DOI:
10.1039/C5RA03354C
(Paper)
RSC Adv., 2015,
5, 34173-34183
Diketo acids and their amino acid/dipeptidic analogues as promising scaffolds for the development of bacterial methionine aminopeptidase inhibitors
Received
23rd February 2015
, Accepted 2nd April 2015
First published on 7th April 2015
Abstract
Using diketoesters as the template, various derivatives were designed and the selected compounds were synthesized as bacterial methionine aminopeptidase (MetAP) inhibitors. The results of in vitro antibacterial screening revealed fifteen compounds (1a–c, 1e–h, 1j, 1l, 2a–c, 3d, 5c and 5e) as potent against different bacterial strains. By using the MTT assay on human cell line (HepG2), the viability of cell proliferation was evaluated and nine compounds (1c, 1e, 1j, 1l, 2a,b, 3d, 5c and 5e) showed no cytotoxic effect at the concentration range of 50–450 μg ml−1. In the biochemical evaluation against methionine aminopeptidase (MetAPs) from Streptococcus pneumonia (SpMetAP), Mycobacterium tuberculosis (MtMetAP), Enterococcus faecalis (EfMetAP) and human (HsMetAP), compounds displayed differential behaviour against these four enzymes. Moreover, compound 1g showed 84% inhibition of SpMetAP, while compound 3d selectively inhibited MtMetAP with 79% inhibition and little effect on HsMetAP at 100 μM concentration. At the same concentration, compound 5e exhibited 87% and 85% inhibition of EfMetAP and SpMetAP, respectively. Understanding the mode of binding through modeling at the active site provided the structural basis for the possible mode of inhibition. Together, these data will be useful for further development of diketo acid based inhibitors with improved potency and selectivity.
1. Introduction
Bacterial infections are responsible for some of the most deadly diseases and widespread epidemics in the world.1–3 Due to the rise in resistance of bacteria to current antibacterial chemotherapeutics, it is necessary to develop novel approaches and new inhibitors against resistant bacterial pathogens.1,3 To overcome this problem, new enzyme targets must be identified which can be targeted with small molecules. MetAPs represent one such important enzyme family, which is essential for bacteria.4 MetAPs cleave the initiator methionine from about 70% of all ribosome assisted newly synthesized proteins. The cleavage of N-terminal methionine by MetAP is an important event during protein synthesis and maturation.5,6 Since, the N-terminal methionine excision process is crucial for the metabolism and cell survival, MetAP is an ideal drug target for designing new inhibitors against bacterial pathogens.
MetAPs are first-row transition metallo-enzymes with five conserved metal ion-binding residues in the active site. Therefore, we have designed organic scaffolds that can serve as the metal chelators. Compounds with β-diketo pharmacophoric motif are believed to function as chelators of dinuclear Mn(II) or Mg(II) active site of HIV-1 integrase enzyme.7 Very recently, a diketo acid coupled with L-alanine methyl ester has been reported as EcMetAP inhibitor.8 Considering the large pharmacological importance of β-diketo acids and in continuation to our efforts to explore novel biologically active molecules,9,10 in-house database of 201 compounds virtual library was screened against Escherichia coli MetAP (EcMetAP). Based on this preliminary data, diketoesters (1a–m), diketo acids (2a–f, 2h, 2m) were selected and synthesized in good yields. Selected diketo acids were also coupled with methyl ester of L-Ala, L-Phe and a dipeptide to get their novel amino acid/dipeptidic analogues (5a–e). All the synthetic compounds were well characterized using various spectroscopic techniques. Antibacterial activity was carried out against gram positive Staphyloccocus aureus (S. aureus) and gram negative Escherichia coli (E. coli), Klebsiella pneumoniae (K. pneumoniae) and Salmonella typhimurium (S. typhimurium). MIC and MBC values were calculated. The cytotoxicity by MTT assay on active compounds was also performed on a human cell line (HepG2). Most active compounds against bacteria were screened against SpMetAP, MtMetAP, EfMetAP and HsMetAP.
2. Results and discussion
2.1. Screening using molecular docking
We modelled 201 diketo esters, acids and their amino acid/peptidic analogues and did a virtual screening using docking against EcMetAP. Not all ligands showed binding and a great variation in the binding affinities was observed using Auto dock Vina11 and Gilde docking.12 Appreciable binding affinity in kcal mol−1 was observed for 14 compounds (1a (−6.6), 1b (−6.1), 1c (−6.5), 1e (−7.0), 1f (−7.0), 1g (−6.2), 1h (−6.4), 1j (−7.0), 2a (−7.1), 2b (−8.1), 2c (−7.0), 3d (−6.4), 5c (−6.7) and 5e (−8.6)). The main active site residues which take part in H-bonding interactions and ionic interactions are Glu204, His171, His178, His79, Asp97, and Asp108 while several residues were involved in hydrophobic interactions. Compounds showing good binding affinity and interactions were further tested for antibacterial potential and their ability to inhibit various MetAPs.
2.2. Chemistry
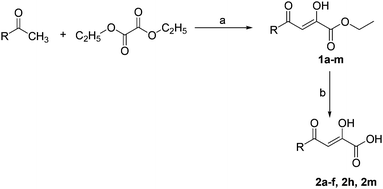
The synthesis of diketoesters (1a–m) and diketo acids (2a–f, 2h, 2m) was accomplished as outlined in Scheme 1. The oxalylation of variously substituted aryl, heteroaryl or alicyclic methyl ketones by diethyl oxalate in the presence of freshly prepared sodium ethoxide afforded β-diketoesters (1a–m).13 These compounds were purified by column chromatography (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate: 9:1) to give desired compounds in moderate to high yields as confirmed by spectroscopic analysis (Table 1).
:ethyl acetate: 9:1) to give desired compounds in moderate to high yields as confirmed by spectroscopic analysis (Table 1).
 |
| | Scheme 1 Reagents and conditions: (a) Na metal, C2H5OH, 0 °C-r.t., 3–4 h; (b) LiOH (2 M), THF:H2O, r.t., 2 h. | |
Table 1 Structure of various diketoesters (1a–m)
| Compound |
R |
Chemical structure |
Mol. formula |
Ref. |
| 1a |
 |
 |
C12H11ClO4 |
18 |
| 1b |
 |
 |
C12H11ClO4 |
19 |
| 1c |
 |
 |
C12H11BrO4 |
18 |
| 1d |
 |
 |
C9H12O4 |
— |
| 1e |
 |
 |
C13H14O4 |
19 |
| 1f |
 |
 |
C12H12O4 |
19 |
| 1g |
 |
 |
C10H10O4S |
19 |
| 1h |
 |
 |
C13H14O5 |
— |
| 1i |
 |
 |
C11H11NO4 |
19 |
| 1j |
 |
 |
C12H10Cl2O4 |
— |
| 1k |
 |
 |
C10H10O5 |
19 |
| 1l |
 |
 |
C10H11NO4 |
— |
| 1m |
 |
 |
C14H14O6 |
— |
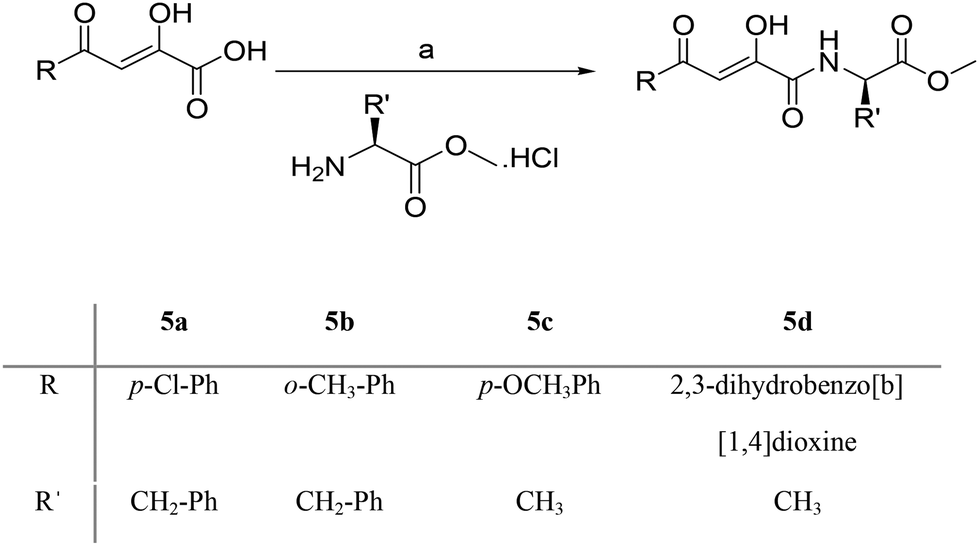
