
Gallic acid: a versatile antioxidant with promising therapeutic and industrial applications
Bharti Badhani,
Neha Sharma and
Rita Kakkar*
Computational Chemistry Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: rkakkar@chemistry.du.ac.in; Tel: +91-1127666313
First published on 3rd March 2015
Abstract
Oxidative stress, a result of an overproduction and accumulation of free radicals, is the leading cause of several degenerative diseases such as cancer, atherosclerosis, cardiovascular diseases, ageing and inflammatory diseases. Polyphenols form an important class of naturally occurring antioxidants, having innumerable biological activities such as anticancer, antifungal, antibacterial, antiviral, antiulcer and anticholesterol, to name a few. Among various polyphenols, gallic acid (3,4,5-trihydroxybenzoic acid), a naturally occurring low molecular weight triphenolic compound, has emerged as a strong antioxidant and an efficient apoptosis inducing agent. Starting from the bioavailability and the biosynthetic pathway of gallic acid, this review includes various in vitro, in vivo and in silico studies providing the mode of action, radical scavenging activity, ability to inhibit lipid peroxidation, maintenance of endogenous defense systems and metal ion chelation by this triphenolic molecule, along with a comprehensive overview of factors responsible for its high antioxidant activity. Gallic acid derivatives have also been found in a number of phytomedicines with diverse biological and pharmacological activities, including radical scavenging, interfering with the cell signaling pathways and apoptosis of cancer cells. The diverse range of applications of this simple polyphenol is due to a fine amalgam between its antioxidant and prooxidant potential. The existing literature on this dual behavior of gallic acid and its derivatives is reviewed here. This is followed by an account of their potential clinical and industrial applications.
1. Introduction
There is rapidly growing literature on potential applications of antioxidants from natural sources. Naturally occurring antioxidants have the potential to protect cells from oxidative stress via a number of pathways. Since oxidative stress is responsible for a variety of degenerative diseases, these antioxidants can contribute significantly towards human welfare.1 Determining and optimizing the antioxidant properties of a compound is therefore one of the most important areas of research.Free radical generation, an inevitable and continuous process, is a part of our normal metabolism. Nevertheless, an overproduction and accumulation of reactive oxygen species (ROS), due to internal and/or external factors, may create an imbalance resulting in oxidative stress. In general, oxidative stress increases the production of superoxide radicals (O2˙−) and hydrogen peroxide (H2O2). In the presence of a suitable transition metal catalyst, these reactive species can further interact to form highly toxic hydroxyl radicals (HO˙). Hence, an excess of H2O2 can be detrimental to cells. Under normal circumstances, these free radicals are counteracted by the endogenous antioxidant defense system, involving enzymatic systems such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GRx) and glutathione-S-transferase (GST).2 However, an unchecked rampant free radical production can alter cell viability by disrupting biological macromolecules such as DNA, proteins and lipids. Oxidative damage to such macromolecules accumulates with age and has been postulated as a major type of endogenous damage leading to aging, degenerative diseases, brain dysfunction, atherosclerosis, cardiovascular diseases, inflammatory diseases and cancer.3–5
A number of synthetic drugs may provide protection against the deleterious effects of oxidative stress but these are also associated with adverse side effects. The focus has thus shifted towards identifying antioxidant compounds that are clinically potent with low or no side effects, to be used in preventive therapies. There is ample support for the claim that a higher intake of fresh fruits, vegetables or plants rich in natural antioxidants is associated with lower incidences of free radical induced diseases such as atherosclerosis, cardiovascular, neurodegenerative diseases and certain types of cancer.6–10 Consequently, special attention is being given to antioxidants of natural origin.11–13
These preventive properties of plants are attributed to the presence of flavonoids, anthocyanins and phenolic compounds.14 A large number of phenolic compounds possess the ability to scavenge reactive species such as superoxide radicals and hydroxyl radicals; reduce lipid peroxyl radicals, and inhibit lipid peroxidation. These phenolic compounds emerge as strong antiradical agents largely due to their redox properties, which make them efficient hydrogen donors, reducing agents and metal chelators.15
Among various polyphenols, gallic acid (GA) (3,4,5-trihydroxybenzoic acid), a naturally occurring low molecular weight triphenolic compound, has been suggested to possess strong antioxidant activities in many studies.16–23 It provides efficient protection against oxidative damage caused by reactive species often encountered in biological systems including, hydroxyl (HO˙), superoxide (O2˙−), and peroxyl (ROO˙) and the non-radicals, hydrogen peroxide (H2O2) and hypochlorous acid (HOCl). Furthermore, GA has been demonstrated as the chief antioxidant component responsible for the efficient antiradical and anticancer properties of a number of plant extracts.24–28 Similarly, gallic acid derivatives (GADs) have also been found in a number of phytomedicines with diverse biological and pharmacological activities, such as ROS scavenging, interfering the cell signaling pathways, and apoptosis of cancer cells.29–32 This latter property i.e. induction of apoptosis, is mainly associated with its prooxidant, rather than antioxidant behavior. GA can thus exhibit both antioxidant as well as prooxidant characteristics,33 displaying a dual-edge sword behavior.
There is a diverse variety of bioactive natural molecules possessing the GA moiety, imparting several industrial and pharmacological applications such as being used as a vital building block for various pharmaceutical leads, including combretastatin A-4, podophyllotoxin and colchicine.34–36 In analytical research, GA is used as a standard for determining the phenol content of various analytes.37 Further, some of its ester derivatives (propyl gallate, octyl gallate and lauryl gallate) (Fig. 1) are widely used in cosmetics, processed food and food packing materials to prevent oxidative rancidity and spoilage.38
 | ||
| Fig. 1 Chemical structure of gallic acid and some of its n-alkyl ester derivatives. | ||
In this review, the emphasis would lie on the dual behavior of gallic acid and its derivatives. The resulting biological and industrial applications would then be discussed, focusing majorly on antitumor activities.
1.1. Bioavailability
Gallic acid is widely present in the plant kingdom and largely found in free form or as a derivative in different food sources such as nuts, tea, grapes and sumac (Rhus coriaria L.).39–41 Other sources include gallnuts, oak bark, honey, different berries, pomegranate, mango and other fruits, vegetables and beverages.37,42–47 Gallic acid is found in plant tissues in ester form and diverse esters with sugars glycosides, polyols and phenols have been reported.48Apart from the plant species involved, the amount of GA in plant tissues may also get affected due to some external stimuli such as UV radiation, chemical stressors and microbial infections. On similar lines, the phenolic composition in grape juices and wines gets affected by factors such as grape variety, processing practices, and storage.49 In particular, red wine has a high content of this phenolic acid. Burns et al. analyzed a number of red wines and found the total phenolic content as 1100 to 3165 mg L−1, out of which 35 to 70 mg L−1 and 120 to 360 mg L−1 is allocated to GA and the epicatechin gallate derivatives, respectively.50 GA content is higher in green tea51 but the total gallate content of cocoa is shown to be even higher than that of green tea or red wine.52
The absorption and metabolism information of polyphenols is useful for the design and interpretation of numerous studies investigating their health effects.53 It is known that among the hundreds of dietary polyphenols which are better absorbed and which lead to the formation of active metabolites, GA is extremely well absorbed, compared with other polyphenols.41,54–56 It most likely gets absorbed by the degradation of tannic acid (TA) and gallates in the gastrointestinal tract. Hydrolysable tannins, which are almost invariably present in every food plant, are naturally occurring polymers of galloyl moieties and a glucose molecule. Tannic acid exhibited greater antioxidant activity than α-tocopherol in multiple in vitro assays such as reducing power, DPPH radical, ABTS radical, superoxide anion radical and hydrogen peroxide scavenging and metal chelating activities.57 It was thus proposed to be of use in the preservation of the shelf life and nutritional quality of food products. These hydrolysable tannins may get hydrolyzed in the acidic pH of the stomach, with the release of ten potentially reactive GA residues.58 The major metabolites of GA are products of methylation (unconjugated and conjugated 4-O-methylgallic acid, 2-O-methylgallic acid), decarboxylation (unconjugated and conjugated pyrogallol, 4-O-methylpyrogallol) and dehydroxylation (resorcinol).56 Additionally, some more methyl ether derivatives (3-O-methylgallic acid and 3,4-O-methylgallic acid) have also been reported.59 Accordingly, gallates are first hydrolyzed to GA and then methylation yields 4-O-methylgallic acid as a biotransformation product.60 Among the various derivatives of GA, the metabolism of ester derivatives shows that the octyl gallate (OG) and lauryl gallate (LG) are absorbed and hydrolyzed to a lesser extent than propyl gallate (PG), which is readily absorbed in the gastrointestinal tract.60
1.2. Biosynthesis
Gallic acid is formed in substantial amounts by plants and fungi.61 Because of the importance of GA as an antioxidant in foods, controlling its production and accumulation in plants could significantly increase the nutritional value of many crop species.62 Thus, a number of studies have been reported regarding the elucidation of the route of biosynthesis of gallic acid.Multiple studies have postulated the existence of two potential pathways for GA synthesis (Fig. 2). It could either be formed from phenylalanine (4) or from an early shikimate intermediate i.e. 3-dehydroshikimate (3-DHS) (2).63–65 However, the carboxylic group of GA has been found to be biosynthetically equivalent to the carboxylic group of shikimate, rather than to the phenylalanine side chain.66–68 It thus entails that GA is formed from 3-DHS61,68,69 either by direct dehydrogenation or via protocatechuic acid (3) as an intermediate.70,71 Though Werner et al., using retrobiosynthetic NMR studies with 13C labeled glucose and oxygen isotope ratio mass spectrometry, showed that GA is primarily synthesized by direct dehydrogenation of 3-DHS.61,68 The results align well with the earlier reports of Dewick and Haslam.66,67
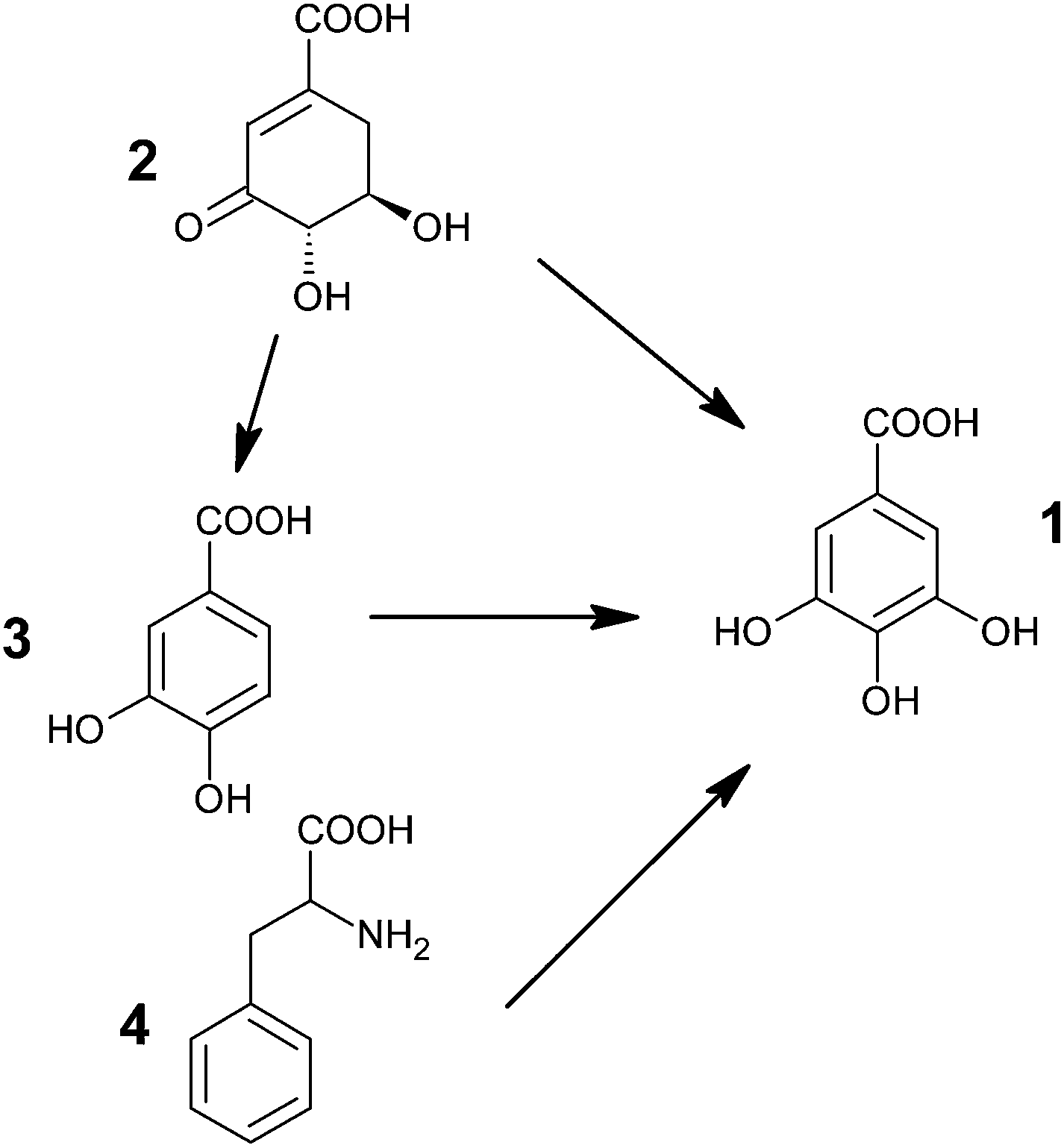 | ||
| Fig. 2 Possible pathways for biosynthesis of gallic acid (1: GA; 2: 3-dehydroshikimic acid; 3: protocatechuic acid; 4: phenylalanine). | ||
Two different pathways could also be effective in the same organism i.e., via early shikimate intermediate and via one of the aromatic amino acid.64,72,73 It has been asserted that the young leaves of Rhus succedanea synthesize gallic acid predominantly by the dehydrogenation of shikimate, whereas the older ones adopt the phenylalanine route.72
Following this, a key enzyme of the shikimate pathway i.e. shikimate dehydrogenase (SDH) has been isolated.68,69 In the presence of NADPH, SDH catalyzes the reduction of 3-DHS to shikimic acid, while in the presence of NADP+ it catalyzes its oxidation to 3,5-didehydroshikimate, an unstable compound that undergoes a spontaneous rearrangement to gallate.62
Hence, the enzyme (SDH) responsible for the reduction of 3-DHS to shikimic acid, which is primarily used in the formation of aromatic amino acids L-phenylalanine, L-tryptophan and L-tyrosine,74 is also required in the biosynthesis of GA.
