
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtificial maturation of [FeFe] hydrogenase in a redox polymer film†
Christina
Felbek
 a,
Steffen
Hardt
b,
Cecilia
Papini
c,
Debajyoti
Pramanik
d,
Vincent
Artero
d,
Marc
Fontecave
c,
Vincent
Fourmond
a,
Nicolas
Plumeré
be and
Christophe
Léger
*a
a,
Steffen
Hardt
b,
Cecilia
Papini
c,
Debajyoti
Pramanik
d,
Vincent
Artero
d,
Marc
Fontecave
c,
Vincent
Fourmond
a,
Nicolas
Plumeré
be and
Christophe
Léger
*a
aCNRS, Aix-Marseille Université, Laboratoire de Bioénergétique et Ingénierie des Protéines, Marseille, France. E-mail: christophe.leger@imm.cnrs.fr
bCenter for Electrochemical Sciences (CES), Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitätsstr. 150, Bochum D-44780, Germany
cLaboratoire de Chimie des Processus Biologiques, Collège de France, Université Pierre et Marie Curie, CNRS UMR 8229, PSL Research University, 11 place Marcelin Berthelot, Paris 75005, France
dUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 17 rue des Martyrs, Grenoble 38000, France
eCampus Straubing for Biotechnology and Sustainability, Technical University Munich, Schulgasse 22, Straubing 94315, Germany
First published on 5th January 2021
Abstract
We demonstrate that the insertion of the dinuclear active site of [FeFe] hydrogenase into the apo-enzyme can occur when the enzyme is embedded in a film of redox polymer, under conditions of mediated electron transfer. The maturation can be monitored by electrochemistry, and is as fast as under conditions of direct electron transfer. This new approach further clears the way to the implementation of hydrogenases in large scale real life processes.
The advantages of using hydrogenases as supported catalysts of H2 oxidation in fuel cells and H2 photoproduction in photoelectrochemical cells have been emphasized. These enzymes use active sites based on transition metals such as Ni and/or Fe, instead of rare and expensive metals, to reversibly catalyze the oxidation and evolution of dihydrogen.
A major obstacle to be removed is the production of hydrogenases at a cost and in quantities compatible with large scale applications. Indeed, hydrogenases are complex metalloenzymes, whose biosynthesis in vivo is limited by the efficiency of a complex biological maturation machinery, which involves a number of other dedicated enzymes. In the particular case of the so-called [FeFe] hydrogenases, whose active site is a [Fe2(adt)(CO)3(CN)2]2− cluster (adt2− = 2-aza-1,3-propanedithiolate) covalently bound to a [Fe4S4] cluster, a major step in the direction of large scale production has been taken by Fontecave, Happe and Lubitz and coworkers.1–3 They developed an artificial maturation process, which involves: (1) producing (potentially in large amounts) hydrogenase protein devoid of any cofactor; (2) reconstituting the accessory FeS clusters with standard non-biological procedures; (3) exposing the resulting solution of the apo-enzyme to a synthetic dinuclear mimic of the active site, [Fe2(adt)(CO)4(CN)2]2−. Spontaneous incorporation of the dinuclear cluster and release of one CO ligand leads to mature and active holo-hydrogenase. This very original strategy has greatly advanced the thematic field.4
The last step of the biosynthesis of the enzyme may also be performed with the apo-enzyme adsorbed onto a rotating electrode, under conditions of direct electron transfer.4,5 After the diiron complex is added in solution, the increase in catalytic current informs on the kinetics of formation of the holo-enzyme (upon incorporation of the complex into the apo-enzyme).6
Regarding the potential applications of hydrogenases, their oxygen sensitivity has also been considered a critical issue,7–12 and various strategies have been proposed to get around this problem. When a redox polymer film is used to support the enzyme onto an electrode and to mediate electron transfer, hydrogen oxidation can occur in the presence of oxygen, because a self-protection mechanism prevents dioxygen from penetrating the film and damaging the enzyme.13–15 It is also possible to prepare polymer films of [FeFe] hydrogenase under aerobic conditions by protecting the anaerobically purified hydrogenase using a sulfide inhibitor,11 and then reactivating it by removing the sulfide under anaerobic conditions.16
Here we show that we can embed the apo-enzyme in a film of redox polymer and mature it by exposing the film to the dinuclear complex. The activation is fast, suggesting that the polymer does not hamper the diffusion of the complex, nor its incorporation into the apo-enzyme. The current that was obtained after artificial maturation in the redox polymer was significantly larger than in equivalent experiments performed under conditions of direct electron transfer, with the apo-enzyme simply adsorbed onto an electrode. We also optimized the film formation procedure to obtain a significant current after transient exposure of the apo-hydrogenase polymer film to air.