Treatment of the selected β-diketoesters with freshly prepared lithium hydroxide solution (2 M) in THF–H2O mixture (1:4) for 2 h at room temperature yielded corresponding β-diketo acids (2a–f, 2h, 2m) in quantitative yields (Table 2).14 All the acids obtained showed a single spot on TLC, therefore used without any further purification. Purity of the acids was also checked by elemental analyses and the structures were confirmed by FT-IR, 1H, 13C-NMR and mass analyses. For the synthesis of dipeptides (3a–e), desired boc-protected amino acid was coupled with methyl ester of L-Phe in acetonitrile at room temperature using EDC·HCl as coupling reagent with HOBt and N-methyl morpholine (NMM) as a base (Scheme 2).15
Table 2 Structure of various diketo acids (2a–f, 2h, 2m)
| Compound |
R |
Structure |
Mol. formula |
Ref. |
| 2a |
 |
 |
C10H7ClO4 |
— |
| 2b |
 |
 |
C10H7ClO4 |
— |
| 2c |
 |
 |
C10H7BrO4 |
— |
| 2d |
 |
 |
C7H8O4 |
— |
| 2e |
 |
 |
C11H10O4 |
— |
| 2f |
 |
 |
C10H8O4 |
20 |
| 2h |
 |
 |
C11H10O5 |
20 |
| 2m |
 |
 |
C12H10O6 |
— |
 |
| | Scheme 2 Reagents and conditions: (a) EDC·HCl, HOBt, NMM, CH3CN, r.t., 2 h; (b) DCM, TFA, r.t., 2 h. | |
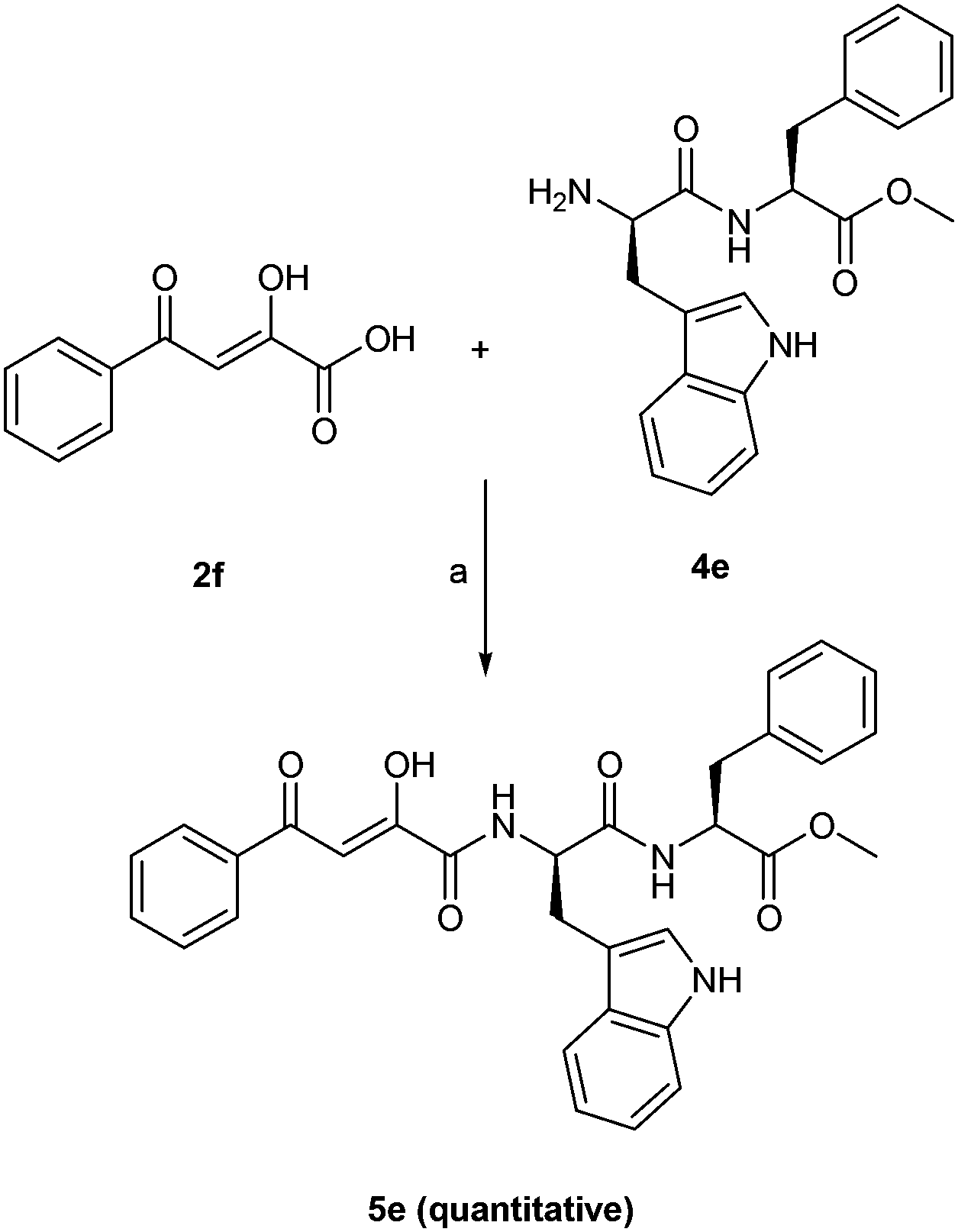
The crude product obtained was purified by column chromatography using ethyl acetate (30–40%) in petroleum ether to give pure dipeptides (3a–e) in quantitative yields. Boc group of a dipeptide (3e) was cleaved using TFA:DCM mixture (1:1). The reaction mixture was stirred for 2 h at room temperature to give the desired dipeptide Trp-Phe-OCH3 (4e) in quantitative yield which was used without further purification.16 As shown in Scheme 3, the coupling of diketo acids (2a, 2e, 2h, 2m) with methyl ester of L-Ala/L-Phe was done in anhyd. DMF using PyBOP and HOBt in presence of triethyl amine to give novel amino acid conjugated to diketo acids (5a–d) in moderate to good yields.17 Compound 2a was coupled with Trp-Phe-OCH3 (4e) using the same methodology to give the dipeptidic analogue of diketo acid (5e) in excellent yield (Scheme 4).
 |
| | Scheme 3 Reagents and conditions (a) HOBt, DMF, Et3N, PyBOP, r.t., 24 h. | |
 |
| | Scheme 4 Reagents and conditions (a) HOBt, DMF, Et3N, PyBOP, r.t., 24 h. | |
Spectral analysis. The structure of β-diketoesters (1a–m) was in accordance to their spectroscopic and analytical data. In the IR spectrum, absorption bands in the region 1520–1605 cm−1 suggested the presence of β-diketoesters in enolic form and the peaks in the region 1721–1739 cm−1 corresponding to α, β-unsaturated ester also indicated the formation of β-diketoesters. In the 1H-NMR spectrum, the proton adjacent to enolic hydroxyl group showed a sharp singlet in the range δ 6.48–7.57 ppm which confirmed that the β-diketoester is present in enolic form. The 13C-NMR spectral data were in good agreement with the assigned structures. The carbons of β-diketone group (in enolic form) exhibited chemical shift values in the range δ 189.22–200.80 ppm and δ 180.38–193.26 ppm. The mass spectra of all the compounds exhibited molecular ion peaks and contain fragments which further confirmed the formation of desired compounds. In case of diketo acids (2a–f, 2h, 2m), peaks for the enolic form occur at their corresponding positions as in the case of β-diketoesters. But the disappearance of peaks for α, β-unsaturated ester group and the appearance of peaks in the region 3410–3494 cm−1 (broad) and 1684–1709 cm−1 corresponding to acid functionality and α, β-unsaturated acid indicate the conversion of diketoesters into their corresponding acids. All the compounds displayed either [M + H]+ or [M − H]− corresponding to their molecular formulae. The coupling between diketo acid with amino acid (L-Ala-OCH3/L-Phe-OCH3) or dipeptide (Trp-Phe-OCH3) can be monitored based on the disappearance of peaks corresponding to free amino and carboxylic groups. Two distinct bands were also observed in the region 1650–1687 cm−1 and 1510–1570 cm−1 related to –CONH for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and N–H bending vibrations, respectively. In the 1H-NMR spectra, broad singlet for the carboxylic group disappeared and peaks for the amino group was also absent while the aromatic protons as well as protons belonging to alicyclic system appeared at their usual chemical shift and integral values. The 13C-NMR spectra clearly showed coupling between diketoacids and amino acids or dipeptide as peaks corresponding to amide bond appeared at the expected chemical shift values δ 169.23–160.85 ppm. The mass spectra of some of the coupled compounds showed either [M + H]+ and [M − H]− or [M − COOCH3]+ and [M − COOCH3]− which further provided evidence for the formation of desired compounds.
O stretching and N–H bending vibrations, respectively. In the 1H-NMR spectra, broad singlet for the carboxylic group disappeared and peaks for the amino group was also absent while the aromatic protons as well as protons belonging to alicyclic system appeared at their usual chemical shift and integral values. The 13C-NMR spectra clearly showed coupling between diketoacids and amino acids or dipeptide as peaks corresponding to amide bond appeared at the expected chemical shift values δ 169.23–160.85 ppm. The mass spectra of some of the coupled compounds showed either [M + H]+ and [M − H]− or [M − COOCH3]+ and [M − COOCH3]− which further provided evidence for the formation of desired compounds.
2.3. Antibacterial activity
Agar well diffusion assay was carried out to access the susceptibility of one gram positive (S. aureus MTCC737) and three gram negative (E. coli MTCC739, K. pneumoniae MTCC109, and S. typhimurium MTCC98) bacterial strains. Twenty two compounds that displayed significant zone of inhibition at 0.5 mg ml−1 concentration were further evaluated to calculate their MIC and MBC values against respective strains by micro dilution method. Ciprofloxacin was used as the positive control and the results are summarized in Table 3. Most of the synthesized compounds showed good antibacterial effect against E. coli and S. aureus. Among all the tested compounds, 1c showed most potent antibacterial effect against all the strains with MIC value in the range 9.9–39.6 μg ml−1. Compounds 1b, 1l, 2a, and 5c selectively inhibited E. coli with MIC value of 9.9 μg ml−1. Moreover 1a–c, 1f,g, 1j, and 1l also showed good activity against S. aureus with MIC value of 19.8 μg ml−1. The 5e was the only compound with MIC value of 19.8 μg ml−1 against S. typhimurium. The MBC values of potent compounds were also calculated which showed their bactericidal nature.