1.3. Structural facet
Gallic acid is a planar molecule, consisting of an aromatic ring, three phenolic hydroxyl groups and a carboxylic acid group (Fig. 3). The three hydroxyl groups are bonded to the aromatic ring in an ortho position with respect to each other. It is this order of arrangement which is the chief determinant for the strong antioxidant capacity of phenolic compounds.75 According to quantum chemical calculations with the Becke3P86 DFT functional and the 6-311G(d,p) basis set, the hydroxyl groups are coplanar and in a bent configuration, whereas the carboxylic group turns out of the plane of the benzene ring.76 However, structure–activity relationship (SAR) studies on ester derivatives of GA revealed that the most stable conformers exhibit a planar geometry.77 This is possibly due to the stabilizing effect of π-electron delocalization between the benzene ring and the carbonyl group, which is favored when they lie in the same plane. Unlike the neutral and the anionic forms, the carboxylate group of the anion radical is slightly turned out of the plane.78 Depending on the orientation of the attached carboxylic and hydroxyl groups, GA may have a number of conformations. Different studies involving crystal structure38,79,80 and conformational81,82 analysis of GA have been reported. Four stable conformers (Fig. 3) were obtained, differing in the orientation of the hydroxyl groups around the ring. GA(I) conformer, having all the three hydrogen atoms of the hydroxyl groups oriented in the same direction, emerged as the most stable conformer.81,82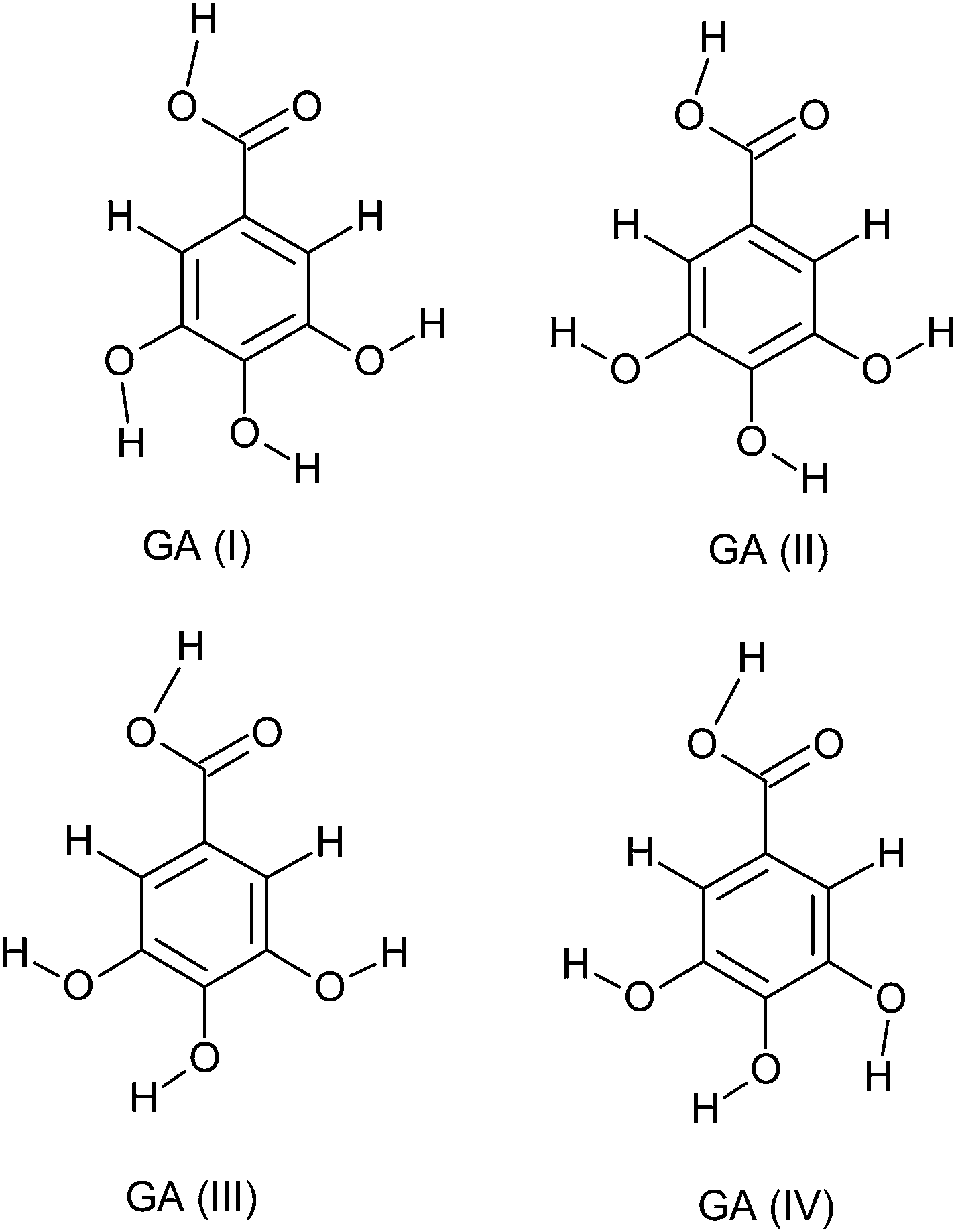 | ||
| Fig. 3 Conformers of gallic acid. | ||
Gallic acid can form both intra (between hydroxyl groups) as well as intermolecular hydrogen bonds.82 GA(I), GA(II), GA(III) and GA(IV) (Fig. 3) can accordingly have two, one, one and two intramolecular hydrogen bonds, respectively. According to a DFT study at the B3LYP/6-311++G(3df,2p) level,83 the hydroxyl groups are involved in two hydrogen bonds (2.196 Å each). The two intramolecular hydrogen bonds between the three hydroxyl groups contribute towards the stability of the molecule.84 For neutral GA, the intramolecular hydrogen bonds are of the moderate type (2.1–2.2 Å).78 With the formation of the anionic or anion radical structure, the negative charge is mainly redistributed over oxygen atoms, resulting in shorter and stronger hydrogen bonds.
The phenolic aromatic arrangement markedly influences the antioxidant activity of phenolic acids. Accordingly, several factors such as the number and the position of hydroxyl group, the presence of other functional groups and their position with respect to hydroxyl groups have been shown to affect the antioxidant and antiradical activity.
Galato et al. have demonstrated, by studying eight phenolic and analogous compounds, that the antioxidant activity of a molecule increases with increase in the number of hydroxyl groups attached to the aromatic ring.85 A positive correlation was further obtained between the number of –OH groups attached to the aromatic ring and the antioxidant as well as the antiradical activity of phenolic acids.27,75 Thus, gallic acid was found to exhibit the highest antioxidant capacity among various polyphenols.27 On similar lines, all the pyrogallol (1,2,3-trihydroxybenzene) derivatives displayed a higher antioxidant activity compared to the catechol (1,2-dihydroxybenzene) analogues and even with the antioxidant used as reference (tocopherol or Trolox).86
SAR studies have shown that the radical scavenging efficiency of gallic acid derivatives is dependent on the presence of hydroxyl groups as well as steric freedom.87 Especially the para substituted –OH group was found to be highly efficient in radical scavenging. Previously, the role of the ortho hydroxyl group in the antioxidant activity of phenolic acids has also been described.88,89 The extra hydroxyl group in trihydroxy phenolic acid provides greater stability along with higher antioxidant activity. Hydroxyl groups not only affect antioxidant ability by intramolecular hydrogen bonding (Fig. 4)89–92 but they also tend to stabilize the antioxidant radical formed.89 According to Wright et al., the –OH group ortho to phenol tends to stabilize the radical formed, resulting in a lower hydrogen bond dissociation enthalpy and hence an increased antioxidant capacity.89
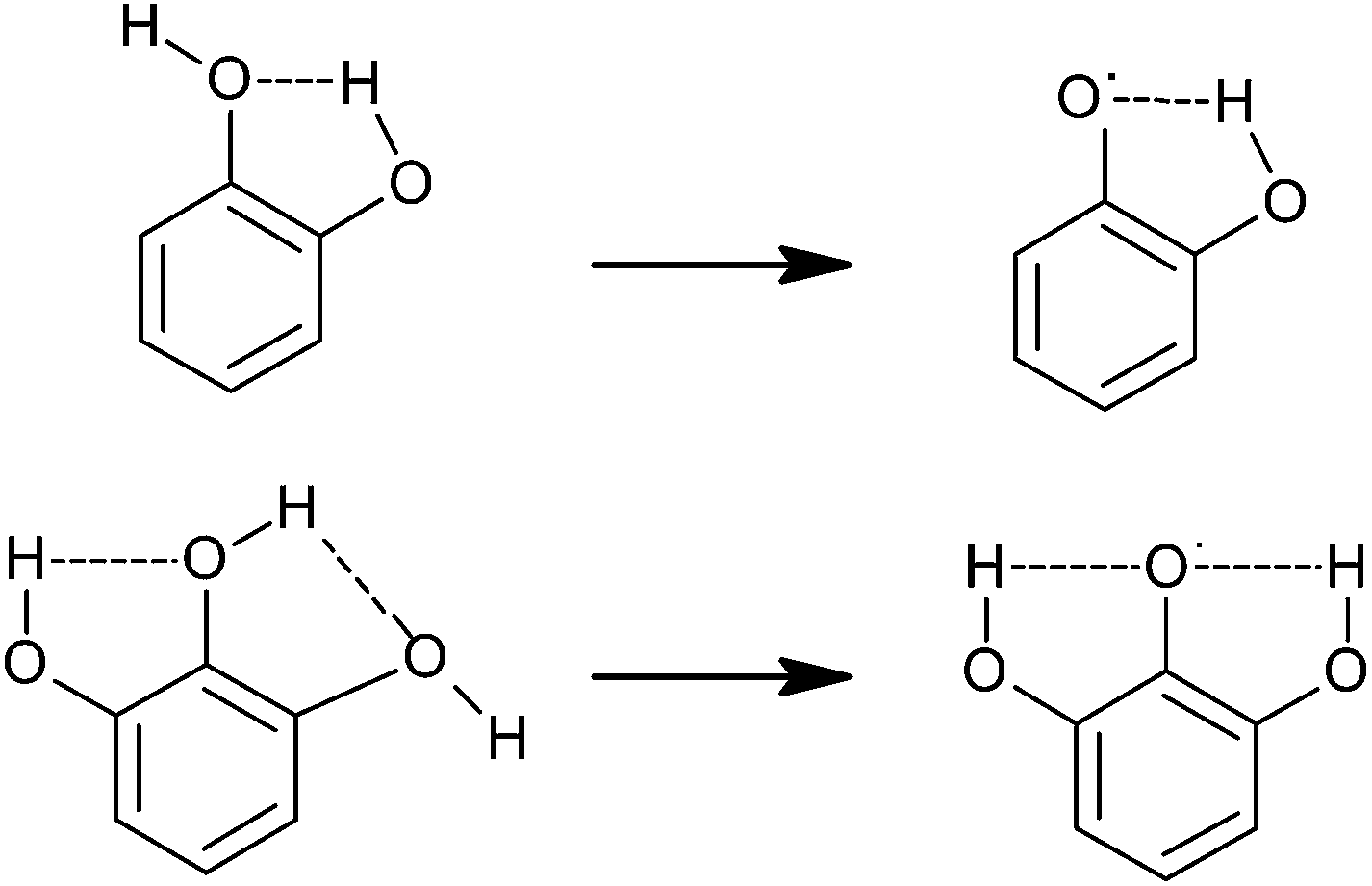 | ||
| Fig. 4 Intramolecular hydrogen bonding in the catechol (top) and pyrogallol (bottom) moieties and radical stabilization.92 | ||
The nature and the position of the substituents with respect to the hydroxyl group also affect the activity of polyphenols. For instance, the easily ionizable carboxylic group contributes toward the efficient hydrogen donation tendency of phenolic acids.83 Gallic acid showed a higher antioxidant activity than pyrogallol,88 thus proving a beneficial influence of carboxylate on the antioxidant activity of phenolic acids. However, Sroka and Cisowski did not observe any influence of carboxylate on the antioxidant or antiradical activities of gallic acid.75
Thus, utilizing the findings of various studies, we can attribute the high antioxidant activity of gallic acid to a blend of multiple factors, as illustrated in Fig. 5.
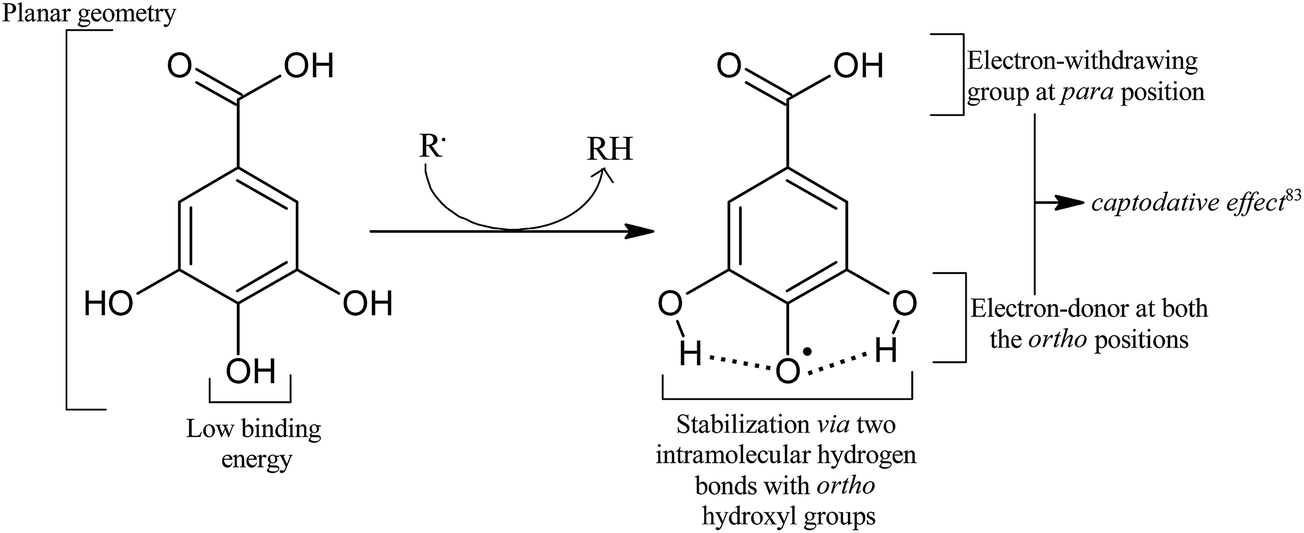 | ||
| Fig. 5 Factors responsible for an efficient antioxidant activity of the gallic acid molecule. | ||
Multiple studies have obtained experimental and simulated IR spectra of GA.76,82,93 The experimental and calculated frequencies differ quite significantly due to strong intermolecular H-bonding (Fig. 6).