The experiments described here were performed with the apo-hydrogenase from Megasphaera elsdenii (Me), prepared as described previously,17,18 either adsorbed onto an electrode under conditions of direct electron transfer (DET),17 or embedded into a redox polymer film allowing mediated electron transfer (MET) between the enzyme and the electrode. The redox polymer, whose structure is drawn in Fig. S1 (ESI†), consisted of a 4,4′-methylviologen redox unit attached to a polyvinyl alcohol backbone (205 kDa) by a previously published isocyanate-based coupling procedure (see methods).
We could make films of this polymer embedding the apo-hydrogenase by depositing onto a glassy carbon (GC) disk electrode a mixture of enzyme solution and polymer, and letting it dry for 24 h at 4 °C in a glove box filled with N2 (and containing a significant amount of H2). The green trace in Fig. 1 shows an example of the current response observed when such an electrode is inserted and rotated in an electrochemical cell under 1 atm. of H2, and poised above the H+/H2 Nernst potential. The current appears after a small amount of [Fe2(adt)(CO)4(CN)2]2− complex is added to the solution (at t = 0). No current is observed in the absence of H2. Hence, this current results from the catalytic oxidation of H2 by the enzyme molecules that become active upon incorporation of the dinuclear complex. The variation in current suggests approximatively first-order kinetics of complex incorporation into the apo-enzyme. Fitting an exponential function to the change in current gives the time constant of maturation as 86 s.
 | ||
| Fig. 1 Electrochemically-monitored time-resolved artificial maturation of anaerobically prepared films of Me apo-hydrogenase, under three distinct conditions: MET in a thin polymer film (solid green, enlarged ten times) or in a thicker polymer film (solid dark blue), and DET (solid orange, enlarged ten times). The maturation was also monitored with another thick polymer film of apo-hydrogenase exposed to air for two hours (dark blue, dashed line, see the optimization procedure in the ESI†). In all experiments, the maturation is detected as an increase in hydrogen oxidation current after 53 nM [Fe2(adt)(CO)4(CN)2]2− complex is added to the electrochemical cell under anaerobic conditions, at t = 0 s. E = −160 mV vs. SHE, 40 °C, pH 7, 1 atm. H2, ω = 3000 rpm. | ||
Maturation under conditions of DET has been reported before,6 although not for Me hydrogenase. We include it here as a control and a reference. The orange trace in Fig. 1 shows the results of the same experiment as that plotted in green, but with the apo-hydrogenase from Me simply adsorbed onto the rotating disc electrode and undergoing direct electron transfer. The time constant of the exponential variation of the current is very similar to that obtained with the polymer film, around 84 s.
Comparing the voltammetric catalytic responses of the films prepared with and without polymer (Fig. S2, ESI,† panels A and B) clearly confirms that electron transfer is mediated by the polymer in the former, and direct in the latter. Indeed, the observed catalytic waveshapes are very different: roughly sigmoidal in the former case (Fig. S2B, ESI,† obtained by subtracting the catalytic and non-catalytic currents), and similar to that obtained before with the holo-enzyme adsorbed on graphite17 in the latter (Fig. S2A, ESI†). The magnitude of the current was similarly low in the two cases, but we found that the MET current is significantly larger if we include a small fraction of holo-hydrogenase in the apo-hydrogenase/polymer mix (about 1/500 holo/apo) when we make the film. In that case, the resulting dry film is deep violet, as a result of catalytic reduction of the viologen in the film, in the presence of H2 in the glove box. When this electrode is poised in the electrochemical cell under H2, the initial current is very small, indicating that the tiny amount of holo-enzyme in the film does not give a significant catalytic current, but a large current appears when the dinuclear complex is added (dark blue trace in Fig. 1). The final current was about 20 times larger than that observed with the film produced without holo-enzyme (the green trace in Fig. 1 was enlarged ten times). The shape of the catalytic signature (Fig. S2C, ESI†) demonstrates that electron transfer is mediated, and not direct. Varying the drying time, we found an optimum in terms of current magnitude after two hours of drying (Fig. S3, ESI†), although maximal stability is seen after 24 h.