Table 3 In vitro antibacterial activity (MIC and MBC) and cytotoxicity of synthesized compounds (in μg ml−1)
| Compound |
E. coli |
S. aureus |
K. pneumonia |
S. typhimurium |
Cytotoxicity |
| MIC |
MBC |
MIC |
MBC |
MIC |
MBC |
MIC |
MBC |
IC50 ± S.D. |
| 1a |
>312.5 |
n.d. |
19.8 |
78.1 |
156.2 |
>312.5 |
>312.5 |
n.d. |
300 ± 0.02 |
| 1b |
9.9 |
9.9 |
19.8 |
78.1 |
>312.5 |
n.d. |
>156.2 |
n.d. |
300 ± 0.01 |
| 1c |
19.8 |
39.6 |
9.9 |
19.8 |
19.8 |
78.1 |
39.6 |
156.2 |
>450 ± 0.0 |
| 1d |
>312.5 |
n.d. |
>312.5 |
n.d. |
>156.2 |
n.d. |
>312.5 |
n.d. |
n.d. |
| 1e |
>312.5 |
n.d. |
39.6 |
78.1 |
>156.2 |
n.d. |
>156.2 |
>312.5 |
>450 ± 0.0 |
| 1f |
>312.5 |
n.d. |
19.8 |
78.1 |
>312.5 |
n.d. |
>312.5 |
n.d. |
350 ± 0.03 |
| 1g |
39.6 |
156.2 |
19.8 |
39.6 |
39.6 |
>312.5 |
78.1 |
>312.5 |
250 ± 0.03 |
| 1h |
78.1 |
>312.5 |
39.6 |
>156.2 |
>312.5 |
n.d. |
39.6 |
>156.2 |
200 ± 0.01 |
| 1j |
>312.5 |
n.d. |
19.8 |
78.1 |
78.1 |
>312.5 |
>312.5 |
n.d. |
>450 ± 0.0 |
| 1l |
9.9 |
19.8 |
19.8 |
19.8 |
>78.1 |
>312.5 |
39.6 |
156.2 |
>450 ± 0.0 |
| 1m |
>312.5 |
n.d. |
>312.5 |
n.d. |
312.5 |
n.d. |
312.5 |
n.d. |
n.d. |
| 2a |
9.9 |
19.8 |
39.6 |
39.6 |
156.2 |
312.5 |
39.6 |
156.2 |
>450 ± 0.0 |
| 2b |
39.6 |
312.5 |
78.1 |
>312.5 |
312.5 |
n.d. |
>312.5 |
n.d. |
>450 ± 0.0 |
| 2c |
19.8 |
78.1 |
78.1 |
>312.5 |
312.5 |
n.d. |
>312.5 |
n.d. |
195 ± 0.02 |
| 2e |
>312.5 |
n.d. |
78.1 |
312.5 |
>312.5 |
n.d. |
156.2 |
312.5 |
n.d. |
| 3b |
39.6 |
312.5 |
78.1 |
>312.5 |
>312.5 |
n.d. |
156.2 |
>312.5 |
n.d. |
| 3c |
>312.5 |
n.d. |
>312.5 |
n.d. |
>312.5 |
n.d. |
>312.5 |
n.d. |
n.d. |
| 3d |
>312.5 |
n.d. |
>312.5 |
n.d. |
78.1 |
156.2 |
39.6 |
39.6 |
>450 ± 0.0 |
| 5a |
>312.5 |
n.d. |
78.1 |
>312.5 |
>312.5 |
n.d. |
>312.5 |
n.d. |
n.d. |
| 5c |
9.9 |
19.8 |
>312.5 |
n.d. |
>312.5 |
n.d. |
>312.5 |
n.d. |
>450 ± 0.0 |
| 5d |
312.5 |
n.d. |
>312.5 |
n.d. |
>312.5 |
n.d. |
>312.5 |
n.d. |
n.d. |
| 5e |
>312.5 |
n.d. |
>312.5 |
n.d. |
39.6 |
312.5 |
19.8 |
19.8 |
>450 ± 0.0 |
| Cip |
<1 |
<1 |
<1 |
<1 |
<1 |
<1 |
<1 |
<1 |
n.d. |
Methionine aminopeptidase inhibition studies. Compounds with good antibacterial properties were screened for inhibition against four selected methionine aminopeptidases (MetAP's); HsMetAP, SpMetAP, EfMetAP, and MtMetAP. IC50 and Ki values were determined for the compounds showing greater than 70% inhibition of MetAP at 100 μM concentration (Tables 4 and 5). The results showed that the enzyme SpMetAP was inhibited by compound 5e (Ki = 35.5) and 1g (Ki = 33.9), EfMetAP was inhibited by compound 5e (Ki = 21.3) and MtMetAP was inhibited by compound 3d (Ki = 56.9).
Table 4 Inhibition of different MetAPs (in % age) at 100 μg ml−1 concentration
| Comp. |
SpMetAP |
MtMetAP |
HsMetAP |
EfMetAP |
| 1a |
52 ± 0.32 |
33 ± 0.3 |
54 ± 3.58 |
34 ± 1.23 |
| 1b |
63 ± 2.52 |
−29 ± 2.21 |
59 ± 3.71 |
26 ± 26.22 |
| 1c |
29 ± 3.00 |
−12 ± 3.76 |
19 ± 5.54 |
20 ± 0.75 |
| 1e |
25 ± 0.79 |
26 ± 3.39 |
24 ± 2.29 |
27 ± 2.97 |
| 1f |
37 ± 0.91 |
47 ± 2.39 |
40 ± 3.20 |
32 ± 2.93 |
| 1g |
84 ± 2.71 |
22 ± 0.87 |
60 ± 3.01 |
57 ± 1.60 |
| 1h |
68 ± 2.24 |
29 ± 2.54 |
50 ± 4.15 |
27 ± 2.80 |
| 1j |
53 ± 1.59 |
20 ± 2.09 |
38 ± 2.76 |
30 ± 2.22 |
| 2a |
35 ± 0.45 |
−3 ± 3.28 |
38 ± 2.83 |
40 ± 0.40 |
| 2b |
30 ± 1.23 |
−2 ± 1.06 |
38 ± 1.22 |
22 ± 2.3 |
| 2c |
49 ± 0.82 |
4 ± 2.22 |
40 ± 4.41 |
31 ± 2.04 |
| 3d |
19 ± 0.41 |
79 ± 1.41 |
9 ± 2.85 |
32 ± 3.73 |
| 5c |
41 ± 1.52 |
49 ± 5.32 |
39 ± 1.71 |
55 ± 2.63 |
| 5e |
85 ± 1.7 |
40 ± 4.53 |
43 ± 2.55 |
87 ± 1.11 |
Table 5 IC50 and Ki values of lead compounds against bacterial MetAPs
| Compound |
Protein |
IC50 |
Ki |
| 1g |
SpMetAP |
28.7 ± 3.97 |
33.9 ± 2.44 |
| 3d |
MtMetAP |
28.5 ± 1.69 |
56.9 ± 3.2 |
| 5e |
SpMetAP |
27.3 ± 3.83 |
35.5 ± 4.76 |
| 5e |
EfMetAP |
21.3 ± 3.00 |
25.9 ± 3.31 |
In vitro cytotoxicity profile. To examine the toxicity of most active compounds based on MIC and MBC on cell proliferation, the cytotoxicity was checked by MTT assay on HepG2 cell line and the results are shown in Table 3. Nine compounds (1c, 1e, 1j, 1l, 2a,b, 3d, 5c and 5e) were found to be non cytotoxic at concentration range 50–450 μg ml−1. Compound 1g, 1h and 2c showed cytotoxicity in the range of 195–250 μg ml−1. The effect of compounds on % viability of cell proliferation versus concentration shown in Fig. 1, indicates toxicity at a very high concentration range to MIC and MBC values of the lead compounds in antibacterial assay. These results encouraged us to further examine enzymatic and in vivo studies to give a better lead in the antibacterial potential.
 |
| | Fig. 1 Cell viability assay on HepG2 cell line. | |
Interactions of lead molecules with MetAPs. Comprehensive inhibitory studies conducted on MetAPs with identified leads (1g, 3d and 5e) showed appreciable inhibition on purified proteins. We did a comprehensive docking of these leads with MtMetAP, EfMetAP and compared it with HsMetAP. The results shown in Fig. 2 indicated high binding affinity and tight binding. Compound 1g showed significant increase in the % inhibition with SpMetAP, however its inhibitory activity against HsMetAP and EfMetAP were comparable, consequent to this it also showed similar binding affinities. Compound 5e was found with best binding affinity with all MetAPs but it showed significant increase in % inhibition with EfMetAP as compared to HsMetAP. It also showed a reduced IC50 value and a very low Ki. Significantly 5e binds to three different sites on EfMetAP, MtMetAP and HsMetAP. Compound 3d interacts with the active site residues (Ser109 and Val111) of MtMetAP and inhibits it with low IC50 (Table 6).
 |
| | Fig. 2 Interactions of 1g, 3d and 5e with different MetAPs. | |
Table 6 Docking score and interaction of lead with MtMetAP (1YJ3), HsMetAP (2G6P), and EfMetAP (3TB5)
| Protein |
Ligand |
| 1g |
3d |
5e |
| 1YJ3 |
−5.4 kcal−1 |
−6.7 kcal−1 |
−7.5 kcal−1 |
| His212 |
Ala58, Arg164 |
Thr203 |
| 2 G6P |
−5.6 kcal−1 |
−6.9 kcal−1 |
−8.9 kcal−1 |
| Tyr196 |
His310 |
Glu128, Thr311, Ala312, Asn314 |
| 3TB5 |
−5.3 kcal−1 |
−5.8 kcal−1 |
−7.6 kcal−1 |
| His108 |
Arg28, Ser109, Val111, Tyr231 |
Glu251 |
However in HsMetAP, its binds away from the active site and there is significant reduction the % inhibition. Based on the binding affinity, MIC values, % inhibition, Ki and IC50 values, 3d and 5e are significant lead and can be explored with much more specific synthetic designs around these two compounds.