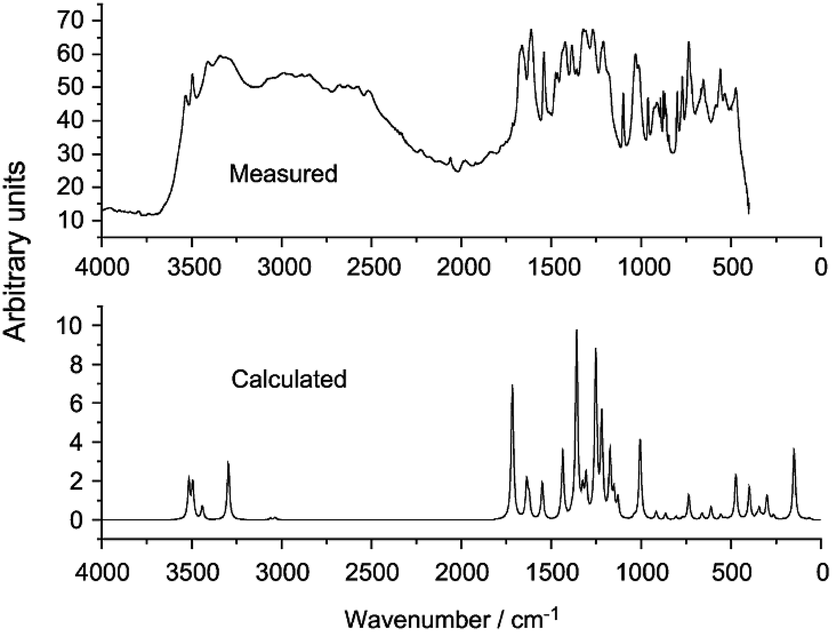 | ||
| Fig. 6 Experimental (top) and calculated (bottom) IR spectra of gallic acid.76 | ||
Gallic acid possesses four acidic protons with pKa values of 4.0 (–COOH group), 8.7, 11.4, and >13 (–OH groups).94 The first three pKa values for gallate free radical, derived from gallic acid, are ∼4 (–COOH group), 5 and 10 (–OH groups).95,96
The three hydroxyl groups attached to the aromatic ring are prone to oxidation, resulting in the formation of hydrogen peroxide, quinones, and semiquinones.96,97 In a spectrophotometric study,98 gallic acid and its analogues were shown to be rapidly oxidized by atmospheric oxygen at pH > 7. Previously, at a pH range of 2–7, the oxidation was found to be irreversible, and the authors proposed a two-electron oxidation scheme leading to the production of quinoid structures in acidic media.99
2. Dual edge sword behavior
2.1. Antioxidant activity
| R˙ + ArOH → RH + ArO˙ | (1) |
| R˙ + ArOH → R− + ArOH+˙ → RH + ArO˙ | (2) |
The two specific molecular properties responsible for deciding which mechanism would be most favored are bond dissociation enthalpy (BDE) for the O–H bond and ionization potential (IP). According to Wright et al., both the mechanisms should always occur in parallel, simply with different rates.89 Analysis of several phenolic antioxidants using density functional theory suggested the prevalence of HAT mechanism.89
Using DFT calculations, the dependence of the mode of action on the nature of the reacting free radical and the polarity of the environment was demonstrated by considering the involvement of five different mechanisms viz. HAT, SET, sequential proton loss electron transfer (SPLET), sequential double proton loss electron transfer (SdPLET) and radical adduct formation (RAF).104 In aqueous medium, the SPLET and SdPLET prevail for the HO˙ and HOO˙ radical scavenging reactions of gallic acid, respectively. While in non-polar medium, the major mechanism is HAT for HOO˙ radical and both HAT and RAF for HO˙ radical, a prior in silico study on propyl gallate supported the dominance of the HAT mechanism, regardless of the polarity of the solvent or the nature of the free radical.105
Theoretical analysis of four phenolic acids i.e. gallic acid (GA), methyl gallate (MG), ethyl gallate (EG), and 4,5-dihydroxy-3-methoxy benzoic acid (OME) yielded the following trend in BDE and IP values: GA > OME > MG > EG (BDE) and GA > MG > EG > OME (IP),100 suggesting a higher antioxidant potential of EG. However, the conversion to carboxylate (by proton dissociation) results in a significant lowering of the BDE value of GA due to electron donating effect of the carboxylate group,94 thus resulting in greater antioxidant efficiency of gallic acid as compared to its ester derivatives.
Radicalization of GA yields gallic acid radical in which the unpaired electron spin density is delocalized over the entire molecule with the largest portion centered on the three carbon atoms bound to the hydroxyl groups.78,83 Two radicals, 3-OH (5-OH) and 4-OH, are possible by the abstraction of a hydrogen atom from the corresponding meta or para hydroxyl groups. The para O–H bond is found to be the weakest of all, with the lowest binding energy value.83,100,106 It is believed that the presence of an electron withdrawing group (–COOH) at the para position and an electron donating group (–OH) at the ortho position augments the stability of the free radical by resonance.83 Moreover, the resulting radical (4-OH radical) is stabilized by two hydrogen bonds (2.152 Å) on either sides. On the contrary, DFT calculations at the hybrid HF/B3LYP/6-31G(d,p) level gave almost the same BDE for meta and para groups.107
At a concentration of 4.17 mM, GA exhibited 43.9% and 60% scavenging effects on DPPH and H2O2 radicals, respectively.108 Moreover, out of various water soluble phenolic acids, the strongest H2O2 and DPPH radical scavenging activity was exhibited by gallic acid and pyrogallol.75 Although GA exhibited a strong H2O2 and HO˙ radical scavenging power,108,75 it showed rather no O2˙− scavenging activity.20 Gallic acid and n-alkyl gallates were shown to scavenge another reactive species, HOCl, at low concentrations.111 Thus, GA is able to protect α1-antiproteinase against inactivation by HOCl molecule. It also decreases the peroxidation of ox brain phospholipids.112
To further support the efficiency of GA in preventing the detrimental effect of hydroxyl radicals, a rate constant of the order of 1010 M−1 s−1 has been obtained for the diffusion-limited reaction of GA with the HO˙ radical, both in polar and non-polar media.104,113,114 Antioxidant capacity of GA and its ester derivatives has also been reported against a number of reactive oxygen and nitrogen species, including peroxyl, azide, and hydroperoxyl radicals.104,105,115
GA and PGA protect peroxidation of lipids, using free radical scavenging activity and lipid peroxidation inhibitory activity. This characteristic has been supported by a study in which the two compounds scavenge the DPPH radical by a hydrogen donating mechanism and are more effective than Vitamin E itself.20 Even though they exhibit strong inhibition against lipid peroxidation, they do not scavenge O2˙− radical. Similar anti-lipid-peroxidative effect with no antiradical potential has been reported for a flavonol compound as well.20
The shelf life of processed food is of prime importance, both for the producer as well as the consumer. It is dramatically affected by the action of oxidative rancidity. Oxidative rancidity involves the autoxidation of fatty acids in the oxygenated environment, releasing off-flavors and undesirable compounds that can alter the sensory and nutritional characteristics of food, proving injuries to health.118 Since the preservation of shelf life as well as nutritional qualities is of vital importance, this deterioration is a major problem. Autoxidation, a slow, radical process, begins via a chain reaction with induction, propagation and termination steps. The alkyl radicals, formed in the induction period, start the propagation by reacting with oxygen to form hydroperoxides and peroxide radicals. Ultimately, the association of two radicals results in a stable adduct formation, marking the termination step.109
The use of antioxidants as food additives can arrest the oxidative deterioration of lipids,118 which in turn may lead to prevention of certain diseases such as cancer, hypertension and vascular diseases.8,118 These antioxidants include both natural antioxidants such as Vitamin E, rosemary extracts, flavonoids, as well as synthetic antioxidants such as butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA) and propyl gallate. However, the adverse side effects of synthetic antioxidant food additives, often used by food manufacturers, raise some health concerns. Thus, limitations over the use of synthetic antioxidants in food and the protective behavior of natural antioxidants together give rise to the emerging interest in using natural antioxidants.119,120
In a study by Jacobsen et al.,121 lipid hydroperoxide level in mayonnaises and effect on flavor in the presence and absence of gallic acid was investigated. As compared to the reference mayonnaises, the amount of free radicals and lipid hydroperoxides is much less in the gallic acid containing mayonnaises. These lower levels of free radicals, along with a rather increased intensity of fishy, rancid, and metallic off-flavor, implies an elevated production of secondary oxidation products due to a rapid decay of lipid hydroperoxides.121 In a previous study, Halliwell et al. proposed that because of the ability of GA to reduce metal ions, it is capable of catalyzing the decomposition of lipid hydroperoxide.122 The binding of the gallates to the lipid membrane has also been proposed to be the prime factor affecting their antioxidant activity.41
Zhang et al. proposed that solubility, a parameter that affects the mobility of antioxidants between membranes, plays a vital role in determining the antioxidant properties of phenolic acids with respect to lipid peroxidation.90 Phenolic acids, being more hydrophilic, are thus less effective than Vitamin E (lipid soluble). At the physiological pH, the hydrophilic carboxylic group is easily ionized, hence phenolic acids are believed to exhibit their antioxidant effect in the aqueous phase.123 But since biomembranes suffer a continuous attack by free radicals generated in the aqueous phase of cellular and subcellular fractions, it is essential to prevent the initial reaction between these aqueous radicals and membrane phospholipids. Thus, the aqueous phase antiradical activity of phenolic acids must be considered as a fruitful method for retarding the peroxidation of membrane phospholipids.92
 | ||
| Fig. 7 Naturally occurring derivatives of gallic acid. | ||
The widely used food antioxidant additives i.e. methyl gallate (MG), lauryl gallate (LG), and propyl gallate (PG), are manufactured using gallic acid. GA and its derivatives are therefore present in the diet. PG, MG and LG were shown to scavenge hypochlorous acid (HOCl) and trichloromethyl peroxyl radical (CC13O2˙). All three could also decrease the peroxidation of ox brain phospholipids, better than GA.112
Gallate esters are efficient scavengers of the DPPH radical both in the ethanol solution and in the liposome.87 Along with the importance of the presence of free hydroxyl groups, DPPH scavenging assay suggested that the optimal chain length for gallic acid esters to act as strong antioxidants is four carbon atoms in the aliphatic chain.135,136 In another study,87 a theoretical analysis was able to explain the activity difference between molecules 1 to 4 (Fig. 8) in terms of O–H bond dissociation energy (BDE), but the lower activity of ester derivatives (MG, PG, OG and LG), in spite of having lower BDEs, could not be explained. Furthermore, Kasture et al. demonstrated the poorest ability of triacetoxy gallic acid to scavenge free radicals.135
 | ||
| Fig. 8 Structures of gallic acid derivatives. | ||
Even though phenolic acids have been proven to possess strong antiradical and antioxidant activities, a finite therapeutic success of natural antioxidants might be associated with their limited distribution throughout the body and their inherent difficulties to attain the target sites.86 Thus, the poor bioavailability and solubility of phenolic compounds in the cell membrane render their antioxidant properties less evident in vivo as compared to in vitro.137 Hence, increasing the lipophilicity of water soluble antioxidants holds great therapeutic potential.111 Lauryl gallate is one such example which exhibits antioxidant activity in the prevention of mitochondrial lipid peroxidation.138 Methyl gallate is another naturally occurring antioxidant having anticarcinogenic and antimicrobial properties.
 | ||
| Fig. 9 Structure of phenolic antioxidants derived from gallic acid (1) and caffeic acid (2).86 | ||
Another hybrid compound (GA–LA) was prepared using gallic acid and linoleic acid (LA).141 GA–LA exhibited a higher tyrosinase inhibition than either of the two parent molecules.142 It was found to be very effective against human and mouse melanoma and lung cancer cell, much efficient than either GA or LA.
2.2 Prooxidant behavior: Fenton chemistry
The simultaneous presence of H2O2 and a transition metal in the reduced form (Fenton type systems) may result in the formation of a strong oxidant, hydroxyl radical, according to the following reaction,| Fe2+ + H2O2 → Fe3+ + HO− + HO˙ | (3) |
Hydroxyl radical scavenging efficiency can thus be used as an indicator of antioxidant activity.
In general, depending on the stability constants with Fe(II) and Fe(III), the iron chelators may either boost or inhibit the generation of HO˙ radicals.33 The antioxidant activity of GA is thus considerably influenced by the presence of transition metals. Although it is a free radical scavenger, GA may either inhibit or promote free radical production due to metal chelation.33 It was shown to exhibit a slight prooxidative effect in the presence of Fe(II) or Fe(III).143 In general, several in vitro studies have shown that GA promotes HO˙ radical production in iron containing systems, whereas it acts as an HO˙ radical scavenger in the absence of iron.33,108,144,145
A steady state concentration of Fe(II) has been observed in the Fe(III)/H2O2 system as well.146
| Fe3+ + H2O2 → Fe2+ + HO2˙ + H+ | (4) |
| Fe3+ + HO2˙ → Fe2+ + O2 + H+ | (5) |
Another determinant of the antioxidative activity of GA in Fenton-type systems containing Fe(III)/H2O2 is GA concentration. At low concentration (GA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe < 2:1), prooxidative activity dominates due to reduction of Fe3+ to Fe2+, leading to HO˙ radical production, whereas, at higher concentrations (GA:Fe > 2:1), the overall effect is antioxidative due to marked scavenging of HO˙ radicals by GA.108,33 The prooxidative action of GA at lower concentration was ascribed to a weak metal-chelating effect and a strong reducing ability. However, the reducing power increases with increasing concentration and hence it inhibits the oxidation process by scavenging the hydroxyl radical.108
:Fe < 2:1), prooxidative activity dominates due to reduction of Fe3+ to Fe2+, leading to HO˙ radical production, whereas, at higher concentrations (GA:Fe > 2:1), the overall effect is antioxidative due to marked scavenging of HO˙ radicals by GA.108,33 The prooxidative action of GA at lower concentration was ascribed to a weak metal-chelating effect and a strong reducing ability. However, the reducing power increases with increasing concentration and hence it inhibits the oxidation process by scavenging the hydroxyl radical.108
Direct hydroxyl radical scavenging assay also shows a similar trend i.e. excess gallic acid with respect to iron results in significant radical scavenging, whereas below GA:Fe: 2:1, iron chelation becomes the dominating factor leading to prooxidant behavior.33
With peroxidases, GA again shows a dual behavior depending on the presence or absence of H2O2. In the absence of H2O2, GA causes cell injury via the prooxidative effect, whereas in its presence, GA inhibits the formation of oxidative species in the peroxidase cycle of peroxidases.147
Although multiple in vitro studies have demonstrated the behavior of gallic acid in Fenton-type systems, it is necessary to note that an in vitro analysis alone cannot establish an overall in vivo response.