The difference in magnitude of the current is easily explained by a difference in film thickness. Consistent with this hypothesis, Fig. S4 (ESI†) shows the cyclic voltammograms recorded with the two matured hydrogenase/polymer films under non-catalytic conditions (under an atmosphere of argon). In both cases, the CVs consist of a pair of peaks, at an average potential of −320 ± 10 mV, which result from the oxidation and reduction of the viologen moieties in the films. When the film is made with just apo-enzyme, the peak current (ip) is low and changes in proportion to the scan rate ν (as shown by the green symbols in the plot of ip/√ν against √ν in Fig. 2); this is expected if the film is thin enough that all viologen moieties are oxidized/reduced in a single sweep. When the film of apo-enzyme is made with a small amount of holo-enzyme, the non-catalytic peaks are about ten times larger (dark blue traces in Fig. S4, ESI†), and the change in ip/√ν against √ν deviates from linearity at high scan rate (dark blue symbols in Fig. 2). This indicates a transition towards diffusion limited voltammetry, consistent with the film being thicker. We believe that the reason for this is that the viologen modified polymer is reduced when it is dissolved with holo-hydrogenase under H2, while the same polymer in the presence of the apo-enzyme without holo-hydrogenase is oxidized. The singly charged radical cation of the reduced viologen is more hydrophobic than the oxidized viologen dication. This high hydrophobicity enables the production of thick and adhesive films from the reduced viologen modified polymer; in contrast, the oxidized form leads to thinner films probably because of partial redissolution of the more hydrophilic oxidized polymer.
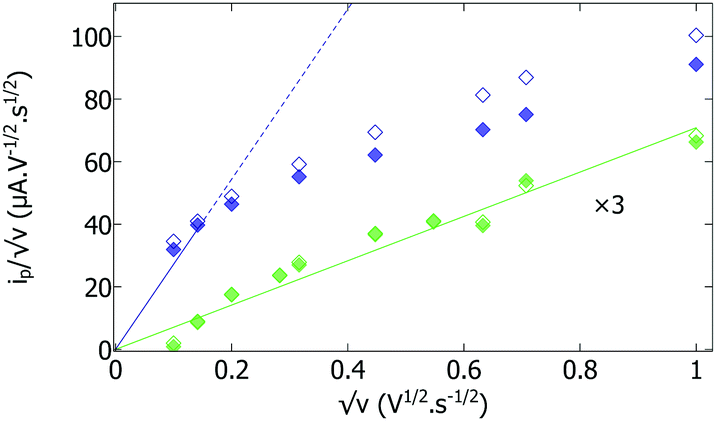 | ||
| Fig. 2 Evaluation of the thin (green, enlarged three times) and thick (dark blue) hydrogenase/polymer films by cyclic voltammetry under non-catalytic conditions (under argon). Plot of anodic (open) and cathodic (full) peak currents divided by the square root of scan rate (√ν), against √ν. A straight line through the origin is expected when the film is so thin that all viologen moieties are oxidized or reduced in each sweep. | ||
The analysis (see methods) of the maturation trace (dark blue trace, Fig. 1) seems to suggest that the MET maturation is faster in the thick film than in the thin film, with a time constant of 67 s, rather than 85 s, when an exponential function is fitted to the current trace (Fig. S5A, ESI†). However, as explained below, this is a misinterpretation of the data. Indeed, a complication of the MET configuration compared to DET is that the interpretation of the magnitude of the current depends on the film thickness. Going from very thin to very thick films, various regimes are predicted,19 with the current being either proportional to the concentration of active enzyme [E] in the film (Bartlett's “regime V”19), proportional to the square root of [E] (regime VII), or independent of [E] (regime III). This corresponds to distinct situations where the current is limited by catalysis, electron transfer, or mass transport inside the film, respectively.19 Therefore, the quantitative interpretation of the change in current observed during the activation of the film requires the previous identification of the operational regime.