3. Experimental protocols
3.1. Screening of ligands by molecular docking studies
AutoDock Vina was used for screening of 201 designed compounds against E. Coli MetAP enzyme. The three dimensional coordinate (2MAT) of EcMetAP was retrieved from PDB library. AutoDock Vina 4.2 is advanced version which is much faster and accurate in binding mode prediction if compared with its older version AutoDock 4.0.10 Default parameters of AutoDock Vina were used with slight modification. The affinity grid box was centred to the whole protein and the grid spacing fixed to 1 Å. The program automatically calculates the grid map and grid centre coordinates (X = 44, Y = 46, Z = 44). After generating receptor and all ligands' PDB in PyMOL AutoDock/Vina plugin, Vina was ran to obtained docking score (binding affinity in kcal mol−1) and RMSD value for all compounds. PyMOL was used for visualization of the polar contacts in between ligands and binding site of protein.21 The best docking result can be considered to be the conformation with the lowest docking score and lowest RMSD. The screening of ligands was also performed by glide docking using GLIDE 5.8 software with standard procedure.22,23
3.2. Materials and methods
All the chemicals purchased from Sigma-Aldrich, Spectrochem and Hi Media were used without further purification. Precoated Aluminium sheets (Silica gel 60 F254, Merck Germany) were used for thin-layer chromatography (TLC) and spots were visualized under UV light. The IR spectra of compounds were taken on Agilent Cary 630 FT-IR spectrometer. 1H and 13C NMR spectra were obtained at ambient temperature using Bruker Spectrospin DPX-300 MHz, 400 MHz, Agilent 500 MHz FT-NMR in CDCl3 using tetramethylsilane (TMS) as an internal standard. Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet. Chemical shift values are given in parts per million (ppm) and coupling constants (J) in Hertz (Hz). Mass spectra were recorded on a Q Star XL hybrid electron spray ionization high resolution mass spectrometer (Applied biosystems) in a scan range of 100 to 1000 atomic mass units (amu). Melting points were recorded on a digital Buchi melting point apparatus (M-560) and were reported uncorrected. Purification of the compounds was done by column chromatography using silica gel (230–400 mesh size) with cyclohexane/ethyl acetate as eluent. The optical rotation of compounds 5a–e was recorded on Anton Paar MCP-200 polarimeter at 20 °C using sodium D light.
3.3. General procedure for the synthesis of diketo esters (1a–m)
In a two neck round bottom flask, sodium metal (21 mmol) was dissolved in anhydrous ethanol at 0 °C to give sodium ethoxide solution. To this freshly prepared solution, a mixture of diethyl oxalate (20 mmol) and ketone (21 mmol) was added slowly with the help of dropping funnel over a period of 20 minutes. Thick precipitate was formed and reaction mixture was stirred for 3–4 h at room temperature. After completion of the reaction checked by TLC, the precipitate obtained was dissolved in 2 N sulphuric acid (72 ml) and the compound was extracted with diethyl ether, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The characterization of compounds 1a, 1b, 1c, 1e, 1f, 1g, 1i, and 1k is previously reported.18,19
(Z)-Ethyl 4-(4-chlorophenyl)-2-hydroxy-4-oxobut-2-enoate (1a). Yield: 72%.
(Z)-Ethyl 4-(2-cholorophenyl)-2-hydroxy-4-oxobut-2-enoate (1b). Yield: 69%.
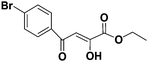
(Z)-Ethyl 4-(4-bromophenyl)-2-hydroxy-4-oxobut-2-enoate (1c). Yield: 54%.
(Z)-Ethyl 4-cyclopropyl-2-hydroxy-4-oxobut-2-enoate (1d). Colourless oil, yield: 52%, Rf (cyclohexane:ethyl acetate = 7:3): 0.47, anal (C9H12O4) calc. C 58.69 H 6.57, found: C 58.72 H 6.54. IR (neat): 2948, 2872, 1719, 1678, 1655, 1579, 1508, 1430, 1350, 1322, 1290, 1207, 1130, 1065, 1013, 901, 879, 843, 767, 707 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 14.62 (s, 1H, OH) 6.48 (s, 1H, CH), 4.34 (q, 2H, J = 2.4 Hz, CH2), 1.92–1.86 (m, 1H, cyclopropyl ring), 1.38 (t, 3H, J = 5.2 Hz, CH3), 1.24 (q, 2H, J = 4.4 Hz, cyclopropyl ring), 1.07 (q, 2H, J = 4.4 Hz, cyclopropyl ring). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 198.89, 185.24, 161.98, 99.76, 62.84, 14.03, 10.04. LC-MS: m/z [M + H]+ 185.34.
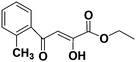
(Z)-Ethyl 2-hydroxy-4-oxo-4-o-tolylbut-2-enoate (1e). Yield: 78%.
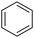
(Z)-Ethyl 2-hydroxy-4-oxo-4-phenylbut-2-enoate (1f). Yield: 43%.
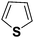
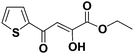
(Z)-Ethyl 2-hydroxy-4-oxo-4-(thiophen-2-yl)but-2-enoate (1g). Yield: 43%.
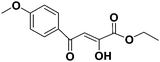
(Z)-Ethyl-2-hydroxy-4-(4-methoxyphenyl)-4-oxobut-2-enoate (1h). Orange solid, M.pt.: 92 °C, yield: 55%, Rf (petroleum ether:ethyl acetate = 7:3): 0.60, anal (C13H14O5) calc. C 62.39 H 5.64, found: C 62.40 H 5.66. IR (neat): 2948, 2844, 2386, 2125, 1898, 1676, 1596, 1512, 1462, 1423, 1361, 1289, 1240, 1175, 1140, 1119, 1020, 851, 830, 778, 696 cm−1. 1H-NMR (300 MHz, CDCl3) (δ, ppm): 7.99–7.90 (m, 2H, Ar-H), 7.18–7.08 (m, 2H, Ar-H), 6.91 (s, 1H, CH), 4.42 (q, 2H, J = 6.9 Hz, CH2), 3.89 (s, 3H, OCH3), 1.42 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 196.94, 190.34, 168.06, 162.47, 130.61, 127.66, 114.21, 97.73, 62.50, 55.45, 14.09. LC-MS: m/z [M + H]+ 251.2.
(Z)-Ethyl 2-hydroxy-4-oxo-4-(pyridin-2-yl)but-2-enoate (1i). Yield: 63%.
(Z)-Ethyl 4-(2,4-dichlorophenyl)-2-hydroxy-4-oxobut-2-enoate (1j). Brown solid, M.pt.: 60 °C, yield: 70%, Rf (cyclohexane:ethyl acetate = 7:3): 0.86, anal (C12H11ClO4) calc. C 49.85 H 3.49, found: C 49.86 H 3.46. IR (neat): 3101, 2983, 1737, 1628, 1583, 1475, 1456, 1391, 1385, 1253, 1225, 1106, 1089, 1039, 1020, 883, 819, 778, 687 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 14.94 (s, 1H, OH) 7.92 (d, 1H, J = 8.4 Hz, Ar-H), 7.76 (s, 1H, Ar-H), 7.68 (d, 1H, J = 8.4 Hz, Ar-H), 7.57 (s, 1H, CH), 4.71 (q, 2H, J = 7.2 Hz, CH2), 1.72 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 194.78, 189.22, 161.88, 140.14, 136.76, 135.45, 133.12, 129.09, 128.73, 98.67, 63.57, 13.93. LC-MS: m/z [M − H]− 288.9.
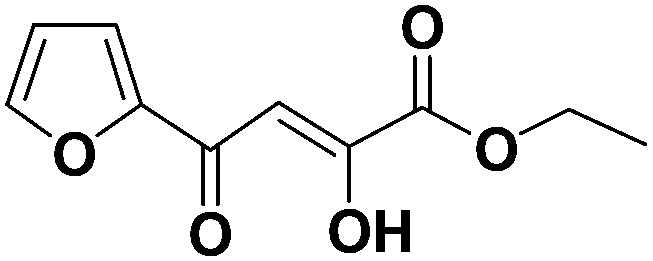
(Z)-Ethyl 4-(furan-2-yl)-2-hydroxy-4-oxobut-2-enoate (1k). Yield: 72%.
(Z)-Ethyl 2-hydroxy-4-oxo-4-(1H-pyrrol-2-yl)but-2-enoate (1l). Black solid, M.pt.: 62 °C, yield: 68%, Rf (petroleum ether:ethyl acetate = 7:3): 0.60, anal (C10H11NO5) calc. C 57.41 H 5.30 N 6.70 found: C 57.42 H 5.32 N 6.71. IR (neat): 3265, 3116, 1721, 1633, 1547, 1510, 1428, 1398, 1324, 1264, 1130, 1078, 1045, 1022, 974, 929, 843, 767, 750 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.03 (d, 1H, J = 6 Hz, Ar-H), 6.91 (d, 1H, J = 6.3 Hz, Ar-H), 6.63 (s, 1H, CH), 6.29 (t, 1H, J = 7.2 Hz, Ar-H), 4.25 (q, 2H, J = 7.23 Hz, CH2), 1.54 (t, 3H, J = 7.23 Hz, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 200.80, 193.26, 161.67, 131.3, 128.9, 125.09, 113.74, 98.67, 61.87, 14.96. LC-MS: m/z [M + 2H]+ 211.3.
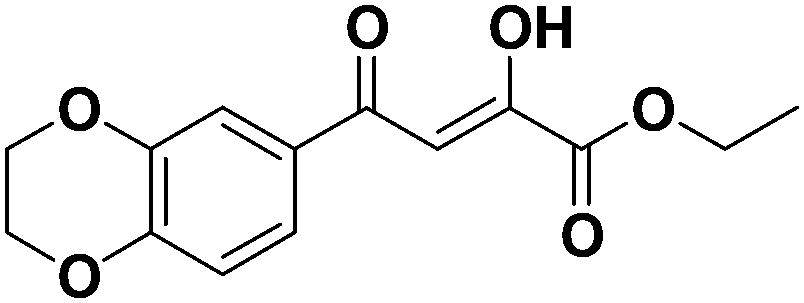
(Z)-Ethyl 4-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-hydroxy-4-oxobut-2-enoate (1m). Light yellow oil, yield: 54%, Rf (cyclohexane:ethyl acetate = 7:3): 0.40, anal (C14H14O6) calc. C 60.43 H 5.07, found: C 60.48 H 5.10. IR: 2958, 2920, 2853, 1736, 1605, 1577, 1508, 1460, 1369, 1330, 1289, 1253, 1184, 1127, 1024, 912, 890, 819, 780, 726 cm−1.1H-NMR (300 MHz, CDCl3) (δ, ppm): 7.49 (s, 1H, CH), 7.21 (s, 1H, Ar-H), 6.91 (d, 2H, J = 9 Hz, Ar-H), 4.93 (q, 2H, J = 15.3 Hz, CH2), 4.21 (d, 4H, J = 17.7 Hz, alicyclic ring), 1.22 (t, 3H, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 190.93, 189.46, 161.24, 153.53, 148.37, 129.88, 121.90, 114.81, 99.84, 64.34, 61.71, 14.52. LC-MS: m/z [M + H]+ 279.3, [M − H]− 277.2.
3.4. General procedure for the synthesis of diketo acids (2a–f, 2h, 2m)
To a solution of diketo ester (1.0 mmol) in THF:water mixture (1:4), freshly prepared 2 M LiOH solution (4.5 mmol) was added with the help of a dropping funnel. The reaction was continued for 2 h until white precipitate is obtained. After completion of the reaction, pH of the reaction was adjusted to 2–3 by adding 1 N HCl. The compound was extracted with diethyl ether, dried over sodium sulphate, concentrated to give diketo acids (2a–f, 2h, 2m) in moderate to excellent yields (42–99%). The characterization of compounds 2f and 2h is previously reported.20
(Z)-4-(4-Cholorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (2a). Light yellow solid, M.pt.: 102 °C yield: 75%, Rf (cyclohexane:ethyl acetate = 7:3): 0.0, anal (C10H7ClO4) calc. C 53.00 H 3.11, found C 53.01 H 3.09. IR (neat): 3410, 2974, 2866, 1684, 1585, 1395, 1358, 1256, 1090, 1010, 956, 907, 823, 762, 620, 584, 519 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 15.07 (s, 1H, OH), 7.90 (d, 2H, J = 8.4 Hz, Ar-H), 7.45 (d, 2H, J = 8.8 Hz, Ar-H), 7.14 (s, 1H, CH). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 197.04, 189.24, 163.12, 138.96, 133.47, 129.27, 128.63, 97.99. LC-MS: m/z [M + H]+ 227.2, [M − H]− 225.0.
(Z)-4-(2-Chlorophenyl)-2-hydroxy-4-oxobut-2-enoic acid (2b). Light yellow solid, M.pt.: 154 °C, yield: 86%, Rf (cyclohexane:ethyl acetate = 7:3): 0.0, anal (C10H7ClO4) calc. C 53.00 H 3.11, found C 53.02 H 3.12. IR: 3457, 2925, 1685, 1585, 1358, 1261, 1127, 1067, 1001, 823, 746, 475 cm−1. 1H-NMR (300 MHz, acetone-D6) (δ, ppm): 8.05 (d, 1H, J = 8.4 Hz, Ar-H), 7.93 (d, 1H, J = 8.7 Hz, Ar-H), 7.79 (d, 1H, J = 8.4 Hz, Ar-H) 7.71 (d, 1H, J = 8.7 Hz, Ar-H), 7.15 (s, 1H, CH). 13C-NMR (75 MHz, acetone-D6) (δ, ppm): 196.32, 189.62, 162.38, 134.00, 132.26, 131.75, 130.06, 129.68, 128.34, 97.54. LC-MS: m/z [M + H]+ 227.45, [M − H]− 225.18.
(Z)-4-(4-Bromophenyl)-2-hydroxy-4-oxobut-2-enoic acid (2c). Orange oil, yield: 82%, Rf (cyclohexane:ethyl acetate = 7:3): 0.0, anal (C10H7BrO4) calc. C 44.31 H 2.60, found C 44.35 H 2.63. IR: 3494, 1721, 1613, 1583, 1485, 1451, 1398, 1233, 1183, 1139, 1107, 1069, 1051, 903, 819, 773 cm−1. 1H-NMR (300 MHz, acetone-D6) (δ, ppm): 8.15 (d, 2H, J = 8.7 Hz, Ar-H), 7.31 (d, 2H, J = 8.7 Hz, Ar-H), 7.02 (s, 1H, CH). 13C-NMR (75 MHz, acetone-D6) (δ, ppm): 205.03, 186.49, 162.24, 142.06, 136.02, 133.53, 129.09, 99.04. LC-MS: m/z [M + H]+ 270.87.
(Z)-4-Cyclopropyl-2-hydroxy-4-oxobut-2-enoic acid (2d). Light yellow oil, yield: 43%, Rf (cyclohexane:ethyl acetate = 7:3): 0.0, anal (C7H8O4) calc. C 53.85 H 5.16, found C 53.88 H 5.20. IR: 3341, 2963, 2920, 2851, 1745, 1669, 1520, 1449, 1438, 1393, 1371, 1300, 1274, 1242, 1222, 1168, 1018, 979, 883, 754, 674 cm−1. 1H-NMR (300 MHz, CDCl3) (δ, ppm): 14.95 (s, 1H, OH), 6.50 (s, 1H, CH), 1.85–1.78 (m, 1H, cyclopropyl ring), 1.22 (q, 2H, J = 4.6 Hz, cyclopropyl ring), 1.08 (q, 2H, J = 4.6 Hz, cyclopropyl ring). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 198.54, 185.96, 161.39, 98.89, 43.09, 11.13. LC-MS: m/z [M − H]− 155.0.
(Z)-2-Hydroxy-4-oxo-4-o-tolylbut-2-enoic acid (2e). Yellow oil, yield: 99%, Rf (cyclohexane:ethyl acetate = 5:5): 0.0, anal (C11H10O4) calc. C 64.07 H 4.89, found C 64.09 H 4.88. IR: 2931, 2507, 1709, 1596, 1492, 1456, 1387, 1233, 1169, 1127, 1028, 914, 832, 763, 728, 693 cm−1. 1H-NMR (400 MHz, acetone-D6) (δ, ppm): 7.66 (d, 1H, J = 7.2 Hz, Ar-H), 7.45 (t, 1H, J = 7.2 Hz, Ar-H), 7.33–7.26 (m, 2H, Ar-H), 6.93 (s, 1H, CH), 2.53 (s, 3H, CH3). 13C-NMR (75 MHz, acetone-D6) (δ, ppm): 194.02, 187.52, 163.74, 138.60, 134.64, 132.46, 132.09, 129.21, 126.13, 100.75, 21.20. LC-MS: m/z [M − H]+ 205.1.
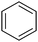
(Z)-2-Hydroxy-4-oxo-4-phenylbut-2-enoic acid (2f). Yield: 83%.
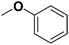
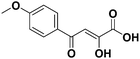
(Z)-2-Hydroxy-4-(4-methoxyphenyl)-4-oxobut-2-enoic acid (2h). Yield: 71%.
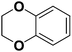
(Z)-4-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2-hydroxy-4-oxobut-2-enoic acid (2m). Lemon yellow oil, yield: 91%, Rf (cyclohexane:ethyl acetate = 5:5): 0, anal (C12H10O6) calc. C 57.60 H 4.03, found: C 57.55 H 4.01. IR: 3408, 2967, 2912, 2847, 1732, 1610, 1575, 1504, 1455, 1364, 1321, 1284, 1247, 1181, 1121, 1021, 908, 887, 816, 765, 723 cm−1. 1H-NMR (300 MHz, CDCl3) (δ, ppm): 7.55 (s, 1H, CH), 7.19 (s, 1H, Ar-H), 6.89 (d, 2H, J = 8.8 Hz, Ar-H), 4.33 (d, 4H, J = 8.7 Hz, alicyclic ring). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 189.97, 189.56, 163.24, 152.53, 148.74, 130.88, 122.90, 115.81, 114.90, 99.84, 64.54, 62.71, 14.52. LC-MS: m/z [M + H]+ 251.97.