3. Chelation with metal ions
Gallic acid and tannic acid affect the bioavailability of certain key minerals such as iron, zinc and calcium, by forming insoluble complexes with them.58,148,149 Among the various metal ions present in the body, iron is the most abundant transition metal, with the dietary intake being in the ferric form.150 Thus, the complex forming tendency of polyphenols may lead to antinutritional effects of iron, causing its deficiency.45 This has led to a number of ongoing in vitro and in vivo studies involving iron biofortification, an approach to counterbalance the inhibitory effect of polyphenols on iron absorption.151–154 In an attempt to distinguish the polyphenols that inhibit iron uptake, Hart and Glahn found that gallic acid is among those polyphenols that enhance iron uptake in Caco-2 cells.155 Nevertheless, extensive data is required to fully decipher the net effect of such polyphenols on iron bioavailability.Accumulation of iron in the pre-frontal cortex and variations in the distribution of ferritin, resulting in enhanced ROS production, evolves into degenerative changes leading to Alzheimer's and Parkinson's disease.156,157 Utilizing the chelation potential of hydroxyl groups, polyphenols can inhibit ferrous ion catalyzed DNA damage either by binding with the Fe(II) ion or by stabilizing the Fe(III)–polyphenol complex formed by autooxidation in the presence of oxygen. Hence, the ferrous ion is either unable to catalyze HO˙ radical generation or is oxidized to ferric which does not promote Fenton reaction. Kinetic study of the autooxidation of Fe(II) complexed with some galloyl containing polyphenols yielded a positive correlation between the rate of oxidation and the antioxidant property of the polyphenol.158 The rate of autooxidation of gallate–Fe(II) was ten times faster than that of the catecholate analogues and is attributed to a stronger binding affinity of gallates. A high chelating potential of galloyl moiety tends to inhibit the ability of Fe to promote ROS formation through catalysis of hydrogen peroxide decomposition and Fenton type reactions,159,160 hence preventing the deleterious effects of oxidative stress. The use of iron chelators, such as gallocatechins, to control the concentration of free iron, can thus help in eliminating the Fenton chemistry contribution to neurodegenerative diseases.156 Hence, it is of vital importance to study the interaction between iron and GA.
Gallic acid, being a strong chelating agent, forms highly stable complexes with Fe(III).161–163 It has also been claimed that the complex formation with Fe(III) leads to the oxidation of GA to hydroxyquinone with a simultaneous reduction of Fe(III) to Fe(II).164 Multiple studies84,165 have shown that the complexation process is pH dependent, proceeding from pH = 3 and continuing till pH = 9 with increasing degree of chelation. In a 1:1 complex, iron is bound to the two adjacent hydroxyl groups of the GA molecule. Additionally, GA and its azo derivatives were found to form stable complexes with ratios varying from 1:1 to 1:4.163 A prior study stated that at a pH range of 4–6, a 1:3 complex is formed with ferric ions.166
In the presence of ferrous sulfate, a bathochromic shift in the UV-Vis spectra of gallic acid has been observed.167,168 The shift is accompanied by the appearance of a new peak in the visible region, indicating the formation of a GA–Fe(II) complex. Kinetic study further indicated the formation of a 1:1 complex following second order kinetics.168
The complexation of Fe(III) with gallocatechins was reported by Jovanovic et al.156 A 1:2 stoichiometry for Fe(III) complexes with gallocatechins and with methyl gallate, and 1:3 for that of Fe(III) and catechin (Table 1) was observed. Further, the value logK ≈ 27 was obtained for the formation of Fe(III)-bis(gallocatechin) at pH 7. Analogously, a molar ratio of 1:2 was shown for Fe(III) complex with gallic acid at pH 7, with the value logK = 34.33 It is because of the high stability of these complexes that intake of tea may hinder the absorption and bioavailability of iron.169 However, a net effect on iron absorption is determined by a number of dietary elements such as ascorbic acid, which can reverse the inhibitory effect of polyphenols on heme iron absorption.170
| Complex | n |
|---|---|
| Fe(III) (catechin) | 3 |
| Fe(III) (epigallocatechin gallate) | 2 |
| Fe(III) (epigallocatechin) | 2 |
| Fe(III) (epicatechin gallate) | 2 |
| Fe(III) (methyl gallate) | 2 |
| Fe(III) (catechol) | 3 |
Other than Fe, GA forms complexes with various other metal ions too.171,172 A complex with neodymium, having a stoichiometry of 1:2, has been obtained as a solid.173 Lanthanide complexes with gallic acid have been obtained with the general formula Ln(C7H5O5)(C7H4O5)·nH2O (n = 2 for La–Ho; n = 0 for Er–Lu).84 Moreover, GA and its azo derivatives form stable complexes with transition metal ions such as Fe(III), Mn(II), Co(II), Ni(II), Cu(II), Zn(II) and Cd(II).84,163 Some of these complexes showed notable antimicrobial activity as well.84 Complexation of Cu(II), both with gallic acid and epigallocatechin gallate, has been demonstrated to be pH dependent with respect to the stoichiometry of the resultant complex.174 Utilizing the efficient chelating potential of gallic acid, GA-capped nanoassemblies have been successfully prepared and tested for the detection of Pb(II) in a green tea sample.175
Recently, Nath et al. synthesized some potential metal based anticancer drugs viz. triorganotin(IV) hydroxycarboxylates and screened them against various cancer cell lines.176 The gallic acid complexes, n-Bu3Sn(GA) and Ph3Sn(GA), emerged as the most potent anticancer and anti-inflammatory agents, respectively. The anticancer activity was primarily imparted to ROS generation by triorganotin(IV) hydroxycarboxylates, ultimately leading to apoptosis in cancer cells.176
The ability to form complexes with transition metals with the advent of intense blue color (with iron) has been utilized in the determination of the gallocatechin content of beverages, like beer and tea.156 Additionally, gallic acid, because of its blue colored complex with iron, proved to be of prime importance in the production of writing inks (iron gall inks), which primarily contain a mixture of gallic acid, ferrous sulfate and gum arabic.167,177 A rich blue color is obtained in the presence of oxygen and an excess of ferrous ions.178
In water systems, including ground waters, surface waters and municipal wastewaters, high concentrations of iron are often associated with organic matter of natural origin. The humic substances in natural waters retard the oxidation of Fe(II) ions,179–181 due to complexation. In addition, it has been observed that the humic materials stabilize this iron and enhance its nutritive availability for the growth of aquatic species.182,183 Similarly, a number of model organic compounds having a similar structural features as these humic substances, can also modify the rate of oxidation of Fe(II) ions.181 In this context, gallic acid and pyrogallol were shown to completely inhibit Fe(II) oxidation, while vanillic acid, vanillin, phenol, resorcinol, synergic acid, and histidine had no effect on its oxidation rate.184 Especially tannic acid could stabilize high levels of Fe(II) for several days. Furthermore, galloyl moiety containing compounds were able to rapidly reduce Fe(III). Thus, the organic species of natural origin such as tannic acid, gallic acid and pyrogallol are capable of maintaining sufficiently high levels of dissolved Fe(II) concentration under aerobic aquatic conditions, due to the formation of oxidation resistant complex with Fe(II).
However on the downside, the presence of humic substances in groundwater can interrupt the water quality management processes. In order to remove iron from groundwater, water treatment plants employ oxidation of Fe(II) ions followed by sedimentation and filtration of ferric hydroxide formed. The existence of oxidation resistant complexes of Fe(II) can thus obstruct these techniques.184
4. Potential clinical applications
4.1 As anticancer agent
A number of studies have demonstrated the potential anticancer activity of gallic acid and its derivatives, both in vivo and in vitro. It is not the antioxidant, but rather the prooxidant action of gallate compounds, which is responsible for their potent anticancer and apoptosis inducing properties. Reactive oxygen species generated by gallates have been asserted to be responsible for the apoptotic and necrotic cell death.185,186 There has been ample evidence claiming that gallates induce apoptosis selectively in fast growing tumor cells, leaving the healthy cells intact.25,29,108,185–192 This selective cytotoxicity towards cancer cells was proposed to be due to the ability of normal cells to resist GA induced apoptosis by the release of some inhibitors, unlike tumor cells which could not produce sufficient amounts of these inhibitors.189 Thus, cancer cells are more sensitive towards GA induced apoptosis than normal healthy cells.Anticancer activity of GA has been reported in a variety of cancer cells, such as, leukemia,29,30,32,188,192–197 prostate cancer,24,190,198–202 lung cancer,203–207 stomach and colon cancer,23,191,192 lymphocyte proliferation,31,208 breast, cervical and esophageal cancer.187,209
Several studies have provided evidence that the inhibitory effect of GA on carcinogenesis is mediated through the regulation of multiple signaling pathways.187 Depending on the cell lines used, GA may induce cell death or cell cycle arrest in different manners.210 But still a common mechanism is associated with oxidative stress, derived from reactive oxygen species, mitochondrial dysfunction and an increase in intracellular Ca2+ level. Considering that GA induced cell death is mediated by ROS and intracellular Ca2+, Isuzugawa et al.189 found that the ROS generated by GA is hydrogen peroxide, which induced cell death in dRLh-84 (rat hepatoma) cells. Since catalase is responsible for H2O2 degradation, the normal cells are insensitive to the mentioned H2O2 generation which can be dealt by the catalase enzyme. However, due to lack of proper protection system in tumor cells, they are incapable of defending themselves from the deleterious effects of H2O2 resulting in cell death.189
Fenton chemistry, involving HO˙ radical production by H2O2 and Cu(II), is generally considered to be the cause of oxidative DNA lesion in tumor cells. This copper mediated DNA damage is initiated by the preferential coordination of the Cu(II) ion to N7 of the guanine base of DNA,211 followed by the reduction of Cu(II) to Cu(I) by gallates.186 Cu(I) ion then reduces molecular O2 to superoxide anion, and then to hydrogen peroxide with the formation of Cu(II). The interaction between hydrogen peroxide and DNA bound copper then results in the production of hazardous hydroxyl radicals,212 which cause DNA strand breaks and pronounced degradation of guanine bases along with the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG), a guanine adduct at the C-8 position, as a marker of oxidative DNA damage.186 Propyl gallate resulted in an increased oxidative DNA lesion in human leukemia cell line HL-60, but not in HP100, which is a hydrogen peroxide resistant cell line derived from HL-60.213 The HP100 cells exhibit 18 times higher catalase activity as compared to that in HL-60 cells.214 Apart from Cu(II), GA has also been shown to induce DNA damage in the presence of Fe(III) complexes such as Fe(III) EDTA or Fe(III) ADP.213 Fe(III) complex-mediated DNA damage is also related to Fenton chemistry. Since serum, tissue and cellular copper levels in cancer cells are much elevated in various malignancies, cancer cells are perhaps more subjected to electron transfer between copper ions and polyphenols, resulting in ROS formation.215
In addition to ROS generation, these compounds can also induce apoptosis in cancer cells by inhibiting protein tyrosinase kinases (PTKs), one of the proteases vital for normal cell processes. Gallates have been shown to have excellent inhibitory activity against PTKs.216 GA derivatives displayed much higher inhibiting activity than widely used PTK inhibitors.217 Lauryl gallate for example proved to be a good inhibitor of human spleen PTK.
GA administration could also inhibit metastasis formation and growth.218 P-selectin is a protein which is expressed by several metastatic pancreatic tumor cells219 and has a role in metastasis by promoting tumor growth. P-selectin deficiency is thus accompanied by reduced implanted carcinoma cells growth and reduced metastasis formation.220 GA has been reported to bind and antagonizing P-selectin under static and dynamic conditions, resulting in beneficial effects in conditions such as coronary artery disease, thrombosis, and cancer. Moreover, the required concentration is easily attainable by moderate wine, green tea, or cocoa consumption.219
The dependence of the action of gallic acid on cell cycle has been argued upon in numerous studies. Sakaguchi et al.,30 using flow cytometric analysis and agarose gel electrophoresis, observed an inter nucleosomal breakdown of chromatin DNA in HL-60RG cells but not in dRLh-84, HeLa, and PLC/PRF/5 cells; and the mechanism was found to be independent of the cell cycle. Whereas in colon adenocarcinoma cells,191 GA was found to hinder with the G2/M phase. Similarly in human bladder carcinoma cells, GA prompted G2/M cell cycle arrest involving molecular alterations in cell cycle regulatory proteins.205 On the contrary, in the studies by Inoue et al.,185 Faried et al.187 and Hsu et al.,221 no such phenomena was observed. Although GA had no effect on the cell cycle profile, it increased the number of apoptotic cells in a time dependent manner.187 According to still another study,195 GA induces cell cycle perturbation in the G1 phase. Meanwhile, multiple studies205,222,223 have demonstrated the effect of GA on cancer cell proliferation by arresting cells at the G2/M phase. GA downregulated the activities of cyclin B1, Cdc2 and Cdc25C, which are essential for G2/M transition. Thus, it is being increasing implied that GA can block proliferation of cancer cells in culture via apoptosis and/or cell cycle arrest.24,198
GA inhibits cell growth of prostate cancer (PCa) cell lines. Highly significant cytotoxicity was shown in PCa DU145 cells by the activation of pre-existing apoptotic processes and cell cycle arrest machinery.199,202 With respect to its potency against prostate cancer, both anticancer and cancer chemopreventive effects in human PCa DU145 cells in culture and TRAMP model, respectively, have been demonstrated.198,201
GA dose dependently decreased the growth and induced cell death in Calu-6 and A549 lung cancer cells.206 Lung cancer cells were much more susceptible to growth inhibition by gallic acid as compared to normal human pulmonary fibroblast (HPF) cells. GA-induced lung cancer cell death has been related to ROS increase as well as GSH depletion.206,207 Similarly, GA-induced HeLa cell death was accompanied by a significant increase in ROS and GSH depletion.209 Introduction of gallic acid into human colon adenocarcinoma COLO 205 cells and stomach cancer KATO III cells resulted in growth inhibition and induction of apoptosis.192
Gallic acid-based indanone derivatives have also been shown to exhibit substantial anticancer activity against various human cancer cell lines, such as KB403 (oral and mouth cancer cells), WRL68 (liver cancer cells), Caco2 (colon cancer cells), HepG2 (liver cells) and MCF7 (hormone dependent breast cancer cells).36
Besides, GA is one of the major phenolic acids present in grape seed extract (GSE). Due to the strong antioxidant activity shown by GSE, it is considered to be highly beneficial for the treatment of various diseased conditions.28,224,225 A number of in vivo and in vitro models have been utilized by numerous studies in order to demonstrate the anticancer effects of GSE.190,201,226–235 Being one of the main phenolic compounds in GSE, gallic acid may be a major contributory factor responsible for these therapeutically beneficial activities. Accordingly, Veluri et al. identified gallic acid as one of the most active GSE constituents contributing significantly in growth inhibition, death and apoptosis in human PCa DU145 cells.24 Following this, Agarwal et al. further investigated the efficacy and associated mechanism of GA.198 Gallic acid treatment resulted in a strong cell growth inhibition, cell cycle arrest, and apoptotic death in DU145 cells. Caspases are a group of proteases which are responsible for programmed cell death. Activation of caspases is thus associated with the process of apoptosis.236 Both caspase-dependent and caspase-independent pathways have been suggested for the antitumor activity of GSE derived gallic acid.24,198,237
Furthermore, GA purified from pomegranate peel extract, was found to induce apoptosis in the human alveolar epithelial A549 cell line, through the elevation of ROS and by the activation of caspase 9.238
GA is also abundantly present in Toona sinensis, one of the most popular vegetarian cuisines in Taiwan.239 It has been demonstrated that T. sinensis exerts antiproliferative effects on cultured human HL-60 cells in vitro and in vivo through the induction of cell-cycle arrest and apoptosis. Later on, using the in vivo bioluminescent imaging technique, Hsiang et al. affirmed the anti-inflammatory potential of both T. sinensis and GA in mice.240 Such potent biological activities of T. sinensis have been attributed to the abundance of gallic acid which acts as its chief bioactive molecule.25,199,239,240
4.2 Other therapeutically beneficial activities
Gallic acid has been recognized to display diverse therapeutically beneficial activities. It has been shown to possess antifungal,241–244 antibacterial,244,245 antiviral,246–250 antiallergic,251 anti-inflammatory,252,253 antimelanogenic,21,254–256 antitubercular,257–259 antimutagenic,20 antiulcer,196,260 anticholesterol,261 antiobesity262 and immunomodulatory activities.263 Additionally, it displays neuroprotective,87,135,264–269 cardioprotective,22 hepatoprotective270–272 and nephroprotective272–274 potential.5. Industrial applications
GA and its esters have a diverse range of applications in industrial uses as well, such as additives in food and in cosmetics. Its ester derivatives (PG, OG and LG) are widely used in cosmetics, processed food and food packing materials to prevent oxidative rancidity and spoilage.38GA is extensively used in tanning as well as in the manufacturing of paper.292 It is used for the stabilization of collagen in the process of leather making.293 Additionally, it is employed as a source material for inks, paints, color developers and pharmaceuticals.177 The iron gall inks were one of the key writing materials used in the preparation of documents, drawings, and other written materials.294,295
In analytical research, gallic acid is being persistently used as a standard antioxidant in order to determine the phenolic content of plant extracts or any other analyte and the results are expressed in gallic acid equivalents (GAE).37,296,297 GA can also act as a non-toxic corrosion inhibitor by adsorbing on the metal surfaces. It has been suggested that it can cover the steel surface by forming an insoluble complex with ferric ions, thus inhibiting corrosion.298 Additionally, propyl gallate has been shown to be an effective stabilizing agent for the protection of biodiesel oxidation.299
6. Conclusions and perspective
Considering the increasing occurrence of oxidative stress mediated degenerative diseases, there is a pressing need to boost the body's defense system with minimal side effects. This latter requirement has popularized the use of naturally occurring compounds as potent antioxidants. In this regard, hydroxybenzoic acid derivatives have shown favorable activities. Among them, gallic acid displays the highest antioxidant potential and wide bioavailability. Moreover, the antioxidant potential can be further improved by forming a hybrid between gallic acid and another phenolic antioxidant like caffeic acid. Gallic acid and its derivatives have been gaining attention, not only because of their antiradical, but also due to their selective apoptosis inducing potential. However, it is the prooxidant aspect of this molecule that gives rise to this latter effect. The feasibility of both anti- and pro-oxidant behavior gives rise to a diverse range of biological activities, including antitumor, antimicrobial, antimelanogenic and anticholesterol. The manifold properties of gallic acid and its derivatives discussed in this review make it a highly promising therapeutic agent in preventive therapies. However, a thorough mechanistic investigation at the molecular level is of prime importance in order to overcome the limited clinical success of natural compounds.Acknowledgements
Two of the authors (B.B. and N.S.) thank the Council of Scientific and Industrial Research (CSIR), New Delhi, for Research Fellowships.References
- H. J. D. Dorman, T. A. Lantto, A. Raasmaja and R. Hiltunen, Food Funct., 2011, 2, 328–337 CAS.
- S. E. Lee, H. T. Shin, H. J. Hwang and H. J. Kim, Phytother. Res., 2003, 17, 1041–1047 CrossRef PubMed.
- K. Braekke, N. K. Harsem and A. C. Staff, Pediatr. Res., 2006, 60, 560–564 CrossRef CAS PubMed.
- M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS PubMed.
- Z. Hracsko, H. Orvos, Z. Novak, A. Pal and I. S. Varga, Redox Rep., 2008, 13, 11–16 CrossRef CAS PubMed.
- A. Trichopoulou, P. Lagiou, H. Kuper and D. Trichopoulos, Cancer Epidemiol., Biomarkers Prev., 2000, 9, 869–873 CAS.
- T. Gao, Y. Ci, H. Jian and C. An, Vib. Spectrosc., 2000, 24, 225–231 CrossRef CAS.
- L. A. Bazzano, J. He and L. G. Ogden, Arch. Intern. Med., 2001, 161, 2573–2578 CrossRef CAS.
- A. C. Argolo, A. E. Sant'Ana, M. Pletsch and L. C. Coelho, Bioresour. Technol., 2004, 95, 229–233 CrossRef CAS PubMed.
- W. B. Grant, Eur. Urol., 2004, 45, 271–279 CrossRef PubMed.
- S. Ghosal, A. Bhattacharya and S. K. Bhattacharya, in Recent Progress in Medicinal Plants, ed. D. K. Majumdar, J. N. Govil and V. K. Singh, Studium Press, 2003, vol. 8, p. 149 Search PubMed.
- İ. Gülçin, Arch. Toxicol., 2012, 86, 345–391 CrossRef PubMed.
- İ. Gülçin and Ş. Beydemir, Mini-Rev. Med. Chem., 2013, 13, 408–430 Search PubMed.
- H. Y. Zhang and L. F. Wang, Bioorg. Med. Chem. Lett., 2002, 12, 229–233 CrossRef.
- A. S. Rao, S. G. Reddy, P. P. Babu and A. R. Reddy, BMC Complementary Altern. Med., 2010, 10, 4 CrossRef PubMed.
- N. S. Khan, A. Ahmad and S. M. Hadi, Chem.-Biol. Interact., 2000, 125, 177–189 CrossRef CAS.
- W. Zheng and S. Y. Wang, J. Agric. Food Chem., 2001, 49, 5165–5170 CrossRef CAS PubMed.
- D. O. Kim, K. W. Lee, H. J. Lee and C. Y. Lee, J. Agric. Food Chem., 2002, 50, 3713–3717 CrossRef CAS PubMed.
- K. Schlesier, M. Harwat, V. Bohm and R. Bitsch, Free Radical Res., 2002, 36, 177–187 CrossRef CAS.
- A. Abdelwahed, I. Bouhlel, I. Skandrani, K. Valenti, M. Kadri, P. Guiraud, R. Steiman, A.-M. Mariotte, K. Ghedira, F. Laporte, M.-G. Dijoux-Franca and L. Chekir-Ghedira, Chem.-Biol. Interact., 2007, 165, 1–13 CrossRef CAS PubMed.
- Y.-J. Kim, Biol. Pharm. Bull., 2007, 30, 1052–1055 CAS.
- D. H. Priscilla and P. S. Prince, Chem.-Biol. Interact., 2009, 179, 118–124 CrossRef CAS PubMed.
- J. S. Giftson, S. Jayanthi and N. Nalini, Invest. New Drugs, 2010, 28, 251–259 CrossRef CAS PubMed.
- R. Veluri, R. P. Singh, Z. Liu, J. A. Thompson, R. Agarwal and C. Agarwal, Carcinogenesis, 2006, 27, 1445–1453 CrossRef CAS PubMed.
- Y. C. Chia, R. Rajbanshi, C. Calhoun and R. H. Chiu, Molecules, 2010, 15, 8377–8389 CrossRef CAS PubMed.
- U. Bhadoriya, P. Sharma and S. S. Solanki, Asian Pac. J. Trop. Dis., 2012, S833–S836 CrossRef CAS.
- H. Palafox-Carlos, J. Gil-Chávez, R. Sotelo-Mundo, J. Namiesnik, S. Gorinstein and G. A. González-Aguilar, Molecules, 2012, 17, 12657–12664 CrossRef CAS PubMed.
- N. González-Abuín, N. Martínez-Micaelo, M. Margalef, M. Blay, A. Arola-Arnal, B. Muguerza, A. Ardévol and M. Pinent, Food Funct., 2014, 5, 2357–2364 Search PubMed.
- M. Inoue, N. Sakaguchi, K. Isuzugawa, H. Tani and Y. Ogihara, Biol. Pharm. Bull., 2000, 23, 1153–1157 CAS.
- N. Sakaguchi, M. Inoue and Y. Ogihara, Biochem. Pharmacol., 1998, 55, 1973–1981 CrossRef CAS.
- K. K. Sohi, N. Mittal, M. K. Hundal and K. L. Khanduja, J. Nutr. Sci. Vitaminol., 2003, 49, 221–227 CrossRef CAS.
- K. Saeki, A. You, M. Isemura, I. Abe, T. Seki and H. Noguchi, Biol. Pharm. Bull., 2000, 23, 1391–1394 CAS.
- M. Strlic, T. Radovic, J. Kolar and B. Pihlar, J. Agric. Food Chem., 2002, 50, 6313–6317 CrossRef CAS PubMed.
- M. C. Bibby, Drugs Future, 2002, 27, 475–480 CrossRef CAS.
- V. Srivastava, A. S. Negi, J. K. Kumar, M. M. Gupta and S. P. S. Khanuja, Bioorg. Med. Chem., 2005, 13, 5892–5908 CrossRef CAS PubMed.
- H. O. Saxena, U. Faridi, S. Srivastava, J. K. Kumar, M. P. Darokar, S. Luqman, C. S. Chanotiya, V. Krishna, A. S. Negi and S. P. S. Khanuja, Bioorg. Med. Chem. Lett., 2008, 18, 3914–3918 CrossRef CAS PubMed.
- E. Damiani, T. Bacchetti, L. Padella, L. Tiano and P. Carloni, J. Food Compos. Anal., 2014, 33, 59–66 CrossRef CAS PubMed.
- J. Zhao, I. A. Khan and F. R. Fronczek, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o316–o317 CAS.
- V. Samanidou, A. Tsagiannidis and I. Sarakatsianos, J. Sep. Sci., 2012, 35, 608–615 CrossRef CAS PubMed.
- V. Schmitzer, A. Slatnar, R. Veberic, F. Stampar and A. Solar, J. Food Sci., 2011, 76, S14–S19 CrossRef CAS PubMed.
- S. Shahrzad, K. Aoyagi, A. Winter, A. Koyama and I. Bitsch, J. Nutr., 2001, 131, 1207–1210 CAS.
- K. Pyrzynska and M. Biesaga, Trends Anal. Chem., 2009, 28, 893–902 CrossRef CAS PubMed.
- J. Ma, X. D. Luo, P. Protiva, H. Yang, C. Ma, M. J. Basile, I. B. Weinstein and E. J. Kennely, J. Nat. Prod., 2003, 7, 983–986 CrossRef PubMed.
- W. Qu, A. P. Breksa III, Z. Pan and H. Ma, Food Chem., 2012, 132, 1585–1591 CrossRef CAS PubMed.
- E. Tako, S. E. Beebe, S. Reed, J. J. Hart and R. P. Glahn, Nutr. J., 2014, 13, 28 CrossRef PubMed.
- Y. Yuan, Y. Song, W. Jing, Y. Wang, X. Yang and D. Liua, Anal. Methods, 2014, 6, 907–914 RSC.
- J. Li, X. He, M. Li, W. Zhao, L. Liu and X. Kong, Food Chem., 2015, 176, 7–11 CrossRef CAS PubMed.
- E. A. Haddock, R. K. Gupta, S. M. K. Al-Shafi, K. Layden, E. Haslam and D. Magnolato, Phytochemistry, 1982, 21, 1049–1062 CrossRef CAS.
- M. A. Amerine and C. S. Ough, in Methods of Analysis of Musts and Wines, Wiley-Interscience, New York, 1980, pp. 175–199 Search PubMed.
- J. Burns, P. T. Gardner, J. O'Neil, S. Crawford, I. Morecroft, D. B. McPhail, C. Lister, D. Matthews, M. R. MacLean, M. E. J. Lean, G. G. Duthie and A. Crozier, J. Agric. Food Chem., 2000, 48, 220–230 CrossRef CAS PubMed.
- S. Chandra and E. De Mejia Gonzalez, J. Agric. Food Chem., 2004, 52, 3583–3589 CrossRef CAS PubMed.
- K. W. Lee, Y. J. Kim, H. J. Lee and C. Y. Lee, J. Agric. Food Chem., 2003, 51, 7292–7295 CrossRef CAS PubMed.
- C. Manach, G. Williamson, C. Morand, A. Scalbert and C. Remesy, Am. J. Clin. Nutr., 2005, 81, S230–S242 Search PubMed.
- R. A. Caccetta, K. D. Croft, L. J. Beilin and I. B. Puddey, Am. J. Clin. Nutr., 2000, 71, 67–74 CAS.
- E. Cartron, G. Fouret, M. A. Carbonneau, C. Lauret, F. Michel, L. Monnier, B. Descomps and C. L. Léger, Free Radical Res., 2003, 37, 1021–1035 CrossRef CAS.
- S. Shahrzad and I. Bitsch, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 705, 87–95 CrossRef CAS.