One strategy to determine which regime is operational involves examining the dependence of the catalytic current on substrate (hydrogen) concentration. Again, going from very thin to very thick films, one expects the current to be proportional to either 1/(1 + Km/[H2]) (regime V, Km is the Michaelis constant), √(1/(1 + Km/[H2])) (regime VII), or [H2] (regime III).19 To accurately examine the dependence of the current on [H2], the effect of H2 depletion in the solution outside the rotating film (which is all the more significant when the current is large) must be taken into account.20 We used a new method where we step the bulk concentration of H2 using a gas mixer, record the current at different rotation rates at each H2 concentration (a portion of the signal is shown in Fig. 3A), and calculate the H2 concentration at the film/solution interface, [H2]0, as described in the methods. Fig. 3B shows the resulting catalytic current against [H2]0; fitting the Michaelis Menten equation (black trace in Fig. 3B) returns a value of Km of around 0.13 atm of H2. This value is significantly lower than the value of 0.40 ± 0.05 atm obtained under conditions of DET (Fig. 3C). In contrast, fitting the square root of the Michaelis Menten equation to the red trace in Fig. 3B returns a value of Km = 0.48 atm, much closer to the expected value. This suggests that the current obtained with the thick film is proportional to the square root of the amount of active enzyme in the film, which is the case if the catalytic current is limited by electron transfer inside the film.19
 | ||
| Fig. 3 Measurement of the Km for H2, under conditions of MET with a thick hydrogenase/polymer film (A and B) and under conditions of DET (C) at 5 °C, pH 7, E = −60 mV and −160 mV vs. SHE, respectively. The H2 concentrations are expressed in units of partial pressure of H2, and can be converted to mM using the Henry constant for H2, 0.78 mM atm−1. Panel A shows a portion of the experiment where we measured the H2 oxidation current at different partial pressures of H2 and different rotation rates. The corresponding change in current against interfacial H2 concentration is shown in panel B (blue squares). We fitted the current traces either by the Michaelis Menten equation (black trace) or the square root of the Michaelis Menten equation (red trace). Panel C shows the DET current against bulk concentration of H2 (changed by setting various partial pressures of H2, Fig. S6A, ESI†). Fig. S6B (ESI†) shows a distinct DET experiment, where we interpret the change in current over time as H2 is flushed away from the cell. From the two types of DET experiments (Fig. 3C and Fig. S6B, ESI†), we obtained Km = 0.40 ± 0.05 atm H2. | ||
The above conclusion implies that the time constant of maturation in the thick film can be obtained from the dark blue trace in Fig. 1 by fitting an exponential function to the square of the current (Fig. S5B, ESI†). Doing so returns a time constant of about 87 s, which is indeed the same as in the thin film (green trace in Fig. 1) or under conditions of DET (orange trace in Fig. 1), showing that the maturation kinetics is independent of polymer thickness. The rate of activation depends on temperature, but remains the same under conditions of direct or mediated ET (see the data at 30 °C in Fig. S8, ESI†). The comparison with data obtained with other [FeFe] hydrogenases6,21 shows that the rates of maturation increase in the order Desulfovibro desulfuricans < Clostridium pasteurianum, M. elsdenii < Chlamydomonas reinhardtii.
Using the above-described method, we could not prepare films of apo-hydrogenase from M. elsdenii under air and then activate them under anaerobic conditions, which could have been a strategy to prevent oxygen damage. This strategy is not possible with M. elsdenii because the apo-enzyme is O2 sensitive (the residual activity after two hours in air is about 1%, Fig. S9, ESI†) and because reductive conditions are required to stabilize thick polymer films. However, embedding the apo-enzyme in the film is advantageous in the case of accidental exposure of the film to O2. Indeed, when a thick film of apo-hydrogenase was prepared with a small amount of holo-enzyme under anaerobic conditions, allowed to dry under anaerobic conditions for ten minutes (during which catalytic oxidation of H2 and reduction of the viologen stabilizes the film), exposed to air at 4 °C for two hours, and finally activated under 1 atm of H2, a significant current was seen (dashed line in Fig. 1, compare also the continuous and dashed blue lines in Fig. S10 (ESI†) for films dried for the same time); it is much more intense than that expected from the residual activity of the enzyme exposed to air for the same time: the final current is not in proportion to the remaining activity, because in these thick films, it depends on the square root of the concentration of active enzyme. If the film is prepared without a small fraction of holo-enzyme and exposed to air, no activation is detected (green dashed line in Fig. S10, ESI†). These experiments show that if the thick film of apo-enzyme is exposed to air for two hours before activation, a large fraction of the apo-enzyme is irreversibly inactivated, but the magnitude of the H2 current that is detected after activation remains significant, as a consequence of the favorable scaling between current and concentration of active enzyme in the thick polymer films.
Last, we note that a very small amount of dinuclear complex (53 nM) was used to activate the apo-hydrogenase films in all experiments shown here. Increasing the concentration of the complex five times does not change the time constant of maturation (Fig. S12, ESI†), which is consistent with previous results obtained under DET conditions.6 This shows that the affinity of the enzyme for the complex is extremely high (as observed before in DET experiments6) even when the enzyme is embedded in the polymer.
From previous studies and this work, we conclude that the redox polymer film not only helps interface the holo-hydrogenase with an electrode and provides very efficient stabilization and protection from oxygen even under conditions of fuel cell operation, but can also embed the apo-enzyme and allow the dinuclear complex to reach the enzyme, without hindering this final step of the maturation process.
We thank Giorgio Caserta and Lorenzo. Mannino for performing some of the preliminary experiments and Gabriel Kopiec for preparing the viologen modified polymer. We acknowledge support from CNRS, Aix Marseille Université, Agence Nationale de la Recherche (ANR-15-CE05-0020, Labex ARCANE, CBH-EUR-GS, ANR-17-EURE-0003), and the Excellence Initiative of Aix-Marseille University – A*MIDEX, a French “Investissements d’Avenir” programme (ANR-11-IDEX-0001-02), RESOLV, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2033 – Projektnummer 390677874, the ERC starting grant 715900, and the ANR-DFG project SHIELDS (PL 746/2-1). The French group is part of the FrenchBIC network (http://www.frenchbic.cnrs.fr).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS.
- H. Li and T. B. Rauchfuss, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, [FeFe]-Hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models, Chem. Soc. Rev., 2021 10.1039/d0cs01089h.
- M. Sensi, M. del Barrio, C. Baffert, V. Fourmond and C. Léger, Curr. Opin. Electrochem., 2017, 5, 135–145 CrossRef CAS.
- C. F. Megarity, J. Esselborn, S. V. Hexter, F. Wittkamp, U.-P. Apfel, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2016, 138, 15227–15233 CrossRef CAS.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS.
- J. Esselborn, L. Kertess, U.-P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS.
- P. Rodríguez-Maciá, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef.
- P. Rodríguez-Maciá, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span and J. A. Birrell, Angew. Chem., 2020, 59(38), 16786–16794 CrossRef.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS.
- H. Li, D. Buesen, S. Dementin, C. Léger, V. Fourmond and N. Plumeré, J. Am. Chem. Soc., 2019, 141, 16734–16742 CrossRef CAS.
- A. A. Oughli, S. Hardt, O. Rüdiger, J. A. Birrell and N. Plumeré, Chem. Commun., 2020, 56, 9958–9961 RSC.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles, D. Pramanik, V. Artero, M. Atta, M. Del Barrio, B. Faivre, V. Fourmond, C. Léger and M. Fontecave, J. Am. Chem. Soc., 2018, 140, 5516–5526 CrossRef CAS.
- G. Caserta, A. Adamska-Venkatesh, L. Pecqueur, M. Atta, V. Artero, S. Roy, E. Reijerse, W. Lubitz and M. Fontecave, Biochim. Biophys. Acta, 2016, 1857, 1734–1740 CrossRef CAS.
- P. N. Bartlett and K. F. E. Pratt, J. Electroanal. Chem., 1995, 397, 61–78 CrossRef.
- M. Merrouch, J. Hadj-Saïd, C. Léger, S. Dementin and V. Fourmond, Electrochim. Acta, 2017, 245, 1059–1064 CrossRef CAS.
- J. A. Birrell, K. Wrede, K. Pawlak, P. Rodriguez-Maciá, O. Rüdiger, E. J. Reijerse and W. Lubitz, Isr. J. Chem., 2016, 56, 852–863 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/d0cc08168j |
| This journal is © The Royal Society of Chemistry 2021 |