3.5. General procedure for the synthesis of boc-dipeptides (3a–e)
In a 50 ml round bottom flask, L-phenylalanine methyl ester hydrochloride (1.0 mmol) and desired boc amino acid (1.2 mmol), were dissolved in acetonitrile (10 ml) and cooled to 0 °C. To this solution, NMM (1.2 mmol), HOBt (2.0 mmol) and EDC·HCl (1.2 mmol) were added in small portions. The reaction mixture was brought to room temperature and the progress of the reaction was checked by TLC. After completion of the reaction, solvent was removed under vacuum and ethyl acetate was added to the residue, washed with 10% citric acid solution, 5% sodium bicarbonate solution, water and then with brine and finally dried over anhydrous MgSO4. The organic layer was concentrated under vacuum. The dipeptide obtained was purified by column chromatography using cyclohexane:ethyl acetate (7:3) as eluent to obtain pure dipeptides in 83–97% yields.
Boc-Ala-Phe-OCH3 (3a). White powder, M.pt.: 80 °C, yield: 91%, Rf (cyclohexane:ethyl acetate = 7:3): 0.24, anal (C18H26N2O5) calc. C 61.70 H 7.48 N 7.99, found C 61.72 H 7.46 N 7.80. IR (neat): 3326, 2978, 2931, 1737, 1691, 1654, 1520, 1447, 1391, 1367, 1335, 1251, 1207, 1162, 1127, 1080, 1056, 1035, 968, 907, 871 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.29–7.22 (m, 3H, Ar-H), 7.10 (d, 2H, J = 6.8 Hz, Ar-H), 6.49 (s, 1H, NH), 4.85 (q, 1H, J = 6 Hz, CH), 4.12 (q, 1H, J = 7.2 Hz, CH), 3.72 (s, 3H, OCH3), 3.12 (d, 2H, J = 10.8 Hz, CH2), 1.43 (s, 9H, CH3), 1.32–1.22 (d, 3H, J = 4.2 Hz, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 172.84, 171.83, 154.90, 137.01, 129.09, 128.11, 126.53, 78.05, 53.39, 51.81, 49.51, 36.66, 28.16, 18.09. LC-MS: m/z [M + H]+ 351.3, [M − H]+ 349.3, [M − Boc]+ 251.3.
Boc-Gly-Phe-OCH3 (3b). Colourless oil, yield: 92%, Rf (cyclohexane:ethyl acetate = 5:5): 0.65, anal (C18H26N2O5) calc. C 60.70 H 7.19 N 8.33, found C 60.72 H 7.18 N 8.34. IR (neat): 3339, 2978, 2933, 1655, 1510, 1439, 1369, 1279, 1249, 1216, 1166, 1052, 1032, 944, 866, 747, 702 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.29–7.26 (m, 3H, Ar-H), 7.09 (d, 2H, J = 7.2 Hz, Ar-H), 6.45 (s, 1H, NH), 5.02 (s, 1H, NH), 4.89 (m, 1H, CH), 3.82–3.76 (m, 2H, CH2), 3.72 (s, 3H, OCH3), 3.11 (d, 2H, J = 8.4 Hz, CH2), 1.44 (s, 9H, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 171.69, 171.30, 155.73, 135.70, 129.23, 128.62, 127.16, 79.89, 53.12, 52.28, 37.99, 30.84, 28.30. LC-MS: m/z [M + H]+ 337.4, [M − H]+ 335.2.
Boc-Val-Phe-OCH3 (3c). White solid, M.pt.: 94 °C, yield: 84%, Rf (cyclohexane:ethyl acetate = 5:5): 0.74, anal (C20H30N2O5) calc. C 63.47 H 7.99 N 7.40, found C 63.48 H 7.98 N 7.41. IR (neat): 3343, 2963, 2929, 2874, 1745, 1669, 1518, 1449, 1371, 1300, 1274, 1244, 1222, 1168, 1121, 1082, 1018, 981, 938, 883, 804, 754, 702, 672 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.31–7.20 (m, 3H, Ar-H), 7.11 (d, 2H, J = 6.8 Hz, Ar-H), 6.26 (s, 1H, NH), 4.99 (s, 1H, NH), 4.88 (t, 1H, J = 5.6 Hz, CH), 3.88 (d, 1H, J = 8 Hz, CH), 3.71 (s, 3H, OCH3), 3.17–3.07 (m, 2H, CH2), 1.44 (s, 9H, CH3). 0.89 (d, 6H, J = 6.8 Hz, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 191.31, 156.43, 145.30, 137.90, 136.17, 133.07, 130.15, 128.69, 128.58, 122.16, 120.82, 118.03, 115.23. LC-MS: m/z [M + H]+ 379.4, [M-Boc]+ 279.4.
Boc-Ile-Phe-OCH3 (3d). White solid, M.pt.: 112 °C, yield: 98%, Rf (cyclohexane:ethyl acetate = 7:3): 0.71, anal (C21H32N2O5) calc. C 64.26 H 8.22 N 7.14, found C 64.27 H 8.24 N 7.16. IR (neat): 3343, 3328, 2965, 1743, 1667, 1514, 1454, 1447, 1367, 1277, 1235, 1168, 1102, 1048, 1022, 889, 752, 700 cm−1. 1H-NMR (500 MHz, CDCl3) (δ, ppm): 7.29–7.25 (m, 3H, Ar-H), 7.11 (d, 2H, J = 9.84 Hz, Ar-H), 6.35 (s, 1H, NH), 5.0 (s, 1H, NH), 4.88 (q, 1H, J = 5.6 Hz, CH), 3.88 (t, 1H, J = 5.6 Hz, CH), 3.17 (s, 3H, OCH3), 3.17 (d, 2H, J = 5.3 Hz, CH2), 2.87 (m, 1H, CH), 1.44 (m, 11H, CH3, CH2), 0.89–0.85 (m, 6H). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 171.62, 171.24, 155.61, 135.63, 129.21, 128.57, 127.12, 77.31, 59.20, 53.05, 52.28, 37.94, 37.15, 28.26, 24.60, 15.38, 11.36. LC-MS: m/z [M + H]+ 393.6, [M-Boc]+ 293.6.
Boc-Trp-Phe-OCH3 (3e). Brown solid, M.pt.: 110 °C, yield: 96%, Rf (petroleum ether:ethyl acetate = 7:3): 0.35, anal (C26H31N3O5) calc. C 67.08 H 6.71 N 9.03, found C 67.10 H 6.72 N 9.04. IR: 3404, 3065, 2959, 2127, 1737, 1665, 1544, 1536, 1501, 1465, 1359, 1141, 1015, 855, 842, 801, 745, 726, 704 cm−1, 1H-NMR (400 MHz, CDCl3) (δ, ppm): 8.08 (s, 1H, NH), 7.66 (d, 1H, J = 7.6 Hz, Ar-H), 7.33 (d, 1H, J = 14.8 Hz, Ar-H), 7.22–7.11 (m, 5H, Ar-H), 7.02 (s, 1H, Ar-H), 6.80 (d, 2H, J = 6.8 Hz, Ar-H), 6.20 (s, 1H, NH), 4.73 (m, 1H, CH), 4.49 (m, 1H, CH), 3.61 (s, 3H, OCH3), 3.30–3.08 (m, 2H, CH2), 2.94 (d, 2H, J = 5.6 Hz, CH2), 1.42 (s, 9H, CH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 172.67, 171.98, 156.61, 139.86, 135.63, 129.64, 128.57, 127.32, 126.56, 122.57, 119.62, 111.42, 78.31, 56.20, 53.05, 51.28, 37.82, 31.15, 28.26. LC-MS: m/z [M + H]+ 466.1, [M − H]− 464.1 [M − Boc]+ 366.0.
3.6. Synthesis of Trp-Phe-OCH3 (4e)
To a solution of 3e (1.0 mmol) in 5 ml of anhydrous dichloromethane was added 5 ml of trifluoroacetic acid and stirred the reaction mixture for 2 h at room temperature. Evaporation to dryness of the mixture led to yellowish oil. This residue was triturated in ether and the precipitate was recovered as a white powder after drying in quantitative yield.
Scarlet brown solid, M.pt.: 108 °C, yield: 99%, Rf (petroleum ether:ethyl acetate = 7:3):0.42, anal (C21H23N3O3) calc. C 69.02 H 6.34 N 11.50, found C 69.05 H 6.37 N 11.47. IR: 1726, 1687, 1655, 1524, 1453, 1426, 1359, 1343, 1292, 1244, 1166, 1099, 1045, 1013, 868, 778, 735, 704 cm−1, 1H-NMR (400 MHz, CDCl3) (δ, ppm): 8.35 (s, 1H, NH), 7.45 (d, 1H, J = 7.6 Hz, Ar-H), 7.31 (d, 1H, J = 8 Hz, Ar-H), 7.15 (s, 4H, Ar-H), 7.06 (t, 2H, J = 7.6 Hz, Ar-H), 6.96–6.90 (m, 2H, Ar-H), 6.74 (s, 1H, NH), 5.48 (s, 2H, NH2), 4.60 (t, 1H, J = 6.8 Hz, CH), 4.28 (t, 2H, J = 7 Hz, CH), 3.63 (s, 3H, OCH3), 3.25 (t, 2H, J = 7.6 Hz, CH2), 2.99 (t, 2H, J = 5.6 Hz, CH2). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 172.89, 171.50, 157.10, 139.56, 136.23, 129.89, 128.91, 127.12, 126.31, 122.52, 120.62, 110.98, 56.45, 53.25, 51.37, 37.76, 31.23. LC-MS: m/z [M + H]+ 366.0, [M − H]− 364.0.
3.7. General procedure for the synthesis of amino acid/dipeptide analogues of diketo acids (5a–e)
To a solution of desired diketo acid (1.0 mmol) in anhydrous DMF under argon were added HOBt (2.0 mmol), PyBOP (1.2 mmol), L-amino acid methyl ester hydrochloride/dipeptide (4e) (1.0 mmol) and Et3N (1.6 mmol) and the reaction mixture was stirred for 24 h at room temperature. Solvent was removed and the residue was dissolved in 50 ml of ethyl acetate. The organic phase was washed with a solution of 10% citric acid, 5% NaHCO3, water and finally with brine solution (each 50 ml). The organic phase then dried over Na2SO4 and evaporated to dryness. Purification by column chromatography on silica gel (cyclohexane:ethyl acetate = 7:3) provided desired compound in 70–99% yields.
(S,Z)-Methyl2-(4-(4-chlorophenyl)-2-hydroxy-4-oxobut-2-enamido)-3-phenyl propanoate (5a). Brown solid, M.pt.: 98 °C yield: 70%, Rf (cyclohexane:ethyl acetate = 5:5): 0.50, [α]20D = −47.19 (c 0.45, C2H5OH), anal (C20H18ClNO5) calc. C 61.94 H 4.68 N 3.61, found C 61.89 H 4.72 N 3.64. IR (neat): 1737, 1655, 1579, 1572, 1558, 1439, 1400, 1363, 1309, 1246, 1222, 1175, 1113, 1091, 1024, 1015, 775, 747, 702 cm−1. 1H-NMR (300 MHz, CDCl3) (δ, ppm): 10.83 (s, 1H, OH), 7.90 (d, 2H, J = 8.4 Hz, Ar-H), 7.70 (d, 2H, J = 8.1 Hz, Ar-H), 7.44 (d, 2H, J = 8.7 Hz, Ar-H), 7.28 (t, 3H, J = 9.6 Hz, Ar-H), 6.96 (s, 1H, CH), 4.76 (t, 1H, J = 5.4 Hz, CH), 3.78 (d, 2H, J = 6.3 Hz, CH2), 3.74 (s, 3H, OCH3). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 191.67, 189.95, 170.97, 166.56, 139.67, 135.41, 129.75, 129.25, 128.91, 128.69, 127.42, 53.23, 52.59, 37.20. LC-MS: m/z [M + H]+ 388.95.
(S,Z)-Methyl2-(2-hydroxy-4-(4-methoxyphenyl)-4-oxobut-2-enamido)-3-phenylpropanoate (5b). Yellow oil, yield: 76%, Rf (cyclohexane:ethyl acetate = 5:5): 0.60, [α]20D = −0.06 (c 0.75, C2H5OH), anal (C21H21NO6) calc. C 65.79 H 5.52 N 3.65, found C 65.83 H 5.58 N 3.67. IR (neat): 2937, 2842, 1671, 1595, 1508, 1456, 1420, 1357, 1309, 1247, 1169, 1114, 1023, 956, 831, 753, 699, 566, 478 cm−1. 1H-NMR (300 MHz, CDCl3) (δ, ppm): 7.93 (d, 2H, J = 8.7 Hz, Ar-H), 7.24–7.15 (m, 5H, Ar-H), 7.04 (d, 2H, J = 8.7 Hz, Ar-H), 6.81 (s, 1H, CH), 4.45 (q, 1H, J = 7.6 Hz, CH), 3.84 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 3.54 (d, 2H, J = 5.6 Hz, CH2). 13C-NMR (75 MHz, CDCl3) (δ, ppm): 196.30, 189.43, 172.02, 163.09, 139.07, 130.46, 129.88, 128.93, 127.53, 126.06, 113.81, 108.41, 55.50, 53.79, 51.83, 38.66. LC-MS: m/z [M + H]+ 384.59.
(Z)-Methyl 2-(2-hydroxy-4-oxo-4-o-tolylbut-2-enamido)propanoate (5c). Yellow oil, yield: 70%, Rf (cyclohexane:ethyl acetate = 5:5): 0.66, [α]20D = −37.65 (c 0.80, C2H5OH), anal (C31H29N3O6) calc. C 61.85 H 5.88 N 4.81, found C 61.86 H 5.89 N 4.82. IR (neat): 2952, 2861, 2872, 1687, 1678, 1605, 1510, 1451, 1439, 1208, 761, 737 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.63 (d, 1H, J = 7.6 Hz, Ar-H), 7.42 (t, 1H, J = 7.6 Hz, Ar-H), 7.31–7.28 (m, 2H, Ar-H), 6.83 (s, 1H, CH), 4.39 (q, 1H, J = 7.6 Hz, CH), 3.57 (s, 3H, OCH3), 2.55 (s, 3H, CH3), 1.41 (t, 3H, J = 7.2 Hz, CH3). 13C-NMR (125 MHz, CDCl3) (δ, ppm): 191.80, 175.82, 172.48, 160.85, 138.10, 134.78, 131.89, 131.82, 129.11, 125.99, 98.28, 52.65, 48.17, 21.12, 18.19. LC-MS: m/z [M − COOCH3]+ 235.2, [M − COOCH3]− 233.1.
(Z)-Methyl 2-(4-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)-2-hydroxy-4-oxobut-2-enamido)propanoate (5d). Golden yellow oil, yield: 75%, Rf (cyclohexane:ethyl acetate = 5:5): 0.47, [α]20D = −11.75 (c 0.40, C2H5OH), anal (C16H17NO7) calc. C 57.31 H 5.11 N 4.18, found C 57.37 H 5.14 N 4.15. IR (neat): 2965, 2868, 1745, 1678, 1607, 1581, 1508, 1462, 1430, 1350, 1322, 1292, 1207, 1132, 1065, 1013, 901, 845, 767, 707 cm−1. 1H-NMR (400 MHz, CDCl3) (δ, ppm): 7.51 (s, 1H, CH), 7.49 (s, 1H, Ar-H), 6.93 (d, 2H, J = 8.7 Hz, Ar-H), 4.87 (q, 1H, J = 5.4 Hz, CH), 4.33 (d, 4H, J = 5.1 Hz, alicyclic ring), 3.59 (s, 3H, OCH3), 1.26 (d, 3H, J = 5.4 Hz, CH3). 13C-NMR (125 MHz, CDCl3) (δ, ppm): 196.60, 189.47, 171.62, 166.56, 147.95, 143.21, 131.05, 122.39, 117.75, 115.88, 110.02, 64.62, 64.06, 48.54, 46.26, 18.61. LC-MS: m/z [M + H]+ 336.42.
(Z)-Methyl 2-(2-(2-hydroxy-4-oxo-4-phenylbut-2-enamido)-3-(1H-indol-3-yl) propanamido)-3-phenylpropanoate (5e). Light yellow solid, yield: 99%, M.pt.: 73 °C, Rf (ethyl acetate:cyclohexane = 6:4): 0.59, [α]20D = −16.47 (c 0.60, C2H5OH), anal (C31H29N3O6) calc. C 69.00 H 5.42 N 7.79, found C 69.01 H 5.43 N 7.78. IR (neat): 2926, 2853, 1743, 1655, 1601, 1510, 1458, 1439, 1343, 1253, 1214, 1182, 1078, 1028, 819, 775, 745, 702 cm−1. 1H-NMR (300 MHz, DMSO) (δ, ppm): 8.67 (d, 1H, J = 7.5 Hz, Ar-H), 8.51 (d, 1H, J = 8.4 Hz, Ar-H), 8.01 (d, 2H, J = 7.5 Hz, Ar-H), 7.68–7.54 (m, 4H, Ar-H), 7.33–7.23 (m, 6H, Ar-H, NH), 7.10–6.93 (m, 4H, Ar-H, CH), 4.68 (t, 1H, J = 5.7 Hz, CH) 4.54 (t, 1H, J = 6.3 Hz, CH), 3.57 (s, 3H, OCH3), 3.15–2.97 (m, 4H, CH2). 13C-NMR (125 MHz, CDCl3) (δ, ppm): 176.92, 170.19, 169.23, 159.28, 135.19, 134.87, 132.28, 132.14, 127.70, 127.50, 126.85, 126.03, 125.22, 122.32, 119.76, 117.10, 116.97, 110.02, 107.89, 92.78, 52.36, 52.19, 50.52, 35.56, 30.22. LC-MS: m/z [M + H]+ 540.4, [M − H]+ 538.3.
3.8. Antibacterial activity
In vitro bacterial susceptibility assay. In vitro antibacterial susceptibility of all the compounds against four different bacterial strains was determined by standard agar well diffusion assay.24 Petri dishes (size 100 mm diameter) containing 18 ml of cool and molten Mueller Hinton Agar (MHA) (at 40 °C) were seeded with 50 μl inoculums of bacterial strain (inoculums size was adjusted so as to deliver a final inoculums of approximately 1.0 × 105 CFU ml−1). Wells of 6 mm diameter were cut into solidified agar media with the help of sterilized cork borer. An aliquot of 50 μl of each concentration was poured in the respective well and the plates were incubated at 37 °C for overnight. DMSO was used as negative control while ciprofloxacin (10 μg ml−1) was used as positive control. The experiment was performed in triplicate under strict aseptic conditions. The antibacterial activity for each of the compound evaluated was expressed in terms of the average of the diameter of zone of inhibition (in mm) produced by the respective concentration of each compound at the end of incubation period.
Determination of MIC and MBC of the susceptible compounds. A micro broth dilution technique was employed to determine the MIC (minimum inhibitory concentration) of synthesized compounds along with positive (ciprofloxacin) and negative (10% DMSO) controls.25 The concentration of DMSO were maintained less than 10% in the final test volume. Various concentrations (312.5 … 2.475 μg ml−1) of test compounds and (512.0 … 1.0 μg ml−1) of ciprofloxacin were dispensed into wells, then inoculated with test organisms with approx. 2.5 × 106 cells per ml (McFarland standard) and incubated at 37 °C for 24 h. The effect of 10% DMSO was also checked on all the strains separately. The MIC values were determined as the lowest concentration resulting in no growth. The MBC of potent compounds were also determined by transferring of 10 μl aliquot on sterile MH agar plate from wells with no growth. The plates were incubated at 37 °C for 24 h. After incubation, the MBC was determined as the lowest concentration of test compound that results no growth. All the experiments were done in triplicate in separate time.
Enzymatic inhibition assay. All compounds were dissolved in DMSO to a stock concentration of 10 mM. Initially the compounds were screened for inhibition with each of the four MetAP's (HsMetAP, MtMetAP, SpMetAP, EfMetAP). Expression, purification and biochemical assays are performed as reported previously.26,27 The enzyme assays were performed using Met-AMC as substrate. IC50 values were determined by using compound concentration range of 1 μM to 120 μM. The Ki values were determined by Dixon method. The reaction mixture contained 50 mM Hepes (pH-7.5 for SpMetAP, EfMetAP, MtMetAP and pH-8.0 for HsMetAP), 150 mM KCl, CoCl2 (three molar equivalents of the enzyme concentration), 4 μM enzymes (HsMetAP, MtMetAP, SpMetAP or EfMetAP) and 50, 100, 150 μM concentrations of L-Met-AMC. All reactions were performed in triplicate. 2,2′ Bipyridine was used as positive control.
In vitro cytotoxicity. HepG2 cells were cultured and maintained as a monolayer in Dulbecco's Modified Eagle's Medium (DMEM, Sigma) supplemented with 10% of fetal calf serum (Sigma) and antibiotics (100 IU ml−1 of penicillin and 100 mg ml−1 of streptomycin, Sigma). All cells were cultured at 37 °C in a 100% humidity atmosphere and 5% CO2. Exponentially growing viable cells were plated at 1.2 × 10−4 cells per well into 96-well plates and incubated for 48 h before the addition of the compounds/metronidazole. Stock solutions of compounds were initially dissolved in 20% (v/v) DMSO and further diluted with fresh complete medium. The growth-inhibitory effects of the compounds were measured using standard tetrazolium MTT assay. After 48 h of incubation at 37 °C, the medium was removed and 25 ml of MTT (5 mg ml−1) in serum free medium was added to each well. The plates were incubated at 37 °C for 4 h. At the end of the incubation period, the medium was removed and 100 μl DMSO added to all wells. The metabolized MTT product dissolved in DMSO was quantified by reading the absorbance at 570 nm with a reference wavelength of 655 nm in an ELISA plate reader. All assays were performed in triplicate. Percent viability was defined as the relative absorbance of treated versus untreated control cells.
4. Conclusion
In this study, we successfully designed and synthesized some diketo acids, their amino acid/dipeptidic analogues as novel antibacterial agents targeting bacterial MetAPs. The results of in silico screening and in vitro antibacterial activity support the above findings and suggest their candidature to act as small molecule inhibitors of MetAPs. Compounds 1c, 1e, 1j, 1l, 2a, 2b, 3d, 5c and 5e showed moderate to excellent antibacterial activity with no cytotoxic effect in the concentration range of 50–450 μg ml−1. In the biochemical evaluation, compounds 1g and 3d showed 84% and 79% inhibition of SpMetAP and MtMetAP, respectively at 100 μM concentration. At the same concentration, compound 5e exhibited 87% and 85% inhibition of EfMetAP and SpMetAP respectively. Furthermore, efforts in modifying these and other lead structures with the aim of improving potency as well as specificity are in progress.
Conflicts of interest
All authors declare no competing interests.
Acknowledgements
Mohammad Abid gratefully acknowledges funding support in the form of Young Scientist from Science & Engineering Research Board (SR/FT/LS-03/2011), New Delhi, India. AA acknowledges the support from DST (SR/SO/BB-55/2008) and DBT (BT-BRB-TF-2-2011), India. VKP, MI and BA acknowledge the fellowship support from UGC, INDIA. The authors are thankful to Prof. Umar Farooq, Faculty of Biotechnology, Shoolini University of Biotechnology and Management Sciences, Solan, H.P. India, for providing laboratory facilities for antibacterial activities.
References
- J. Chin, New Sci., 1996, 2051, 32–35 Search PubMed.
- R. A. Howe, K. E. Bowker, T. R. Walsh, T. G. Feest and A. P. MacGowan, Lancet, 1998, 351, 602 CrossRef CAS.
- S. B. Levy, Sci. Am., 1998, 278, 32–39 CrossRef PubMed.
- R. A. Bradshaw, W. W. Brickey and K. W. Walker, Trends Biochem. Sci., 1998, 23(7), 263–267 CrossRef CAS.
- W. T. Lowther and B. W. Matthews, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1477, 157–167 CrossRef CAS.
- W. T. Lowther and B. W. Matthews, Chem. Rev., 2002, 102, 4581–4608 CrossRef CAS PubMed.
- C. Fossey, A. H. Vu, A. Vidu, I. Zarafu, D. Laduree, S. Schmidt, G. Laumond and A. M. Aubertin, J. Enzyme Inhib. Med. Chem., 2007, 22, 591–607 CrossRef CAS PubMed.
- E. Goemaere, A. Melet, V. R. Larue, A. L. Lieutaud, R. A. De Sousa, J. Chevalier, L. Yimga-Djapa, C. Giglione, F. Huguet and M. Alimi, J. Antimicrob. Chemother., 2012, 67(6), 1392–1400 CrossRef CAS PubMed.
- M. Abid, A. R. Bhat, F. Athar and A. Azam, Eur. J. Med. Chem., 2009, 44(1), 417–425 CrossRef CAS PubMed.
- M. Irfan, B. Aneja, U. Yadav, S. I. Khan, N. Manzoor, C. G. Daniliuc and M. Abid, Eur. J. Med. Chem., 2015, 93, 246–254 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- L. L. C. Schrödinger, Schrödinger Suite 2012 Induced Fit Docking Protocol, Glide Version 5.8, New York, NY, 2012 Search PubMed.
- D. W. Ribbons, New J. Chem., 1999, 23, 437–446 RSC.
- R. N. Patel, L. Chu, R. Chidambaram, J. Zhu and J. Kant, Tetrahedron: Asymmetry, 2002, 13, 349–355 CrossRef CAS.
- S. P. Chakrabarty, R. Ramapanicker, R. Mishra, S. Chandrasekaran and H. Balaram, Bioorg. Med. Chem., 2009, 17, 8060–8072 CrossRef CAS PubMed.
- Y. Zhou, X. C. Guo, T. Yi, T. Yoshimoto and D. Pei, Anal. Biochem., 2000, 280, 159–165 CrossRef CAS PubMed.
- M. Rowley, Prog. Med. Chem., 2008, 46, 1–28 CAS.
- D. Geffken, R. Soliman, F. S. G. Soliman, M. M. Abdel-Khalek and D. A. E. Issa, Med. Chem. Res., 2011, 20, 408–420 CrossRef CAS.
- S. S. Zimmerman, A. Khatri, E. C. Garnier-Amblard, P. Mullasseril, N. L. Kurtkaya, S. Gyoneva, K. B. Hansen, S. F. Traynelis and D. C. Liotta, J. Med. Chem., 2014, 57, 2334–2356 CrossRef CAS PubMed.
- B. Chen, H. F. Yin, Z. S. Wang, J. H. Xu, L. Q. Fan and J. Zhao, Adv. Synth. Catal., 2009, 351, 2959–2966 CrossRef CAS PubMed.
- W. L. Delano, The PyMol Molecular Graphics System, DeLano Scientific, San Carlos, CA, USA, 2002 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- L. L. C. Schrödinger, QikProp Version 3.5, Schrödinger, LLC, New York, 2012 Search PubMed.
- C. Perez, M. Pauli and P. Bazerque, ActaBiol. Med. Exp., 1990, 15, 113–115 Search PubMed.
- M. A. Wikler, Performance standards for antimicrobial susceptibility testing: Seventeenth informational supplement, Clinical and Laboratory Standards Institute, 2007 Search PubMed.
- C. Kishor, T. Arya, R. Reddi, X. Chen, V. Saddanapu, A. K. Marapaka, R. Gumpena, D. Ma, J. O. Liu and A. Addlagatta, J. Med. Chem., 2013, 56(13), 5295–5305 CrossRef CAS PubMed.
- T. Arya, C. Kishor, V. Saddanapu, R. Reddi and A. Addlagatta, PLoS One, 2013, 8(10), 75207 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.