- İ. Gülçin, Z. Huyut, M. Elmastaş and H. Y. Aboul-Enein, Arabian J. Chem., 2010, 3, 43–53 CrossRef PubMed.
- M. Brune, L. Rossander and L. Hallberg, Eur. J. Clin. Nutr., 1989, 43, 547–558 CAS.
- J. M. Hodgson, L. W. Morton, I. B. Puddey, L. J. Beilin and K. D. Croft, J. Agric. Food Chem., 2000, 48, 2276–2280 CrossRef CAS PubMed.
- C. A. Van der Heijden, P. J. C. M. Janssen and J. J. T. W. A. Strik, Food Chem. Toxicol., 1986, 24, 1067–1070 CrossRef CAS.
- I. Werner, A. Bacher and W. Eisenreich, J. Biol. Chem., 1997, 272, 25474–25482 CrossRef CAS PubMed.
- R. M. Muir, A. M. Ibáñez, S. L. Uratsu, E. S. Ingham, C. A. Leslie, G. H. McGranahan, N. Batra, S. Goyal, J. Joseph, E. D. Jemmis and A. M. Dandekar, Plant Mol. Biol., 2011, 75, 555–565 CrossRef CAS PubMed.
- A. C. Neish, G. H. N. Towers, D. Chen, S. Z. El-Basyouni and R. K. Ibrahim, Phytochemistry, 1964, 3, 485–492 CrossRef.
- M. H. von Zenk, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1964, 19, 83–84 Search PubMed.
- N. Kato, M. Shiroya, S. Yoshida and M. Hasegawa, Bot. Mag., Tokyo, 1968, 81, 506–507 CrossRef CAS.
- P. M. Dewick and E. Haslam, Chem. Commun., 1968, 673–675 RSC.
- P. M. Dewick and E. Haslam, Biochem. J., 1969, 113, 537–542 CAS.
- R. A. Werner, A. Rossmann, C. Schwarz, A. Bacher, H.-L. Schmidt and W. Eisenreich, Phytochemistry, 2004, 65, 2809–2813 CrossRef CAS PubMed.
- V. Ossipov, J.-P. Salminen, S. Ossipova, E. Haukioja and K. Pihlaja, Biochem. Syst. Ecol., 2003, 31, 3–16 CrossRef CAS.
- S. Kambourakis, K. M. Draths and J. W. Frost, J. Am. Chem. Soc., 2000, 122, 9042–9043 CrossRef CAS.
- K. Li and J. W. Frost, Biotechnol. Prog., 1999, 15, 876–883 CrossRef CAS PubMed.
- N. Ishikura, S. Hayashida and K. Tazaki, Bot. Mag., Tokyo, 1984, 97, 355–367 CrossRef CAS.
- R. Saijo, Agric. Biol. Chem., 1983, 47, 455–460 CrossRef CAS.
- S. A. Singh and D. Christendat, Biochemistry, 2006, 45, 7787–7996 CrossRef CAS PubMed.
- Z. Sroka and W. Cisowski, Food Chem. Toxicol., 2003, 41, 753–758 CrossRef CAS.
- I. Mohammed-Ziegler and F. Billes, J. Mol. Struct., 2002, 618, 259–265 CrossRef CAS.
- S. M. Fiuza, C. Gomes, L. J. Teixeira, M. T. G. da Cruz, M. N. D. S. Cordeiro, N. Milhazes, F. Borges and M. P. M. Marques, Bioorg. Med. Chem., 2004, 12, 3581–3589 CrossRef CAS PubMed.
- R. Šolc, M. H. Gerzabek, H. Lischka and D. Tunega, Comput. Theor. Chem., 2014, 1032, 42–49 CrossRef PubMed.
- N. Hirun, S. Saithong, C. Pakawatchai and V. Tantishaiyakul, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o787 CAS.
- N. Okabe, H. Kyoyama and M. Suzuki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o764–o766 CAS.
- C. Cappelli, B. Mennucci and S. Monti, J. Phys. Chem. A, 2005, 109, 1933–1943 CrossRef CAS PubMed.
- S. Pardeshi, R. Dhodapkar and A. Kumar, Spectrochim. Acta, Part A, 2013, 116, 562–573 CrossRef CAS PubMed.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 4916–4922 CrossRef CAS.
- M. S. Masoud, S. S. Hagagg, A. E. Ali and N. M. Nasr, J. Mol. Struct., 2012, 1014, 17–25 CrossRef CAS PubMed.
- D. Galato, K. Ckless, M. F. Susin, C. Giacomelli, R. M. Ribeiro-do-Valle and A. Spinelli, Redox Rep., 2001, 6, 243–250 CrossRef CAS PubMed.
- J. Teixeira, T. Silva, S. Benfeito, A. Gaspar, E. M. Garrido, J. Garrido and F. Borges, Eur. J. Med. Chem., 2013, 62, 289–296 CrossRef CAS PubMed.
- Z. Lu, G. Nie, P. S. Belton, H. Tang and B. Zhao, Neurochem. Int., 2006, 48, 263–274 CrossRef CAS PubMed.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Free Radical Biol. Med., 1996, 20, 933–956 CrossRef CAS.
- J. S. Wright, E. R. Johnson and G. A. DiLabio, J. Am. Chem. Soc., 2001, 123, 1173–1183 CrossRef CAS PubMed.
- H.-Y. Zhang, Y.-M. Sun and X.-L. Wang, Chem.–Eur. J., 2003, 9, 502–508 CrossRef CAS PubMed.
- N. Nenadis, H.-Y. Zhang and M. Z. Tsimidou, J. Agric. Food Chem., 2003, 51, 1874–1879 CrossRef CAS PubMed.
- C. Siquet, F. Paiva-Martins, J. L. F. C. Lima, S. Reis and F. Borges, Free Radical Res., 2006, 40, 433–442 CrossRef CAS PubMed.
- F. Billes, I. Mohammed-Ziegler and P. Bombicz, Vib. Spectrosc., 2007, 43, 193–202 CrossRef CAS PubMed.
- H.-F. Ji, H.-Y. Zhang and L. Shen, Bioorg. Med. Chem. Lett., 2006, 16, 4095–4098 CrossRef CAS PubMed.
- S. V. Jovanovic, Y. Hara, S. Steenken and M. G. Simic, J. Am. Chem. Soc., 1995, 117, 9881–9888 CrossRef CAS.
- A. C. Eslami, W. Pasanphan, B. A. Wagner and G. R. Buettner, Chem. Cent. J., 2010, 4, 15–18 CrossRef PubMed.
- J. F. Severino, B. A. Goodman, C. W. M. Kay, K. Stolze, D. Tunega, T. G. Reichenauer and K. F. Pirker, Free Radical Biol. Med., 2009, 46, 1076–1088 CrossRef CAS PubMed.
- M. Friedman and H. S. Jürgens, J. Agric. Food Chem., 2000, 48, 2101–2110 CrossRef CAS PubMed.
- S. Gunckel, P. Santander, G. Cordano, J. Ferreira, S. Munoz, L. J. Nunez-Vergara and J. A. Squella, Chem.-Biol. Interact., 1998, 114, 45–59 CrossRef CAS.
- D. Kalita, R. Kar and J. G. Handique, J. Theor. Comput. Chem., 2012, 11, 391–402 CrossRef CAS.
- M. Leopoldini, F. Rondinelli, N. Russo and M. Toscano, J. Agric. Food Chem., 2010, 58, 8862–8871 CrossRef CAS PubMed.
- N. Nenadis, L.-F. Wang, M. Z. Tisimidou and H.-Y. Zhang, J. Agric. Food Chem., 2005, 53, 295–299 CrossRef CAS PubMed.
- M. Reis, B. Lobato, J. Lameira, A. S. Santos and C. N. Alves, Eur. J. Med. Chem., 2007, 42, 440–446 CrossRef CAS PubMed.
- T. Marino, A. Galano and N. Russo, J. Phys. Chem. B, 2014, 118, 10380–10389 CrossRef CAS PubMed.
- M. E. Medina, C. Iuga and J. R. Alvarez-Idaboy, Phys. Chem. Chem. Phys., 2013, 15, 13137–13146 RSC.
- M. Leopoldini, N. Russo and M. Toscano, Food Chem., 2011, 125, 288–306 CrossRef CAS PubMed.
- C. Giacomelli, F. S. Miranda, N. S. Gonçalves and A. Spinelli, Redox Rep., 2004, 9, 263–269 CrossRef CAS PubMed.
- G.-C. Yen, P.-D. Duh and H.-L. Tsai, Food Chem., 2002, 79, 307–313 CrossRef CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Lebensm.-Wiss. Technol., 1995, 28, 25–30 CrossRef CAS.
- N. Nenadis, O. Lazaridou and M. Z. Tsimidou, J. Agric. Food Chem., 2007, 55, 5452–5460 CrossRef CAS PubMed.
- R. Rosso, T. O. Vieira, P. C. Leal, R. J. Nunes, R. A. Yunes and T. B. Creczynski-Pasa, Bioorg. Med. Chem., 2006, 14, 6409–6413 CrossRef CAS PubMed.
- O. I. Aruoma, A. Murcia, J. Butler and B. Halliwell, J. Agric. Food Chem., 1993, 41, 1880–1885 CrossRef CAS.
- P. Dwibedy, G. R. Dey, D. B. Naik, K. Kishore and P. N. Moorthy, Phys. Chem. Chem. Phys., 1999, 1, 1915–1918 RSC.
- F. J. Benitez, F. J. Real, J. L. Acero, A. I. Leal and C. Garcia, Phys. Chem. Chem. Phys., 2013, 15, 13137–13146 RSC.
- W. Bors and C. Michel, Free Radical Biol. Med., 1999, 27, 1413–1426 CrossRef CAS.
- M. K. Hundal, S. Jnagal and K. L. Khanduja, J. Clin. Biochem. Nutr., 1997, 22, 155–161 CrossRef CAS.
- L. Li, T. B. Ng, W. Gao, W. Li, M. Fu, S. M. Niu, L. Zhao, R. R. Chen and F. Liu, Life Sci., 2005, 77, 230–240 CrossRef CAS PubMed.
- S. Mannino, S. Buratti, M. S. Cosio and N. Pellegrini, Analyst, 1999, 124, 1115–1118 RSC.
- İ. Gülçin, Innovative Food Sci. Emerging Technol., 2010, 11, 210–218 CrossRef PubMed.
- İ. Gülçin, E. Bursal, M. H. Şehitoğlu, M. Bilsel and A. C. Gören, Food Chem. Toxicol., 2010, 48, 2227–2238 CrossRef PubMed.
- C. Jacobsen, K. Hartvigsen, M. K. Thomsen, L. F. Hansen, P. Lund, L. H. Skibsted, G. Hølmer, J. Adler-Nissen and A. S. Meyer, J. Agric. Food Chem., 2001, 49, 1009–1019 CrossRef CAS PubMed.
- B. Halliwell, R. Aeschbach, J. Löliger and O. I. Aruoma, Food Chem. Toxicol., 1995, 33, 601–617 CrossRef CAS.
- E. Niki, Methods Enzymol., 1990, 186, 100–108 CAS.
- J. Relat, A. Blancafort, G. Oliveras, S. Cufí, D. Haro, P. F. Marrero and T. Puig, BMC Cancer, 2012, 12, 280 CrossRef CAS PubMed.
- G.-J. Du, Z. Zhang, X.-D. Wen, C. Yu, T. Calway, C.-S. Yuan and C.-Z. Wang, Nutrients, 2012, 4, 1679–1691 CrossRef CAS PubMed.
- G.-J. Du, C.-Z. Wang, L. W. Qi, Z. Y. Zhang, T. Calway, T. C. He, W. Du and C. S. Yuan, Phytother. Res., 2013, 27, 272–277 CrossRef CAS PubMed.
- E. Lecumberri, Y. M. Dupertuis, R. Miralbell and C. Pichard, Clin. Nutr., 2013, 32, 894–903 CrossRef CAS PubMed.
- A. L. Gray, C. A. Stephens, R. L. Bigelow, D. T. Coleman and J. A. Cardelli, PLoS One, 2014, 9, e109208 Search PubMed.
- L. Zeng, J. M. Holly and C. M. Perks, Front. Endocrinol., 2014, 5, 61 CrossRef PubMed.
- J. I. Sonoda, R. Ikeda, Y. Baba, K. Narumi, A. Kawachi, E. Tomishige, K. Nishihara, Y. Takeda, K. Yamada, K. Sato and T. Motoya, Exp. Ther. Med., 2014, 8, 59–63 CAS.
- J. H. Weisburger, A. Rivenson, C. Aliaga, J. Reinhardt, G. J. Keloff, C. W. Boone, V. E. Steele, D. A. Balentine, B. Pittman and E. Zang, Proc. Soc. Exp. Biol. Med., 1998, 217, 104–108 CrossRef CAS.
- E. S. Fiala, R. S. Sodum, M. Bhattacharya and H. Li, Experientia, 1996, 52, 922–926 CrossRef CAS.
- F. Takabayashi, N. Harada, S. Tahara, T. Kaneko and Y. Hara, Pancreas, 1997, 15, 109–112 CrossRef CAS PubMed.
- F. Takabayashi, S. Tahara, T. Kaneko and N. Harada, Biofactors, 2004, 21, 335–337 CrossRef CAS.
- V. S. Kasture, S. A. Katti, D. Mahajan, R. Wagh, M. Mohan and S. B. Kasture, Pharmacologyonline, 2009, 1, 385–395 Search PubMed.
- L. A. Savi, P. C. Leal, T. O. Vieira, R. Rosso, R. J. Nunes, R. A. Yunes, T. B. Crec-zynski-Pasa, C. R. Barardi and C. M. Simoes, Arzneimittelforschung, 2005, 55, 66–75 CAS.
- W. Mullen, B. A. Graf, S. T. Caldwell, R. C. Hartley, G. G. Duthie, C. A. Edwards, M. E. J. Lean and A. Crozier, J. Agric. Food Chem., 2002, 50, 6902–6909 CrossRef CAS PubMed.
- I. Kubo, N. Masuoka, P. Xiao and H. Haraguchi, Agric. Food Chem., 2002, 50, 3533–3539 CrossRef CAS PubMed.
- V. D. Kancheva, P. V. Boranova, J. T. Nechev and I. I. Manolov, Biochimie, 2010, 92, 1138–1146 CrossRef CAS PubMed.
- J. Milde, E. F. Elstner and J. Grassmann, Mol. Nutr. Food Res., 2007, 51, 956–961 CAS.
- N. Y. Lee, Ph. D. thesis, Chonbuk National University, Korea, 2007.
- C. Jo, J. C. Jeong, I. Y. Lee, N. Y. K. S. Kim and M. W. Byun, Food Sci. Biotechnol., 2006, 15, 317–320 CAS.
- M. Kumamoto, T. Sonda, K. Nagayama and M. Tabata, Biosci., Biotechnol., Biochem., 2001, 65, 126–132 CrossRef CAS PubMed.
- H. Iwahashi, H. Morishita, T. Ishii, R. Sugata and R. Kido, J. Biochem., 1989, 105, 429–434 CAS.
- Y. Yoshiki, K. Okubo, Y. Akiyama, K. Sato and M. Kawanari, Luminescence, 2000, 15, 183–187 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Biochem. J., 1984, 219, 1–14 CAS.
- J. Serrano, A. Cipak, J. Baoda, H. Gonzalo, D. Cacabelos, A. Cassanye, R. Pamplona, N. Zarkovic and M. Portero-Otin, Acta Biochim. Pol., 2010, 57, 193–198 CAS.
- R. P. Glahn, G. M. Wortley, P. K. South and D. D. Miller, J. Agric. Food Chem., 2002, 50, 390–395 CrossRef CAS PubMed.
- R. M. Welch and R. D. Graham, J. Exp. Bot., 2004, 55, 353–364 CrossRef CAS PubMed.
- M. J. Hynes and M. O. Coincenainn, J. Inorg. Biochem., 2001, 85, 131–142 CrossRef CAS.
- E. Tako, M. Laparra, R. P. Glahn, R. M. Welch, X. Lei, S. Beebe and D. Miller, J. Nutr., 2009, 139, 305–309 CrossRef CAS PubMed.
- E. Tako and R. P. Glahn, Int. J. Vitam. Nutr. Res., 2011, 81, 1–14 Search PubMed.
- E. Tako, M. W. Blair and R. P. Glahn, Nutr. J., 2011, 10, 113 CrossRef CAS PubMed.
- E. Tako, O. A. Hoekenga, L. V. Kochian and R. P. Glahn, Nutr. J., 2013, 12, 3 CrossRef CAS PubMed.
- J. J. Hart and R. P. Glahn, FASEB J., 2013, 27, 634.13 Search PubMed.
- S. V. Jovanovic, M. G. Simic, S. Steeken and Y. Hara, J. Chem. Soc., Perkin Trans. 2, 1998, 11, 2365–2369 RSC.
- Q. He, N. Song, F. Jia, H. Xu, X. Yu, J. Xie and H. Jiang, Int. J. Biochem. Cell Biol., 2013, 45, 1019–1030 CrossRef CAS PubMed.
- N. R. Perron, H. C. Wang, S. N. DeGuire, M. Jenkins, M. Lawson and J. L. Brumaghim, Dalton Trans., 2010, 9982–9987 RSC.
- G. K. Lopes, H. M. Schulman and M. Hermes-Lima, Biochim. Biophys. Acta, 1999, 1472, 142–152 CrossRef CAS.
- R. G. Andrade, J. S. Ginani, G. K. Lopes, F. Dutra, A. Alonso and M. Hermes-Lima, Biochimie, 2006, 88, 1287–1296 CrossRef CAS PubMed.
- A. S. Li, B. Bandy, S. S. Tsang and A. J. Davison, Free Radical Res., 2000, 33, 551–566 CrossRef CAS.
- R. B. Sorkaz and I. Mazol, J. Biosci., 2000, 49, 881–894 Search PubMed.
- M. S. Masoud, A. E. Ali, S. S. Haggag and N. M. Nasr, Spectrochim. Acta, Part A, 2014, 120, 505–511 CrossRef CAS PubMed.
- M. J. Hynes, J. Inorg. Biochem., 2001, 85, 131–142 CrossRef CAS.
- D. Knockaert, K. Raes, K. Struijs, C. Wille and J. V. Camp, LWT--Food Sci. Technol., 2014, 55, 335–340 CrossRef CAS PubMed.
- S. A. Kazmi, M. S. Qureshi and Z. Maqsood, Inorg. Chim. Acta, 1987, 137, 151–154 CrossRef CAS.
- V. Jančovičová, M. Čeppan, B. Havlínová, M. Reháková and Z. Jakubíková, Chem. Pap., 2007, 61, 391–397 Search PubMed.
- L.-L. Lu, Y.-H. Li and X.-Y. Lu, Spectrochim. Acta, Part A, 2009, 74, 829–834 CrossRef PubMed.
- Q. Ma, E. Y. Kim and O. Han, J. Nutr., 2010, 140, 1117–1121 CrossRef CAS PubMed.
- Q. Ma, E.-Y. Kim, E. A. Lindsay and O. Han, J. Food Sci., 2011, 76, H143–H150 CrossRef CAS PubMed.
- P. J. Saines, H. H.-M. Yeung, J. R. Hester, A. R. Lenniec and A. K. Cheetham, Dalton Trans., 2011, 6401–6410 RSC.
- S. Taşcioğlu, O. Şendil and S. Beyreli, Anal. Chim. Acta, 2007, 590, 217–223 CrossRef PubMed.
- M. D. Agarwal, C. S. Bhandari, M. K. Dixit and N. C. Sogani, J. Inst. Chem., 1977, 49, 124–126 Search PubMed.
- K. F. Pirker, M. C. Baratto, R. Basosi and B. A. Goodman, J. Inorg. Biochem., 2012, 112, 10–16 CrossRef CAS PubMed.
- C. Jiang, M. Ma and Y. Wang, Anal. Methods, 2012, 4, 3570–3574 RSC.
- M. Nath, M. Vats and P. Roy, Inorg. Chim. Acta, 2014, 423, 70–82 CrossRef CAS PubMed.
- M. A. Lutui, F. Gilard and M. Sablier, J. Mass Spectrom., 2008, 43, 1123–1131 CrossRef CAS PubMed.
- V. Jančovičová, M. Ciglanská, B. Havlínová and M. Čeppan, Chem. Listy, 2008, 102, s1010–s1012 Search PubMed.
- J. Ghosh, J. T. O'Connor and R. S. Engelbrecht, J.–Am. Water Works Assoc., 1967, 59, 878–896 Search PubMed.
- K. Komolrit, MS thesis, University of Illinois, Urbana, IL, 1962.
- T. L. Theis and P. C. Singer, in Trace Metals and Metal–Organic Interactions in Natural Waters, ed. A. Arbor, Science Publishers, Ann Arbor, 1973 Search PubMed.
- A. G. Davies, J. Mar. Biol. Assoc. U. K., 1970, 50, 65–86 CrossRef CAS.
- A. Prakash and M. A. Rashid, Limnol. Oceanogr., 1968, 13, 598–606 CrossRef CAS.
- T. L. Theis and P. C. Singer, Environ. Sci. Technol., 1974, 8, 569–573 CrossRef CAS.
- M. Inoue, R. Suzuki, T. Koide, N. Sakaguchi, Y. Ogihara and Y. Yabu, Biochem. Biophys. Res. Commun., 1994, 204, 898–904 CrossRef CAS PubMed.
- M. Yoshino, M. Haneda, M. Naruse, H. H. Htay, S. Iwata, R. Tsubouchi and K. Murakami, Toxicol. In Vitro, 2002, 16, 705–709 CrossRef CAS.
- A. Faried, D. Kurnia, L. S. Faried, N. Usman, T. Miyazaki, H. Kato and H. Kuwano, Int. J. Oncol., 2007, 30, 605–613 CAS.
- C. A. Gomes, T. G. da Cruz, J. L. Andrade, N. Milhazes, F. Borges and M. P. M. Marques, J. Med. Chem., 2003, 46, 5395–5401 CrossRef CAS PubMed.
- K. Isuzugawa, M. Inoue and Y. Ogihara, Biol. Pharm. Bull., 2001, 24, 1022–1026 CAS.
- M. Kaur, B. Velmurugan, S. Rajamanickam, R. Agarwal and C. Agarwal, Pharm. Res., 2009, 26, 2133–2140 CrossRef CAS PubMed.
- M. Salucci, L. A. Stivala, G. Maiani, R. Bugianesi and V. Vannini, Br. J. Cancer, 2002, 86, 1645–1651 CrossRef CAS PubMed.
- K. Yoshioka, T. Kataoka, T. Hayashi, M. Hasegawa, Y. Ishi and H. Hibasami, Oncol. Rep., 2000, 7, 1221–1223 CAS.
- A. J. Hou, L. Y. Peng, Y. Z. Liu, Z. W. Lin and H. D. Sun, Planta Med., 2000, 66, 624–626 CrossRef CAS PubMed.
- C. Locatelli, R. Rosso, M. S. Silva, C. A. Souza, M. A. Licinio, P. Leaf, M. L. Bazzo, R. A. Yunes and T. B. C. Pasa, Bioorg. Med. Chem., 2008, 16, 3791–3799 CrossRef CAS PubMed.
- S. Madlener, C. Illmer, Z. Horvath, P. Saiko, A. Losert, I. Herbacek, M. Grusch, H. L. Elford, G. Krupitza, A. Bernhaus, M. F. Szekeres and T. Szekeres, Cancer Lett., 2007, 245, 156–162 CrossRef CAS PubMed.
- M. N. Siddaraju and S. M. Dharmesh, Mol. Nutr. Food Res., 2007, 51, 324–332 CAS.
- W. Zhang, K. Hashimoto, G. Y. Yu and H. Sakagami, Anticancer Res., 2002, 22, 219–224 CAS.
- C. Agarwal, A. Tyagi and R. Agarwal, Mol. Cancer Ther., 2006, 5, 3294–3302 CrossRef CAS PubMed.
- H.-M. Chen, Y.-C. Wu, Y.-C. Chia, F.-R. Chang, H.-K. Hsu, Y.-C. Hsieh, C.-C. Chen and S.-S. Yuan, Cancer Lett., 2009, 286, 161–171 CrossRef CAS PubMed.
- S. Gupta, N. Ahmad, A. L. Nieminen and H. Mukhtar, Toxicol. Appl. Pharmacol., 2000, 164, 82–90 CrossRef CAS PubMed.
- K. Raina, S. Rajamanickam, G. Deep, M. Singh, R. Agarwal and C. Agarwal, Mol. Cancer Ther., 2008, 7, 1258–1267 CrossRef CAS PubMed.
- S. Yasuhara, Y. Zhu, T. Matsui, N. Tipirneni, Y. Yasuhara, M. Kaneki, A. Rosenzweig and J. A. J. Martyn, J. Histochem. Cytochem., 2003, 51, 873–885 CrossRef CAS PubMed.
- B.-C. Ji, W.-H. Hsu, J.-S. Yang, T.-C. Hsia, C.-C. Lu, J.-H. Chiang, J.-L. Yang, C.-H. Lin, J.-J. Lin, L.-J. W. Suen, W. G. Wood and J.-G. Chung, J. Agric. Food Chem., 2009, 57, 7596–7604 CrossRef CAS PubMed.
- M. Kawada, Y. Ohno, Y. Ri, T. Ikoma, H. Yuuqetu, T. Asai, M. Watanabe, N. Yasuda, S. Akao, G. Takemura, S. Minatoquchi, K. Gotoh, H. Fujiwara and K. Fukuda, Anti-Cancer Drugs, 2001, 12, 847–852 CrossRef CAS PubMed.
- T.-T. Ou, C.-J. Wang, Y.-S. Lee, C.-H. Wu and H.-J. Lee, Mol. Nutr. Food Res., 2010, 54, 1781–1790 CAS.
- B. R. You and W. H. Park, Toxicol. In Vitro, 2010, 24, 1356–1362 CrossRef CAS PubMed.
- B. R. You, S. Z. Kim, S. H. Kim and W. H. Park, Mol. Cell. Biochem., 2011, 357, 295–303 CrossRef CAS PubMed.
- H. C. Li, S. Yashiki, J. Sonoda, H. Lou, S. K. Ghosh, J. J. Byrnes, C. Lema, T. Fujiyoshi, M. Karasuyama and S. Sonoda, Jpn. J. Cancer Res., 2000, 91, 34–40 CrossRef CAS PubMed.
- B. R. You, H. J. Moon, Y. H. Han and W. H. Park, Food Chem. Toxicol., 2010, 48, 1334–1340 CrossRef CAS PubMed.
- C. D. Pellegrina, G. Padovani, F. Mainente, G. Zoccatelli, G. Bissoli, S. Mosconi, G. Veneri, A. Peruffo, G. Andrighetto, C. Rizzi and R. Chignola, Cancer Lett., 2005, 226, 17–25 CrossRef PubMed.
- Y. G. Gao, M. Sriram and A. H. Wang, Nucleic Acids Res., 1993, 21, 4093–4101 CrossRef CAS PubMed.
- Y. Li, P. Kuppusamy, J. L. Zweier and M. A. Trush, Chem.-Biol. Interact., 1995, 94, 101–120 CrossRef CAS.
- H. Kobayashi, S. Oikawa, K. Hirakawa and S. Kawanishi, Mutat. Res., 2004, 558, 111–120 CAS.
- I. Kasugai and M. Yamada, Leuk. Res., 1992, 16, 173–179 CrossRef CAS.
- S. M. Hadi, S. H. Bhat, A. S. Azmi, S. Hanif, U. Shamim and M. F. Ullah, Semin. Cancer Biol., 2007, 17, 370–376 CrossRef CAS PubMed.
- C. Palacios, C. Cespon, C. Martin de la Vega, G. Roy, A. Serrano, M. Salinas and P. Gonzalez-Porque, J. Enzyme Inhib. Med. Chem., 2002, 16, 527–533 CrossRef.
- I. Làzaro, C. Palacios, M. Gonzàlez and P. Gonzàlez-Porquè, Anal. Biochem., 1995, 225, 180–183 CrossRef.
- T. Ohno, M. Inoue and Y. Ogihara, Anticancer Res., 2001, 21, 3875–3880 CAS.
- C. C. M. Appeldoorn, A. Bonnefoy, B. C. H. Lutters, K. Daenens, T. J. C. van Berkel, M. F. Hoylaerts and E. A. L. Biessen, Circulation, 2005, 111, 106–112 CrossRef CAS PubMed.
- Y. J. Kim, L. Borsig, N. M. Varki and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9325–9330 CrossRef CAS.
- C. L. Hsu, S. L. Huang and G. C. Yen, J. Agric. Food Chem., 2006, 54, 4191–4197 CrossRef CAS PubMed.
- C. E. Caldon, R. J. Daly, R. L. Sutherland and E. A. Musgrove, J. Cell. Biochem., 2006, 97, 261–274 CrossRef CAS PubMed.
- M. Motwani, C. Rizzo, F. Sirotnak, Y. She and G. K. Schwartz, Mol. Cancer Ther., 2003, 2, 549–555 CAS.
- D. Bagchi, M. Bagchi, S. J. Stohs, S. D. Ray, C. K. Sen and H. G. Preuss, Ann. N. Y. Acad. Sci., 2002, 957, 260–270 CrossRef CAS PubMed.
- Y. Yoshimura, H. Nakazawa and F. Yamaguchi, J. Agric. Food Chem., 2003, 51, 6409–6412 CrossRef CAS PubMed.
- C. Agarwal, R. P. Singh and R. Agarwal, Carcinogenesis, 2002, 23, 1869–1876 CrossRef CAS PubMed.
- C. Agarwal, R. P. Singh, S. Dhanalakshmi and R. Agarwal, Oncol. Rep., 2004, 11, 681–685 Search PubMed.
- I. Durak, R. Cetin, E. Devrim and I. B. Erguder, Life Sci., 2005, 76, 2995–3000 CrossRef CAS PubMed.
- M. Kaur, R. P. Singh, M. Gu, R. Agarwal and C. Agarwal, Clin. Cancer Res., 2006, 12, 6194–6202 CrossRef CAS PubMed.
- I. Kijima, S. Phung, G. Hur, S. L. Kwok and S. Chen, Cancer Res., 2006, 66, 5960–5967 CrossRef CAS PubMed.
- M. C. Kowalczyk, Z. Walaszek, P. Kowalczyk, T. Kinjo, M. Hanausek and T. J. Slaga, Carcinogenesis, 2009, 30, 1008–1015 CrossRef CAS PubMed.
- H. Nomoto, M. Iigo, H. Hamada, S. Kojima and H. Tsuda, Nutr. Cancer, 2004, 49, 81–88 CrossRef CAS PubMed.
- K. Raina, R. P. Singh, R. Agarwal and C. Agarwal, Cancer Res., 2007, 67, 5976–5982 CrossRef CAS PubMed.
- R. P. Singh, A. K. Tyagi, S. Dhanalakshmi, R. Agarwal and C. Agarwal, Int. J. Cancer, 2004, 108, 733–740 CrossRef CAS PubMed.
- K. W. Singletary and B. Meline, Nutr. Cancer, 2001, 39, 252–258 CrossRef CAS PubMed.
- C. Pop and G. S. Salvesen, J. Biol. Chem., 2009, 284, 21777–21781 CrossRef CAS PubMed.
- S. W. G. Tait and D. R. Green, Oncogene, 2008, 27, 6452–6461 CrossRef CAS PubMed.
- S. Elango, R. Balwas and V. V. Padma, J. Cancer Ther., 2011, 2, 638–645 CrossRef CAS.
- P.-J. Huang, Y.-C. Hseu, M.-S. Lee, K. J. S. Kumar, C.-R. Wu, L.-S. Hsu, J.-W. Liao, I.-S. Cheng, Y.-T. Kuo, S.-Y. Huang and H.-L. Yang, Food Chem. Toxicol., 2012, 50, 3489–3497 CrossRef CAS PubMed.
- C.-Y. Hsiang, Y.-C. Hseu, Y.-C. Chang, K. J. S. Kumar, T.-Y. Ho and H.-L. Yang, Food Chem., 2013, 136, 426–434 CrossRef CAS PubMed.
- F. D. D'Auria, M. Tecca, R. Strippoli and N. Simonetti, Lett. Appl. Microbiol., 2001, 32, 220–223 CrossRef.
- P. F. Dowd, J. P. Duvick and T. Rood, Nat. Toxins, 1997, 5, 180–185 CrossRef CAS.
- J. González, J. M. Cruz, H. Domínguez and J. C. Parajó, J. Food Chem., 2004, 84, 243–251 CrossRef.
- I. Kubo, P. Xiao and K. Fujita, Bioorg. Med. Chem. Lett., 2001, 11, 347–350 CrossRef CAS.
- I. Kubo, P. Xiao and K. Fujita, Bioorg. Med. Chem. Lett., 2002, 12, 113–116 CrossRef CAS.
- M. J. Ahn, C. Y. Kim, J. S. Lee, T. G. Kim, S. H. Kim, C. K. Lee, B. B. Lee, C. G. Shin, H. Huh and J. Kim, Planta Med., 2002, 68, 457–459 CrossRef CAS PubMed.
- C. J. M. Kane, J. H. Menna, C.-C. Sung and Y.-C. Yeh, Biosci. Rep., 1988, 8, 95–102 CrossRef CAS.
- M. Uozaki, H. Yamasaki, Y. Katsuyama, M. Higuchi, T. Higuti and A. H. Koyama, Antiviral Res., 2006, 73, 85–91 CrossRef PubMed.
- H. Yamasaki, M. Uozaki, Y. Katsuyama, H. Utsunomiya, T. Arakawa, M. Higuchi, T. Higuti and A. H. Koyama, Int. J. Mol. Med., 2007, 19, 685–688 CAS.
- J. M. Kratz, C. R. Andrighetti-Fröhner, D. J. Kolling, P. C. Leal, C. C. Cirne-Santos, R. A. Yunes, R. J. Nunes, E. Trybala, T. Bergström, I. C. P. P. Frugulhetti, C. R. M. Barardi and C. M. O. Simões, Mem. Inst. Oswaldo Cruz, 2008, 103, 437–442 CrossRef CAS PubMed.
- S.-H. Kim, C.-D. Jun, K. Suk, B.-J. Choi, H. Lim, S. Park, S. H. Lee, H.-Y. Shin, D.-K. Kim and T.-Y. Shin, Toxicol. Sci., 2006, 91, 123–131 CrossRef CAS PubMed.
- N. C. Desai, A. M. Bhavsar, M. D. Shah and A. K. Saxena, Indian J. Chem., 2008, 47(B), 579–589 Search PubMed.
- M. A. Indap, S. Radhika, L. Motiwale and K. Rao, Indian J. Pharm. Sci., 2006, 68, 470–474 CrossRef CAS PubMed.
- L.-G. Chen, W.-L. Chang, C.-J. Lee, L.-T. Lee, C.-M. Shih and C.-C. Wang, Biol. Pharm. Bull., 2009, 32, 1447–1452 CAS.
- C. Locatelli, P. C. Leal, R. A. Yunes, R. J. Nunes and T. B. Creczynski-Pasa, Chem.-Biol. Interact., 2009, 181, 175–184 CrossRef CAS PubMed.
- U. Panich, T. Onkoksoong, S. Limsaengurai, P. Akarasereenont and A. Wongkajornsilp, J. Photochem. Photobiol., B, 2012, 108, 16–22 CrossRef CAS PubMed.
- S. Arunkumar, K. Ilango, R. S. Manikandan, M. Sudha and N. Ramalakshmi, Int. J. ChemTech Res., 2009, 1, 1094–1099 CAS.
- P. Sengupta, D. K. Dash, V. C. Yeligar, K. Murugesh, D. Rajalingam, J. Singh and T. K. Maity, Indian J. Chem., 2008, 47(B), 460–462 Search PubMed.
- H. M. Vagadevi, S. D. Joshi, V. P. Vaidya and G. S. Gadaginamath, Eur. J. Med. Chem., 2008, 43, 1989–1996 CrossRef PubMed.
- S. I. Abdelwahab, Int. Immunopharmacol., 2013, 16, 296–305 CrossRef CAS PubMed.
- I. Abe, T. Seki and H. Noguchi, Biochem. Biophys. Res. Commun., 2000, 27, 137–140 CrossRef PubMed.
- C.-L. Hsu, W.-H. Lo and G.-C. Yen, J. Agric. Food Chem., 2007, 55, 7359–7365 CrossRef CAS PubMed.
- D. K. Yadav, F. Khan and A. S. Negi, J. Mol. Model., 2012, 18, 2513–2525 CrossRef CAS PubMed.
- S. F. Nabavi, S. Habtemariam, M. Jafari, A. Sureda and S. M. Nabavi, Bull. Environ. Contam. Toxicol., 2012, 89, 73–77 CrossRef CAS PubMed.
- S. Bastianetto, Z. X. Yao, V. Papadopoulos and R. Quirion, Eur. J. Neurosci., 2006, 23, 55–64 CrossRef PubMed.
- I. J. Kade and J. B. T. Rocha, Neurochem. Res., 2013, 38, 761–771 CrossRef CAS PubMed.
- M. T. Mansouri, Y. Farbood, M. J. Sameri, A. R. Sarkaki, B. Naghizadeh and M. Rafeirad, Food Chem., 2013, 138, 1028–1030 CrossRef CAS PubMed.
- M. T. Mansouri, B. Naghizadeh, B. Ghorbanzadeh, Y. Farbood, A. Sarkaki and K. Bavarsad, Pharmacol., Biochem. Behav., 2013, 111, 90–96 CrossRef CAS PubMed.
- J. Sun, Y.-Z. Li, Y.-H. Ding, J. Wang, J. Geng, H. Yang, J. Ren, J.-Y. Tang and J. Gao, Brain Res., 2014, 1589, 126–139 CrossRef CAS PubMed.
- S. Jagan, G. Ramakrishnan, P. Anandakumar, S. Kamaraj and T. Devaki, Mol. Cell. Biochem., 2008, 319, 51–59 CrossRef CAS PubMed.
- S. F. Nabavi, S. M. Nabavi, S. Habtemariam, A. H. Moghaddam, A. Sureda, M. Jafari and A. M. Latifi, Ind. Crops Prod., 2013, 44, 50–55 CrossRef CAS PubMed.
- V. V. Padma, P. Sowmya, T. A. Felix, R. Baskaran and P. Poornima, Food Chem. Toxicol., 2011, 49, 991–998 CrossRef PubMed.
- S. M. Nabavi, S. Habtemariam, S. F. Nabavi, A. Sureda, M. Daglia, A. H. Moghaddam and M. A. Amani, Mol. Cell. Biochem., 2013, 372, 233–239 CrossRef CAS PubMed.
- L. Prasad, T. H. Khan, T. Jahangir and S. Sultana, Hum. Exp. Toxicol., 2006, 25, 523–529 CAS.
- Y. Kato, A. Nagao, J. Terao and T. Osawa, Biosci., Biotechnol., Biochem., 2003, 67, 1136–1139 CrossRef CAS PubMed.
- S. Arunkumar, N. Ramalakshmi, T. Saraswathi and L. Aruloly, Indian J. Heterocycl. Chem., 2006, 16, 29–32 CAS.
- V. Strippoli, F. D. D'Auria, M. Tecca, A. Callari and G. Simonetti, Int. J. Antimicrob. Agents, 2000, 16, 73–76 CrossRef CAS.
- R. Díaz-Gómez, R. López-Solís, E. Obreque-Slier and H. Toledo-Araya, LWT--Food Sci. Technol., 2013, 54, 331–335 CrossRef PubMed.
- B. M. Basavaraja, H. M. Vagdevi, L. P. Srikrishna, B. S. Shubha and V. P. Vaidya, Indian J. Heterocycl. Chem., 2008, 18, 5–8 CAS.
- B. Kalluraya, N. R. Jyothi and K. V. Sujith, Indian J. Heterocycl. Chem., 2008, 17, 359–362 CAS.
- S. Briganti, E. Camera and M. Picardo, Pigm. Cell Res., 2003, 16, 101–110 CrossRef.
- V. J. Hearing, J. Dermatol. Sci., 2005, 37, 3–14 CrossRef CAS PubMed.
- H. J. Choi, J. H. Song, L. R. Bhatt and S. H. Baek, Phytother. Res., 2010, 24, 1292–1296 CrossRef CAS PubMed.
- X. L. Qi, J. Xiu, K. R. Shan, Y. Xiao, R. Gu, R. Y. Liu and Z. Z. Guan, Neurochem. Int., 2005, 46, 613–621 CrossRef CAS PubMed.
- X. Liu, N. Yamada, W. Maruyama and T. Osawa, J. Biol. Chem., 2008, 283, 34887–34895 CrossRef CAS PubMed.
- J. A. G. Crispo, M. Piché, D. R. Ansell, J. K. Eibl, I. T. Tai, A. Kumar, G. M. Ross and T. C. Tai, Biochem. Biophys. Res. Commun., 2010, 393, 773–778 CrossRef CAS PubMed.
- C. L. Hsu and G. C. Yen, Br. J. Nutr., 2007, 98, 727–735 CrossRef CAS PubMed.
- O. Nerya, R. Musa, S. Khatib, S. Tamir and J. Vaya, Phytochemistry, 2004, 64, 1389–1395 CrossRef PubMed.
- A. Kumaran and R. J. Karunakaran, Plant Foods Hum. Nutr., 2006, 61, 1–5 CrossRef CAS PubMed.
- I. Abe, T. Seki, K. Umehara, T. Miyase, H. Noguchi, J. Sakakibara and T. Ono, Biochem. Biophys. Res. Commun., 2000, 268, 767–771 CrossRef CAS PubMed.
- V. R. Punithavathi, P. S. M. Prince, R. Kumar and J. Selvakumari, Eur. J. Pharmacol., 2011, 650, 465–471 CrossRef CAS PubMed.
- G. Ramamurthy, G. Krishnamoorthy, T. P. Sastry and A. B. Mandal, Clean Technol. Environ. Policy, 2014, 16, 647–654 CrossRef CAS.
- J. K. Jackson, J. Zhao, W. Wong and H. M. Burt, J. Mater. Sci.: Mater. Med., 2010, 21, 1435–1443 CrossRef CAS PubMed.
- M. Budnar, J. Simčič, Z. Rupnik, M. Uršič, P. Pelicon, J. Kolar and M. Strlič, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 219, 41–47 CrossRef PubMed.
- M. Budnar, M. Ursic, J. Simcic, P. Pelicon, J. Kolar, V. S. Selih and M. Strlic, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 243, 407–416 CrossRef CAS PubMed.
- A. A. Karaçelik, M. Küçük, Z. İskefiyeli, S. Aydemir, S. D. Smet, B. Miserez and P. Sandra, Food Chem., 2015, 175, 106–114 CrossRef PubMed.
- J. E. Ramirez, R. Zambrano, B. Sepúlveda, E. J. Kennelly and M. J. Simirgiotis, Food Chem., 2015, 176, 106–114 CrossRef CAS PubMed.
- J. A. Jaén, L. González, A. Vargas and G. Olave, Hyperfine Interact., 2003, 148/149, 227–235 CrossRef.
- Y.-H. Chen and Y.-M. Luo, Fuel Process. Technol., 2011, 92, 1387–1393 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |