whilst this process overall is not strictly metal-free, as the synthesis of the five-membered cyclic carbonates required a metal catalyst, this research is very impressive, as it provides a rare example of forming six-membered cyclic carbonates organocatalytically at room temperature.
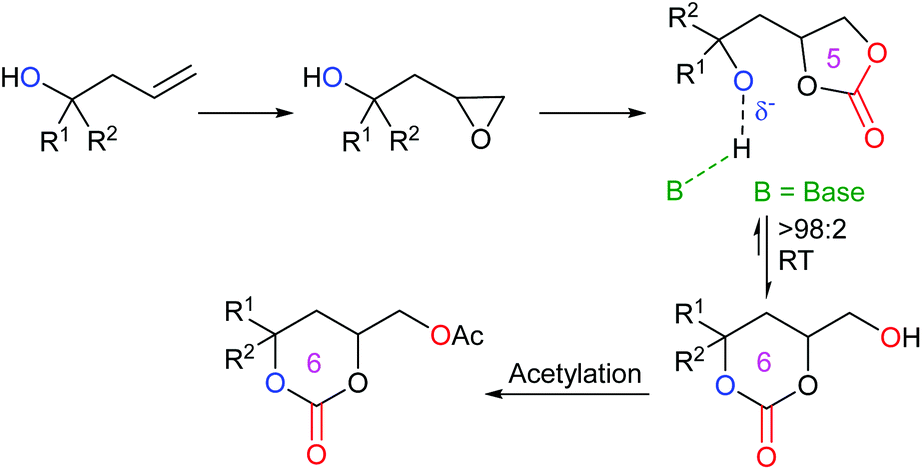 | ||
| Scheme 12 Overview of the synthetic route employed by Kleij et al. in the synthesis of six-membered cyclic carbonates. | ||
In 2017, Leveneur et al. performed a kinetic study of the ability of tetraethylammonium bromide to catalyse the conversion of vegetable oils, and in particular cottonseed oil, into epoxides and then cyclic carbonates.54 After performing a mass transfer analysis, a kinetic model was developed which led to the hypothesis that the rate of carbonation was dependent on the concentrations of catalyst and epoxide and on the carbon dioxide solubility. The conversion of complex vegetable oils into cyclic carbonates is an area of high interest, as it provides a greener route to polymers such as polyurethanes but requires very active catalysts. The knowledge gained from this study is therefore vital to developing catalysts capable of producing cyclic carbonates from epoxidised vegetable oils. In a similar study, Isbell et al. studied the epoxidation and then carbonation of castor oil using tetrabutylammonium bromide.55 Harsh reaction conditions were required to obtain 95% conversion to the carbonate product (100 °C and 100 bar of carbon dioxide for 45 hours), but considering that only 1.4 mol% of tetrabutylammonium bromide was used with respective to epoxidised vegetable oil, this is not surprising.
In 2017, Shirakawa et al. developed a simple bifunctional triethylamine hydroiodide catalyst, which could ring-open epoxides by providing a Brønsted acidic and nucleophilic site within the same catalyst (in a similar manner to Scheme 1).56 This catalyst was particular promising as it was capable of ring-opening twelve terminal, enantiomerically pure epoxides, with no loss of enantiomeric purity under near-ambient conditions (10 mol% of catalyst, 40 °C, 24 h, 1 bar of carbon dioxide) in 87–99% isolated yields (Scheme 13). Unfortunately, this catalyst gave low yields when disubstituted epoxides were used as substrates. The catalyst could however, be used on a large scale, forming 6.4 g of styrene carbonate in 84% isolated yield under the same reaction conditions. Catalyst activity gradually declined over just four uses of the catalyst.
 | ||
| Scheme 13 Triethylamine hydroiodide catalyst developed by Shirakawa et al. | ||
Multi-hydroxylated bis-(quaternary ammonium) ionic liquids were developed in 2018 by Lei et al., with the most active catalyst 14 (Fig. 7) capable of ring-opening eleven epoxides, three of which were internal or compounds with two epoxide rings, via an epoxide activation mechanism (Scheme 1).57 Most of these substrates gave cyclic carbonates in 91–99% isolated yields, though only 67% yield was reported for styrene carbonate and some tricky substrates gave yields as low as 10%. Only a low catalytic loading (0.25 mol%) was required at 20 bar of carbon dioxide and 120 °C for 3, 10 or 20 h. All six catalysts tested were highly active, though other catalysts have reported higher yields (especially for styrene carbonate) under milder conditions.
 | ||
| Fig. 7 Ammonium salt based catalysts 14–15. | ||
In 2018, twenty novel scorpionate organocatalysts were developed by Kim et al., with the most active catalyst giving 90–99% yields for six terminal epoxides (using 2 mol% of catalyst, 10 bar of carbon dioxide and 25 °C for 6 or 12 h). The catalyst could be reused five times (Scheme 14).58 This paper highlights how a methodical approach to catalyst structure optimisation can lead to a highly active catalyst, an approach which should be employed by more researchers in this field. It would have been interesting though to see how active this catalyst was against internal epoxides and against a wider range of substrates.
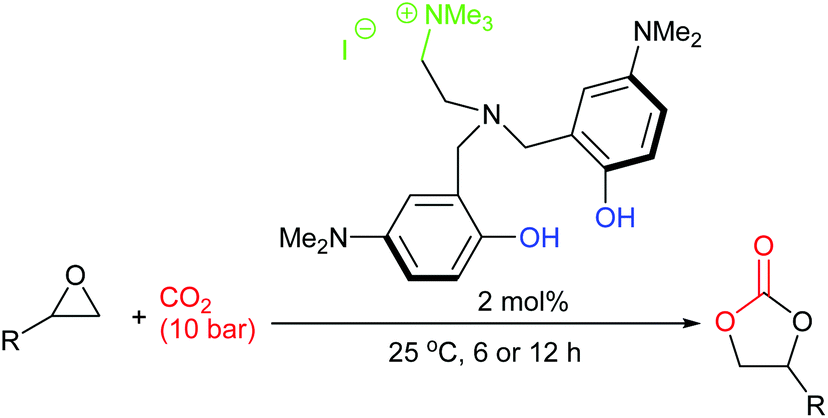 | ||
| Scheme 14 Scorpionate catalysts developed by Kim et al. | ||
Dual-ionic ammonium salts were found by Liu et al. to give 88–99% GC yields for six terminal epoxides via an epoxide activation mechanism (Scheme 1).59 A high catalytic loading was however required (15 mol% of catalyst 15 at 15 bar of carbon dioxide at 30 or 40 °C for 15 h). Only 65% yield was reported when cyclohexene oxide 1 was used as substrate, but this is still impressive for an organocatalyst. The substrate scope could have been more extensively investigated.
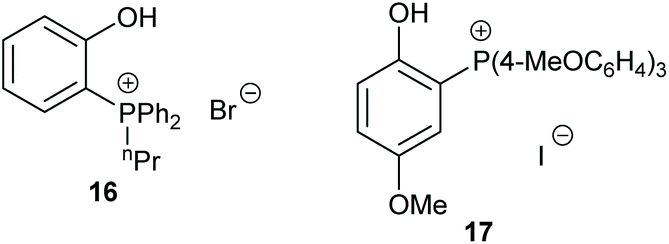 | ||
| Fig. 8 Phosphonium salt based catalysts 16–17. | ||
The following year, Werner et al. performed a mechanistic study into the use of ammonium and phosphonium salts as catalysts for the formation of cyclic carbonates.61 By using a combination of kinetic and infrared studies, Werner determined that in the case of monofunctional catalysts, the reaction followed first-order kinetics and that the interaction of the salt cation and anion influenced the catalytic activity. Interestingly, bifunctional catalysts exhibited the opposite trend in cation and anion interactions and thus the catalytic activity did not follow first-order kinetics. In this case, infrared analysis was helpful in identifying intermediates formed during cyclic carbonate synthesis. This study highlights the importance of performing detailed kinetic studies whilst developing novel catalysts, rather than just assuming that the system follows a traditional reaction mechanism.
Over twenty bifunctional tetraarylphosphonium salt catalysts were investigated by Suga et al. in 2019, obtaining 72–96% yields from seven terminal epoxides with the most active catalyst (17) under mild conditions (2–15 mol% of catalyst, 0.3–10 M in chlorobenzene at 60 or 120 °C for 12 or 24 h at 1 bar of carbon dioxide) via an epoxide activation mechanism (Scheme 1).62 Although the substrate scope used in this study was rather narrow, an extensive experimental and theoretical study into the reaction mechanism was performed, unlike the majority of studies in this area.
A computational kinetic study of the use of carbonyl-stabilised phosphonium ylides to catalyse the conversion of ethylene oxide into ethylene carbonate was carried out by Norozi-Shad in 2020.63 This study concluded that phosphonium ylides bearing electron donating groups were the more effective catalysts and that the carbon dioxide activation mechanism (Scheme 5) was more favourable than epoxide activation (Scheme 1). However, no experimental results were performed to confirm these computational predictions.
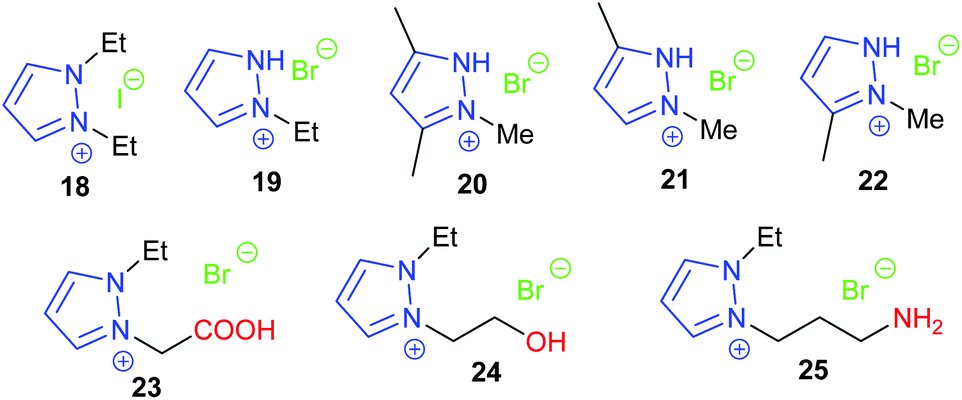 | ||
| Fig. 9 Pyrazolium salt catalysts. | ||
In 2018, the same research group investigated six protic pyrazolium ionic liquids, with the most active 19 giving 74–94% isolated yields of cyclic carbonates derived from six terminal epoxides, via the epoxide activation mechanism (Scheme 1). Again, the catalyst could be reused up to five times.65 However, no internal epoxides were tested and the reaction conditions were still harsh (1 mol% of catalyst, 20 bar of carbon dioxide and 140 °C for 4 h). The substrate scope investigated in both these studies is rather limited, though the combination of computational and experimental methods was good to see. In the same year, three protic pyrazolium ionic liquids 20–22 were also studied by Zheng et al. for the synthesis of propylene carbonate from propylene oxide and carbon dioxide.66 Both theoretical calculations and experimental results showed that the catalytic activity of these three catalysts was 20 ≈ 22 > 21. The theoretical calculations indicated that the reaction mechanism and catalytic activity were more reliably predicted using a model involving two molecules of ionic liquid catalyst, rather than a model based on use of a single molecule of ionic liquid. In 2019, Zheng et al. further studied the effect of clusters of protic pyrazolium ionic liquids in ring-opening propylene oxide.67 Whether the ionic liquid or propylene oxide was used as solvent in the reaction was found to significantly affect the reaction mechanism. The same group have also investigated the effect of cluster formation in quaternary ammonium ionic liquids (such as Et4NBr, Et3N(CH2CH2OH)Br, Et3NHBr, and Et2NH(CH2CH2OH)Br), on the catalyst performance in the reaction between carbon dioxide and propylene oxide via molecular dynamics simulations and quantum chemistry calculations.68
In 2018, Zhang again built on previous studies into pyrazolium ionic liquids by investigating with DFT calculations how changing the functional groups on the catalysts affects the conversion of propylene oxide69 in a similar manner to Werner's work.60 By studying three different ionic liquids, it was determined that both electrostatic interactions and hydrogen bonding with propylene oxide promote the ring-opening step of the reaction mechanism. The catalyst and substrate scope would ideally have been larger and no experimental results were reported within this study. Following on from this work, Zhang synthesised ten novel carboxy-functionalised pyrazolium ionic liquid catalysts.70 All ten catalysts were highly active, with the most active (23) giving 53–99% GC yields of cyclic carbonates derived from six terminal epoxides. This reaction again required harsh conditions (1.0 mol% of catalyst, 20 bar of carbon dioxide at 110 °C for 4 h) but the catalyst could be reused up to eight times before catalytic activity started to significantly decline. No internal epoxides were tested as substrates however.
Hydroxy functionalised pyrazolium salts were found to catalyse the ring-opening of six terminal and one internal epoxide, with the most active (24) out of five catalysts studied giving 65–95% isolated yields using only 1 mol% of catalyst.71 High temperatures and pressures (10 bar of carbon dioxide and 110 °C for 4 h) were however required. Amino functionalised versions of these catalyst were also tested by Zhang et al., with the most active catalyst 25 out of four studied giving 66–96% yields for six terminal epoxides.72 Harsh reaction conditions were again required (110 or 140 °C with 15 or 20 bar of carbon dioxide for 4 h), although only 1 mol% of catalyst was used. Interestingly these catalysts formed cyclic carbonate via carbon dioxide activation (Scheme 5) rather than epoxide activation (Scheme 1). Both of these classes of catalysts could have been tested against a broader substrate scope. The hydroxyl-based catalysts were determined to be one of the most active pyrazolium salts, due to the hydrogen bond it could form with epoxide substrates.
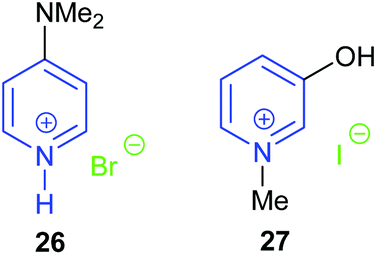 | ||
| Fig. 10 Pyridinium salt catalysts as reported by Zhang and Rostami. | ||
Another pyridinium based catalyst was hydroxy functionalised pyridinium iodide 27 reported by Rostami et al. in 2018.74 The hydroxy group could act as a hydrogen bond donor during the ring-opening of the epoxide, thus improving the catalytic activity of the pyridinium iodide. The position of the hydroxyl group on the pyridine ring dramatically affected the catalytic performance, as 2-hydroxy-N-methylpyridinium iodide was much less active than 27. On the basis of analysis of 1H NMR spectra, the authors suggested that the hydrogen in the 2-position of catalyst 27 together with the hydroxyl group stabilised the key transition states. Nine terminal epoxides could be converted into the corresponding cyclic carbonates in 81–98% yield using 1 bar of carbon dioxide at 50 °C for 6 h with 5 mol% catalyst. However, the yields of cyclic carbonate obtained from trans-stilbene oxide and cyclohexene oxide 1 were only 25% and 15%, respectively.
 | ||
| Fig. 11 Nitrogen-salt based catalysts. | ||
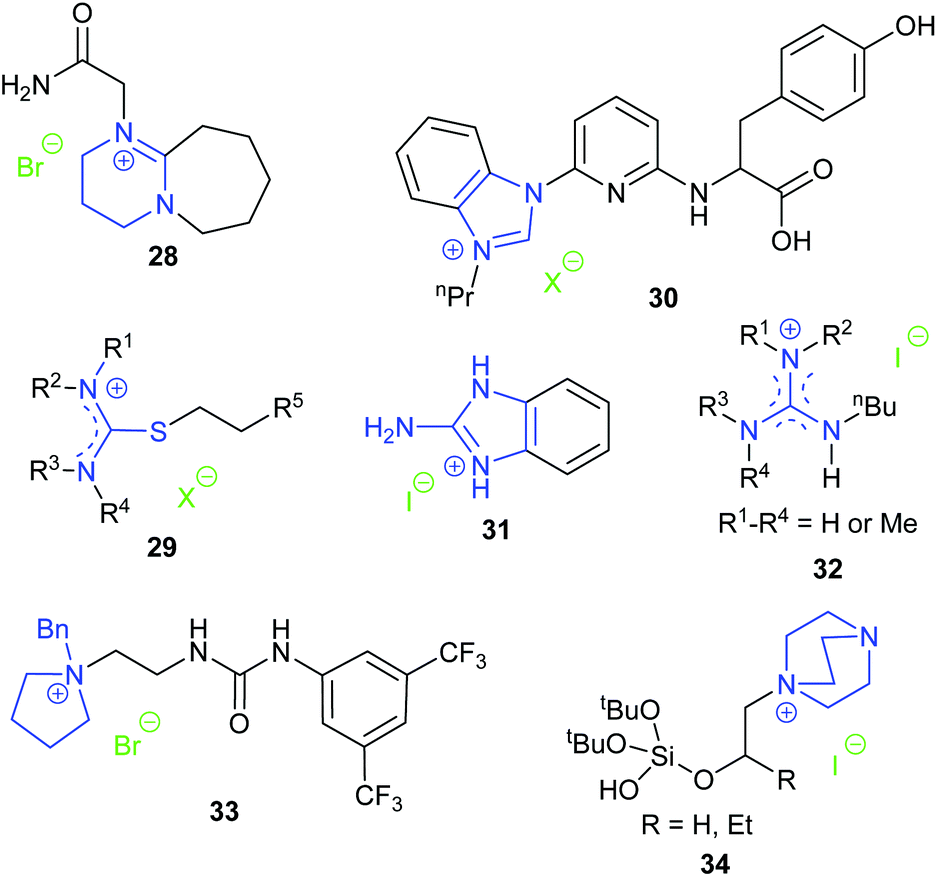
The organic superbase 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is frequently used as an additive for the metal catalysed synthesis of cyclic carbonates from carbon dioxide and epoxides. In 2018, the DBU derived salt 28 was shown by Zhang et al. to be effective for the conversion of carbon dioxide and epoxides into cyclic carbonate at 20 bar of carbon dioxide and 120 °C.75 NMR spectra and DFT calculations verified the formation of a hydrogen bond between the amide hydrogen in this catalyst and the epoxide, which was hypothesised to facilitate the reaction. Catalyst 28 could be separated by centrifugation and be reused at least five times.
Endo et al. screened salts of DBU and acids, such as acetic, trifluoroacetic, trifluoromethanesulfonic and hydrogen halides, finding that the hydroiodide salt of DBU was the most active catalyst at 1 bar of carbon dioxide and 25 °C with 2-MeTHF as the reaction solvent.76 The catalytic activity of DBU hydroiodide was much higher than that of N-Me-DBU iodide and N-Bn-DBU iodide, indicating that the proton on the amidinium group was crucial for the reaction. Ten hydroiodides of tertiary amidines and amines were then examined as catalysts, highlighting that the high basicity of the amidine moiety is a prerequisite for forming catalysts with high activity. The DBU hydroiodide catalyst was also efficient for the reaction between epoxides and carbon disulphide to form five-membered cyclic dithiocarbonates. In another study, Endo et al. showed that the DBU hydroiodide catalyst could effectively catalyse the reaction of carbon dioxide and oxetane containing epoxide reagents at 1 bar of carbon dioxide to give the corresponding oxetane containing five-membered cyclic carbonate, which could then be used as a monomer in a copolymerisation reaction to form polymers bearing a cyclic carbonate pendant group.77
Luo et al. prepared a series of functional isothiouronium ionic liquids 29. Those that were hydroxyl- or carboxyl-functionalised catalysed formation of cyclic carbonates with 100% selectivity at 20 bar of carbon dioxide and 140 °C for 2 h.78 The cyclic carbonates of terminal epoxides were obtained in 50–99% yield after 1.5–3 h, and cyclohexene carbonate was obtained in 73% yield after 22 h. The catalytic activity increased with the basicity of the nitrogen atom within the catalyst and on this basis, the authors suggested that the tertiary nitrogen atom of the cation could coordinate with carbon dioxide to afford a carbamate salt as an activated form of carbon dioxide. When forming propylene carbonate from propylene oxide, the catalyst could be separated from the reaction mixture by distillation of the product under reduced pressure and could be reused at least five times. However, this separation method is only feasible when the cyclic carbonate has a relatively low boiling point.
Dai et al. synthesised a series of compounds containing an N-heterocyclic carbene precursor and a carboxylic acid group, and explored their application as catalysts for the reaction of epoxides and carbon dioxide.79 Thirteen terminal epoxides were examined and the most active catalyst (30) gave 62–95% yields of cyclic carbonates with 99% selectivity at 1 bar of carbon dioxide and room temperature or 40 °C. A large range of internal epoxides were tested at a higher reaction temperature or with tetrabutylammonium iodide as a co-catalyst, with most of them giving good activity and high stereoselectivity. Since catalyst 30 possessed multiple functional groups that may all contribute to the activity of the catalyst, a series of control experiments were performed, using analogues in which some of the functional groups were removed. These experiments verified the necessity of the benzimidazolium ring, the carboxylic acid group and the pincer-type structure for the high activity of this catalyst. Further combining these results with DFT calculations indicated that an intramolecular, synergistic activation mechanism was operating.
Kim et al. investigated the activity of guanidinium iodide 31 for catalysing the transformation of epoxides and carbon dioxide into cyclic carbonates at 1 bar of carbon dioxide and 40 °C for 24 h.80 As the catalyst dosage was relatively high (10 mol%), the TOF values reported were fairly low; approximately 0.4 h−1 for various terminal epoxides and 0.046 h−1 for cyclohexene oxide 1. More recently, Endo et al. demonstrated that guanidinium iodides 32 were catalysts for the synthesis of cyclic carbonates under mild reaction conditions (1 bar carbon dioxide, 25 °C, 24 h, 5 mol% catalyst).81
In 2019, Wang et al. prepared ten achiral phase-transfer catalysts and showed that compound 33 with a quaternary ammonium centre, a urea group as a hydrogen bond donor and a 3,5-bis-trifluoromethylphenyl substituent was the most active catalyst for cyclic carbonate synthesis at 1 bar of carbon dioxide and 80 °C for 24 h.82 Many terminal epoxides were converted into cyclic carbonates in 65–91% yields. When enantiomerically pure epoxides were used, cyclic carbonates were obtained with 67–99% enantiomeric excess. The performance of internal epoxides was not examined and no conversion occurred when oxetanes were used as a substrate. Three types of chiral bifunctional phase-transfer catalysts: urea-onium salts, thiourea-onium salts, and squaramide-ammonium salts, were further studied in an attempt to accomplish the kinetic resolution of epoxides by cyclic carbonate formation. However, amongst the fourteen chiral catalysts studied, only one catalyst provided a non-racemic cyclic carbonate and it had an enantiomeric excess of just 22%.
In 2019, Jancik et al. reported bifunctional silanol-based hydrogen bond donor catalysts 34.83 The hydroxyl group acted as a hydrogen bond donor to facilitate the ring-opening of the epoxide. The optimised reaction conditions for this reaction were 5 bar of carbon dioxide and 70 °C for 10 h, giving yields of 72–98% with various terminal epoxides as substrates, but less than 4% for reaction of cyclohexene oxide 1. The recyclability of catalyst 34 (where R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H) was examined for the transformation of styrene oxide, as the catalyst could be precipitated from the reaction mixture using diethyl ether. The yields of cyclic carbonate in the first three runs were 87%, 57% and 29%, respectively, indicating poor recyclability.
H) was examined for the transformation of styrene oxide, as the catalyst could be precipitated from the reaction mixture using diethyl ether. The yields of cyclic carbonate in the first three runs were 87%, 57% and 29%, respectively, indicating poor recyclability.
2.2. Two-component systems
Two-component catalytic systems usually consist of a nucleophile, such as an organic halide salt, or an organic base combined with an additive which possesses functional groups capable of activating carbon dioxide or the epoxide. Quaternary ammonium halides are inexpensive and commercially available chemicals which are often used as the nucleophilic component of the catalyst system. Amongst these, tetrabutylammonium halides (TBAX or Bu4NX, where X = Cl, Br or I) are the most commonly used, though bis(triphenylphosphine)iminium (PPNX), tetraethylammonium (TEAX) and imidazolium (ImX) halides have also been shown to be effective nucleophiles for two-component catalyst systems.In 2018, Khan et al. investigated the ability of deep eutectic solvents composed of choline chloride and hydrogen bond donors, to form previously unreported spiro-cyclic carbonates derived from 2-oxindole derivatives (Scheme 15). The deep eutectic solvent obtained from choline chloride and urea (1:2 molar ratio) converted spiro-epoxy oxindoles 35a–j into cyclic carbonates 36a–j at 1 bar of carbon dioxide and 70 °C with isolated yields of 67–98% after 2–8 h.84 The authors proposed that the enhanced reactivity of this system was due to the combination of choline chloride and the hydrogen bond donor increasing the solubility of carbon dioxide in the deep eutectic solvent. Green metrics were considered during this research, a factor that is often omitted from other studies. Cyclic carbonates 36a–j were isolated by simply washing with 2-MeTHF, rather than using column chromatography. Hence, the authors reported a low of E-factor of only 0.40 kg kg−1, highlighting the small amount of waste produced per kg of product.85 This catalyst system could also be recycled four times giving consistent yields and the reaction could be scaled up to mmol scale. Internal epoxides were however still problematic substrates for this system and care must be taken with choline chloride, as it is hydroscopic and fairly expensive. This catalyst system is nonetheless still extremely promising.
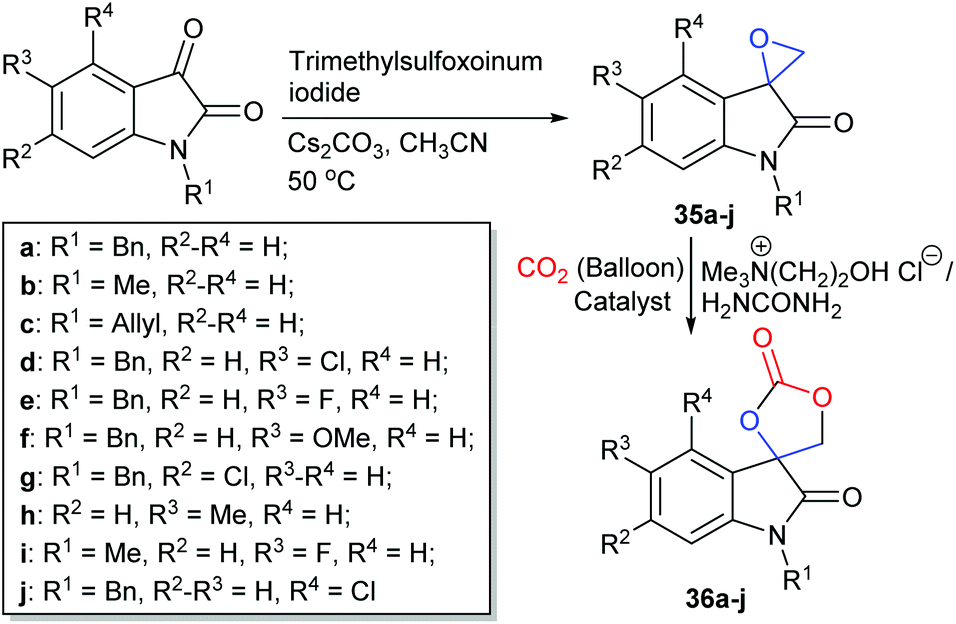 | ||
| Scheme 15 Synthesis of spiro-cyclic carbonates 36a–j, using a choline chloride and urea based deep eutectic solvent. | ||
In a similar project, Lü et al. studied a bifunctional deep eutectic solvent, obtained from a 1:2 ratio of choline chloride and PEG200 in the formation of cyclic carbonates via an epoxide activation mechanism (Scheme 1).86 Under the optimum reaction conditions, using 17.5 mmol of epoxide and 2 mol% of deep eutectic solvent at 150 °C and 8 bar of carbon dioxide for 5 h, conversions of 75–99% were obtained for five terminal epoxides, but only 43% conversion was achieved for cyclohexene oxide 1. Whilst this catalyst system could be recycled five times without any drop in propylene oxide conversion, the substrate scope investigated was poor.
Water is the simplest and least expensive hydrogen bond donor, providing a sustainable alternative to traditional hydrogen bond donors such as phenol, gallic acid or ascorbic acid, and can improve the performance of nucleophilic organic halide salts in the formation of cyclic carbonates. Water was first applied in this reaction in 200887 and the role of water was studied further between 200988 and 2010.89 However, care must be taken with the addition of water as this can lead to diol formation and poor selectivity. In 2019, Pescarmona and Alassmy90 revisited the role of water in cyclic carbonate formation under mild reaction conditions (25–45 °C and 10 bar of carbon dioxide). When the reaction was catalysed by tetrabutylammonium iodide (3 mol%) at room temperature at 10 bar of carbon dioxide for 48 h, the addition of water increased the conversion of propylene oxide from 17 to 85%, whilst retaining 99% selectivity for cyclic carbonate formation. The authors noted that water not only acted as a hydrogen bond donor in this reaction, but also increased catalyst solubility in the reaction mixture. Whilst the reaction conditions are very mild, the type and amount of catalyst used in this work, along with the water loading, are not substantially different from previous literature. There was no discussion of how by-product formation was inhibited in the presence of water.
The relationship between the pKa of hydrogen bond donor additives and their catalytic activity has been investigated by Hirose et al. and D'Elia et al.91,92 Three hydroxypyridine isomers (5 mol%) were investigated by Hirose91 in the reaction between 1,2-epoxyhexane and carbon dioxide, in the presence of tetrabutylammonium iodide (5 mol%) at 40 °C and 1 bar of carbon dioxide for 24 h. Cyclic carbonate yields increased with hydroxypyridine acidity, as yields of 67%, 86% and 95% were reported for 2-hydroxy-, 4-hydroxy- and 3-hydroxypyridine, which have pKa values of 11.7, 11.1 and 8.8, respectively. It was also shown that steric hindrance due to substituent groups near the hydrogen bond donor group could lower the catalytic activity, as 2-methyl-3-hydroxypyridine and 2,4-dimethyl-3-hydroxypyridine gave lower conversions than 3-hydroxypyridine.
In 2019, D'Elia et al. further studied the catalytic activities of eighteen hydroxyl hydrogen bond donor additives, with pKa values of 2.5–15.5.92 The relationship between the additive's pKa value and the relative initial reaction rate for the conversion of epichlorohydrin and carbon dioxide into 3-chloropropylene carbonate is shown in Fig. 12. It was shown that the catalytic activity is strongly dependent on the pKa value of the most acidic proton in the hydrogen bond donor. No correlation was found between the number of hydroxyl groups and the catalytic activity, but multiple hydroxyl groups in the hydrogen bond donor might improve the overall performance. It was concluded that the ideal pKa range for hydroxyl hydrogen bond donors was 9–11. The high catalytic activity of phenol and a simple ascorbic acid derivative supported this conclusion. This is a simple method to predict the activity of hydroxyl hydrogen bond donor additives.
 | ||
| Fig. 12 Relationship between pKa and relative initial rate of reaction (krel) for the cycloaddition of CO2 with epichlorohydrin. Red dots: phenols; green diamonds: carboxylic acids; yellow dots: mono- and polyalcohols; blue squares: ascorbic acid and its derivatives. The blue dashed line is a guide to highlight the pKa-dependent change of catalytic activity among ascorbic acid and its analogues. Reproduced with permission from D'Elia et al.92 | ||
When the additive has functional groups in addition to a hydrogen bond donor, the catalytic system often shows enhanced activity. In 2017, Takaishi et al. developed chiral macrocycles (37a–q, Scheme 16).93 These macrocycles contained several amides and were employed in the enantioselective synthesis of cyclic carbonates, via epoxide activation (Scheme 1) from mono- and di-substituted epoxides and carbon dioxide. The kinetic resolution of trans-stilbene oxide with tetrabutylammonium iodide (3 mol%) and 37 (3 mol%) was performed at 75 °C and 1 bar of carbon dioxide for 72 h. Additive 37m gave a high conversion (41%) and the highest degree of enantioselectivity. The kinetic resolution of nine internal and nine terminal epoxides was investigated using additive 37m, with selectivity values of 9.5–13 and 2.5–4.3, respectively.
 | ||
| Scheme 16 Macrocycles 37a–q used in the enantioselective synthesis of cyclic carbonates. | ||
The X-ray crystal structure of 37m indicated that it had a well-defined chiral cavity and could form two hydrogen bonds between the amide NH groups and the epoxide. This highlighted why this macrocycle was effective for the enantioselective activation of epoxides. In addition to reporting high enantioselectivity, the impressive catalytic activity reported both for mono- and di-substituted epoxides, using only 1 bar of carbon dioxide, highlights of the efficiency of this catalytic system.
In a similar study, Ema et al. investigated a different kind of macrocyclic organocatalyst: calix[4]pyrroles, with four pyrrolic NH groups, to act as hydrogen bond donors.94 Their catalytic ability was also compared to related macrocycles (Fig. 13). An isolated yield of 98% was obtained for butylene carbonate, after 15 h at 100 °C and 17 bar of carbon dioxide, using 1 mol% of calix[4]pyrrole 38a and 1 mol% of tetrabutylammonium iodide. DFT calculations suggested that among all possible hydrogen bond formations, the most stable was that in which two opposite NH groups formed a hydrogen bond with the epoxide and the iodide anion of tetrabutylammonium iodide, respectively, thus facilitating the ring-opening of the epoxide. DFT calculations also suggested epoxide activation was key to the reaction mechanism (Scheme 1).
 | ||
| Fig. 13 Macrocycles employed by Ema et al. in the formation of cyclic carbonates. | ||
Not all hydrogen bond donors accelerate cyclic carbonate synthesis via epoxide activation. In 2017, Kleij et al. investigated the catalytic activity of squaramide organocatalysts, combined with quaternary ammonium halides, for the synthesis of cyclic carbonates from epoxides and carbon dioxide.95 The optimal squaramide derivative, of nine symmetrical and twelve non-symmetrical di-substituted squaramides, was highly efficient in the transformation of terminal and internal epoxides (Scheme 17). Control experiments suggested that the predominant role of the two NH groups of the squaramide was to stabilise the intermediate oxo and carbonato anions, rather than forming hydrogen bonds with the epoxide during the ring-opening step. In 2018, Wang et al. studied the same catalytic system via DFT calculations, considering the reaction of carbon dioxide with propylene oxide to form propylene carbonate.96 Their results supported the reaction mechanism proposed by Kleij et al.
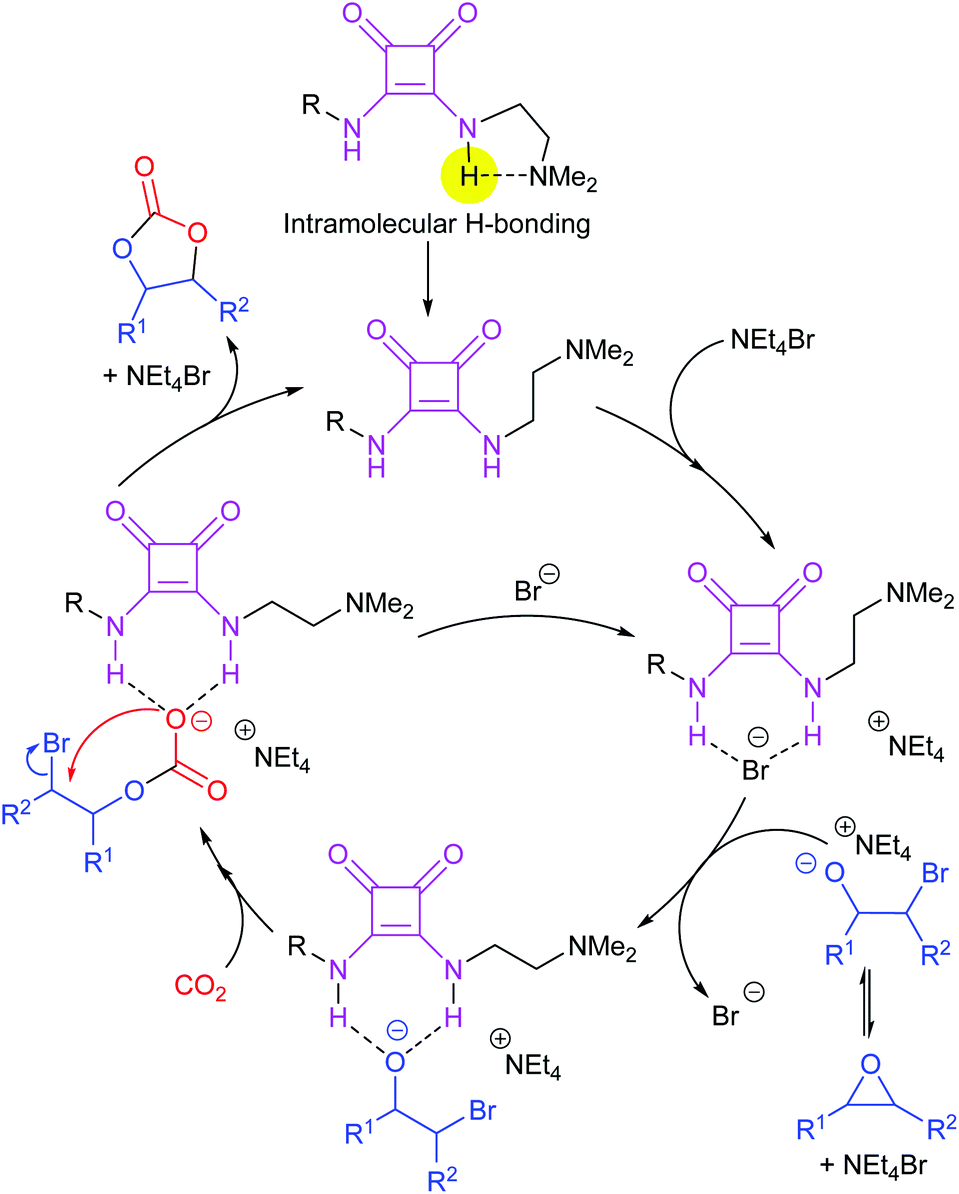 | ||
| Scheme 17 Reaction mechanism for the formation of cyclic carbonates catalysed by disubstituted squaramides. | ||
 | ||
| Fig. 14 Ionic liquids used by Bhanage and Saptal et al. in the formation of cyclic carbonates. | ||
In 2018, Hirose et al. investigated the combination of tetrabutylammonium halide salts and organic bases for the synthesis of di-substituted (internal) cyclic carbonates from nine internal epoxides.98 Seven strong organic bases, with planar and/or rigid structures were studied, displaying good to excellent catalytic activity (73–80% isolated yield), with DBU the most active base reported. Unlike other reaction systems, in which tetrabutylammonium salts with a bromide or iodide anion showed optimal activity, tetrabutylammonium chloride gave the most active catalyst. It was speculated that the smaller chloride anion aided the reaction with the sterically hindered internal epoxides. When the reaction of cyclohexene oxide 1 with carbon dioxide was carried out with tetrabutylammonium chloride (10 mol%) and DBU (5 mol%) for 24 h at 120 °C and 1 bar of carbon dioxide, an 85% yield of cyclic carbonate was obtained via a dual activation mechanism (Scheme 7).
In 2019, Zhong et al. performed experimental and theoretical studies on carbon dioxide activation catalysed by tetrabutylammonium bromide and seven nitrogen-based organic bases.99 The screening of organic bases showed that pKa values, as well as steric hindrance, influenced catalytic activity. The DFT calculation results illustrated that triethylamine could activate carbon dioxide by electrostatic interactions, thereby reducing the reaction energy remarkably and promoting the reaction. Overall it was proposed that triethylamine activated carbon dioxide whilst the tetrabutylammonium bromide ring-opened the epoxide (via the mechanism shown in Scheme 7).
In 2017, Wang et al. employed DBU and N-iodosuccinimide to form butylene carbonate in excellent yields, using 5 mol% of both catalysts and 1 bar of carbon dioxide at 100 °C for 10 h.100 High yields were still obtained when the reaction temperature was reduced to 60 °C, and the optimum catalytic system (DBU and N-iodosuccinimide) could be used five times without any drop in conversion. This system was capable of ring-opening simple terminal epoxides (via a mechanism similar to that shown in Scheme 1), but required harsher reaction conditions when cyclohexene oxide 1 or isobutylene oxide were used as substrates. This system though was only tested on seven epoxides, with little difference between the functional groups present in the epoxides.
In 2018, Gao et al. combined imidazolium ionic liquids with various organic bases; with 1 mol% of BnBimBr and diethanolamine proving to be the most active combination. GC yields of 82–95% were recorded for six simple terminal epoxides (at 1–5 bar of carbon dioxide and 80–110 °C for 3 h).101 Although this catalyst could work in the presence of water via a dual activation mechanism (Scheme 7), no recyclability studies were performed and no internal epoxides were screened. In a similar study, Kim et al. synthesised 3-(2-hydroxyethyl)-1-vinyl-1H-imidazol-3-ium [EvimOH] ionic liquids and used them in combination with bases. [EvimOH] chloride and DBU provided the most active system.102 This binary catalyst gave 76–99% yields for six simple terminal epoxides, and even gave 76% yield when ring-opening cyclohexene oxide 1, using <2 mol% of ionic liquid and base, at 20 bar of carbon dioxide at 120 °C for 1–8 h. This catalyst also maintained consistent activity after five uses and it was proposed that the reaction occurred via carbon dioxide activation (Scheme 5).
The combination of eight naturally abundant amino acids with twelve superbases was researched by Zhang et al., with the synergistic system of (S)-histidine and DBU exhibiting the highest activity in propylene oxide conversion (97% with 99% selectivity).103 The reaction is proposed to occur via a dual activation mechanism (Scheme 7). Under the optimum conditions (2 mol% (S)-histidine and 10 mol% DBU at 20 bar of carbon dioxide and 120 °C for 2 h), excellent conversions were obtained for terminal and internal epoxides, although no isolated yields were reported and the substrate scope was limited. It is notable that histidine was the most effective amino acid in both the work of Bhanage and Saptal96 and that of Zhang.
 | ||
| Scheme 18 Proposed initiation steps in forming cyclic carbonates from epoxides and CO2, using boronic acids and pyridinic bases. | ||
In 2019, Kerton and Andrea investigated the use of triarylboranes with bis(triphenylphosphine)iminium chloride (PPNCl) in both cyclic and polycarbonate synthesis via epoxide activation (Scheme 1).106 Extremely low catalytic loadings were required for this catalyst system (0.025 or 0.1 mol% of borane and 0.1 mol% of PPNCl) under mild reaction conditions (1 or 20 bar of carbon dioxide at 100 °C for 3–24 h). A thorough study into the reaction kinetics and mechanism was performed, but only two terminal epoxides (propylene oxide and epichlorohydrin) were converted into cyclic carbonates with 92% and 99% conversion, respectively (no yields were reported). In the same year, Zhang et al. combined triethylboranes with Lewis bases to give catalysts for the synthesis of polycarbonates and cyclic carbonates.107 By using PPNCl as a co-catalyst, 100% conversions were obtained for styrene oxide and phenyl glycidyl ether, using 0.2–1.2 mol% of borane, 0.2 mol% of PPNCl and 1.4 g of carbon dioxide at 60 °C for 8 h. A solvent (THF) was however required and more emphasis was placed on synthesising polycarbonates, with only three reaction conditions out of 25 producing cyclic carbonate (via a similar mechanism to that shown in Scheme 6).
Boric oxide also exhibited good performance in catalysing the transformation of epoxides and carbon dioxide into cyclic carbonates with tetrabutylammonium bromide as co-catalyst.108 Ball milling of the boric acid could significantly improve its catalytic activity by enhancing its ability to adsorb propylene oxide. At 100 °C and 20 bar of carbon dioxide, after 2 h, a 95% yield of propylene carbonate was obtained after ball-milling but only a 40% yield using untreated boric acid. On the basis of DRIFTS analysis, the authors elucidated that epoxide ring-opening occurred on the surface of the boric acid and was induced by the boron atoms. The boric acid could be separated by centrifugation and reused at least five times. The performance of other epoxide substrates was examined at 100–120 °C and 20 bar of carbon dioxide for 2–7 h. Yields of 63–98% for cyclic carbonates derived from terminal epoxides and 95% for cyclohexene carbonate were obtained.
2.3. Halide-free systems
Whilst most catalytic systems employ a nucleophilic halide to induce ring-opening of the epoxide, there are systems that operate in the absence of halides.109 This can often be troublesome and require harsher conditions. However, halides lead to the corrosion of reactor vessels and can pose risks to the environment when disposed of as waste.110 Therefore, in this section, halide-free homogeneous systems will be discussed.In 2017, Chung et al. developed seven halide-free multifunctional alkanolamines to catalyse the formation of propylene carbonate.111 The most active of these catalysts (39, Fig. 15) achieved 90% yield and >99% selectivity (GC analysis) of propylene carbonate, using 5.6 mol% of catalyst at 100 °C and 5 bar of carbon dioxide for 8 h. A synergistic effect was proposed involving the tertiary amine and hydroxyl groups in the catalysts, as increasing the number of both of these groups created a more active catalyst depending on their proximity to one another. The proposed reaction mechanism was supported by DFT calculations. Whilst catalyst 39 shows promise, it would be interesting to test its ability to convert internal epoxides into cyclic carbonates. Toxic amines and carcinogenic reagents were required to synthesise the catalysts, but the absence of halides perhaps provides a sustainable counterbalance.
 | ||
| Fig. 15 Structures of halide-free catalysts 39–41. | ||
DFT calculations were performed by Ryu in 2019 to investigate the ability of amines to catalyse the conversion of propylene oxide into propylene carbonate.112 Interestingly, Ryu reported that tertiary amines make more reactive catalysts, due to lower steric hindrance during the catalytic cycle rather than their nucleophilicity. Ryu's work highlighted in a similar manner to Chung's that amine structure is extremely important in catalyst structure, and that amines form cyclic carbonates via epoxide activation (Scheme 1).
In 2017, Seddon et al. tested five azolate ionic liquids, with the more nucleophilic catalysts giving higher conversions, under the optimum conditions (5 mol% of ionic liquid at 100 °C and 10 bar of carbon dioxide for 2 h).113 The catalysts were recyclable and the most active catalyst (40) gave 71% yield of cyclohexene carbonate; which is impressive considering this catalyst is metal- and halide-free. However, it was only screened against one internal epoxide and eight epoxides overall. The reaction mechanism was proposed to occur via carbon dioxide activation (Scheme 5).
Use of amino acid derived ionic liquids as catalysts for cyclic carbonate synthesis has been reported by numerous groups. In 2017, Zang et al. synthesised dual amino-functionalised imidazolium ionic liquids, with 3-aminopropyl-butylimidazolium cations and aspartic acid, glutamic acid or glycine amino acid anions.114 All these ionic liquids could achieve >96% conversion of epichlorohydrin into 3-chloropropylene carbonate (via a dual activation type mechanism, Scheme 7) using 0.3 mol% of ionic liquid at 105 °C and 5 bar of carbon dioxide for 13 h. The glutamic acid containing ionic liquid was tested against five simple internal epoxides and could be recycled five times. Similar work was performed by Jain et al. with a histidine derived ionic liquid, obtaining excellent isolated yields (91–98%) for eight terminal epoxides under mild and green conditions (5 mol% of ionic liquid at 80 °C and 1 bar of carbon dioxide for 5–6 h) using dimethyl carbonate as solvent. The catalyst could be recycled six times.115 This reaction was again predicted to occur via a dual activation mechanism (Scheme 7). Likewise, Li et al. found that an aspartic acid based ionic liquid could synthesise five cyclic carbonates from terminal epoxides in 96–99% yields via an epoxide activation mechanism (Scheme 1). The catalyst could be recycled 5 times and required fairly mild reaction conditions (5 bar of carbon dioxide at 130 °C for 12 h) and had similar reactivity to halide analogues.116 No internal epoxides were tested in these studies and the substrate scope in all cases was rather limited.
Pyridine based ionic liquids (optimally 41) were tested by Liu et al. as catalysts for the conversion of five terminal epoxides into cyclic carbonates with carbon dioxide via a dual activation mechanism (Scheme 7), using 10 mol% of ionic liquid at 30 or 80 °C and 1–20 bar of carbon dioxide for 4 or 20 h.117 These catalysts were, however, less active than previously reported ionic liquids, especially in the synthesis of styrene carbonate, where they gave only 59% conversion using a high (50 mol%) catalytic loading.
Imidazolium ionic liquids based on glycerol were capable of converting four terminal epoxides and one internal epoxide into cyclic carbonates via carbon dioxide activation (Scheme 5), as reported by Kim et al. in 2019, and were more active than similar halide containing ionic liquids.118 This system however required 20 mol% of DBU in addition to 10 mol% of ionic liquid, at 20 bar of carbon dioxide and 110 °C for 4 h, to obtain good conversions. Considering the reported success of DBU salts as catalysts,75 the addition of an ionic liquid may not be beneficial from a sustainability point of view. A good yield (65%) of cyclohexene carbonate was however reported.
Guazzelli et al. studied some halide-free dicationic ionic liquids, with the acetate-based ionic liquid 42 (Fig. 16) giving a better conversion (at 80 °C and 10 bar carbon dioxide with 2 mol% of catalyst for 2 h) of epichlorohydrin into 3-chloropropylene carbonate compared to the bromide analogue.119 Epichlorohydrin was however the only epoxide included in this study.
 | ||
| Fig. 16 Structures of halide-free catalysts 42, 43 and 46. | ||
In 2018, four novel polymeric multifunctional alkanolamines were synthesised and tested by Lim et al.; successfully converting simple terminal epoxides into cyclic carbonates in 54–96% yields (25 wt% of catalyst, at 120 °C and 10 bar of carbon dioxide for 3 h).120 Despite the recyclability of the most active catalyst (43) and their simple synthesis, conversions were poor for cyclohexene oxide 1 and no yields were reported for the synthesis of the catalysts.
A unique and versatile carbodicarbene organocatalyst 44 was developed in 2018 by Lu et al.37 Carbon dioxide adducts of 44 were capable of converting epoxides, aziridines and propargylic alcohols into heterocycles (Scheme 19). This system was extremely effective in ring-opening internal epoxides, with an impressive tolerance to numerous functional groups. 27 cyclic carbonates were formed under relatively mild conditions (5 mol% of catalyst, 20 bar of carbon dioxide and 80 °C for 12 h). Unfortunately, dichloromethane solvent was required for oxazolidinone synthesis, DMSO was needed for some epoxide conversions and the catalyst was synthesised using benzene. Developing greener solvents to prepare and use the catalyst would therefore be beneficial. Creating a recyclable version of this catalyst would also increase its sustainability.
 | ||
| Scheme 19 Proposed mechanisms for heterocycle formation using catalyst 44. | ||
In a similar study in 2020, Bischoff et al. reported that aminopyridines could form stabilised adducts with carbon dioxide and could form cyclic carbonates from epoxides via hydrogen bond formation (Scheme 20).121 These catalysts could also work on a kilogram scale and this is one of the few organocatalysts to give good yields in reactions carried out below 100 °C (46–100% yield, using 10 mol% of catalyst and 20 bar of carbon dioxide at 60–85 °C for 16 h). Like many catalysts though, this system was not tested in ring-opening internal epoxides and a larger substrate scope could have been evaluated. A drawback of the reaction is the toxic chemicals required to synthesise the aminopyridines via the Chichibabin reaction,122 but this is still an impressive system.
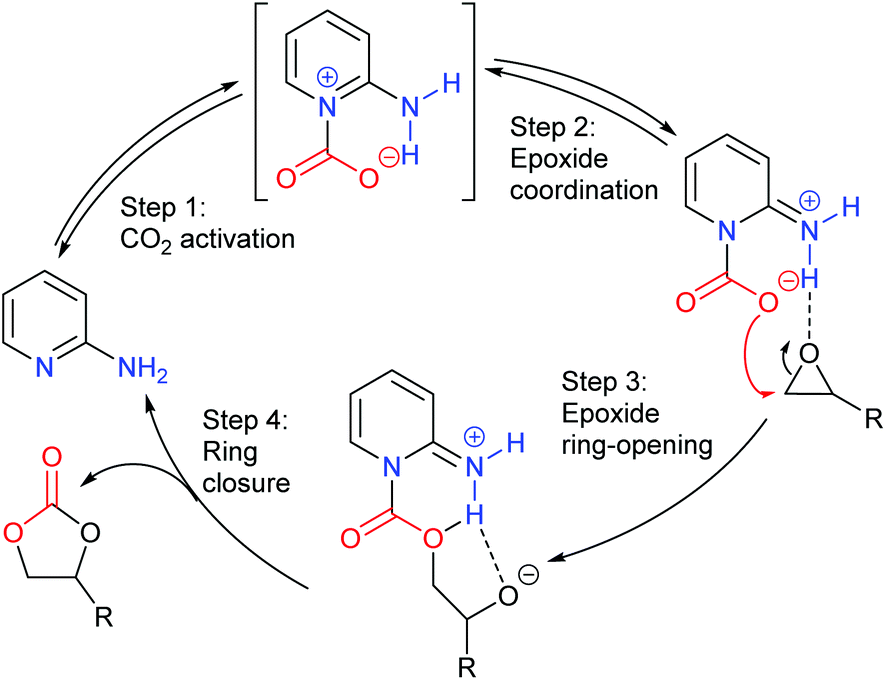 | ||
| Scheme 20 Proposed mechanism for forming cyclic carbonates using aminopyridines. | ||
The first metal- and halide-free salophen ligand based catalysts were reported by North et al. in 2019, with the most active of eleven tested ligands (45), capable of converting six terminal epoxides into cyclic carbonates in good isolated yields (57–89%). This reaction required relatively mild reaction conditions (1–10 mol% of catalyst, 120 °C and 10 bar of carbon dioxide).123 Experiments using deuterated epoxides highlighted that two different catalytic cycles occur with catalyst 45, one involving retention and the other involving inversion of the epoxide stereochemistry. This indicated that this reaction does not simply occur via the typical Lewis or Brønsted acid catalysed reaction mechanism (Scheme 1). It was therefore proposed that the two phenolic components of the salophen catalyst play different roles, with one interacting with the epoxide, increasing its susceptibility for ring-opening, and the other forming a carbonic acid intermediate from carbon dioxide (Scheme 21).
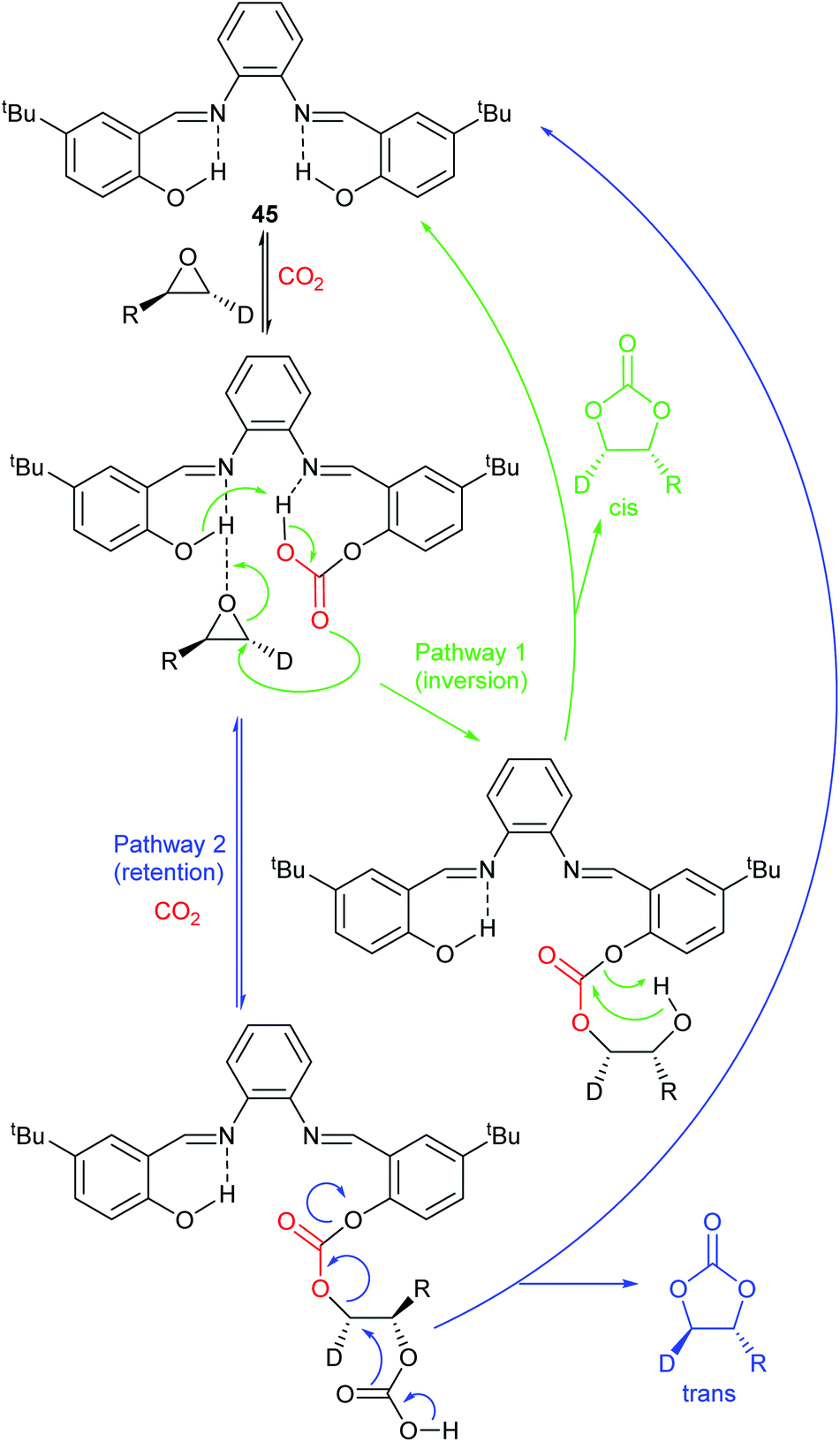 | ||
| Scheme 21 Proposed mechanisms for cyclic carbonate formation using salophen 45. | ||
In 2020, Jia et al. developed a Schiff base acen catalyst 46 (Fig. 16) able to form cyclic carbonates from epoxides.124 Catalyst 46 converted eight terminal epoxides into cyclic carbonates, but no reaction occurred with cyclohexene oxide. Epichlorohydrin was converted into 3-chloropropylene carbonate with 99% conversion using 0.2 mol% of catalyst at 110 °C and 10 bar of carbon dioxide for 4 h. The synthesis of these catalysts involves a simple two-step synthetic route and no toxic solvents.
2.4. Miscellaneous
In 2017, Belokon et al. discovered that triarylmethane dyes, acting as carbocations, could catalyse the formation of cyclic carbonates in the presence of polyols.125 The iodide salts of the commercially available dyes: malachite green, brilliant green and crystal violet, acted as bifunctional catalysts in which the iodide was the nucleophile and the carbocation provided a Lewis acidic centre. Low to moderate conversions (<1–45%) were reported for styrene carbonate synthesis using the dyes alone, albeit with high selectivity (>99%) under moderate conditions (50 °C, 50 bar of carbon dioxide and 1–2 mol% of catalyst for 24 h). By adding polyols to the dye carbocations, under identical conditions with 0.5–2.5 mol% of polyol, conversions significantly increased (48–100%). This was postulated to be due to the polyols acting as anion complexing agents and separating the iodide anion from the carbocation centre. The most active catalytic system (crystal violet with BINOL) was extremely effective in ring-opening ten terminal epoxides under the same conditions (48–79% isolated yields). Internal epoxides; cyclohexene oxide 1 and cyclopentene oxide gave lower yields of 10 and 31% respectively. This system could also be recycled five times with no loss in activity. A mechanistic study was performed with deuterated epoxides, suggesting that the carbocations increase the Brønsted acidity of a BINOL OH group, increasing its ability to hydrogen bond with the epoxide, thus triggering the ring-opening of the epoxide by the iodide anion. The other OH group activates carbon dioxide and thus promotes intramolecular carbonate formation (Scheme 22). This is a particularly impressive and simple catalytic system, although the iodide salts of the dyes were made using the toxic solvent dichloromethane and some of the dyes are suspected carcinogens and teratogenic agents. | ||
| Scheme 22 Proposed catalytic cycle for cyclic carbonate synthesis catalysed by triarylmethyl iodides and polyols. | ||
In 2019, Kerton et al. used oxidised biochar, from hard and soft wood residues, to convert epoxides into cyclic carbonates via an epoxide activation mechanism (Scheme 1).126 This is a very attractive catalyst as the pyrolysis of biomass from waste streams offers the ability to not only reduce the need for fossil fuel combustion, but can also be used to mitigate global warming, by acting as a carbon dioxide capture and storage agent. By treating biochar with nitric acid, chemically modified biochars were created and then screened in their ability to ring-open epoxides. The most active catalyst was able to achieve 70–99% conversions for six terminal epoxides, and showed no loss in conversion after five uses, at 10 bar of carbon dioxide and 100 °C for 6 h, using 100 mg of catalyst and 10 mol% of tetrabutylammonium bromide. The catalytic activity of these complexes was hypothesised to be due to the increase in the hydroxyl and carboxyl groups on the biochar surface. This catalyst however was not very active in ring- opening the internal epoxide cyclohexene oxide, giving only 22% conversion to cyclohexene carbonate.
3. Heterogeneous catalysts
Heterogeneous catalysts can be developed by the immobilisation of a catalytically active unit onto a solid support; by the use of a solid material that has intrinsic catalytic activity; or by combining these two approaches.127 Heterogeneous catalysts generally have advantages associated with ease of catalyst separation and recycling and an ability to be used in flow reactors. The latter is an important consideration in cyclic carbonate synthesis, given the highly exothermic nature of the reaction (Fig. 3) and the highly toxic nature of low molecular weight epoxides. When used in cyclic carbonate synthesis with epoxides with a large molecular size, the activity of heterogeneous materials can be much lower than that of their homogeneous counterparts, due to slow diffusion of the epoxide into small pores in the solid support. For example MOFs have pores about 1 nm in diameter and PMO-UDFs have pores about 2 nm in diameter.1283.1. Immobilised systems
In this section the main solid supports, such as silica, polystyrene and graphene oxide will be discussed. Interesting miscellaneous heterogeneous examples will also be reviewed.Microporous and mesoporous materials are defined by the size of their pores, with pore sizes smaller than 2 nm and between 2 and 50 nm, respectively.129 Mesoporous silica based supports are often preferred as supports for catalysts for cyclic carbonate synthesis as they contain pores which are large enough to accommodate large catalyst structures and have high BET surface areas.
Silicas are one of the most frequently used solid supports, as they are inexpensive, readily available, easy to modify (chemically or in terms of porosity) and versatile. Silica gel, amorphous silica, fumed silica and micro- or mesoporous ordered silicas, including SBA-15 and MCM-41, are some of the most widely used supports.130 They offer useful textural properties such as high specific surface areas which potentially allow a high immobilisation capacity. The presence of silanol surface groups, allows the formation of a covalent bond between the catalyst and the support. Thus, it is quite straightforward to functionalise a silica support with organic groups and catalysts. Therefore, these supports can provide recyclable catalysts for the formation of cyclic carbonates.129
Li et al. studied a mixture of ionic liquids and quaternary ammonium salts as heterogeneous catalysts using SBA-16 as the solid support (Fig. 17).132 The SBA-16 support has a cubic structure consisting of non-interpenetrating 3D channels with cavities with a width of 5–15 nm.133 Even though functionalising SBA-16 with tetrabutylammonium iodide and imidazolium based groups decreased the surface area, volume and pore size of the support, the carbon dioxide adsorption capacities of these supports were greater than that of SBA-16 itself. This was hypothesised to be due to the introduction of iodide and amine groups, which were described as “CO2-philic” groups. By combining these supported catalysts together in a 3:1 ratio of iodide to amine, NMR yields of 86–95% were obtained for four terminal epoxides at 5 bar of carbon dioxide and 50 °C after 48 h. However, only 34% yield was obtained when cyclohexene oxide was used as substrate. The catalyst could also be recycled up to five times and was hypothesised to be acting via a dual activation type mechanism (Scheme 7). Whilst this work is extremely promising, a broader substrate scope could have been tested, in order to further investigate the versatility of the SBA-16 support. Tetrabutylammonium iodide impregnated SBA-16 alone gave a 68% yield of 3-chloropropylene carbonate compared to 93% using the dual acting catalyst. It is therefore also questionable in terms of costs and sustainability whether the added imidazolium component is worth the increase in conversion.
 | ||
| Fig. 17 SBA-16-supported two-component catalyst system. | ||
In 2019, Dong et al. synthesised triethanolamine-modified SBA-15. A 94% yield was obtained for propylene carbonate synthesis at 100 °C and 20 bar of carbon dioxide after 4 h, using 0.2 g of catalyst with 34.5 mmol of propylene oxide.134 This catalyst was able to convert four terminal epoxides into cyclic carbonates in good yields (85–96%) via an epoxide activation mechanism (Scheme 1). Recyclability tests showed that the SBA-15-supported triethanolamine catalyst retained most of its catalytic activity after five uses in propylene carbonate synthesis.
Another silica support that has been studied for cyclic carbonate formation is MCM-41: a porous support from the M41S family with an ordered hexagonal array of unidirectional and non-interconnecting pores.135 In 2018, Aprile et al. developed a bi-functional MCM-41, containing two Lewis acidic sites; a metal centre and a nucleophilic anion component.136 Although the main catalysts studied were tin and zinc embedded in MCM-41 supports, the metal-free imidazolium functionalised support was also tested under reaction conditions of 125 °C and 40 bar of carbon dioxide for 3 h, using 1 g of catalyst and 1.5 mL of ethanol. Intriguingly, the metal-free imidazolium functionalised MCM-41 catalyst gave 54% conversion of styrene oxide to styrene carbonate, which was similar to the 60% conversion reported with the zinc embedded MCM-41 catalyst. Only one epoxide was however screened with this mesoporous organocatalyst.
Eng-Poh Ng et al. grafted a bromide functionalised ionic liquid onto MCM-41, interestingly using rice husk ash as the silica source.137 Conversions of 14–58% were obtained using 1.6 wt% of this catalyst (with respect to epoxide), at 15 bar of carbon dioxide and 150 °C for 8 h. The reaction was proposed to proceed via the dual activation of epoxide and carbon dioxide (Scheme 7). A much more thorough study into the activity of these catalysts could have been performed. The synthetic route employed to synthesise the catalyst also used benzene as a solvent, which is not ideal from a green chemistry perspective. In a similar work, Al-Lohedan et al. made a bromide functionalised MCM-41 catalyst from rice husk ash.138 However, this was not as extensive a study as Ng's work, as only 1,2-epoxyhexane was converted into the corresponding cyclic carbonate. This catalyst gave 100% GC yields using 20 bar of carbon dioxide at 100 °C for 4 h, using 300 mg of catalyst and 30 mmol of epoxide. The reaction was suggested to occur via carbon dioxide activation (Scheme 5).
In 2017, Fontaine et al. investigated grafting tripropylammonium iodide salts onto the surface of SBA-15 and MCM-41 silica supports, forming mesoporous catalysts that could act as hybrid adsorbents.139 Styrene oxide was then used as the solvent, as well as a substrate, in order to increase carbon dioxide solubility in the support prior to forming cyclic carbonate. Both supported catalysts could form styrene carbonate under ambient conditions (1 bar of carbon dioxide at 25 °C for 24 h) in high yields (86% and 99%, for SBA-15 and MCM-41 catalysts, respectively) using only 10 mol% of iodide with respect to epoxide. The MCM-41-supported catalyst was more stable and could be used up to four times with no drop in activity, whereas the activity of the SBA-15-supported catalyst dropped significantly after only two uses. This was hypothesised to be due to the MCM-41 support having a high absorption capacity for carbon dioxide compared to the SBA-15 support,140 as well as to leaching of iodide and the grafted ammonium species from the SBA-15 catalyst. Only two epoxides were tested, therefore the scope of this study is rather limited.
In 2018, zwitterionic mesoporous silica-supported catalysts were created by Arai et al., containing imdizaole-urea frameworks attached onto silica.141 By changing the anions coordinating to the imidazole component, the efficiency of these catalysts to form cyclic carbonate varied in line with the nucleophilicity of the anions tested (I− > Br− > Cl− > NO3−). Under the optimum reaction conditions (110 °C, 25 bar carbon dioxide, 7.5 wt% of catalyst relative to epoxide after 4 h) conversions of 82–98% and ≥98% selectivity were obtained for four terminal epoxides. This catalyst could also be recycled four times and was proposed to follow a hydrogen bonding epoxide activation reaction mechanism (similar to Scheme 1). However, only GC yields were reported, reaction conditions were rather harsh and only 34% conversion was reported for cyclohexene oxide 1, even at 140 °C.
In 2017, bifunctional resorcinarenes were found to be extremely active catalysts for cyclic carbonate synthesis, with 47a (Fig. 18) giving excellent isolated yields: 87–99% for terminal epoxides and 41–69% for di-substituted epoxides.143 This system required only 1 mol% of catalyst at 80 °C and 5 bar of carbon dioxide for 18 h. Polystyrene-supported versions of these catalysts, such as 47b, were found to maintain catalytic activity after twelve uses, when converting 1,2-epoxyhexane into the corresponding cyclic carbonate.
 | ||
| Fig. 18 Bifunctional resorcinarene 47a and polystyrene-supported analogue 47b. | ||
Also in 2017, Wu et al. supported protic ionic liquids onto polystyrene and showed that they were capable of converting simple terminal epoxides (71–98% yield) and cyclohexene oxide (66% yield) into cyclic carbonates via an epoxide activation mechanism (Scheme 1).142 This catalyst could be used in a continuous flow reactor for over 120 h, maintaining a consistent yield of 90% for conversion of epichlorohydrin into 3-chloropropylene carbonate. It is therefore a promising recyclable catalyst. Polystyrene-supported TBD-based ionic liquids were developed in 2019 by Zou et al., by alkylating polystyrene supported TBD; optimally with 3-bromo propionic acid.144 This catalyst was recyclable over five runs. Under the optimum reaction conditions of 0.24 mol% of catalyst, 140 °C at 20 bar of carbon dioxide for 2 h, conversions of 77–100% for five terminal epoxides and 57% conversion of cyclohexene oxide 1 were obtained.
In 2019, Werner et al. synthesised polystyrene-supported bifunctional ammonium and phosphonium salts, which gave 95–99% isolated yields for converting glycidyl methacrylate into the corresponding cyclic carbonate, an important monomer in polymer synthesis.145 By using 2 mol% of catalyst, at 90 °C and 10 bar of carbon dioxide for 2 h over 12 runs, isolated yields remained higher than 60% but slightly declined after each use. The activity of the catalyst against other epoxides was not tested. Interestingly, a life cycle analysis was performed for this work, estimating that transforming glycidyl methacrylate into its cyclic carbonate slightly reduced the overall carbon footprint of this process. This life cycle analysis also highlighted the importance of using bio-based starting materials and that all the reactants used in the reaction must be considered, and not just the reaction itself, as it was estimated that total carbon dioxide emissions could be reduced further (by 47% overall) if epichlorohydrin from the bio-based Epicerol process was used.
In 2018, Sobral et al. created a heterogeneous chitosan graphene oxide-supported organocatalyst, termed as a nanocomposite material.147 This catalyst could transform simple epoxides (propylene oxide, epichlorohydrin and styrene oxide) into cyclic carbonates, at 10 bar of carbon dioxide and 100 °C for 6 h using 50 mg of catalyst and 50 mg of tetrabutylammonium bromide, and could be recycled up to five times. The catalyst though was not particularly active, giving only 61% conversion for propylene oxide. It would be interesting to determine its activity against more sterically hindered epoxides. This catalyst also uses a 1:1 ratio of catalyst and tetrabutylammonium bromide, making it questionable whether the activity reported is due to the catalyst or co-catalyst.
In 2018, Li et al. immobilised ionic liquids onto graphene oxide, by employing epoxy and hydroxyl groups in the support, in order to increase the ionic liquid loading.148 The heterogeneous catalyst was more active than homogeneous ionic liquids, giving 96% conversion of propylene oxide using 0.5 g of catalyst at 20 bar of carbon dioxide and 140 °C for 4 h. This catalyst was effective in ring-opening five terminal epoxides via an epoxide activation mechanism (Scheme 1), as well as an aziridine substrate, and could be recycled up to five times. No internal epoxides were tested though.
In 2017, Sobral et al. developed a chitosan based meso-tetrakis(4-sulfonatophenyl)porphyrin catalyst 48 (Fig. 19) for cyclic carbonate formation.151 The conversion of propylene oxide into propylene carbonate doubled (from 31% to 66%) when solid-supported catalyst 48 was used instead of pure chitosan (using 50 mg of catalyst and 50 mg of tetrabutylammonium bromide with 3.5 mL of propylene oxide, at 10 bar of carbon dioxide and 75 °C for 6 h). However, no other epoxides were screened and no recycling experiments were performed.
 | ||
| Fig. 19 Chitosan-supported meso-tetrakis(4-sulfonatophenyl) porphyrin catalyst 48. | ||
In 2018, Shchukarev et al. immobilised ionic liquids onto cross-linked chitosan, reporting 96–99% conversion for simple terminal epoxides, via dual activation of carbon dioxide and the epoxide (Scheme 7), at 20 bar of carbon dioxide and 120 °C for 5 h using 150 mg of catalyst with 8.73 mmol of epoxide.149 This catalyst was recyclable, giving similar activities after five cycles, but when tested against cyclohexene oxide only gave 27% conversion, even when the reaction was left for 17 h.
In 2019, Einloft et al. immobilised ionic liquids onto chemically modified cellulose extracted from rice husk waste.152 However, this system needed zinc bromide as a co-catalyst in order to reach conversions >70%, giving less than 60% conversion in the absence of the zinc salt. Conversions also gradually dropped when the catalyst was reused over five cycles. Whilst the source of the sugar support is good in terms of green credentials and should be considered more often, the activity and substrate scope reported was not ideal. Only propylene oxide and styrene oxide were tested (styrene oxide required the presence of zinc bromide) at 110 °C and 40 bar carbon dioxide for 6 h using 1.0 or 1.5 g of catalyst.
In 2019, Lei et al. were the first to study amino-bridged bis-β-cyclodextrin (derived from naturally available biomass) as catalysts for cyclic carbonate formation, using the hydroxyl groups of cyclodextrin as hydrogen bond donor sites and potassium iodide as a nucleophilic co-catalyst.150 By using 0.125–0.14 mol% of the catalyst and 1.25 mol% of co-catalyst, at 20–40 bar of carbon dioxide and 100–120 °C for 2–4 h, eight out of eleven catalysts synthesised achieved >95% conversion of propylene oxide into propylene carbonate via an epoxide activation mechanism (Scheme 1). Five of the catalysts required no co-catalyst. Good conversions were obtained when screening these catalysts against eight simple epoxides, but the catalysts struggled to ring-open cyclohexene oxide 1 (giving only 3% conversion) and substrates with two epoxide rings. The catalyst could be recycled five times with no loss in activity.
In 2018, Dupont et al. created silica based ionic liquid materials, termed as silica-supported ionic liquid phase catalysts.154 Using the most active catalysts developed, 99% conversion of propylene oxide into propylene carbonate was reported and the catalyst could be recycled up to five times. However, rather harsh reaction conditions were required (5 bar of carbon dioxide at 150 °C for 30 min) considering that propylene oxide is one of the easiest epoxides to ring-open. These silica-based catalysts were also tested using carbon dioxide from a waste exhaust stream, rather than pure carbon dioxide, and gave similar epoxide conversions. Another system, designed by Bica et al. in 2018, also used solid-supported ammonium-, phosphonium-, and imidazolium ionic liquids, this time in a continuous flow reactor.155 However, all catalysts tested in the flow reactor showed a decline in catalytic activity, perhaps due to the formation of polymeric by-products, and were only screened against propylene oxide. Testing more systems in continuous flow reactors needs to be implemented in future studies.
In 2019, Chen et al. studied silanol-enriched viologen- (1,1′-disubstituted-4,4′-bipyridilium-) based ionic porous hybrid polymers (named V-iPHPs), containing polyhedral oligomeric silsesquioxane (POSS) groups, with the intention of combining hydrogen bond donating silanol groups and bromide nucleophiles into the same catalyst.156 The catalysts developed were active under fairly mild conditions (1 bar of carbon dioxide and 30–80 °C over 72–120 h) using 0.5 g of catalyst for 2 mmol of epoxide, giving 83–99% conversions for eight terminal epoxides. The catalyst was recyclable over five runs and a catalytic cycle was proposed (Scheme 23). The mild conditions reported for this catalyst are promising, compared to other heterogeneous supported catalysts.
 | ||
| Scheme 23 Proposed reaction mechanism for cyclic carbonate synthesis using V-iPHPs catalysts. | ||
Following on from this work, in 2020 Chen et al. reported new POSS materials, using imidazolium instead of viologen ionic liquid linkers, which could be used under mild conditions (1 bar of carbon dioxide at 60–80 °C for 72 h), giving 84–99% conversions for eight aromatic and aliphatic epoxides.157 This catalyst was hypothesised to work via the same reaction mechanism suggested for Chen's earlier studies (Scheme 23).156 In a similar vein, in 2019, Aprile et al. also developed recyclable imidazolium modified POSS materials158 that gave high turnover frequencies and turnover numbers for the conversion of epoxides into cyclic carbonates.159 Although harsh reaction conditions were required relative to those often seen with metal-based complexes (40 bar of carbon dioxide at 150 °C for 3 or 16 h), only 0.133–0.020 mol% of catalyst was required, which is impressive. Even at these low loadings, conversions of 30% and 16% were observed for the difficult substrates cyclohexene oxide 1 and oxetane, respectively, which is particularly promising. It would be interesting to test both Aprile's and Chen's catalysts under the same reaction conditions to effectively judge the differences in reactivity between the catalysts. No isolated yields were reported in Chen and Aprile's work and only Aprile tested these systems against internal epoxides. Future studies into these catalysts would therefore be interesting.
In 2019, Wang et al. developed amino-incorporated organosilica nanotubes as hydrogen bond donors, which in conjunction with 10 mol% of tetrabutylammonium iodide were capable of converting nine epoxides, including two internal epoxides, into cyclic carbonates in 74–99% conversions, by activating the epoxide and carbon dioxide (Scheme 7).128 These results were obtained using 65 mg of catalyst for 10 mmol of epoxide, at 10 bar of carbon dioxide and 70 °C for 12 h. The catalyst could be recycled up to six times.
Heinrichs et al. developed a route to silica-immobilised tributylammonium salts (Scheme 24).129,160 Interestingly, the majority of these catalysts had similar activity to a commercial fumed silica sample used in conjunction with tetrabutylammonium iodide, and were only slightly better than using tetrabutylammonium iodide alone (at 80 °C and 100 bar for 4 h). These catalysts also had problems with recyclability. It should be noted that this catalyst was tested in forming a bis-cyclic carbonate from polyethylene glycol diglycidyl ether. It would therefore be interesting to determine how effective these catalysts are at ring-opening traditionally used epoxide substrates and more challenging internal epoxides.
 | ||
| Scheme 24 Formation of silica-supported ammonium salts via the quaternisation method as reported by Heinrichs et al. | ||
3.2. Polymeric systems
One class of ionic polymers often used for cyclic carbonate synthesis are vinyl-functionalised ionic liquid polymers, obtained from the radical polymerisation/copolymerisation of vinyl-based monomers using azodiisobutyronitrile as initiator. Porous structures can be formed during the polymerisation/copolymerisation step, which can improve catalytic activity. In 2017, Xiao et al. synthesised a family of quaternary phosphonium-based ionic polymers (PIPs) with nanoporous structures and large surface areas via the polymerisation of vinyl-functionalised quaternary phosphonium salts (Scheme 25).161 The surface areas of PIP-Me-I, PIP-Et-Br, and PIP-Bn-Cl were as high as 402, 625, and 758 m2 g−1, respectively, which is beneficial in terms of mass transfer during cyclic carbonate synthesis. The polymers exhibited a strong affinity for carbon dioxide, which was 40 times better than the corresponding unpolymerised quaternary phosphonium salts and so were also more catalytically active. Cyclic carbonate synthesis could be achieved at atmosphere pressure, which meant that the use of an autoclave could be avoided. When 0.46 mol% of PIP-Bn-Cl was used to catalyse the reaction of epichlorohydrin with carbon dioxide, a 95% yield of 3-chloropropylene carbonate was obtained at 1 bar pressure and 25 °C. When the reaction temperature was raised to 100 °C, the yield rose to 99% after 3 h. In addition to the high activity, these catalysts also exhibited excellent stability, as no loss in activity for the PIP-Bn-Cl catalyst was observed after ten uses.
 | ||
| Scheme 25 Synthesis of quaternary phosphonium-based ionic polymers. | ||
In 2019, Gai et al. developed a series of poly(ionic liquids) via the copolymerisation of bis(1-vinylimidazolium) chloride 49 (Fig. 20) with ethylene glycol dimethacrylate162 to form catalysts, termed PIL-X where X is the molar ratio of 49 to ethylene glycol dimethacrylate. All the PIL-X catalysts showed abundant meso- or macroporous structures and the BET surface area of the polymer gradually decreased from 22.8 m2 g−1 to 2.0 m2 g−1 as the monomer 49 content in the polymer increased. When the same quantity of each PIL-X catalyst was used as a catalyst, the highest yield of cyclic carbonate was obtained with PIL-4 as catalyst, as the ionic liquid content in PIL-4 was the highest out of all the catalysts. However, comparison of turnover frequencies showed that PIL-0.5, which had the largest surface area and lowest number of ionic sites, was the most active catalyst.
 | ||
| Fig. 20 Structure of bis(1-vinylimidazolium) chloride 49. | ||
As discussed above for homogeneous catalysts, introducing hydrogen bond donating functional groups into ionic polymers can increase their catalytic activity. Wang et al. synthesised mesoporous poly(ionic liquids) by the copolymerisation of vinyl- and epoxy-containing imidazolium-based ionic liquid monomers with divinylbenzene, followed by hydrolysis of the epoxy group to form vicinal hydroxyl groups.163 The resulting materials possessed high levels of mesoporosity, halogen anions as nucleophiles and 1,2-dihydroxy groups as hydrogen bond donors. As a result, they showed excellent performance for the transformation of epoxides into cyclic carbonates under low carbon dioxide pressure. When 1.9 mol% of catalyst was used, 98% conversion of epichlorohydrin into 3-chloropropylene carbonate was achieved after 4 h at 70 °C and 4 bar of carbon dioxide. When the carbon dioxide pressure was reduced to 1 bar, the conversion of epichlorohydrin was still 91% after 24 h. When styrene oxide was used as a substrate, 93% and 90% conversions were obtained, at 4 bar of carbon dioxide after 16 h and at 1 bar pressure after 96 h, respectively. This catalytic activity was equivalent to that of the homogeneous analogue of the polymer. In addition to the high catalytic activity, the polymer could be easily recovered and reused at least ten times. Quantum chemistry calculations were carried out to expose the role of the vicinal hydroxyl groups in the reaction mechanism, highlighting that in addition to these groups acting as hydrogen bond donors, they could also enhance the leaving group ability of the halogen anions during the catalytic cycle.
In 2017, Dyson et al. developed highly active poly(ionic liquids) for the synthesis of cyclic carbonates from epoxides and carbon dioxide.164 Eleven cross-linked ionic polymers were synthesised by the polymerisation of dicationic styrene-functionalised imidazolium-based ionic liquids (Scheme 26), and polymer 50e-Br, which contained vicinal hydroxyl groups within its structure, was the most active. When 5 mol% of 50e-Br was used to catalyse the reaction of styrene oxide with carbon dioxide, 99% yield of styrene carbonate was obtained after 24 h at 80 °C and 4 bar of carbon dioxide, and 68% yield was obtained when the pressure was reduced to 1 bar. No loss of activity was observed after four uses of the polymeric catalyst.
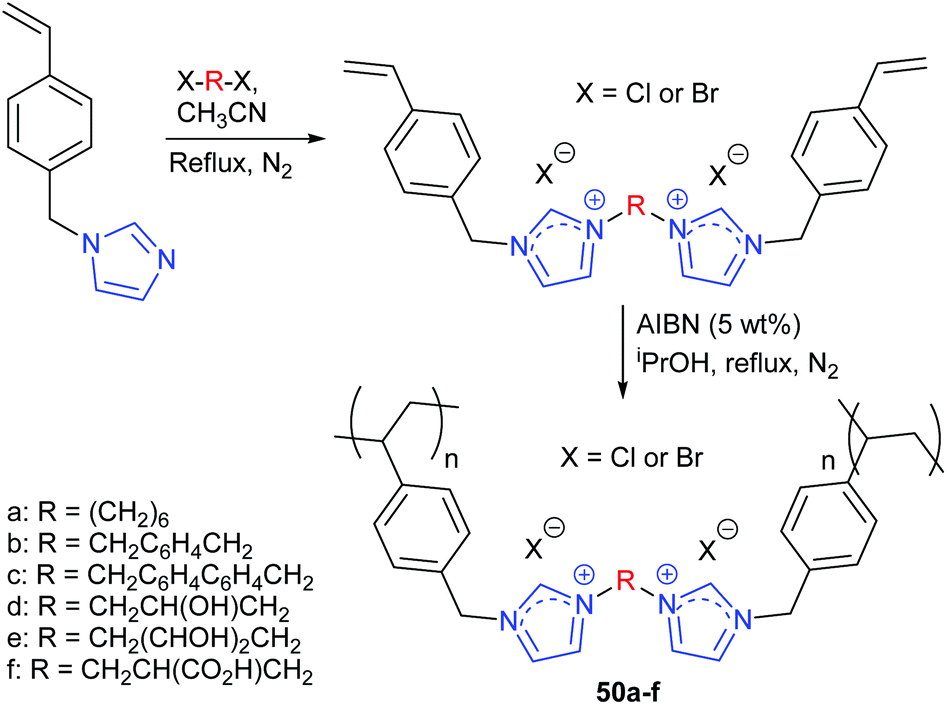 | ||
| Scheme 26 Synthesis of vinyl-functionalised di-imidazolium salts and their subsequent polymerisation to form cross-linked polymers 50. | ||
Another benefit of the copolymerisation synthesis method is that functional groups or special porous structures can be introduced into the polymer using non-ionic liquid-based monomers. In 2017, Yang et al. developed ionic liquid-based hydroxyl-functionalised porous polymers 51via the copolymerisation of three monomers (Scheme 27).165 In polymers 51, the 1-alkyl-3-vinylimidazolium bromide provided the ionic liquid component, 4-vinylbenzyl alcohol provided hydroxyl groups and 1,4-divinylbenzene shaped the porous structure. By comparing different catalysts, it shown that the catalyst with the highest hydroxyl group content and largest BET surface area was the most active.
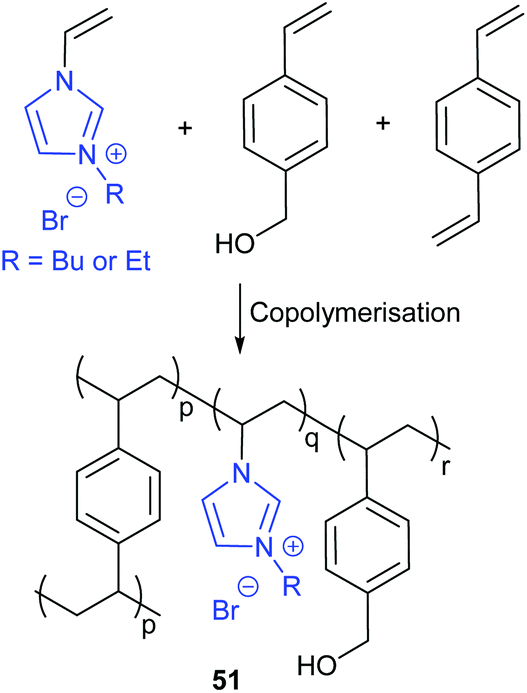 | ||
| Scheme 27 Synthesis of porous polymers 51 functionalised with both ionic liquids and intramolecular hydroxyl groups. | ||
In the same year, Dyson et al. described a series of imidazolium-based ionic polymers 52a–d which were synthesised by the reaction of arene-bridged bis- or tris-alkyl halides with trimethylsilylimidazole (Scheme 28).166 Various terminal epoxides could be converted into cyclic carbonates at 1 bar of carbon dioxide and 100 °C in 24–48 h, using 5 mol% of ionic polymers 52. Polymer 52c could be recycled and reused at least ten times.
 | ||
| Scheme 28 Synthesis of ionic polymers 52a–d. | ||
Chen et al. used the Menschutkin reaction to synthesise pyridinium-based porous ionic polymers from bipyridine and 4,4′-bis(bromomethyl/chloromethyl)biphenyl monomers.167 The conversion of various epoxides into cyclic carbonates could be achieved at 1 bar of carbon dioxide and 60–120 °C, but required a reaction time of 48–72 h. When the bromide containing polymer was used to catalyse the reaction of epichlorohydrin with carbon dioxide, the yield of cyclic carbonate slightly decreased from 99% to 94% after five uses.
A series of quaternary ammonium-based ionic polymers synthesised via the Menschutkin reaction were also developed by Waser et al. (Fig. 21).168 They exhibited good activity for the synthesis of cyclic carbonates from aromatic and aliphatic epoxides and carbon dioxide at 1 bar pressure and 120 °C using 0.5 mol% of ionic sites with reaction times of 4–10 h. In 2018, a carboxylic acid functionalised pyridinium-based porous organic framework (POF) based catalyst, POF-PNA-X (X = Cl, Br or I), was developed by Shi et al.169 SEM images showed that both POF-PN and POF-PNA-Br were composed of regular sphere-shaped agglomerates. As the acid groups functioned as hydrogen bond donor sites during the reaction of epoxides with carbon dioxide, the reaction occurred under mild conditions (40 °C, 1 bar of carbon dioxide, 48 h).
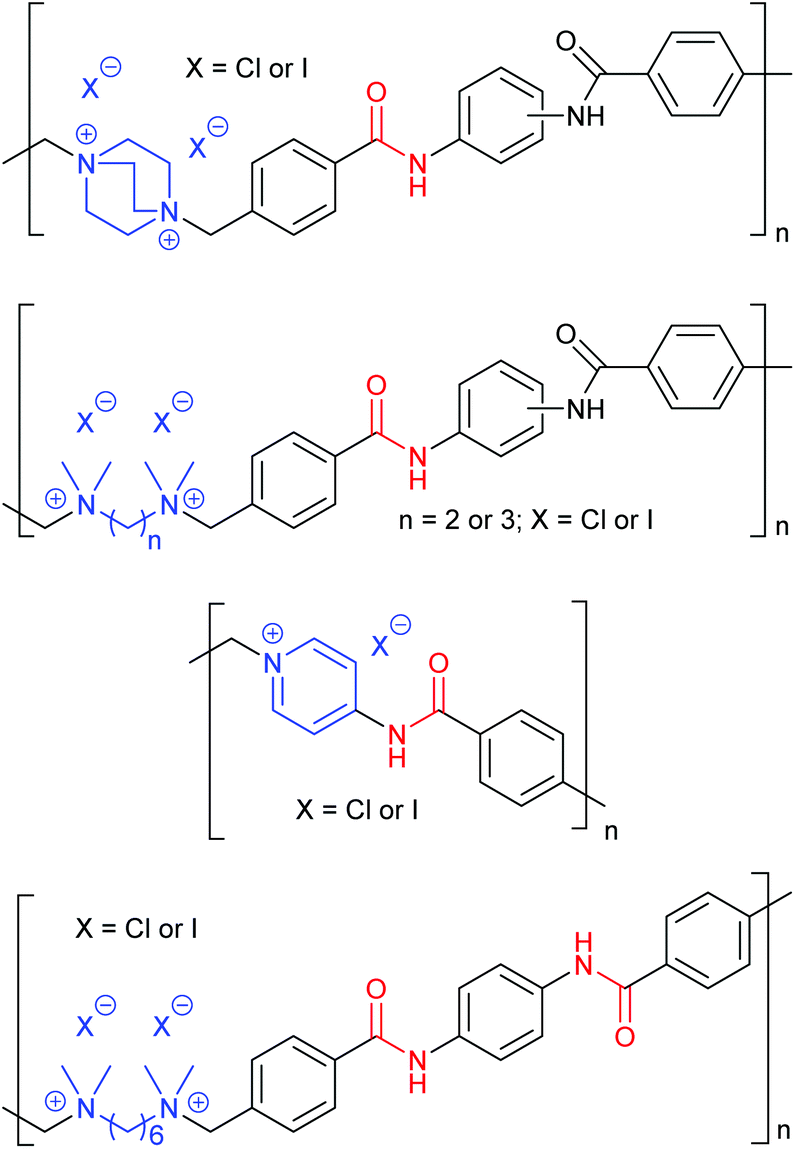 | ||
| Fig. 21 Ionic polymers developed by Waser et al. | ||
In 2019, Wang et al. reported a urea-functionalised imidazolium-based ionic polymer (53), which was obtained through a urea-forming condensation reaction (Scheme 29).170 Epoxides could be converted into cyclic carbonates using 0.39 mol% of ionic sites and 10 bar of carbon dioxide, at 90–110 °C for 1–14 h. The catalyst could be used at least six times. As a comparison, the activity of an imidazolium-based ionic polymer (54), synthesised via the Menschutkin reaction of tris(bromomethyl)-2,4,6-trimethylbenzene and 1,4-bis((1H-imidazol-1-yl)methyl)benzene, was investigated. The superior catalytic activity of 53 compared to 54 indicated that the urea groups functioned as hydrogen bond donors and facilitated the reaction. This hypothesis was also supported via DFT calculations.
 | ||
| Scheme 29 Synthesis of catalysts 53 and 54. | ||
In 2017, Wang et al. carried out the reaction of 1,4-bis(chloromethyl/bromomethyl)benzene with 2-phenylimidazoline in a one-pot synthesis, involving a simultaneous Menschutkin reaction and Friedel–Crafts alkylation reaction, to form imidazolinium-based porous hypercrosslinked ionic polymers with high surface areas (SBET = 534–1114 m2 g−1, Scheme 30).171 These materials exhibited high carbon dioxide capture abilities of 2.9–3.8 mmol g−1 at 273 K and 1.9–2.3 mmol g−1 at 298 K. The catalytic activity of the polymers for the reaction between styrene oxide and carbon dioxide (84–99% yields using 0.2 mol% of ionic sites, 10 bar carbon dioxide, 120 °C, 6 h) were superior to that of the homogeneous ionic liquid 2-phenyl-3-butylimidazolinium chloride (69% yield) and heterogeneous polystyrene resin-supported 2-phenylimidazolinium salt (59% yield). This suggested that the high surface area of the hypercrosslinked ionic polymer played a vital role in the catalysis. The catalytic activity for a series of terminal epoxides was investigated at 1 bar carbon dioxide (4 mol% bromide polymer, 4 mol% ZnBr2, 4 mL DMF, 25 °C, 96–120 h) giving yields of 73–99%. High yields of 85–95% were also obtained when a 1 bar mixture of 15% carbon dioxide and 85% nitrogen was used as the carbon dioxide source at 55 °C for 120 h. However, the requirement for metal salts, toxic solvents and long reaction times at atmospheric pressure are not advantageous. In 2020, the same research group used 1,4-bis(chloromethyl)benzene to synthesise a series of imidazolinium-based porous ionic polymers by reacting it with phenyl-functionalised imidazolinium ionic liquids via Friedel–Crafts alkylation.172 This type of catalyst had a high surface area (the highest SBET reported was 1017 m2 g−1), high carbon dioxide adsorption capacity (3.05 mmol g−1) and a good performance for the synthesis of cyclic carbonates, using carbon dioxide in a mixture of 15% carbon dioxide and 85% nitrogen (with 0.05 mol% ionic sites, 120 °C, 30 bar carbon dioxide). This catalyst also had good reusability as it could be reused five times.
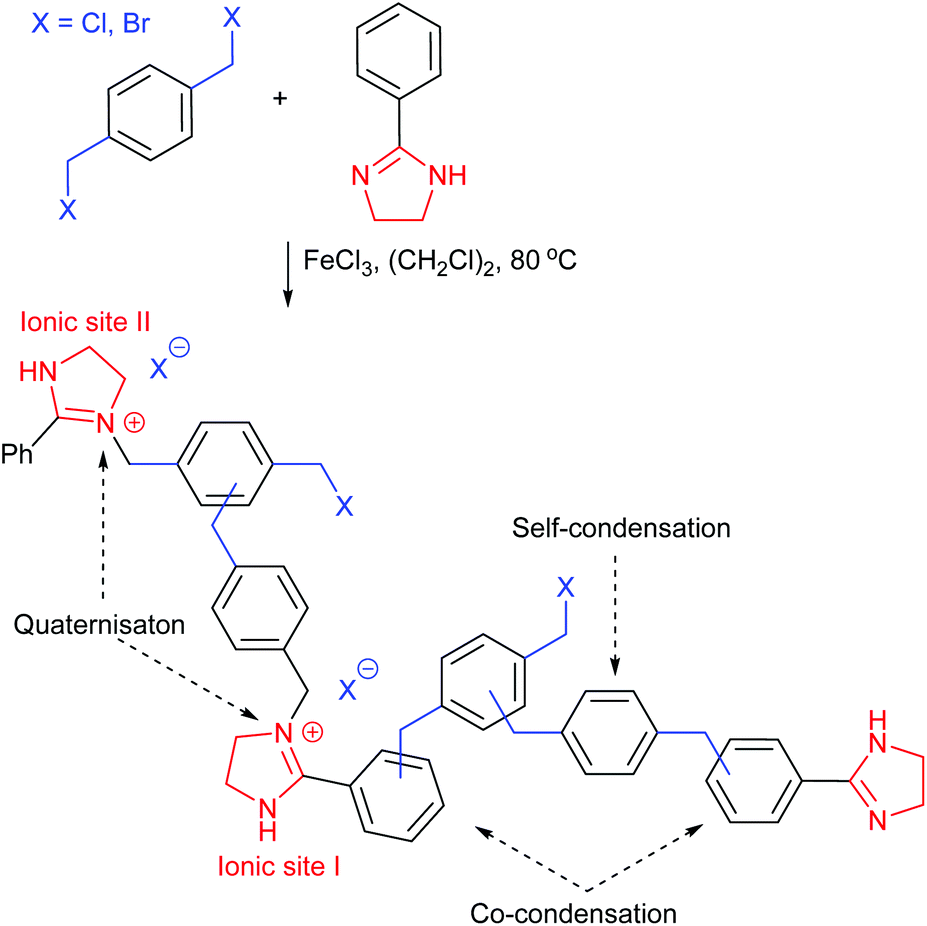 | ||
| Scheme 30 Synthesis of 2-phenylimidazolinium based hypercrosslinked ionic polymers. | ||
Ionic polymers synthesised by other methods have also been reported. N-Methyimidazole was grafted onto the surface of a polymeric benzyl chloride to form an imidazolium-based ionic polymer.173 The ionic liquid content in this polymer was lower than that of other ionic polymers and the required reaction temperature for the synthesis of cyclic carbonates (140 °C) was higher than that used with other catalysts.
Ji et al. presented the synthesis of an imidazolium-based functional ionic polymer 55via a phenol–formaldehyde condensation process (Scheme 31).174 Catalyst 55 with a high density of phenolic groups showed good activity for the transformation of terminal epoxides into cyclic carbonates, with 5 mol% catalyst loading at 10 bar of carbon dioxide and 80 °C for 10–24 h. When cyclohexene oxide 1 was employed as a substrate, 21% yield of cyclohexene carbonate was obtained after 48 h. When the carbon dioxide pressure and reaction temperature were reduced to 1 bar and 25 °C, 86% conversion of epichlorohydrin was achieved after 72 h. Polymer 55 could be further functionalised with quaternary ammonium groups to obtain polymer 56. In the presence of dilute carbon dioxide (15% carbon dioxide and 85% nitrogen), both 55 and 56 exhibited good catalytic activity for the synthesis of cyclic carbonates. Recycling experiments showed that both 55 and 56 could be reused at least six times.
 | ||
| Scheme 31 Synthesis of polymers 55 and 56. | ||
In 2019, Mecerreyes et al. investigated poly(azomethine-pyridinium) salts, synthesised from the condensation of dialdehydes and diamines via microwave irradiation.175 This polymer exhibited high catalytic activity for the reaction between epichlorohydrin and carbon dioxide, giving 100% yield using 0.5 mol% of polymer at 7 bar carbon dioxide and 100 °C for 4 h. The polymer could be reused four times.
In 2020, Wang et al. reported the synthesis of imidazolium-based porous ionic polymers 57via a Click reaction (Scheme 32)176 and their application in the capture and catalytic conversion of carbon dioxide. Polymer 57, with an SBET value of 1128 m2 g−1, could efficiently catalyse the reaction of epichlorohydrin with carbon dioxide, giving 90% yield of 3-chloropropylene carbonate at 1 bar of carbon dioxide and 100 °C after 24 h, without any co-catalyst. DFT calculations demonstrated that the triazole nitrogen in the 3-position (Scheme 32) interacted with the less sterically hindered carbon atom of epichlorohydrin and that a hydrogen bond was formed between a hydrogen atom in the adjacent benzene ring and the oxygen atom of epichlorohydrin. The amount of ionic liquid in the catalyst affected both its textural properties and its catalytic performance in the capture and transformation of carbon dioxide. Amongst the four catalysts tested, that with just 5% ionic liquid and a SBET of 994 m2 g−1, showed the best performance for the capture of carbon dioxide: 141.2 mg g−1 at 273 K and 97.8 mg g−1 at 298 K. This polymer also had the best catalytic activity, giving 99% yield at 1 bar and 100 °C after 24 h. Catalyst surface areas decreased as the amount of ionic liquid incorporated increased, as the polymer containing 25% ionic liquid had a SBET of only 7 m2 g−1, but still gave a reasonable cyclic carbonate yield of 84%. These results indicated that the incorporated ionic liquid and the porous properties of the polymer synergistically contributed to the catalysis of cyclic carbonate synthesis.
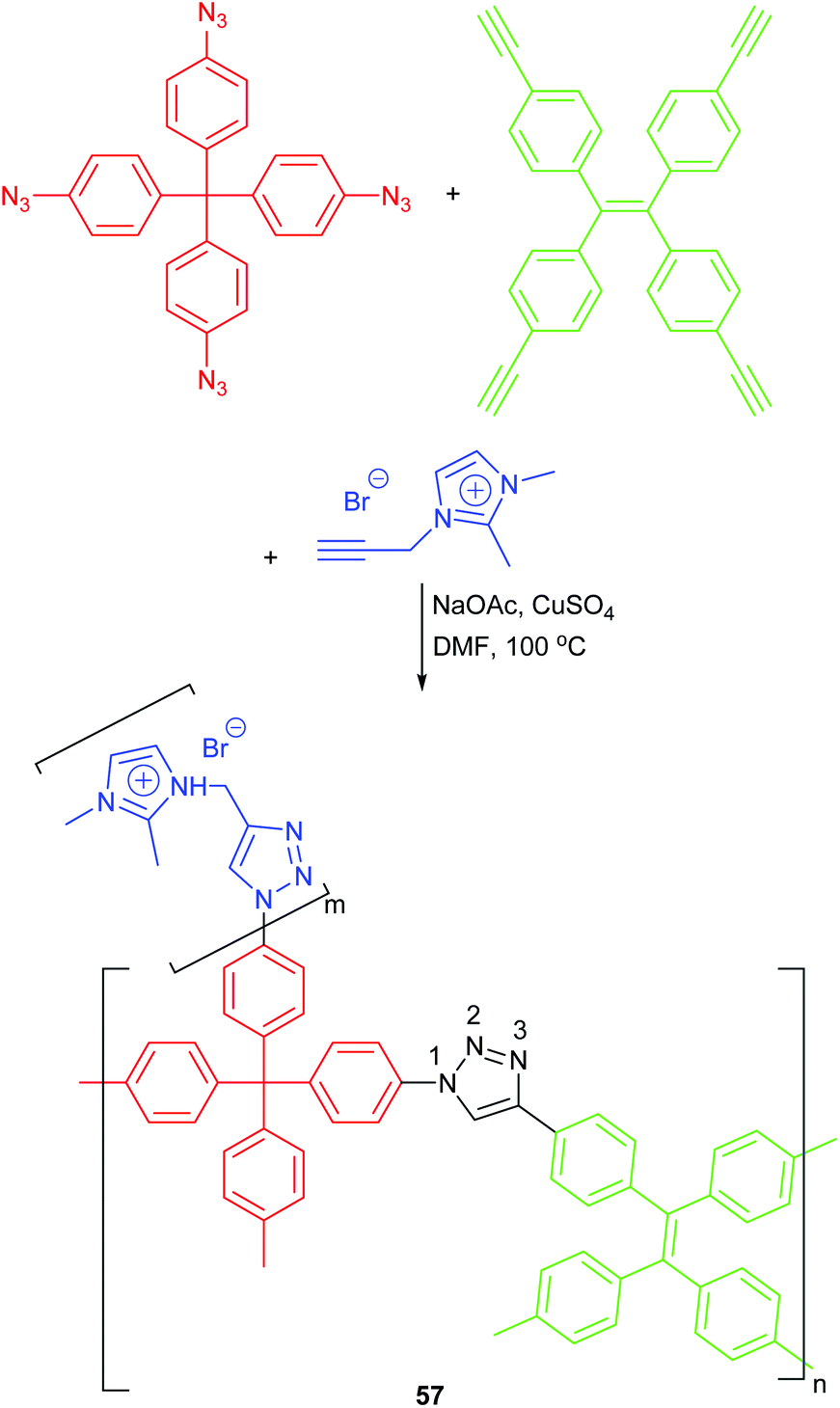 | ||
| Scheme 32 Synthesis of polymers 57. | ||
Arai et al. developed tri-s-triazine-linked imidazolium salts 58 from various urea-based ionic liquids and urea derivatives (Scheme 33).180 The most active of these compounds was the iodine doped derivative of the polymer prepared by adding 60 wt% of urea to the ionic liquid and calcining the mixture at 500 °C. This polymer gave a 94% yield of propylene carbonate, from a reaction carried out at 120 °C and 15 bar of carbon dioxide after 2.5 h, using 5 wt% of catalyst. There was a slight reduction in the catalytic activity after the polymer was used five times. In addition to four terminal epoxides, the use of cyclohexene oxide 1 as substrate was investigated and it gave a 45% yield of cyclohexene carbonate from a reaction carried out at 130 °C and 15 bar of carbon dioxide for 9 h. Xiao et al. also explored the use of melem-based multifunctional catalysts for the conversion of epoxides and carbon dioxide into cyclic carbonates.181 As expected, melem oligomers grafted with both hydroxyl and quaternary ammonium groups exhibited the best catalytic performance. At 120 °C and 20 bar of carbon dioxide after 2–15 h, 87–98% yields were obtained from reactions involving terminal epoxides, and 9% yield was obtained using cyclohexene oxide 1. The yield of propylene carbonate decreased from 97% to 59% after five runs.
 | ||
| Scheme 33 Synthesis and proposed structure of tri-s-triazine-linked ionic liquid catalysts 58. | ||
A hydrazine-bridged covalent triazine polymer 59 was prepared (Scheme 34) and investigated for carbon dioxide capture and catalytic conversion, as hydrazine could play the role of hydrogen bond donor.182 With 5 mol% of tetrabutylammonium bromide as co-catalyst, eighteen terminal epoxides were transformed into cyclic carbonates in 67–99% yields, using 20 bar of carbon dioxide at 80 °C for 12 h. It is worth highlighting that 90% conversion of cyclohexene oxide 1 into cyclohexene carbonate could be achieved under the same reaction conditions. Polymer 59 could also catalyse the synthesis of cyclic carbonates from epichlorohydrin, epibromohydrin and glycidol in the absence of tetrabutylammonium bromide at the higher temperature of 120 °C.
 | ||
| Scheme 34 Synthesis of hydrazine containing polymer 59. | ||
In 2017, Coskun et al. reported bipyridinium-based charged covalent triazine frameworks for carbon dioxide capture and conversion.183 The catalyst created from 1,1′-bis(4-cyanophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride and anhydrous ZnCl2 at 500 °C, had a uniform spherical morphology, high surface area (SBET = 1247 m2 g−1) and high carbon dioxide uptake (133 mg g−1 at 273 K). The carbon dioxide uptake capacities reported for these polymers were higher than those of previously reported covalent triazine frameworks with similar surface areas and nitrogen content. Thus, the authors concluded that the presence of cationic moieties facilitated the absorption of carbon dioxide, owing to the additional electrostatic interactions between carbon dioxide and the charged centres of the pyridinium units. Four terminal epoxides were investigated as substrates for cyclic carbonate synthesis, giving yields of 36–95% from reactions carried out at 90 °C and 10 bar of carbon dioxide for 12 h. The polymeric catalyst could be reused four times with no change in its catalytic activity. In 2018, Cao et al. created an imidazolium-based covalent triazine framework184via a similar synthetic route to that reported by Coskun. 10 wt% of this polymer could give 95% conversion of epichlorohydrin into 3-chloropropylene carbonate at 1 bar of carbon dioxide and 120 °C after 24 h and could be reused at least five times.
Yu et al. reported another bipyridinium-based ionic porous triazine framework, catalyst 60, which contained a high nitrogen content of 20 wt% (Fig. 22).185 The nitrogen atoms and chloride anions in catalyst 60 were proposed to contribute to the absorption and transformation of carbon dioxide, giving an initial isosteric heat of adsorption for carbon dioxide of up to 53 kJ mol−1. This value is high for this type of absorbent and indicates that a strong interaction occurs between the adsorbate and carbon dioxide gas.186,187 The transformation of epoxides and carbon dioxide into cyclic carbonates could be achieved at room temperature and ambient pressure, giving 63 and 80% yields for propylene carbonate after 12 and 48 h, respectively, using only 1 mol% of chloride. The recyclability of catalyst 60 was tested over three runs, showing only a slight drop in propylene carbonate yield from 99 to 95%.
 | ||
| Fig. 22 Structure of polymer 60. | ||
Liu et al. synthesised triazine-based covalent organic frameworks via the Schiff-base condensation of (1,3,5-triazine-2,4,6-triyl)trianiline or 1,3,5-tris(4-aminophenoxy)benzene with 2,5-dihydroxyterephthalaldehyde, respectively.188 Both of these materials showed high surface areas (SBET = 1450 m2 g−1 and 1392 m2 g−1 respectively), excellent thermal and chemical stability and large adsorption capacities for carbon dioxide (129 mg g−1 and 151 mg g−1, respectively). By utilising hydroxyl groups as hydrogen bond donor sites, both frameworks exhibited efficient catalytic activity for the conversion of epichlorohydrin into 3-chloropropylene carbonate under ambient conditions in the presence of tetrabutylammonium bromide (5 mol%), using 0.5 mol% of polymer for 48 h. Both catalysts could be reused at least five times.
In 2019, Mandal et al. explored a triazine-based benz-bis(imidazole)-bridged covalent organic framework catalyst, synthesised via the polycondensation reaction of tri(4-formylphenoxy)cyanurate with aromatic amines, 1,2,4,5-tetraminobenzene and imidazole.189 This polymer also exhibited a high surface area (SBET = 1424 m2 g−1), excellent thermal and chemical stability and a large adsorption capacity for carbon dioxide (135 mg g−1). The performance of this covalent organic framework for the conversion of epoxides and carbon dioxide into cyclic carbonates was better than the frameworks of Liu discussed above. In the presence of tetrabutylammonium bromide (0.5 mol%), 54–98% conversion of various epoxides, including cyclohexene oxide 1, could be achieved under ambient conditions (using 0.1 mol% of polymer for 24 h).
 | ||
| Scheme 35 Synthesis of polymeric catalyst 61. | ||
Han et al. synthesised a biomass-based porous organic polymeric catalyst 62 with multifunctional sites and hydroxyl and carboxylic acid groups obtained from tannic acid (Scheme 36).191 In the presence of silver nanoparticles, catalyst 62 could efficiently catalyse the carboxylative cyclisation of propargyl alcohols with carbon dioxide. In addition, after adsorbing iodine (consequently forming I3− which acts as a nucleophile), catalyst 62 catalysed the reaction of epoxides with carbon dioxide to produce cyclic carbonates, but required harsh reaction conditions (150 °C and 30 bar of carbon dioxide). The requirement for silver, albeit in small quantities, makes this not strictly an organocatalyst and may not be desirable in terms of sustainability.
 | ||
| Scheme 36 Synthesis of polymeric catalyst 62. | ||
Another interesting example was the use of a sulfonated porous polymer as a solid acid catalyst for the conversion of carbon dioxide and epoxides into cyclic carbonates by Islam in 2019.192 In the presence of tetrabutylammonium bromide, both terminal and internal epoxides could be converted into their respective cyclic carbonates in 93–99% yields, at 50 °C and 1 bar of carbon dioxide in 10 h.
3.3. Halide-free systems
Nitrogen-rich triazine-based materials have been used extensively in the reaction of epoxides with carbon dioxide. In 2017, melamine-based mesoporous organic polymers (MOPs) were synthesised via polycondensation of low-cost and readily available melamine and monoaldehydes. The MOPs contained zero to three hydroxyl groups in the phenyl ring (termed MOP-X, where X = number of hydroxyl groups on the aldehyde) and a high density of aminal groups (Scheme 37).193 The performance of these MOPs in carbon dioxide adsorption and the transformation of carbon dioxide into cyclic carbonates was investigated. Although MOP-3 showed the best capability for carbon dioxide adsorption (114 mg g−1 under 1 bar of carbon dioxide at 0 °C) among the four MOPs tested, its catalytic activity for the reaction of carbon dioxide with epichlorohydrin (58% yield of cyclic carbonate, 10 bar of carbon dioxide, 100 °C, 24 h) was lower than the other three MOPs. Interestingly, MOP-0 with no hydroxyl group showed excellent activity for the reaction of carbon dioxide with epichlorohydrin (89% yield of cyclic carbonate, 10 bar of carbon dioxide, 100 °C, 24 h), and moderate capacity for carbon dioxide adsorption (82 mg g−1 under 1 bar carbon dioxide at 273 K). The secondary amine groups acted as hydrogen bond donors and could form multiple hydrogen bonds with the epoxide, thereby activating the epoxide during the ring-opening step (Scheme 1). For halide substituted epoxides, no additive was needed for the reaction, whereas for aromatic or aliphatic epoxides, potassium iodide or tetrabutylammonium iodide (5 mol%) was added to achieve similar catalytic efficiency. It is unfortunate that the authors did not report the results of reacting aromatic or aliphatic epoxides with carbon dioxide in the absence of potassium iodide or tetrabutylammonium iodide, and thus the difference in the catalytic activity of MOP-0 with or without potassium iodide/tetrabutylammonium iodide is unknown.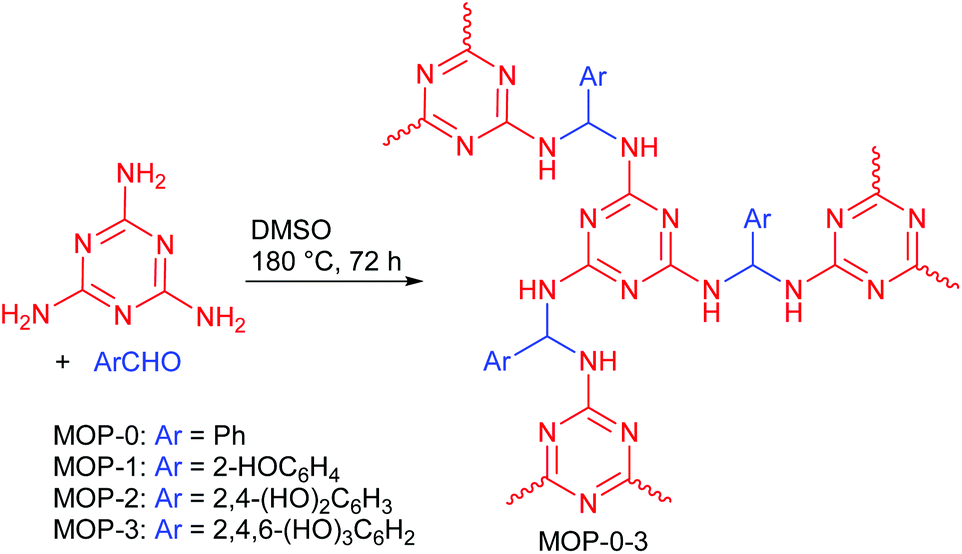 | ||
| Scheme 37 Synthesis of MOPs used in the formation of cyclic carbonates from epoxides and CO2. | ||
A cyanuric–urea polymer (CUP), synthesised from cyanuric chloride and urea (Scheme 38), displayed good catalytic activity for cyclic carbonate synthesis under additive and solvent free conditions.194 The CUP catalyst had a high density of urea groups, in which the NH groups could activate both epoxides, by forming multiple hydrogen bonds, and carbon dioxide (similar to Scheme 7). A total of 18 epoxides were studied and the conversion of epoxides varied from 28–99%, using 10 wt% catalytic loading at 60 bar carbon dioxide and 120 °C for 12 h. The synthesis of bis-cyclic carbonates from resorcinol diglycidyl ether with 20 wt% catalyst was also investigated, achieving 94% conversion. One defect of this catalyst was the high pressure requirements, as when the carbon dioxide pressure was reduced from 60 to 40 to 20 bar, the conversion of propylene oxide dropped from 99 to 72 to 35% respectively. The CUP catalyst could be separated from the reaction mixture by simple filtration, and could be reused at least seven times in the transformation of propylene oxide into propylene carbonate.
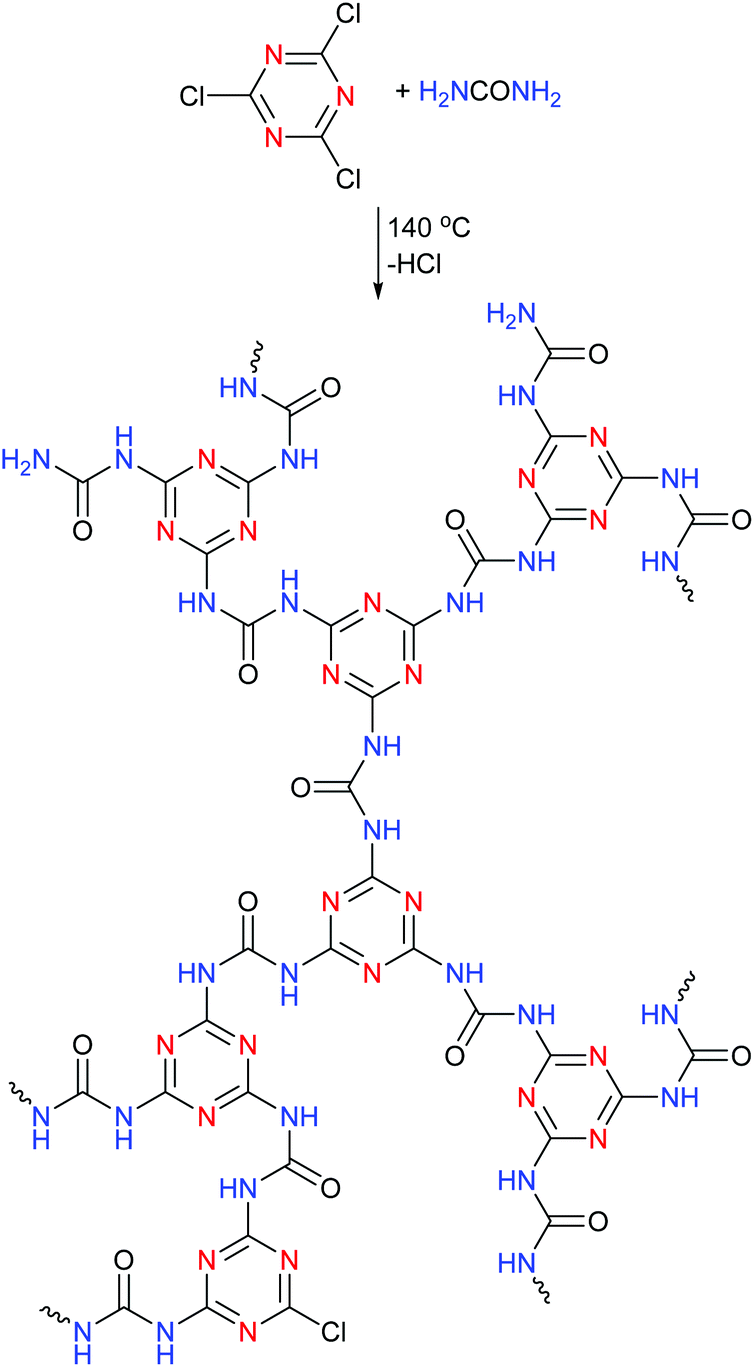 | ||
| Scheme 38 Synthesis of CUP catalyst reported by Khan et al. in 2017. | ||
Another class of triazine-based catalysts were reported by Li et al. in 2019.195 These nitrogen-rich covalent triazine frameworks (CTFs) were synthesised by the trimerisation of 2,5-dicyanopyridine 63 (Scheme 39). A total of five CTFs were synthesised by changing the amount of zinc chloride and reaction temperature used for their synthesis. The catalyst prepared by heating at 400 °C for 4 h with a 1:10 mass ratio of zinc chloride:63, showed the best catalytic performance. When it was applied for the reaction between epichlorohydrin and carbon dioxide, the conversion and selectivity were 99% and 96%, respectively. Higher reaction temperatures for the synthesis of the CTFs might lead to nitrile cleavage and decomposition of the triazine ring and thus destroy the catalytic site. The nitrogen content of the most active catalyst (16.5%) was higher than that of the other CTFs (9.4–12.2%). Therefore, although the other CTFs had higher BET surface areas and pore volumes, their catalytic activities were relatively low. Furthermore, the two-dimensional layer structures and smaller pore sizes present in the most active CTF might further facilitate the transformation of carbon dioxide. The most active CTF could be recycled six times without any change in its activity.
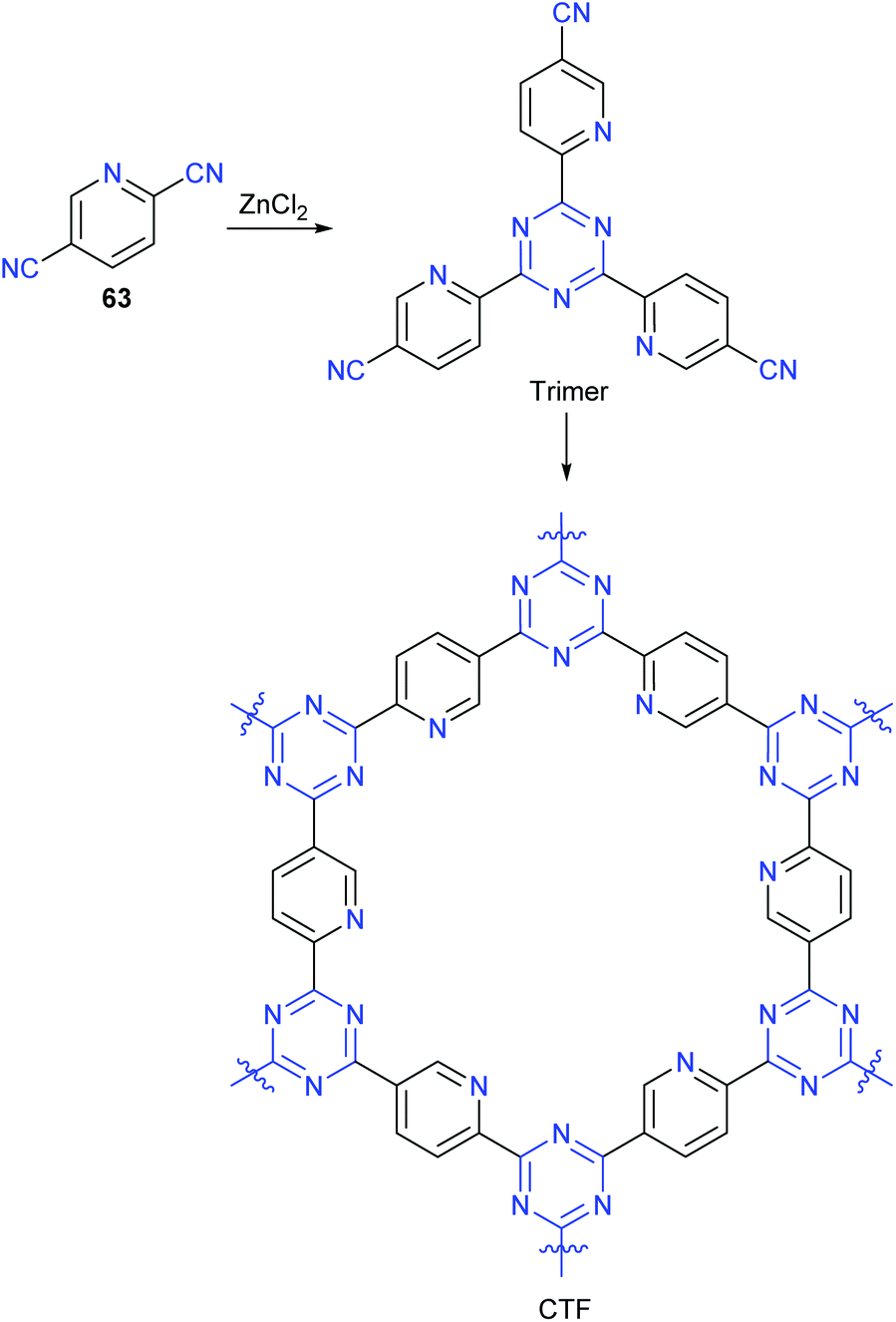 | ||
| Scheme 39 Synthesis of CTFs reported by Li et al. in 2019. | ||
Graphitic carbon nitride (g-C3N4), which is based on the tri-s-triazine structure, is also a nitrogen-rich material. In 2007, it was shown that mesoporous g-C3N4 can chemically activate carbon dioxide,196 and several applications of g-C3N4 in catalysing the reaction of epoxides with carbon dioxide were reported in 2011–2016.197–204 In 2017, Zhao et al. combined g-C3N4 with boric acid groups, by synthesising boron doped carbon nitride (BCN) from dicyandiamide and boric acid.205 The basic secondary amine sites in BCN could activate carbon dioxide and the acidic sites could activate the epoxide (similar to the dual activation mechanism in Scheme 7). This mechanism was supported by DFT calculations. A small amount of boron could significantly improve the catalytic activity of g-C3N4, since 74% conversion of styrene oxide was obtained for B0.03CN compared to 20% using only g-C3N4 (at 130 °C and 30 bar of carbon dioxide for 24 h, using 30 mg of catalyst with 1 mL of styrene oxide). However, using an excess of boron leads to degradation of the layered structure, so the optimal BCN material was B0.03CN. The introduction of SBA-15, which can increase surface area, further enhanced the catalytic activity, as styrene oxide conversions increased to 88% using B0.03CN/SBA-15. Due to the high surface area of BCN/SBA-15, the dispersion of boron in BCN/SBA-15 was sparser than that in the non-silica-supported BCN. Therefore, more boron atoms were required to introduce a similar surface density and the B0.1CN/SBA-15 catalyst showed optimal performance; a styrene oxide conversion of 95%. The catalytic stability of B0.1CN/SBA-15 is not ideal, as when this catalyst was recycled four times, the SO conversion declined gradually.
In 2017, Xiao et al. studied the catalytic activity of nitrogen doped carbon nitride materials; u-C3N4, m-C3N4, and s-C3N4, prepared from urea, melamine and carbon tetrachloride/ethylenediamine, respectively.206 The order of catalytic activity of these catalysts in the reaction of epichlorohydrin with carbon dioxide was u-C3N4 > m-C3N4 > s-C3N4. It was speculated that the poor performance of s-C3N4 was due to the lack of primary and secondary amine groups, as suggested by FT-IR spectra of the materials. However, it would be better to measure the nitrogen content to confirm if the catalytic activity was affected by nitrogen loading. Slight decreases in activity were exhibited, with the same level of selectivity, after the u-C3N4 catalyst was used in five cycles. Although u-C3N4 was effective for the reaction between epichlorohydrin and carbon dioxide without any additive, u-C3N4 alone gave low catalytic activity (5% conversion) in converting propylene oxide into propylene carbonate. The addition of a halide salt such as tetrabutylammonium bromide was necessary to achieve 98% conversion in this case. When propylene oxide with mixed 1:1 with epichlorohydrin, the conversion of propylene oxide was 21%. Therefore, it is speculated that epichlorohydrin reacts with u-C3N4 to form a quaternary ammonium salt, releasing chloride anions, which are more nucleophilic than nitrogen-based organic bases for the ring-opening of epoxides. A dual activation mechanism was also predicted to occur (Scheme 7). More evidence would be valuable to further support this mechanism. It would also be worthwhile proving the existence of chloride anions by suitable characterisation methods. A reaction with a mixture of propylene oxide and epibromohydrin to observe if the propylene oxide conversion is further improved would be informative, since bromide anions are usually more active than chloride anions in ring-opening epoxides. A control reaction with a mixture of propylene oxide and a haloalkane would also be useful, as haloalkanes can easily react with u-C3N4 to form halide anions.
In 2018, Qu et al. studied the catalytic activity of graphene oxide combined with DMF, in which surface hydroxyl groups of graphene oxide activate the epoxide and DMF activates carbon dioxide in a dual activation mechanism (Scheme 7).207 Graphene oxide could be separated from the reaction mixture by centrifugation and the catalytic activity remained at similar levels after recycling four times. This heterogeneous catalytic system had a number of advantageous features. Its performance was excellent since the reaction of epoxides and carbon dioxide was carried out under mild reaction conditions, of 1 bar carbon dioxide, low catalytic loading (2.5 mg graphene oxide for 5 mmol of epoxide) and with short reaction times (6–12 h). The conversion of styrene oxide was not affected by the pressure of carbon dioxide in the range of 1 to 20 bar. For most other catalysts, catalytic activity and thus conversions increase with carbon dioxide pressure. The conversion of styrene oxide was linearly linked to the number of surface hydroxyl groups in the graphene oxide catalyst.
Nitrogen-doped active carbons synthesised from biomass were used to catalyse the reaction of epoxides with carbon dioxide by Mikkola et al. in 2019.208 Lewis basic sites within the carbon material contained pyridinic, pyridonic and quaternary nitrogens, which could activate carbon dioxide and thus promote the transformation of carbon dioxide into cyclic carbonates. Hydroxyl groups in the carbon material also activated the epoxide, and thus formed cyclic carbonates via a dual activation mechanism (Scheme 7). Nitrogen-free multi-walled carbon nanotubes (MWCNT) or commercially available activated carbon, which lack basic sites, were ineffective as catalysts for the synthesis of cyclic carbonates. In addition to the basicity, the porosity of the catalyst was also an important factor for its activity, since the catalytic activities of the activated carbons with the highest surface areas, were greater than an activated carbon which contained more basic nitrogen sites.
Solid nitrogen-based organocatalysts with organic nucleophiles/bases are often supported by porous materials, such as porous polymers or silica. These solids have emerged as favourable heterogeneous catalysts for a number of reactions, due to their convenient separation from the reaction mixture and ability to be recycled. In 2018, three tetramethylguanidine functionalised porous polyphenylenes 64–66 (Fig. 23) were explored for the synthesis of cyclic carbonates from epoxides and carbon dioxide by Iglesias et al.209 Catalyst 66, with a methylene group between the tetramethylguanidine and the polymeric skeleton, showed the best catalytic activity. For the reaction of epichlorohydrin with carbon dioxide catalysed by 66, 100% epichlorohydrin conversion was achieved, after 4 h at 7 bar carbon dioxide and 100 °C using 0.1 mol% of catalyst, with no by-product formation. The reaction was hypothesised to occur by carbon dioxide activation (Scheme 5). Control experiments showed that the tetramethylguanidine groups were essential to the catalytic activity. Catalyst recycling experiments showed that the activity of catalyst 66 decreased gradually during each reaction, but it could be reactivated by treatment with a dilute solution of sodium hydroxide.
 | ||
| Fig. 23 Tetramethylguanidine based catalysts 64–66. | ||
In 2018, Ogura et al.210 investigated the application of methylated nitrogen-substituted mesoporous silica catalysts in the reaction of propylene oxide with carbon dioxide. The activities of two homogeneous species, tetrabutylammonium bromide and trimethylamine, were also investigated for comparison with the heterogeneous catalysts. Under reaction conditions of 30 bar carbon dioxide and 100 °C, N-methylated SBA-15 exhibited the highest turnover frequency (6.4 h−1) and selectivity (99%) of the heterogeneous catalysts, but had a lower turnover frequency than tetrabutylammonium bromide (7.4 h−1). This result, combined with X-ray diffraction analysis of the N-methylated SBA-15, implied that neighbouring methylated nitrogen atoms were formed in the N-methylated SBA-15 and worked as pairs in the catalytic reaction mechanism. Unmethylated, nitrogen doped SBA15 was ineffective as a catalyst for cyclic carbonate formation. The reported changes in the reaction turnover frequency when changing the carbon dioxide pressure or amount of propylene oxide used, implied that carbon dioxide and propylene oxide compete to be adsorbed on the methylated nitrogen atoms of N-methylated SBA-15. Hence, the authors suggest that the reaction follows Langmuir–Hinshelwood kinetics and involves two basic sites. A bimolecular reaction mechanism was thus proposed (Scheme 40), which starts with the activation of the epoxide (similar to Scheme 1). The role of the hydroxyl groups was to stabilise the intermediate by forming hydrogen bonds. Unfortunately, the N-methylated SBA-15 was not stable enough to recycling, as product yields dropped significantly, to 63% and 43% of the initially obtained yield in the second and third use.
 | ||
| Scheme 40 Proposed reaction mechanism for the formation of cyclic carbonates using N-methylated SBA-15 as reported by Ogura et al. | ||
In 2019, picolinic acid and 4,4′,4′′4′′′-porphyrin-5,10,15,20-tetrabenzoic acid were supported on periodic mesoporous organosilica by Van Der Voort et al. (Scheme 41).211 Both supported catalysts were capable of ring-opening epichlorohydrin via epoxide activation (similar to Scheme 1), using 10 mg of catalyst in dichloromethane (2 mL) with 0.5 mmol of epichlorohydrin, at six bar carbon dioxide and 120 °C for 2 h. Conversions of 75–80% were obtained. The addition of 1 mg of 4-dimethylaminopyridine further improved the catalytic activity, giving conversions of 89–91%. Both catalysts showed good stability and could be reused five times.
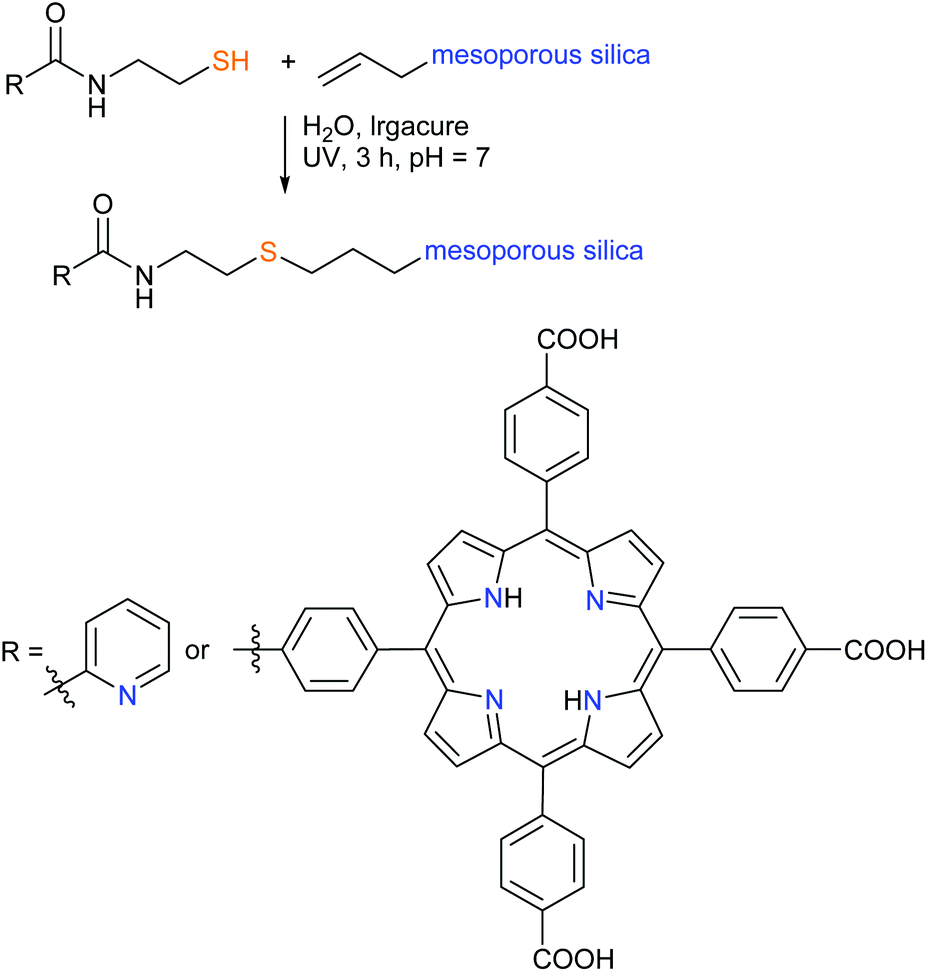 | ||
| Scheme 41 Synthesis of silica-supported catalysts as reported by Van Der Voort et al. | ||
In 2017, five inclusion complexes 67a–e between DBU-phenolates and β-cyclodextrin were prepared (Scheme 42) and used to catalyse the reaction between epoxides and carbon dioxide.212 Phenolates 67a–e could efficiently ring-open epoxides using 1.5 mol% of catalyst with respect to epoxide, at 130 °C and 30 bar carbon dioxide for 10 h. The β-cyclodextrin complexed phenolates had higher catalytic activity and were easier to separate from the reaction mixture and recycle than non-complexed DBU-phenolates. Kinetic studies showed that the activation energy (Ea) for cyclic carbonate synthesis was lower for the process catalysed by 67a–e, compared to that catalysed by the corresponding non-complexed DBU-phenolates. The authors suggested that cyclic carbonates were formed by activating both the epoxide and carbon dioxide (similar to the mechanism shown in Scheme 7).
 | ||
| Scheme 42 Synthesis of inclusion complexes 67a–e as reported by Hou et al. | ||
In 2019, a bis(trifluoromethylsulfonyl)imide imidazolium ionic liquid was immobilised on periodic mesoporous organosilica to form nanomaterials 68 (Scheme 43).213 When applied to the reaction of propylene oxide with carbon dioxide, cyclic carbonate yields of 99% were reported after a reaction time of only 1 h, using the catalyst prepared from a 1:1 ratio of periodic mesoporous organosilica and ionic liquid. In contrast, only 81% yield was obtained after 3 h using the ionic liquid component alone. Relatively mild reaction conditions were required, with only 0.2 g of 68 required with 0.1 mol of propylene oxide, at 6 bar of carbon dioxide and 90 °C. The improvement of catalytic activity on immobilisation of the ionic liquid might be attributed to the introduction of silanol groups within the organosilica support. X-ray diffraction analysis of 68 indicated that the ordered mesoporous structure of the silica was maintained during the synthesis of the nanomaterial. Catalyst 68 was stable and reusable, as negligible loss in catalytic activity was observed after six runs, and the morphology and highly ordered nanostructure was not destroyed. The catalyst was predicted to activate carbon dioxide and epoxide separately (similar to Scheme 7) in order to form cyclic carbonates.
 | ||
| Scheme 43 Synthesis of a nanomaterial as reported by Hu et al. and its use in cyclic carbonate formation. | ||
4. Final thoughts/perspective
Whenever new catalytic processes are reported, reaction conditions are usually optimised to either achieve the fastest rates, and thus the highest turnover frequencies, reported for the reaction; or to obtain high conversions under ambient (and arguable greener) reaction conditions. This, twinned with the ability to change many reaction conditions, makes it extremely difficult to truly compare, or ‘benchmark’, different catalytic systems with one another, as rarely are two systems reported under identical conditions, in the search for the highest turnover frequencies or best yields. Whilst research has been performed by Kleij et al. into comparing different homogeneous aluminium based catalysts for cyclic carbonate synthesis under identical reaction conditions,214 the same type of study is yet to be performed for organocatalysts.A study by Gruttadauria et al. in 2020 suggested that care must be used when calculating turnover frequencies, especially when using Lewis acids and Lewis bases together to form cyclic carbonates.215 Gruttadauria et al. highlighted that when studies used Lewis acids in conjunction with halide salts as Lewis bases, turnover frequencies (and/or turnover numbers) reported were often related only to the amount of Lewis acid used, even when the amount of halide salt (or Lewis base) used was far greater than the Lewis acid. By performing a thorough mathematical investigation into the principles of the turnover frequency equation, when considering the conversion of epoxides and carbon dioxide into cyclic carbonates in the presence of a Lewis acid and a Lewis base, Gruttadauria argued that many turnover frequencies in the literature were reported higher than their true values. By taking the Lewis base into account, all turnover frequencies analysed in the paper were determined to be lower than previously reported; completely changing how catalysts compared to one another in terms of reactivity. Gruttadauria concluded that performing normalisation of turnover frequencies calculated via this method, led to a more consistent comparison of turnover frequencies between different catalytic systems. Gruttadauria therefore emphasised three important points:
(1) Approximate TOF values should be calculated “by considering the reaction yields in the presence or absence of the Lewis acid catalyst”,
(2) Control, or blank experiments, using the Lewis base and Lewis acid alone under the optimum or operational reaction conditions must be performed, in order to ascertain the contribution of both components and the Lewis base only catalysed pathway to the overall process; and
(3) TOFs should not be used in “an absolute way but in a comparative manner”.
Whilst turnover frequencies and yields are important when forming cyclic carbonates, these values should not be the primary factor to consider when forming cyclic carbonates, as the reaction conditions required to report the highest turnover frequencies may lead to poor sustainability. For example, catalytic systems often required high catalytic loading and harsh reaction conditions. This can have the knock-on effect of increasing the overall carbon footprint of the reaction and may create an overall carbon dioxide emitting process, rather than one which utilises carbon dioxide. If toxic, scare and expensive starting materials and solvents are required to make the catalysts, scaling up the synthesis could create problems and hinder the applicability of the catalysts on an industrial scale. The bigger picture must therefore be considered, as even though the catalyst developed may lead to promising conversions and yields, the green credentials of the process may be extremely poor. Only a handful of papers in this area have seriously considered the sustainability of the process, with only a few papers reporting green metrics such as the E-factor and life cycle analysis. Much more thorough utilisation of green metrics and green toolkits should be employed in future studies, as well as considering all reaction aspects and not just the cyclic carbonate formation step. Whilst it is important to get high yields and to create a sustainable system; it is a delicate balancing act to satisfy both of these demands at the same time.
5. Conclusions
It is apparent from this and numerous other reviews and book chapters that a vast number of catalysts have now been developed for the laboratory scale synthesis of cyclic carbonates from epoxides and carbon dioxide. These catalysts include metal containing systems (with just about every metal in the periodic table) and organocatalytic systems. They also include both homogeneous and heterogeneous systems. It is therefore worth asking if further work on this reaction is worthwhile? We would argue that the answer is yes, but that future work must focus on addressing unsolved challenges of which there appear to be three.Firstly, virtually all catalysts are screened against petrochemically derived terminal epoxides. Very few are tested against internal and/or bio-derived epoxides. These are much more challenging substrates due both to steric factors and the additional functionalities they possess. The development of catalysts capable of converting particularly bio-based epoxides (such as those derived from terpenes and fatty acids) into cyclic carbonates under green reaction conditions would be a significant advance in sustainable chemistry as it would facilitate the switch from petrochemicals to biomass derived chemicals and would facilitate the establishment of a carbon dioxide refinery (Fig. 4) which uses waste biomass as reactants for the conversion of carbon dioxide into valuable chemicals. It should also be noted that some progress is being made in developing routes to traditionally petrochemically derived epoxides from biomass. This is exemplified by the Croda route to ethylene oxide by dehydration and epoxidation of bioethanol.
Secondly, the carbon dioxide source and purity needs to be considered. Virtually all work is carried out using high pressures of high purity carbon dioxide. This is not realistic from a sustainability perspective as considerable energy is required to purify and pressurise carbon dioxide. There have been only a handful of papers published which used real (or even simulated) waste carbon dioxide (such as power plant flue gas) and even fewer which did so under mild reaction conditions. The direct utilisation of atmospheric carbon dioxide at ca. 400 ppm concentration and one bar pressure is a challenge no existing catalyst can address.
Finally, there has been very little progress in moving newly developed catalysts out of the laboratory and towards industrial utilisation. Carbon dioxide utilisation is only worthwhile if it can be carried out on an industrial scale. So in future, there needs to be closer collaboration between research chemists and chemical/process engineers, to ensure that key issues relating to the industrial applicability of catalysts are addressed early in the catalyst development process and that key data needed by process engineers to design a process is acquired in a form that is useful to them.
Provided these issues are addressed, then the synthesis of cyclic carbonates from (waste) carbon dioxide and sustainably sourced epoxides is a reaction with a bright future and one which could be central to the development of a sustainable chemicals industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Program for Public Technology of Zhejiang Province (No. LGF18B060003) and the State Scholarship Fund from the China Scholarship Council (No. 201908330607) for financial support.Notes and references
- R. Lindsay, Climate Change: Atmospheric Carbon Dioxide, https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide, (accessed 16/06/2020).
- Global Monitoring Laboratory: Trends in Atmospheric Carbon Dioxide, https://www.esrl.noaa.gov/gmd/ccgg/trends/, (accessed 16/06/2020).
- Climate Change: How Do We Know?, https://climate.nasa.gov/evidence/, (accessed 16/06/2020).
- C. Zahasky and S. Krevor, Energy Environ. Sci., 2020, 13, 1561–1567 RSC.
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- M. Abotalib, F. Zhao and A. Clarens, ACS Sustainable Chem. Eng., 2016, 4, 4743–4751 CrossRef CAS.
- K. Armstrong and P. Styring, Front. Energy Res., 2015, 3, 8 Search PubMed.
- I. Sreedhar, Y. Varun, S. A. Singh, A. Venugopal and B. M. Reddy, Catal. Sci. Technol., 2019, 9, 4478–4504 RSC.
- A. Alvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef CAS PubMed.
- CRI – Carbon Recycling International, https://www.carbonrecycling.is/, (accessed 23/06/2020).
- X. Zhao, L. J. Du, B. You and Y. J. Sun, Catal. Sci. Technol., 2020, 10, 2711–2720 RSC.
- X. Li, J. G. Yu, M. Jaroniec and X. B. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Wongsakulphasatch and D. V. N. Vo, Catal. Sci. Technol., 2020, 10, 35–45 RSC.
- G. B. Chen, G. I. N. Waterhouse, R. Shi, J. Q. Zhao, Z. H. Li, L. Z. Wu, C. H. Tung and T. R. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- Carbon Dioxide Utilisation Volume 1: From Fundamentals to Production Processes, ed. M. North and P. Styring, Walter de Gruyter GmbH, Berlin, 2019 Search PubMed.
- Carbon Dioxide Utilisation Volume 2: Transformations, ed. M. North and P. Styring, Walter de Gruyter GmbH, Berlin, 2019 Search PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, J. CO2 Util., 2013, 3–4, 65–73 CrossRef CAS.
- X. F. Wu and F. Zheng, Top. Curr. Chem., 2017, 375, 4 CrossRef PubMed.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- J. Huang, J. C. Worch, A. P. Dove and O. Coulembier, ChemSusChem, 2020, 13, 469–487 CrossRef CAS PubMed.
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS PubMed.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- M. North, ChemSusChem, 2019, 12, 1763–1765 CrossRef CAS PubMed.
- S. Subramanian, Y. Song, D. Kim and C. T. Yavuz, ACS Energy Lett., 2020, 5, 1689–1700 CrossRef CAS.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- F. Della Monica and A. W. Kleij, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- C. Beattie, M. North, P. Villuendas and C. Young, J. Org. Chem., 2013, 78, 419–426 CrossRef CAS PubMed.
- J. Martinez, J. A. Castro-Osma, A. Earlam, C. Alonso-Moreno, A. Otero, A. Lara-Sanchez, M. North and A. Rodriguez-Dieguez, Chem. – Eur. J., 2015, 21, 9850–9862 CrossRef CAS PubMed.
- J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Muller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS PubMed.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS PubMed.
- A. H. Liu, Y. L. Dang, H. Zhou, J. J. Zhang and X. B. Lu, ChemCatChem, 2018, 10, 2686–2692 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- M. North, P. Villuendas and C. Young, Chem. – Eur. J., 2009, 15, 11454–11457 CrossRef CAS PubMed.
- I. D. V. Ingram, M. North and X. Wu, Chapter 15 in Chemistry Beyond Chlorine, Halide-Free Synthesis of Cyclic and Polycarbonates, ed. P. Tundo, L.-N. He, E. Lokteva and C. Mota, Springer, Switzerland, 2016 Search PubMed.
- F. Xu, W. G. Cheng, X. Q. Yao, J. Sun, W. Sun and S. J. Zhang, Catal. Lett., 2017, 147, 1654–1664 CrossRef CAS.
- S. Wu, Y. Y. Zhang, B. S. Wang, E. H. M. Elageed, L. Z. Ji, H. H. Wu and G. H. Gao, Eur. J. Org. Chem., 2017, 753–759 CrossRef.
- C. Chen, Y. Ma, D. N. Zheng, L. Wang, J. F. Li, J. L. Zhang, H. Y. He and S. J. Zhang, J. CO2 Util., 2017, 18, 156–163 CrossRef CAS.
- T. F. Wang, D. N. Zheng, Y. Ma, J. Y. Guo, Z. P. He, B. Ma, L. H. Liu, T. G. Ren, L. Wang and J. L. Zhang, J. CO2 Util., 2017, 22, 44–52 CrossRef CAS.
- S. Yue, Q. Song, S. L. Zang, G. C. Deng and J. Li, Aust. J. Chem., 2018, 71, 407–415 CrossRef CAS.
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS.
- J. A. Castro-Osma, J. Martinez, F. de la Cruz-Martinez, M. P. Caballero, J. Fernandez-Baeza, J. Rodriguez-Lopez, A. Otero, A. Lara-Sanchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC.
- Y. H. Li, B. Dominelli, R. M. Reich, B. P. Liu and F. E. Kühn, Catal. Commun., 2019, 124, 118–122 CrossRef CAS.
- J. Chen, H. Gao, T. Ding, L. Z. Ji, J. Z. H. Zhang, G. H. Gao and F. Xia, Front. Chem., 2019, 7, 615 CrossRef CAS PubMed.
- Q. Y. Liu, L. Shi and N. Liu, J. Chem. Res., 2019, 43, 248–256 CrossRef CAS.
- T. F. Wang, X. R. Zhu, L. M. Mao, Y. Liu, T. G. Ren, L. Wang and J. L. Zhang, J. Mol. Liq., 2019, 296, 111936 CrossRef CAS.
- C. Qiao, A. Villar-Yanez, J. Sprachmann, B. Limburg, C. Bo and A. W. Kleij, Angew. Chem., 2020, 59, 18446–18451 CrossRef CAS PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- X. S. Cai, J. L. Zheng, J. Wärnå, T. Salmi, B. Taouk and S. Leveneur, Chem. Eng. J., 2017, 313, 1168–1183 CrossRef CAS.
- K. M. Doll, S. C. Cermak, J. A. Kenar, E. L. Walter and T. A. Isbell, Ind. Crops Prod., 2017, 104, 269–277 CrossRef CAS.
- Y. Kumatabara, M. Okada and S. Shirakawa, ACS Sustainable Chem. Eng., 2017, 5, 7295–7301 CrossRef CAS.
- J. Peng, S. Wang, H. J. Yang, B. R. Ban, Z. D. Wei, L. H. Wang and B. Lei, Fuel, 2018, 224, 481–488 CrossRef CAS.
- M. Hong, Y. Kim, H. Kim, H. J. Cho and M. H. Baik, J. Org. Chem., 2018, 83, 9370–9380 CrossRef CAS PubMed.
- F. S. Liu, Y. Q. Gu, P. H. Zhao, J. Gao and M. S. Liu, ACS Sustainable Chem. Eng., 2019, 7, 5940–5945 CrossRef CAS.
- H. Büttner, J. Steinbauer, C. Wulf, M. Dindaroglu, H. G. Schmalz and T. Werner, ChemSusChem, 2017, 10, 1076–1079 CrossRef PubMed.
- J. Steinbauer, C. Kubis, R. Ludwig and T. Werner, ACS Sustainable Chem. Eng., 2018, 6, 10778–10788 CrossRef CAS.
- Y. Toda, Y. Komiyama, H. Esaki, K. Fukushima and H. Suga, J. Org. Chem., 2019, 84, 15578–15589 CrossRef CAS PubMed.
- H. Sabet-Sarvestani, M. Izadyar, H. Eshghi and N. Norozi-Shad, Phys. Chem. Chem. Phys., 2020, 22, 223–237 RSC.
- Y. Ma, C. Chen, T. F. Wang, J. S. Zhang, J. J. Wu, X. D. Liu, T. G. Ren, L. Wang and J. L. Zhang, Appl. Catal., A, 2017, 547, 265–273 CrossRef CAS.
- T. F. Wang, D. M. Zheng, J. S. Zhang, B. W. Fan, Y. Ma, T. G. Ren, L. Wang and J. L. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 2574–2582 CrossRef CAS.
- D. N. Zheng, T. F. Wang, X. R. Zhu, C. Chen, T. G. Ren, L. Wang and J. L. Zhang, Mol. Syst. Des. Eng., 2018, 3, 348–356 RSC.
- D. N. Zheng, L. Wang, J. M. Jiang, F. Liu and J. L. Zhang, J. Mol. Liq., 2019, 295, 111652 CrossRef.
- X. R. Zhu, F. Liu, J. M. Jiang, L. Wang and J. L. Zhang, J. Mater. Sci., 2020, 55, 2419–2428 CrossRef CAS.
- C. Chen, Y. Ma, D. N. Zheng, J. S. Zhang, T. G. Ren, L. Wang and J. L. Zhang, Tetrahedron, 2018, 74, 1776–1784 CrossRef CAS.
- J. S. Zhang, X. R. Zhu, Y. M. Zhang, F. Liu, T. G. Ren, L. Wang and J. L. Zhang, J. CO2 Util., 2019, 34, 422–429 CrossRef CAS.
- T. F. Wang, Y. Ma, J. M. Jiang, X. R. Zhu, B. W. Fan, G. Y. Yu, N. N. Li, S. S. Wang, T. G. Ren, L. Wang and J. L. Zhang, J. Mol. Liq., 2019, 293, 111479 CrossRef CAS.
- J. S. Zhang, X. R. Zhu, B. W. Fan, J. Guo, P. Ning, T. G. Ren, L. Wang and J. L. Zhang, Mol. Catal., 2019, 466, 37–45 CrossRef CAS.
- Z. G. Zhang, F. J. Fan, H. B. Xing, Q. W. Yang, Z. B. Bao and Q. L. Ren, ACS Sustainable Chem. Eng., 2017, 5, 2841–2846 CrossRef CAS.
- A. Rostami, M. Mahmoodabadi, A. H. Ebrahimi, H. Khosravi and A. Al-Harrasi, ChemSusChem, 2018, 11, 4262–4268 CrossRef CAS PubMed.
- W. Li, W. G. Cheng, X. Yang, Q. Su, L. H. Dong, P. Zhang, Y. N. Yi, B. Li and S. J. Zhang, Chin. J. Chem., 2018, 36, 293–298 CrossRef CAS.
- N. Aoyagi, Y. Furusho and T. Endo, Tetrahedron, 2019, 75, 130781 CrossRef.
- N. Aoyagi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2606–2615 CrossRef CAS.
- W. L. Dai, W. Y. Yang, Y. Zhang, D. Wang, X. B. Luo and X. M. Tu, J. CO2 Util., 2017, 17, 256–262 CrossRef CAS.
- N. Liu, Y. F. Xie, C. Wang, S. J. Li, D. H. Wei, M. Li and B. Dai, ACS Catal., 2018, 8, 9945–9957 CrossRef CAS.
- H. S. Seo and H. J. Kim, Bull. Korean Chem. Soc., 2019, 40, 169–172 CrossRef CAS.
- N. Aoyagi, Y. Furusho and T. Endo, Synthesis, 2020, 52, 150–158 CrossRef CAS.
- Y. D. Li, D. X. Cui, J. C. Zhu, P. Huang, Z. Tian, Y. Y. Jia and P. A. Wang, Green Chem., 2019, 21, 5231–5237 RSC.
- J. Perez-Perez, U. Hernandez-Balderas, D. Martinez-Otero and V. Jancik, New J. Chem., 2019, 43, 18525–18533 RSC.
- R. K. Tak, P. Patel, S. Subramanian, R. I. Kureshy and N. U. H. Khan, ACS Sustainable Chem. Eng., 2018, 6, 11200–11205 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- K. Wu, T. Su, D. M. Hao, W. P. Liao, Y. C. Zhao, W. Z. Ren, C. L. Deng and H. Y. Lü, Chem. Commun., 2018, 54, 9579–9582 RSC.
- J. Sun, S. J. Zhang, W. G. Cheng and J. Y. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- J. Sun, J. Y. Ren, S. J. Zhang and W. G. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- L. P. Guo, C. M. Wang, X. Y. Luo, G. K. Cui and H. R. Li, Chem. Commun., 2010, 46, 5960–5962 RSC.
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12, 3856–3863 CrossRef CAS PubMed.
- X. Y. Wang, L. Wang, Y. Y. Zhao, K. Kodama and T. Hirose, Tetrahedron, 2017, 73, 1190–1195 CrossRef CAS.
- P. Yingcharoen, C. Kongtes, S. Arayachukiat, K. Suvarnapunya, S. V. C. Vummaleti, S. Wannakao, L. Cavallo, A. Poater and V. D'Elia, Adv. Synth. Catal., 2019, 361, 366–373 CrossRef CAS.
- T. Ema, M. Yokoyama, S. Watanabe, S. Sasaki, H. Ota and K. Takaishi, Org. Lett., 2017, 19, 4070–4073 CrossRef CAS PubMed.
- C. Maeda, S. Sasaki, K. Takaishi and T. Ema, Catal. Sci. Technol., 2018, 8, 4193–4198 RSC.
- S. Sopena, E. Martin, E. C. Escudero-Adan and A. W. Kleij, ACS Catal., 2017, 7, 3532–3539 CrossRef CAS.
- X. Hu, P. Wang, W. Liu and Y. Wang, ChemistrySelect, 2018, 3, 6531–6535 CrossRef CAS.
- V. B. Saptal and B. M. Bhanage, ChemSusChem, 2017, 10, 1145–1151 CrossRef CAS PubMed.
- T. Hirose, S. J. Qu, K. Kodama and X. Y. Wang, J. CO2 Util., 2018, 24, 261–265 CrossRef CAS.
- J. W. Gao, L. Y. Li, C. Y. Cui, M. A. Ziaee, Y. Q. Gong, R. J. Sa and H. Zhong, RSC Adv., 2019, 9, 13122–13127 RSC.
- R. Ma, L. N. He, X. F. Liu, X. Liu and M. Y. Wang, J. CO2 Util., 2017, 19, 28–32 CrossRef CAS.
- L. Z. Ji, Z. J. Luo, Y. Y. Zhang, R. Wang, Y. Y. Ji, F. Xia and G. H. Gao, Mol. Catal., 2018, 446, 124–130 CrossRef CAS.
- R. B. Mujmule, M. P. R. Rao, P. V. Rathod, V. G. Deonikar, A. A. Chaugule and H. Kim, J. CO2 Util., 2019, 33, 284–291 CrossRef CAS.
- Y. Q. Qi, W. G. Cheng, F. Xu, S. L. Chen and S. J. Zhang, Synth. Commun., 2018, 48, 876–886 CrossRef CAS.
- L. P. da Silva, J. Phys. Chem. C, 2017, 121, 16300–16307 CrossRef.
- L. P. da Silva, Mol. Catal., 2019, 474, 110425 CrossRef.
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- Z. Chen, J. L. Yang, X. Y. Lu, L. F. Hu, X. H. Cao, G. P. Wu and X. H. Zhang, Polym. Chem., 2019, 10, 3621–3628 RSC.
- L.-Y. Zhao, J.-Y. Chen, W.-C. Li and A.-H. Lu, J. CO2 Util., 2019, 29, 172–178 CrossRef CAS.
- A. W. Kleij, Curr. Opin. Green Sustainable Chem., 2020, 24, 72–81 CrossRef.
- B. Zou and C. W. Hu, Curr. Opin. Green Sustainable Chem., 2017, 3, 11–16 CrossRef.
- H. G. Kim, C. S. Lim, D. W. Kim, D. H. Cho, D. K. Lee and J. S. Chung, Mol. Catal., 2017, 438, 121–129 CrossRef CAS.
- S. Ryu, Bull. Korean Chem. Soc., 2019, 40, 1033–1038 CrossRef CAS.
- P. Goodrich, H. Q. N. Gunaratne, J. Jacquemin, L. L. Jin, Y. T. Lei and K. R. Seddon, ACS Sustainable Chem. Eng., 2017, 5, 5635–5641 CrossRef CAS.
- S. Yue, P. P. Wang, X. J. Hao and S. L. Zang, J. CO2 Util., 2017, 21, 238–246 CrossRef CAS.
- P. Kumar, M. Varyani, P. K. Khatri, S. Paul and S. L. Jain, J. Ind. Eng. Chem., 2017, 49, 152–157 CrossRef CAS.
- S. Yue, X. J. Hao, P. P. Wang and J. Li, Mol. Catal., 2017, 433, 420–429 CrossRef CAS.
- G. F. Yuan, Y. F. Zhao, Y. Y. Wu, R. P. Li, Y. Chen, D. M. Xu and Z. M. Liu, Sci. China: Chem., 2017, 60, 958–963 CrossRef CAS.
- A. A. Pawar and H. Kim, J. CO2 Util., 2019, 33, 500–512 CrossRef CAS.
- L. Guglielmero, A. Mezzetta, C. S. Pomelli, C. Chiappe and L. Guazzelli, J. CO2 Util., 2019, 34, 437–445 CrossRef CAS.
- H. G. Kim, H. J. Son and C. S. Lim, Korean J. Chem. Eng., 2019, 36, 197–202 CrossRef CAS.
- R. Azzouz, V. C. Moreno, C. Herasme-Grullon, V. Levacher, L. Estel, A. Ledoux, S. Derrouiche, F. Marsais and L. Bischoff, Synlett, 2020, 31, 183–188 CrossRef CAS.
- A. E. Chichibabin and O. A. Zeide, Zh. Russ. Fiz.-Khim. O-va. (J. Russ. Phys.-Chem. Soc.), 1914, 46, 1216–1236 CAS.
- X. Wu, C. T. Chen, Z. Y. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- Z. X. Yue, M. Pudukudy, S. Y. Chen, Y. Liu, W. B. Zhao, J. Y. Wang, S. Y. Shan and Q. M. Jia, Appl. Catal., A, 2020, 601, 117646 CrossRef CAS.
- Y. A. Rulev, Z. T. Gugkaeva, A. V. Lokutova, V. I. Maleev, A. S. Peregudov, X. Wu, M. North and Y. N. Belokon, ChemSusChem, 2017, 10, 1152–1159 CrossRef CAS PubMed.
- J. L. Vidal, V. P. Andrea, S. L. MacQuarrie and F. M. Kerton, ChemCatChem, 2019, 11, 4089–4095 CrossRef CAS.
- A. A. Marciniak, K. J. Lamb, L. P. Ozorio, C. J. A. Mota and M. North, Curr. Opin. Green Sustainable Chem., 2020, 26, 100365 CrossRef.
- S. B. Zhang, X. Liu, M. Li, Y. Wei, G. F. Zhang, J. Y. Han, X. L. Zhu, Q. F. Ge and H. Wang, Catal. Today, 2019, 324, 59–65 CrossRef CAS.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. H. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739–1758 CAS.
- G. L. M. Leonard, A. Belet, B. Grignard, C. Calberg, B. Gilbert, C. Jerome and B. Heinrichs, Catal. Today, 2019, 334, 140–155 CrossRef CAS.
- R. Khatun, P. Bhanja, R. A. Molla, S. Ghosh, A. Bhaumik and S. M. Islam, Mol. Catal., 2017, 434, 25–31 CrossRef CAS.
- L. J. Shi, S. B. Xu, Q. R. Zhang, T. T. Liu, B. H. Wei, Y. F. Zhao, L. X. Meng and J. Li, Ind. Eng. Chem. Res., 2018, 57, 15319–15328 CAS.
- G. Gonzalez, A. Sagarzazu, A. Cordova, M. E. Gomes, J. Salas, L. Contreras, K. Noris-Suarez and L. Lascano, Microporous Mesoporous Mater., 2018, 256, 251–265 CrossRef CAS.
- M. Zhang, B. X. Chu, G. Y. Li, J. Z. Xiao, H. W. Zhang, Y. J. Peng, B. Li, P. Xie, M. G. Fan and L. H. Dong, Microporous Mesoporous Mater., 2019, 274, 363–372 CrossRef CAS.
- S. Bhattacharyya, G. Lelong and M. L. Saboungi, J. Exp. Nanosci., 2006, 1, 375–395 CrossRef CAS.
- A. Comes, X. Collard, L. Fusaro, L. Atzori, M. G. Cutrufello and C. Aprile, RSC Adv., 2018, 8, 25342–25350 RSC.
- L. Muniandy, F. Adam, N. R. A. Rahman and E. P. Ng, Inorg. Chem. Commun., 2019, 104, 1–7 CrossRef CAS.
- R. J. Ramalingam, J. N. Appaturi, T. Pulingam, N. Ibrahim and H. A. Al-Lohedan, Mater. Chem. Phys., 2019, 233, 79–88 CrossRef CAS.
- M. V. Zakharova, F. Kleitz and F. G. Fontaine, ChemCatChem, 2017, 9, 1886–1890 CrossRef CAS.
- N. L. Ho, J. Perez-Pellitero, F. Porcheron and R. J. M. Pellenq, J. Phys. Chem. C, 2012, 116, 3600–3607 CrossRef CAS.
- M. S. Liu, X. Y. Lu, Y. C. Jiang, J. M. Sun and M. Arai, ChemCatChem, 2018, 10, 1860–1868 CrossRef CAS.
- X. Zhang, D. Su, L. F. Xiao and W. Wu, J. CO2 Util., 2017, 17, 37–42 CrossRef CAS.
- T. Jose, S. Canellas, M. A. Pericas and A. W. Kleij, Green Chem., 2017, 19, 5488–5493 RSC.
- W. L. Dai, J. Mao, Y. Liu, P. Mao, X. B. Luo and J. P. Zou, Catal. Lett., 2019, 149, 699–712 CrossRef CAS.
- H. Büttner, C. Kohrt, C. Wultia, B. Schäffner, K. Groenke, Y. Y. Hu, D. Kruse and T. Werner, ChemSusChem, 2019, 12, 2701–2707 CrossRef PubMed.
- Z. Q. Yi, D. H. Lan, Y. Wang, L. Chen, C. T. Au and S. F. Yin, Sci. China: Chem., 2017, 60, 990–996 CrossRef CAS.
- S. Kumar, M. Y. Wani, J. Koh, J. M. Gil and A. Sobral, J. Environ. Sci., 2018, 69, 77–84 CrossRef PubMed.
- J. Zhu, S. Q. Wang, Y. K. Gu, B. Xue and Y. X. Li, Mater. Chem. Phys., 2018, 208, 68–76 CrossRef CAS.
- M. Taheri, M. Ghiaci and A. Shchukarev, New J. Chem., 2018, 42, 587–597 RSC.
- J. Peng, S. Wang, H. J. Yang, B. R. Ban, Z. D. Wei, L. H. Wang and B. Lei, Catal. Today, 2019, 330, 76–84 CrossRef CAS.
- S. Kumar, J. D. E. Silva, M. Y. Wani, J. M. Gil and A. Sobral, Carbohydr. Polym., 2017, 175, 575–583 CrossRef CAS PubMed.
- D. M. Rodrigues, L. G. Hunter, F. L. Bernard, M. F. Rojas, F. Dalla Vecchia and S. Einloft, Catal. Lett., 2019, 149, 733–743 CrossRef CAS.
- C. Calabrese, L. F. Liotta, E. Carbonell, F. Giacalone, M. Gruttadauria and C. Aprile, ChemSusChem, 2017, 10, 1202–1209 CrossRef CAS.
- O. Martinez-Ferraté, G. Chacón, F. Bernardi, T. Grehl, P. Brüner and J. Dupont, Catal. Sci. Technol., 2018, 8, 3081–3089 RSC.
- A. S. Martinez, C. Hauzenberger, A. R. Sahoo, Z. Csendes, H. Hoffmann and K. Bica, ACS Sustainable Chem. Eng., 2018, 6, 13131–13139 CrossRef.
- Y. D. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. X. Zhu, M. M. Tong, Z. Y. Long and G. J. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16907–16916 CrossRef CAS.
- G. J. Chen, Y. D. Zhang, J. Y. Xu, X. Q. Liu, K. Liu, M. M. Tong and Z. Y. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- C. Calabrese, L. Fusaro, L. F. Liotta, F. Giacalone, A. Comes, V. Campisciano, C. Aprile and M. Gruttadauria, ChemPlusChem, 2019, 84, 1536–1543 CrossRef CAS PubMed.
- C. Calabrese, L. F. Liotta, F. Giacalone, M. Gruttadauria and C. Aprile, ChemCatChem, 2019, 11, 560–567 CrossRef CAS.
- G. L. M. Leonard, S. L. Pirard, A. Belet, B. Grignard, C. Detrembleur, C. Jerome and B. Heinrichs, Chem. Eng. J., 2019, 371, 719–729 CrossRef CAS.
- Q. Sun, Y. Y. Jin, B. Aguila, X. J. Meng, S. Q. Ma and F. S. Xiao, ChemSusChem, 2017, 10, 1160–1165 CrossRef CAS PubMed.
- H. B. Song, Y. J. Wang, M. Xiao, L. Liu, Y. L. Liu, X. F. Liu and H. J. Gai, ACS Sustainable Chem. Eng., 2019, 7, 9489–9497 CrossRef CAS.
- Z. J. Guo, Q. W. Jiang, Y. M. Shi, J. Li, X. N. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- F. D. Bobbink, A. P. Van Muyden, A. Gopakumar, Z. F. Fei and P. J. Dyson, ChemPlusChem, 2017, 82, 144–151 CrossRef CAS PubMed.
- S. Jayakumar, H. Li, Y. P. Zhao, J. Chen and Q. H. Yang, Chem. – Asian J., 2017, 12, 577–585 CrossRef CAS PubMed.
- W. Zhong, F. D. Bobbink, Z. F. Fei and P. J. Dyson, ChemSusChem, 2017, 10, 2728–2735 CrossRef CAS.
- Y. D. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Y. Long, M. M. Tong and G. J. Chen, RSC Adv., 2020, 10, 3606–3614 RSC.
- M. Tiffner, M. Häring, D. D. Díaz and M. Waser, Top. Catal., 2018, 61, 1545–1550 CrossRef CAS.
- D. X. Ma, K. Liu, J. X. Li and Z. Shi, ACS Sustainable Chem. Eng., 2018, 6, 15050–15055 CrossRef CAS.
- M. A. Ziaee, Y. J. Tang, H. Zhong, D. X. Tian and R. H. Wang, ACS Sustainable Chem. Eng., 2019, 7, 2380–2387 CrossRef CAS.
- J. Li, D. G. Jia, Z. J. Guo, Y. Q. Liu, Y. N. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- W. L. Zhang, F. P. Ma, L. Ma, Y. Zhou and J. Wang, ChemSusChem, 2020, 13, 341–350 CrossRef CAS PubMed.
- D. Xing, B. Lu, H. X. Wang, J. X. Zhao and Q. H. Cai, New J. Chem., 2017, 41, 387–392 RSC.
- Y. J. Chen, R. C. Luo, J. H. Bao, Q. H. Xu, J. Jiang, X. T. Zhou and H. B. Ji, J. Mater. Chem. A, 2018, 6, 9172–9182 RSC.
- E. M. Maya, E. Verde-Sesto, D. Mantione, M. Iglesias and D. Mecerreyes, Eur. Polym. J., 2019, 110, 107–113 CrossRef CAS.
- C. Y. Cui, R. J. Sa, Z. X. Hong, H. Zhong and R. H. Wang, ChemSusChem, 2020, 13, 180–187 CrossRef CAS PubMed.
- B. Wang, A. P. Côté, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nature, 2008, 453, 207–206 CrossRef CAS PubMed.
- S. Dey, A. Bhunia, I. Boldog and C. Janiak, Microporous Mesoporous Mater., 2017, 241, 303–315 CrossRef CAS.
- H. Zhong, Y. Q. Su, X. W. Chen, X. J. Li and R. H. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed.
- M. S. Liu, J. W. Lan, L. Liang, J. M. Sun and M. Arai, J. Catal., 2017, 347, 138–147 CrossRef CAS.
- X. Song, Y. Wu, D. Pan, R. Wei, L. Gao, J. Zhang and G. Xiao, J. CO2 Util., 2018, 24, 287–297 CrossRef CAS.
- A. H. Liu, J. J. Zhang and X. B. Lv, Chin. J. Catal., 2018, 39, 1320–1328 CrossRef CAS.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS.
- T. T. Liu, R. Xu, J. D. Yi, J. Liang, X. S. Wang, P. C. Shi, Y. B. Huang and R. Cao, ChemCatChem, 2018, 10, 2036–2040 CrossRef CAS.
- X. Y. Yu, J. K. Sun, J. Y. Yuan, W. J. Zhang, C. Y. Pan, Y. N. A. Liu and G. P. Yu, Chem. Eng. J., 2018, 350, 867–871 CrossRef CAS.
- S. Builes, S. I. Sandler and R. C. Xiong, Langmuir, 2013, 29, 10416–10422 CrossRef CAS PubMed.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- Y. F. Zhi, P. P. Shao, X. Feng, H. Xia, Y. M. Zhang, Z. Shi, Y. Mu and X. M. Liu, J. Mater. Chem. A, 2018, 6, 374–382 RSC.
- P. Das and S. K. Mandal, Chem. Mater., 2019, 31, 1584–1596 CrossRef CAS.
- S. Ravi, P. Puthiaraj and W. S. Ahn, ACS Sustainable Chem. Eng., 2018, 6, 9324–9332 CrossRef CAS.
- C. Xie, J. L. Song, H. R. Wu, Y. Hu, H. Z. Liu, Y. D. Yang, Z. R. Zhang, B. F. Chen and B. X. Han, Green Chem., 2018, 20, 4655–4661 RSC.
- T. K. Dey, P. Bhanja, P. Basu, A. Ghosh and S. M. Islam, ChemistrySelect, 2019, 4, 14315–14328 CrossRef CAS.
- N. Zhang, B. Zou, G. P. Yang, B. Yu and C. W. Hu, J. CO2 Util., 2017, 22, 9–14 CrossRef CAS.
- S. Verma, G. Kumar, A. Ansari, R. I. Kureshy and N. U. Khan, Sustainable Energy Fuels, 2017, 1, 1620–1629 RSC.
- Y. M. Li, L. Yang, L. Sun, L. Ma, W. Q. Deng and Z. Li, J. Mater. Chem. A, 2019, 7, 26071–26076 RSC.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717–2720 CrossRef CAS PubMed.
- Q. Su, J. Sun, J. Q. Wang, Z. F. Yang, W. G. Cheng and S. J. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 RSC.
- T. Zhang, X. F. Wang, X. L. Huang, Y. N. Liao and J. Z. Chen, RSC Adv., 2016, 6, 2810–2818 RSC.
- Z. J. Huang, F. B. Li, B. F. Chen, T. Lu, Y. Yuan and G. Q. Yuan, Appl. Catal., B, 2013, 136, 269–277 CrossRef.
- J. Xu, F. Wu, Q. Jiang and Y. X. Li, Catal. Sci. Technol., 2015, 5, 447–454 RSC.
- M. B. Ansari, B. H. Min, Y. H. Mo and S. E. Park, Green Chem., 2011, 13, 1416–1421 RSC.
- J. Xu, J. K. Shang, Q. Jiang, Y. Wang and Y. X. Li, RSC Adv., 2016, 6, 55382–55392 RSC.
- Z. J. Huang, F. B. Li, B. F. Chen and G. Q. Yuan, Catal. Sci. Technol., 2016, 6, 2942–2948 RSC.
- J. Xu, F. Wu, Q. Jiang, J. K. Shang and Y. X. Li, J. Mol. Catal. A: Chem., 2015, 403, 77–83 CrossRef CAS.
- J. J. Zhu, T. T. Diao, W. Y. Wang, X. L. Xu, X. Y. Sun, S. A. C. Carabineiro and Z. Zhao, Appl. Catal., B, 2017, 219, 92–100 CrossRef CAS.
- X. H. Song, Y. F. Wu, D. H. Pan, F. F. Cai and G. M. Xiao, Mol. Catal., 2017, 436, 228–236 CrossRef CAS.
- S. Zhang, H. Zhang, F. X. Cao, Y. Y. Ma and Y. Q. Qu, ACS Sustainable Chem. Eng., 2018, 6, 4204–4211 CrossRef CAS.
- A. Samikannu, L. J. Konwar, P. Maki-Arvela and J. P. Mikkola, Appl. Catal., B, 2019, 241, 41–51 CrossRef CAS.
- E. M. Maya, E. Rangel-Rangel, U. Diaz and M. Iglesias, J. CO2 Util., 2018, 25, 170–179 CrossRef CAS.
- K. Yamazaki, T. Moteki and M. Ogura, Mol. Catal., 2018, 454, 38–43 CrossRef CAS.
- L. Bourda, H. S. Jena, R. Van Deun, A. M. Kaczmarek and P. Van der Voort, J. Mater. Chem. A, 2019, 7, 14060–14069 RSC.
- K. Li, X. H. Wu, Q. W. Gu, X. G. Zhao, M. M. Yuan, W. B. Ma, W. X. Ni and Z. S. Hou, RSC Adv., 2017, 7, 14721–14732 RSC.
- T. Jin, F. Dong, Y. Liu and Y. L. Hu, New J. Chem., 2019, 43, 2583–2590 RSC.
- J. Rintjema and A. W. Kleij, ChemSusChem, 2017, 10, 1274–1282 CrossRef CAS PubMed.
- V. Campisciano, C. Calabrese, F. Giacalone, C. Aprile, P. Lo Meo and M. Gruttadauria, J. CO2 Util., 2020, 38, 132–140 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |