
Glucan particles loaded with a NIRF agent for imaging monocytes/macrophages recruitment in a mouse model of rheumatoid arthritis†
Francesca Garello‡
a,
Francesca Arena‡a,
Juan Carlos Cutrinab,
Giovanna Espositoa,
Luca D'Angelia,
Federico Cesanoc,
Miriam Filippia,
Sara Figueiredoad and
Enzo Terreno*a
aMolecular & Preclinical Imaging Centres, Department of Molecular Biotechnology and Health Sciences, University of Torino, Via Nizza 52, I-10126, Torino, Italy. E-mail: enzo.terreno@unito.it; Fax: +39-011-6706487; Tel: +39-011-6706452
bININCA, UBA-CONICET, Buenos Aires, Argentina
cDepartment of Chemistry, University of Torino, Via P.Giuria 7, Torino, Italy
dDepartment of Life Sciences, FCTUC and Center for Neurosciences and Cell Biology, University of Coimbra, Coimbra, Portugal
First published on 3rd April 2015
Abstract
Glucan Particles (GPs) are hollow pseudo-microspheres (average diameter 3–5 μm) obtained from common baker's yeast Saccharomyces cerevisiae, in which mannan, lipids and proteins are removed through a process of chemical extraction. GPs shell is mostly made of 1,3-β-D-glucan that allows for their prompt in vivo uptake by immune system cells. In this work, the inner cavity of the particles has been loaded with two amphiphilic fluorescent dyes (based on cyanine for in vivo imaging purposes and rhodamine for ex vivo microscopy experiments) through a sudden change in solvent polarity that allowed the entrapment of the molecules as microemulsion. The ability of fluorescent GPs to label immune cells in vivo and report on their recruitment in inflamed sites has been successfully demonstrated in a mouse model of rheumatoid arthritis (CIA) by NIRF imaging. Besides providing the visualization of the mononuclear cell infiltration in the lesion, the fluorescent signal well correlated with the clinical score associated with the disease. Very interestingly, the signal detected in lesions with the same clinical score allowed the assessment of the time evolution (progression or remission) of the pathology.
Introduction
The development of efficient drug delivery systems represents an important challenge to deal with issues such as drug solubility, targeting, in vivo stability, clearance, and cytotoxicity.1–6Nanotechnology-based drug delivery systems are suitable candidates to overcome the need for innovative delivery, bioavailability of hydrophobic drugs, high drug payload, extended drug half-life, improved therapeutic index, and controlled release with reduced immunogenicity and toxicity.7–11
A primary challenge to define the full promise of nanoparticles-based drug delivery is the lack of optimal strategies to achieve a selective and efficient cellular targeting.12–14 The mechanism of nanoparticles uptake is dependent on many variables including the particle size, shape, outer surface composition, and the targeting approach used.15–18
Typically, the targeting strategies are classified as passive (commonly referred to the EPR effect)19,20 or active. In both cases, when the cellular target is extravascular, the carrier (if administered systemically) needs to cross the endothelium, whose permeability is normally increased by the occurrence of the disease. One of the most important issues associated with the active targeting deals with the high selectivity with which the carrier must recognize the cellular target. This step is often the crucial one in defining the therapeutic index of a drug. In almost all the pathologies, the lesions are quite heterogeneous, and different cell populations are present, thus often jeopardizing the selectivity. A possible solution can be envisaged for specific target cells. A representative example is offered by the bone marrow-derived blood-circulating cells of the immune system that have a pivotal role in both, acute/chronic and regenerative, cellular phases of inflammation. Such cells are mainly represented by monocytes/macrophages and neutrophils, both displaying phagocytic activity. These cells are recruited by inflammation sites to modulate the inflammatory response. In particular, the infiltrated monocytes exert their effect after differentiating in macrophages.
As inflammation is a common process in many diseases, targeting circulating monocytes/macrophages and using them as “Trojan Horses” to deliver drugs to treat (or diagnose by in vivo imaging modalities) diseases with a high grade of inflammation (e.g. arthritis, atherosclerosis, some cancers, neurodegenerative diseases) may be a valuable approach to deliver diagnostic/therapeutic agents in a specific way.
By virtue of the growing interest to design improved delivery systems for targeting macrophages,21 labelling blood-circulating monocytes could be a feasible approach to achieve this goal.
Several drugs/imaging agents have been already developed with the aim to target phagocytic cells; some of them act as agonist or antagonist of specific epitopes exposed on the cell surface,21–24 whereas others are based on nanosystems that can be phagocytized in vivo.25,26
However, in both cases the targeting agent can itself distribute in the inflamed lesion due to the associated leaky endothelium, thus reducing the macrophage targeting selectivity. Negative outcomes could be the diminution of the therapeutic index in case of a drug, whereas for a diagnostic agent, the extravasation of the free imaging probe may bias the outcome of the procedure owing to the generation of a unspecific signal due to the non cell-internalized agent and/or to the agent taken up by cells in situ, thereby not reporting on cell recruitment.
A possible solution to this issue, is the use of a carrier that can target circulating monocytes/macrophages, but is unable to cross the leaky endothelium. In such a way, the therapeutic/diagnostic agent will be guided into the lesion by circulating cells only.
We surmised that Glucan Particles (GPs) were potential candidates to achieve this goal. GPs are porous, hollow microspheres that are prepared from Saccharomyces cerevisiae (baker's yeast).27 They are composed of 1,3-β-D-glucan and trace amounts of chitin and have an average diameter of 2–4 microns that prevent them to pass through leaky endothelium.19,27–29
1,3-β-D-glucan acts as a strong ligand for receptor-mediated phagocytosis of monocytes/macrophages (as well as dendritic cells) expressing β-glucan receptors (dectin-1 receptor (D1) and complement receptor 3 (CR3)).27,29–33 GPs uptake has been demonstrated to be dectin-1 dependent in vitro.34–38 The ability of GPs to serve as carrier relies upon the possibility to fill up the cavity with chemicals with different physico-chemical properties. In fact, procedures for the encapsulation of charged macromolecules, amphiphilic or lipophilic molecules, and also hydrophilic small-sized chemicals (through their pre-encapsulation in liposomes) have been developed.29,39–41 Though not yet reported, the peculiar properties of GPs could be exploited to entrap both a therapeutic and a diagnostic agent. We reported that a macrophage population labelled ex vivo with Gd-loaded GPs and then i.v. injected in a mouse model of acute liver inflammation allowed the MRI visualization of the macrophage recruitment in the inflamed region.42
Differently, the motivation of this work was to test the feasibility of the approach consisting in labeling monocytes/macrophages in vivo (e.g. after i.v. injection of GPs), and tracking their migration in the inflammation site by Near InfraRed Fluorescence (NIRF) imaging.
It has been demonstrated in some recent examples that this imaging technique has the potential to allow the detection of immune cells labeled/targeted with fluorescent dyes.43–46
NIR fluorophores work in the spectrum of 700 to 900 nm, where the light absorption by tissue chromophores is minimal, thus reducing background interferences.47–50
We loaded GPs with two water insoluble fluorescent dyes: one, Cy-5(C16)2, based on cyanine fluorophore, for NIRF imaging, and the other, Rho-DOPE (commercially available), based on rhodamine structure for ex vivo microscopy validation. The particles were tested in vivo on an autoimmune model of rheumatoid arthritis in mice (Collagen Induced Arthritis, CIA), which is a widely used model of arthritis induced by immunization with an emulsion of complete Freund's adjuvant and type II collagen (CII).51 This model is mainly characterized by proliferative synovitis with acute and chronic infiltration of polymorphonuclear and mononuclear cells, pannus formation, and fibrosis.52–55 The mononuclear cell infiltration that characterizes the chronic inflammatory reaction results from the migration of lymphocytes and monocytes through the vascular endothelium.54 Furthermore, this model is very suitable for the purposes of this work, either because the lesions are superficial (thus overcoming the limited tissue penetration of NIRF) or for the extensive recruitment of immune cells. The success of labeling phagocytic cells in vivo with imaging agents could open the way to new macrophage-targeted drug delivery to attenuate/suppress inflammatory processes, where the effective delivery of the drug can be visualized through the co-encapsulation of the imaging agent and the therapeutic drug (theranostic approach).24,56
Materials and methods
Chemicals
All reagents and solvents, unless otherwise indicated, were purchased from Sigma-Aldrich (Milano, Italy). The cyanine-based fluorescent probe (Cy5-(C16)2) was synthesized following a previously reported procedure.57Animal model
Male DBA/1J mice, 7–9 weeks of age and free of murine-specific pathogens, were purchased from Harlan (Harlan Italy, San Pietro al Natisone (UD)). To induce CIA, mice were immunized intradermally at the base of the tail with 0.2 mL of an emulsion containing 0.4 mg of bovine type II collagen (Morwell Diagnostics, Zurich, Switzerland) in Complete Freund's Adjuvant (CFA, Difco, Detroit, U.S.A.) containing 0.4 mg of Mycobacterium tuberculosis. Immunization with CFA + CII resulted, starting approximately from day 18–20 p.i., in the appearance of signs of inflammation affecting one or more limbs. The number of arthritic limbs were quantitated and each limb assigned a severity score according to the criteria reported elsewhere.58 All the procedures involving mice were conducted in agreement with Italian and European guidelines and policies on animal testing, and were approved by the local animal experiments ethical committee.Preparation and characterization of glucan particles
According to a previously reported procedure,27 Saccharomyces cerevisiae were suspended in 1 M NaOH solution and heated at 80 °C for 1 h under continuous magnetic stirring. The insoluble material was then collected by centrifugation and suspended in acidic water (pH 5) and stirred for 1 h at 55 °C. The insoluble material was collected again by centrifugation and washed in water, isopropanol and then acetone. The product obtained was dried at room temperature to a fine powder. The so-obtained dry glucan particles (100 mg) were incubated overnight at 20 °C with 2 mL of a chloroform solution containing Cy5-(C16)2 (50 μg mL−1 corresponding to 37.0 μM) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (rhodamine-DOPE) (50 μg mL−1 corresponding to 38.5 μM) (Avanti® Polar Lipids, Inc. Alabaster, Alabama, USA). The resulting labeled particles (Cy5-(C16)2/Rho-GPs) were washed four times in HEPES isotonic buffer, re-suspended in the same buffer and counted with the haemocytometer.To characterize the microcarrier obtained, IR spectroscopy and Atomic Force Microscopy (AFM) were used (see ESI†). The excitation and emission spectra were acquired with a spectrofluorometer (Fluoromax-4-Horiba Scientific). The concentration of entrapped Rho-DOPE and Cy5(C16)2, respectively, were calculated using a spectrophotometer (Hitachi U-28000), according to calibration curves previously prepared from standard solutions (see ESI†). Drug Loading Content (DLC) and Drug Loading Efficiency (DLE) for the two dyes in glucan particles were estimated as follows:


The suspension of Cy5-(C16)2/Rho-GPs was then analysed by means of confocal microscopy to check the double fluorescence labelling with cyanine- and rhodamine-based dyes (laser1 excitation 561 nm, acquisition window 571–644 nm, laser2 excitation 633 nm, acquisition window 650–720 nm). To verify the detection sensitivity of the system, different concentrations of Cy5-(C16)2/Rho-GPs were imaged using a Pearl Imager (Near-infrared In Vivo Animal Imaging System, LI-COR Biosciences-Lincoln, Nebraska, USA, λexc 685 nm, λem 720 nm). More in details, particles were counted with the hemocytometer and then diluted in PBS at different concentrations ranging from 18 × 106 to 3.5 × 104 GPs per μL. A drop of each sample was dripped on a non-absorbent support and soon after imaged. PBS acted as control (CTRL). A region of interest (ROI) was drawn around each drop and the signal intensity of each sample was calculated. Finally the SI/S0 value was determined as follows:
| SI/S0 = signal intensity sample/signal intensity CTRL |
Cell labeling with Cy5-(C16)2/Rho-GPs
J774A.1 cells were obtained from the American Type Culture Collection (ATCC®) and tested for mycoplasma (MycoAlert™ PLUS Mycoplasma Detection Kit, Lonza). Cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 2 mM glutamine, 100 UI mL−1 penicillin, 100 μg mL−1 streptomycin and 10% v/v Fetal Bovine Serum (FBS) in a humidified 5% CO2 atmosphere at 37 °C (DMEM, glutamine, penicillin and FBS were purchased from Lonza). Cytotoxicity of Cy5-(C16)2/Rho-GPs was evaluated by the following procedure: 1 × 106 cells were seeded in 60 mm cell culture dishes, filled up with 3 mL of complete medium, and left for 24 h at 37 °C in a humidified 5% CO2 atmosphere. The medium was then discarded and cells were incubated with Cy5-(C16)2/Rho-GPs at concentrations ranging between 0 and 2.7 × 106 GPs per μL DMEM (corresponding to 15.5 μM of Cy5-(C16)2 and 18.6 μM of Rho-DOPE) for 90 min. Cells were then washed profusely, detached, and the amount of dead/living cells in each plate was determined using 0.09% trypan blue. Cell viability was calculated as follows:
The experiment was run in triplicate. Thereafter, to investigate the labelling efficiency of GPs toward phagocytic cells, confocal microscopy, Fluorescence Activated Cell Sorting (FACS) analysis and Optical Imaging of cells incubated with Cy5-(C16)2/Rho-GPs were performed (see ESI†). For Optical Imaging 2 × 106 J774A.1 cells were seeded into T75 tissue flasks and left in complete DMEM for 48 h at 37 °C in a humidified 5% CO2 atmosphere. The medium was then discarded, and cells were incubated with 350 μL of Cy5-(C16)2/Rho-GPs in 5 mL of complete medium (5.95 × 105 GPs per μL DMEM corresponding to 3.20 μM of Cy5-(C16)2 and 3.85 μM of Rho-DOPE) for 90 min. Afterwards, cells were washed four times in Phosphate Buffered Saline (PBS), harvested, counted with the haemocytometer, and centrifuged. The pellet was then suspended in PBS at different concentrations from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 down to 300 cells per μL. PBS was used as control. The samples obtained were then analysed under the Pearl imager to evaluate the detection sensitivity of the system. To evaluate the limit of detection, a ROI was drawn around each sample and the signal intensity was calculated. SI/S0 was determined as reported in paragraph “Preparation of glucan particles”.
000 down to 300 cells per μL. PBS was used as control. The samples obtained were then analysed under the Pearl imager to evaluate the detection sensitivity of the system. To evaluate the limit of detection, a ROI was drawn around each sample and the signal intensity was calculated. SI/S0 was determined as reported in paragraph “Preparation of glucan particles”.
NIRF imaging
5 naïve and 15 DBA/1J CIA-affected male mice at eleven weeks of age were anesthetized (1.5% isoflurane inhalation) and placed inside the NIRF scanner. The animals were irradiated with filter light of wavelength 685 nm, and an image of emission intensity was collected at 720 nm by a charge-coupled device (CCD) camera during a 30 s acquisition period (resolution = 170 μm, field of view = 112 mm × 84 mm). Mice were imaged before, immediately after the injection of 100 μL of Cy5-(C16)2/Rho-GPs (corresponding to 45 μM of Cy5-(C16)2), and then 1, 3, 5, 24, and 48 h post injection.The percentage of relative enhancement of fluorescence signal intensity (FSI) was calculated as:

Histochemistry and immunofluorescence
Naïve and CIA-affected DBA/1J mice were sacrificed 3 h p.i. of Cy5-(C16)2/Rho-GPs. Fore and hind legs were excised and left 48 hours in formalin at 4 °C, in rotation. The day after, formalin was removed, the legs were washed with PBS, and decalcified at room temperature in a 1:1 solution A (2.5 g sodium citrate tribasic dehydrate dissolved in 125 mL of distilled water and 102.5 mL of tap water) and B (147.5 mL of formic acid 96% diluted in 102.5 mL of distilled water). When the process was completed, sagittal and coronal sections of fore and hind legs were carved. The sections obtained were cryo-preserved in Optimum Cutting Temperature (O.C.T.) compound (VWR International) (5 min in liquid nitrogen before cooling down to −80 °C). Slices (thickness 5 μm) were cut using a cryostat (Leica CM 1950). Three sections from the center of naïve and CIA affected mice paws were stained with Hematoxylin and Eosin (H&E). Other three sections were stained with periodic acid–Schiff (PAS) to highlight polysaccharides belonging to the glucan shell. Image acquisition was performed with an Olympus BH2 microscope, magnification 10, 20 and 40×.
Immunofluorescence techniques were exploited to visualize monocytes/macrophages in regions of inflammation and investigate co-localization with Cy5-(C16)2/Rho labeled glucan particles.
Macrophages/monocytes were stained by means of a rat anti-mouse F4/80 antibody, clone CI:A3-1 (AbD Serotec). To obtain antigen retrieval, sections were put into sodium citrate 10 mM buffer solution at pH 6.0, and then heated into a microwave oven at full power for 60 s followed by 9 min in defrost modality. Sections were allowed to cool for 30 min and then rinsed in Tris-Buffered Saline (TBS) for 10 min. Serum blocking was performed with 10% goat serum (Invitrogen), 60 min. Overnight incubation with the primary antibody rat anti-mouse F4/80 10 μg mL−1 in blocking solution was performed in a humidified chamber. Blocking solution was used as control. The day after, sections were washed in TBS and incubated 1 h at room temperature with the Alexa Fluor® 647 Goat Anti-Rat IgG (H + L) secondary antibody (Life Technologies) diluted 1:500 in blocking solution. After profuse washing in TBS, sections were stained for 10 min with Hoechst 8 × 10−5 mg mL−1 in TBS, rinsed in distilled water, mounted with Fluoroshield™ Mounting Medium and imaged through confocal microscope, magnification 20× (laser1 excitation 405 nm, acquisition window 415–485 nm, laser2 excitation 458 nm, acquisition window 500–544 nm, laser3 excitation 633 nm, acquisition window 648–798 nm, to visualize Hoechst, rhodamine, and the secondary antibody, respectively).
Results and discussion
Amphiphilic dyes Cy5-(C16)2 and rhodamine-DOPE (Rho-DOPE, Chart 1) were loaded in the inner core of GPs through a sudden change in solvent polarity.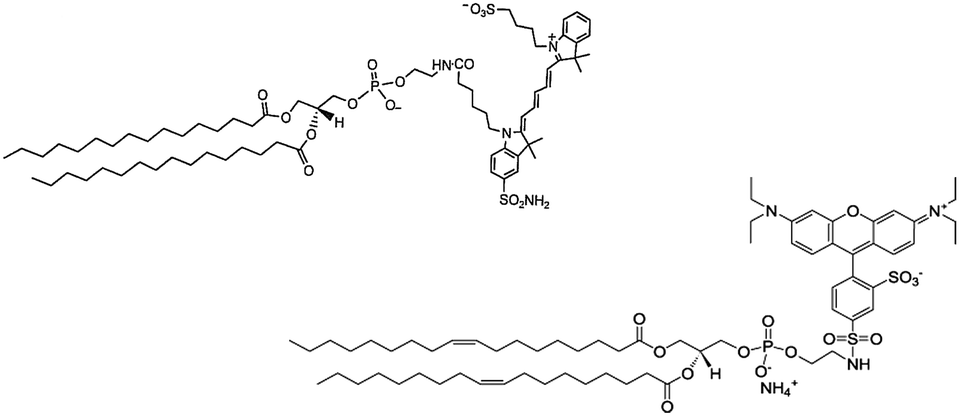 | ||
| Chart 1 Structural formulas of the two amphiphilic dyes entrapped in glucan particles. Left: Cy5-(C16)2, right: Rho-DOPE. | ||
Briefly, the procedure relied upon the addition of HEPES isotonic buffer to a chloroform suspension containing GPs and the amphiphilic fluorescent molecules previously incubated overnight. Following the change in polarity, a microemulsion containing the amphiphiles formed either outside or inside the particles. The external fraction was removed by repeated washing/centrifugation cycles to obtain the GPs entrapping the fluorescent dyes. The loading was stable for at least one month at 4 °C. Morphology and surface properties of modified glucan particles (Cy5-(C16)2/Rho-GPs) have been investigated by AFM. Particles with diameters in the 3–5.5 μm range (with a mean size of 4.4 ± 0.2 μm), were detected. The particle wall structure, as an interwoven network of β-glucan (see ESI†), well corroborates with the atomic structure imaged by STM.59 Fluorescence confocal microscopy demonstrated the occurred internalization and co-localization of the two fluorescent dyes inside GPs cores (Fig. 1). The DLE was about 72% for Rho-DOPE, with an estimated concentration of 550 ± 15 pmol mg−1, and 62% for Cy5-(C16)2 with an estimated concentration of 460 ± 15 pmol mg−1 GPs, respectively (corresponding to 3.7 × 106 Rho-DOPE molecules/GP and 3.1 × 106 Cy5-(C16)2 molecules/GP). DLC attested to around 0.06% and 0.07% for Cy5-(C16)2 and Rho-DOPE, respectively. The sensitivity in the NIRF detection of GPs loaded with Cy5-(C16)2 was assessed by preparing a phantom containing solutions at different particles concentrations starting from 18 × 106 GPs per μL (45 μM of Cy5-(C16)2) down to 3.5 × 104 GPs per μL (87.5 nM). The image reported in Fig. 2 shows that a detection limit of ca. 70000 particles per μL (175 nM of Cy5-(C16)2) was obtained.
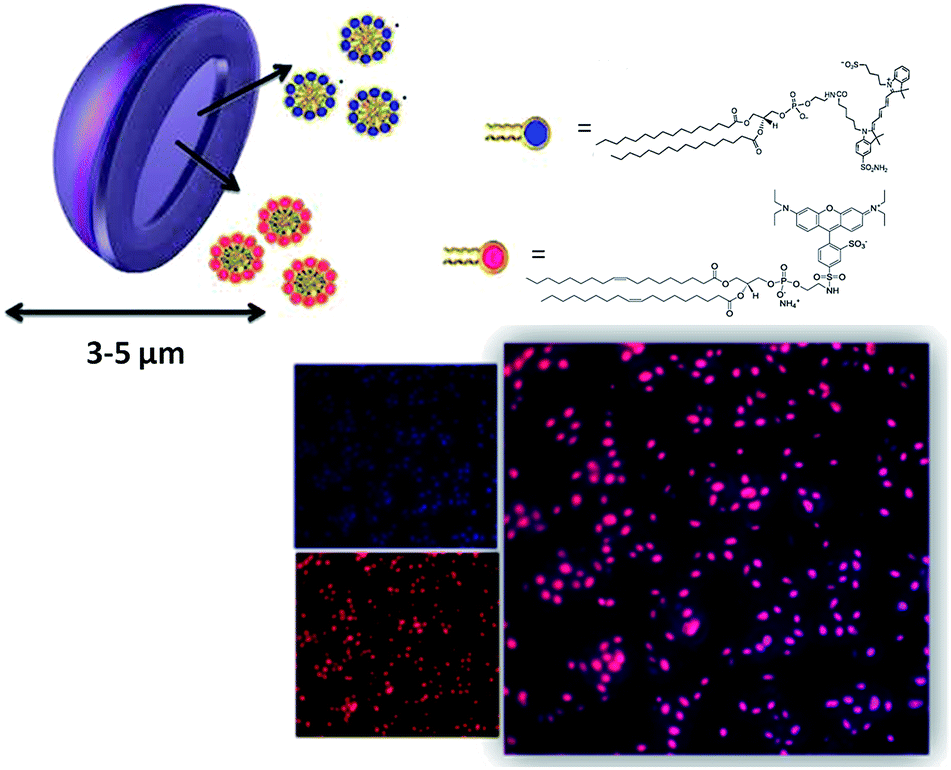 | ||
| Fig. 1 Top: schematic illustration of GPs loaded with fluorescence probes Cy5-(C16)2 (blue), and Rho-DOPE (red). Right: confocal microscopy image of Cy5-(C16)2/Rho-GPs sample, obtained by merging acquisitions at 571–644 nm (red) and 650–720 nm (blue), respectively. | ||
 | ||
| Fig. 2 NIRF imaging of glucan particles diluted in PBS at different concentrations ranging from 18 × 106 GPs per μL to 3.5 × 104 GPs per μL. (1) 18 × 106 GPs per μL, (2) 9 × 106 GPs per μL, (3) 4.5 × 106 GPs per μL, (4) 2.2 × 106 GPs per μL, (5) 1.1 × 106 GPs per μL, (6) 5 × 105 GPs per μL, (7) 2.5 × 105 GPs per μL, (8) 1.25 × 105 GPs per μL, (9) 7 × 104 GPs per μL, (10) 3.5 × 104 GPs per μL. Sample 11 corresponded to PBS (control). | ||
Then, the cytotoxicity of fluorescently labelled GPs was determined using J774A.1 murine macrophages, taken as example of phagocytic cells. To that purpose, different amounts of labelled GPs, between 0 and 2.7 × 106 GPs per μL of medium, were incubated for 90 min in the presence of about 2 million phagocytic cells.
Cell viability was very high, being comprised between 90% and 100%, even at the highest concentration.
The uptake efficiency of Cy5-(C16)2/Rho-GPs by macrophages was analysed by confocal microscopy and FACS (see ESI†). In addition, to define the minimum number of Cy5-(C16)2/Rho-GPs labeled monocytes/macrophages detectable by OI, 10 millions of cells were incubated with 3 × 109 Cy5-(C16)2/Rho-GPs suspended in 5 mL of complete medium corresponding to 3.20 μM of Cy5-(C16)2. After 90 min of incubation, cells were washed in PBS, harvested using a cell scraper, and counted. Then, cells were diluted with PBS and imaged in vitro by NIRF. The image reported in Fig. 3 indicates that the sample containing 300 cells per μL was still detectable, thus representing an in vitro detection limit of the herein presented labeling method under the experimental conditions described above. In vitro detection limit of the same order of magnitude was reported in literature for cells labeled with NIRF fluorophores.60
 | ||
| Fig. 3 NIRF imaging of J774A.1 cells labeled with Cy5-(C16)2/Rho-GPs and diluted in PBS at different concentrations ranging from 20000 to 300 cells per μL. (1) 20000 cells per μL, (2) 10000 cells per μL, (3) 5000 cells per μL, (4) 2500 cells per μL, (5) 1250 cells per μL, (6) 600 cells per μL, (7) 300 cells per μL. Sample 8 corresponded to PBS (control). | ||
To validate the proposed approach, in vivo NIRF imaging of arthritic joints of the mice enrolled in the study was carried out. Collagen-induced rheumatoid arthritis animal model (DBA/1J strain) was obtained by intradermal injection of type II collagen with complete Freund's adjuvant. Three weeks post-injection, erythema and swelling of joints and digits were observed. Then, both diseased and naïve mice were i.v. injected (tail vein) with a suspension of Cy5-(C16)2/Rho-GPs containing 9 millions of fluorescent β-glucan particles per μL (corresponding to 0.22 μmol of Cy5-(C16)2 per kg bw) for in vivo NIRF imaging analysis. Mice were scanned at 5 min, 1 h, 3 h, 5 h, 24 h, and 48 h post injection, and a qualitative in vivo image analysis was performed on each group of mice. The mice were grouped according to the clinical score index value. For each animal, a region of interest was automatically drawn around the inflamed paw. The pixels included in the shape were considered with equal intensity and the threshold intensity value at which the shape was drawn was calculated as follows:
| Threshold intensity value = background mean + (background standard deviation × standard deviation multiplier) |
A total mean intensity signal (in Relative Fluorescence Unit-RFU) was measured over time (post 5 min, 1 h, 3 h, 5 h, 24 h and 48 h) for the four paws of each animal in both frontal and ventral sides. The average of these values, including the animals with different clinical score, was compared with the signal intensity obtained from the analysis of naïve mice. The fluorescence intensity signal in the inflamed paws was higher than the corresponding value measured for the naïve mice. A statistically significant difference was observed 3 h post injection (1.7 ± 0.1 vs. 1.3 ± 0.1; p < 0.05, Fig. 4).
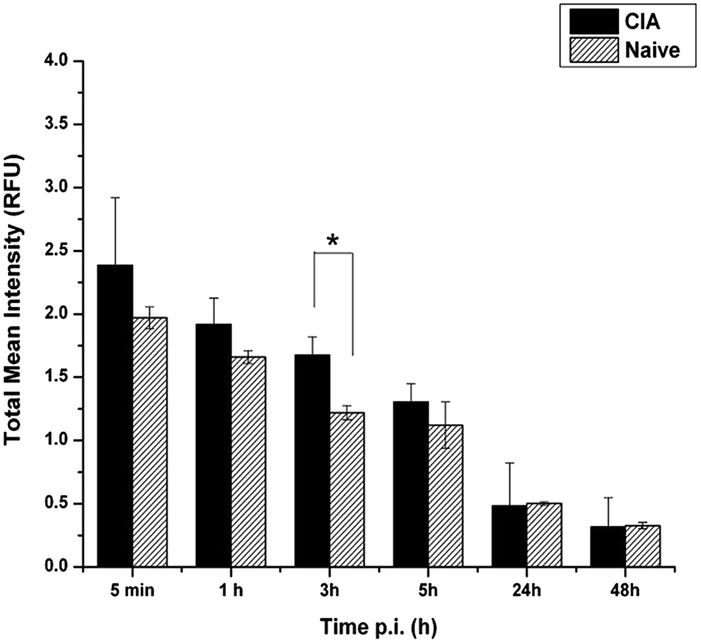 | ||
| Fig. 4 Bar graph showing the difference in the total mean fluorescence intensity signal, in Relative Fluorescence Units (RFU), measured on paws of CIA animals and naïve mice. The maximum difference was obtained 3 hours post injection of the fluorescent GPs (*p < 0.05). | ||
Fig. 5 reports, as representative example, the comparison between a CIA-mouse (top) and a naïve one (bottom) at three different times after GPs injection. A clearly detectable NIRF signal was detected in the inflamed paws, especially in the images acquired 3 h post-injection.
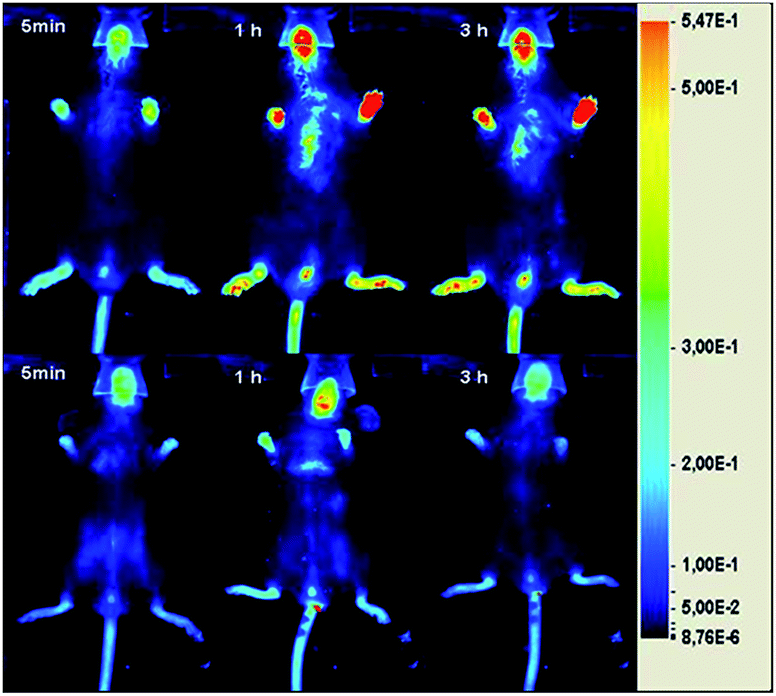 | ||
| Fig. 5 Representative ventral NIRF images of a CIA-mouse (top, clinical score 4.5) and a naïve one (bottom) acquired 5 min, 1 h, and 3 h after the intravenous injection (via tail vein) of Cy5-(C16)2/Rho-GPs at the dose of 220 nmol Cy5-(C16)2 per kg bw. | ||
The short time requested for the detection of the maximum signal in the lesion is likely the result of a fast uptake by blood monocytes. This finding parallels well with the very rapid uptake of GPs (ca. 1 h) reported in vitro by J774A.1 macrophages.42 Fig. 6 indicates that the % enhancement of fluorescence signal intensity (in RFU) calculated at 3 h post injection displayed a good correlation with the severity of the lesion expressed according to the values of the clinical score index.
 | ||
| Fig. 6 The fluorescence intensity signal enhancement (in RFU) measured in paws of mice having different clinical score values (from 1 to 6) 3 h post injection of Cy-(16)2/Rho-GPs. | ||
One peculiarity of the CIA model is due to the remission of the pathology that usually occurs within thirty days.61 In order to assess if the imaging response could depend on the phase (progression or remitting) of the inflammation process, the fluorescence signal of mice having the same clinical score value (4.5), but in ascending, stable, or descending trend, were compared. The result is shown in Fig. 7. Very interestingly, it was possible to detect subtle variations in the imaging response. Animals with an increasing score index displayed a higher signal. Such animals were supposedly into a state of joints bursting inflammation, where the degree of monocyte infiltration was extremely high. On the other hand, the lower signal observed for the animals with a descending score index was correlated to the remitting state of the inflammation, characterized by a far lower tissue degradation and infiltration of immune cells.62
 | ||
| Fig. 7 Comparison of the enhancement of fluorescence signal intensity (In RFU) measured 3 hours post injection in mice having the same clinical score value (4.5) at the moment of GPs injection and scanning, but differing in the score index during the week before GPs administration. ↑ stands for score increasing along the week, = stands for score stable along the week, and ↓ stands for score decreasing along the week (**p < 0.01). | ||
To characterize the arthritic model obtained and prove GPs recruitment in inflamed regions, histological studies were carried out. Cryopreserved paws from naïve and CIA-affected DBA/1J mice were cut through a cryostat. The slices obtained underwent H&E staining to investigate the presence of phagocytic cells in the inflamed regions. In CIA mice, H&E staining highlighted the high degree of inflammation, which was mainly located in the periarticular region, associated with diffuse hemorrhage and widespread edema (Fig. 8). Conversely, the phagocytic population in naïve mice was not relevant and neither edema nor hemorrhage were detected.
 | ||
| Fig. 8 H&E staining of CIA mice (a)–(c) and naïve mouse (d) fore limbs. Black arrows indicate regions with high degree of inflammation characterized by a diffuse presence of mononuclear cells (dark purple spots). In (b) newly formed hemorrhagic foci are highlighted, while in (c) the high degree of edema is appreciable. | ||
In arthritic mice, PAS staining highlighted the presence of GPs in the periarticular region, as well as in blood vessels and in arthritic paws, mainly associated with phagocytic cells (Fig. 9).
 | ||
| Fig. 9 PAS staining of CIA (a)–(b) and naïve (c) mice hind limbs. Black arrows indicate regions with high amount of glucan particles and mononuclear cells. | ||
By fluorescence confocal microscopy, it was possible to appreciate the localization of Cy5-(C16)2/Rho GPs (red) in the periarticular region, while in naïve mice no GPs were detected (Fig. 10). Monocytes/macrophages were stained by anti-F4/80 antibody with the purpose of proving the presence of immune cells and their co-localization with GPs. F4/80 is a well-characterized and extensively referenced membrane protein that is likely the best-known marker for mature mouse macrophages and blood monocytes.63 Unfortunately, F4/80 staining was not perfectly clear, probably due to antigen damage following the procedure of decalcification. Moreover, the emission wavelength of secondary antibody and Cy5-(C16)2 partially overlapped. Therefore, the result was the observation of a diffuse signal around glucan particles, owing to weak bond of the antibody, combined with bright spots corresponding to GPs content. In naïve mice, neither inflammation nor GPs recruitment were detected.
 | ||
| Fig. 10 F4/80 staining of periarticular region in naïve (a) and CIA (b)–(c) mice hind limbs. Blue represents nuclei, red corresponds to GPs and gray to F4/80 antigen staining (image c). | ||
Conclusions
In this study, the diagnostic potential of glucan particles loaded with an amphiphilic NIRF agent to detect inflammation sites was successfully tested on a murine model of collagen-induced arthritis. It has been demonstrated that the NIRF signal detected in the inflammation paws arose from the infiltration of blood monocytes/macrophages labeled after the systemic administration of the GPs. The goodness of the rationale of using highly biocompatible microcarriers for targeting the cellular infiltration in inflammation, without the extravasation of the untargeted agent, was validated by ex vivo histology, which confirmed that the presence of labeled macrophages in the lesion was the result of the rapid uptake of the particles by circulating immune cells. Importantly, in addition to the good correlation with the clinical score of the disease, the fluorescence imaging response allowed to discriminate between animals with a progressive or remitting phase of the pathology. The results presented in this work highlight the clinical potential of glucan particles either as diagnostic agents or as therapeutic carriers. The possibility to entrap both the diagnostic and the therapeutic companions in the same carrier could allow the design of theranostic procedures where the imaging probe would provide information about both the effective delivery of the drug and its therapeutic effects.List of abbreviations
| AFM | Atomic Force Microscopy |
| CCD | Charge-Coupled Device |
| CFA | Complete Freund's Adjuvant |
| CIA | Collagen Induced Arthritis |
| CII | Collagen type II |
| CLSM | Confocal Laser Scanning Microscopy |
| CR3 | Complement Receptor 3 |
| CTRL | Control |
| DLC | Drug Loading Content |
| DLE | Drug Loading Efficiency |
| DMEM | Dulbecco's Modified Eagle Medium |
| D1 | Dectin-1 Receptor |
| EPR | Enhanced Permeability and Retention |
| FACS | Fluorescence Activated Cell Sorting |
| FBS | Fetal Bovine Serum |
| FSI | Fluorescence Signal Intensity |
| GPs | Glucan Particles |
| HEPES | (4-(2-HydroxyEthyl)-1-PiperazineEthaneSulfonic acid) |
| H&E | Hematoxylin and Eosin |
| MRI | Magnetic Resonance Imaging |
| NIR | Near InfraRed |
| NIRF | Near InfraRed Fluorescence |
| PAS | Periodic Acid–Schiff |
| PBS | Phosphate Buffered Saline |
| RFU | Relative Fluorescence Unit |
| Rho-DOPE | 1,2-DiOleoyl-sn-glycero-3-PhosphoEthanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) |
| ROI | Region Of Interest |
| STM | Scanning Tunneling Microscopy |
| TBS | Tris-Buffered Saline |
| TEM | Transmission Electron Microscopy |
Acknowledgements
The financial support of University of Torino/Compagnia di San Paolo (Innovative Nanosized Theranostic Agents project, grant D15E11001710003) and European Union's FP7/2007–2013 under grant agreement no. HEALTH-F2-2011-278850 (INMiND) are gratefully acknowledged. We thank Dr. Marta Gai for confocal microscope images, Alessandro Fioravanti and Marta Mossino for tissue sectioning.Notes and references
- M. L. Kang and G. I. Im, Drug delivery systems for intra-articular treatment of osteoarthritis, Expert Opin. Drug Delivery, 2014, 11(2), 269–282 CrossRef CAS PubMed.
- B. J. Crielaard, T. Lammers, R. M. Schiffelers and G. Storm, Drug targeting systems for inflammatory disease: one for all, all for one, J. Controlled Release, 2012, 161(2), 225–234 CrossRef CAS PubMed.
- K. Park, Controlled drug delivery systems: 560 past forward and future back, J. Controlled Release, 2014, 190, 3–8 CrossRef CAS PubMed.
- A. S. Vanniasinghe, V. Bender and N. Manolios, The potential of liposomal drug delivery for the treatment of inflammatory arthritis, Semin. Arthritis Rheum., 2009, 39(3), 182–196 CrossRef CAS PubMed.
- A. G. Tzakos, E. Briasoulis, T. Thalhammer, W. Jäger and V. Apostolopoulos, Novel Oncology Therapeutics: targeted drug delivery for cancer, J. Drug Delivery, 2013, 2013, 918304 Search PubMed.
- N. Gerwin, C. Hops and A. Lucke, Intraarticular drug delivery in osteoarthritis, Adv. Drug Delivery Rev., 2006, 58(2), 226–242 CrossRef CAS PubMed.
- A. D. Friedman, S. E. Claypool and R. Liu, The smart targeting of nanoparticles, Curr. Pharm. Des., 2013, 19(35), 6315–6329 CrossRef CAS.
- W. Ulbrich and A. Lamprecht, Targeted drug-delivery approaches by nanoparticulate carriers in the therapy of inflammatory diseases, J. R. Soc., Interface, 2010, 7(S1), S55–S66 CrossRef CAS PubMed.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, Designing the nanoparticle–biomolecule interface for “targeting and therapeutic delivery”, J. Controlled Release, 2012, 161(2), 164–174 CrossRef CAS PubMed.
- K. J. Narendra, M. Vijay and K. M. Neelesh, Targeted drug delivery to macrophages, Expert Opin. Drug Delivery, 2013, 10(3), 353–367 CrossRef PubMed.
- J. Patel, B. Jigar, H. Shah and D. Patel, Novel drug delivery technologies for the treatment of rheumatoid arthritis, Internet J. Med. Tech., 2008, 5, 1 Search PubMed.
- J. R. Baker Jr, Why I believe nanoparticles are crucial as a carrier for targeted drug delivery, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5(5), 423–429 Search PubMed.
- E. P. Boot, G. A. Koning and G. Storm, et al., CD134 as target for specific drug delivery to autoaggressive CD4 + T cells in adjuvant arthritis, Arthritis Res. Ther., 2005, 7, R604–R615 CAS.
- R. A. Bader, The development of targeted drug delivery systems for rheumatoid arthritis treatment, in Rheumatoid arthritis – Treatment, part 3, ed. Dr. Andrew Lemmey, InTech, vol. 111–132, ch. 6, 2012 Search PubMed.
- J. J. Haringman, R. L. Oostendorp and P. P. Tak, Targeting cellular adhesion molecules, chemokines and chemokine receptors in rheumatoid arthritis, Expert Opin. Emerging Drugs, 2005, 10(2), 299–310 CrossRef CAS PubMed.
- C. M. Dawidczyk, C. Kim and J. H. Park, et al., State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines, J. Controlled Release, 2014, 187, 133–144 CrossRef CAS PubMed.
- P. Couvreur and C. Vauthier, Nanotechnology: intelligent design to treat complex disease, Pharm. Res., 2006, 23(7), 1417–1450 CrossRef CAS PubMed.
- W. J. Mulder, G. J. Strijkers, G. A. Van Tilborg, A. W. Griffioen and K. Nicolay, Lipid-based nanoparticles for contrast-enhanced MRI and molecular imaging, NMR Biomed., 2006, 19(1), 142–164 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, Long-circulating and target-specific nanoparticles:theory to practice, Pharmacol. Rev., 2001, 53(2), 283–318 CAS.
- J. M. Van den Hoven, S. R. Van Tomme, J. M. Metselaar, B. Nuijen, J. H. Beijnen and G. Storm, Liposomal drug formulations in the treatment of rheumatoid arthritis, Mol. Pharm., 2011, 8(4), 1002–1015 CrossRef CAS PubMed.
- N. K. Jain, V. Mishra and N. K. Mehra, Targeted drug delivery to macrophages, Expert Opin. Drug Delivery, 2013, 10(3), 367 Search PubMed.
- F. Chellat, Y. Merhi, A. Moreau and L. Yahia, Therapeutic potential of nanoparticulate systems for macrophage targeting, Biomaterials, 2005, 26, 7260–7275 CrossRef CAS PubMed.
- C. Kelly, C. Jefferies and S. A. Cryan, Targeted liposomal drug delivery to monocytes and macrophages, J. Drug Delivery, 2011, 2011, 727241 Search PubMed.
- S. K. Patel and J. M. Janjic, Macrophage Targeted Theranostics as Personalized Nanomedicine Strategies for Inflammatory Diseases, Theranostics, 2015, 5(2), 150–172 CrossRef CAS PubMed.
- U. Flögel, Z. Ding and H. Hardung, et al., In vivo monitoring of inflammation after cardiac and cerebral ischemia by fluorine magnetic resonance imaging, Circulation, 2008, 118(2), 140–148 CrossRef PubMed.
- C. Van De Wiele, M. Sathekge and A. Maes, Targeting monocytes and macrophages by means of SPECT and PET, Q. J. Nucl. Med. Mol. Imaging, 2014, 58(3), 269–275 CAS.
- E. R. Soto and G. R. Ostroff, Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery, Bioconjugate Chem., 2008, 19(4), 840–848 CrossRef CAS PubMed.
- E. R. Soto, A. C. Caras, L. C. Kut, M. K. Castle and G. R. Ostroff, Glucan particles for macrophage targeted delivery of nanoparticles, J. Drug Delivery, 2012, 2012, 143524 Search PubMed.
- S. Figueiredo, J. N. Moreira, C. F. Geraldes, S. Rizzitelli, S. Aime and E. Terreno, Yeast cell wall particles: a promising class of nature-inspired microcarriers for multimodal imaging, Chem. Commun., 2011, 47(38), 10635–10637 RSC.
- A. L. Fuentes, L. Millis, J. Vapenik and L. Sigola, Lipopolysaccharide-mediated enhancement of zymosan phagocytosis by RAW 264.7 macrophages is independent of opsonins, laminarin, mannan, and complement receptor 3, J. Surg. Res., 2014, 189(2), 304–312 CrossRef CAS PubMed.
- R. De Smet, T. Demoor and S. Verschuere, et al., β-Glucan microparticles are good candidates for mucosal antigen delivery in oral vaccination, J. Controlled Release, 2013, 172(3), 671–678 CrossRef CAS PubMed.
- H. Huang, G. R. Ostroff and C. K. Lee, et al., Relative contributions of dectin-1 and complement to immune responses to particulate β-glucans, J. Immunol., 2012, 189(1), 312–317 CrossRef CAS PubMed.
- M. De Jesus, G. R. Ostroff, S. M. Levitz, T. R. Bartling and N. J. Mantis, A population of Langerin positive dendritic cells in murine Peyer's patches involved in sampling β-glucan microparticles, PLoS One, 2014, 9(3), e91002 Search PubMed.
- G. D. Brown and S. Gordon, Immune recognition. A new receptor for beta-glucans, Nature, 2001, 413, 36–37 CrossRef CAS PubMed.
- G. D. Brown, P. R. Taylor and D. M. Reid, et al., Dectin-1 is a major beta-glucan receptor on macrophages, J. Exp. Med., 2002, 196, 407–412 CrossRef CAS.
- H. S. Goodridge, C. N. Reyes and C. A. Becker, et al., Activation of the innate immune receptor Dectin-1 upon formation of a 'phagocytic synapse', Nature, 2011, 472(7344), 471–475 CrossRef CAS PubMed.
- R. A. Drummond and G. D. Brown, The role of Dectin-1 in the host defence against fungal infections, Curr. Opin. Microbiol., 2011, 14, 392–399 CrossRef CAS PubMed.
- H. Huang, G. R. Ostroff, C. K. Lee, J. P. Wang, C. A. Specht and S. M. Levitz, Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and toll-like receptor agonists, Infect. Immun., 2009, 77(5), 1774–1781 CrossRef CAS PubMed.
- E. Soto, Y. S. Kim, J. Lee, H. Kornfeld and G. R. Ostroff, Glucan Particle Encapsulated Rifampicin for Targeted Delivery to Macrophages, Polymers, 2010, 2, 681–689 CrossRef CAS PubMed.
- F. Garello, R. Stefania, S. Aime, E. Terreno and D. Delli Castelli, Successful entrapping of liposomes in glucan particles: an innovative micron-sized carrier to deliver water-soluble molecules, Mol. Pharm., 2014, 11(10), 3760–3765 CrossRef CAS PubMed.
- E. Soto and G. R. Ostroff, Glucan Particles as Carriers of Nanoparticles for Macrophage-Targeted Delivery, in Nanomaterials for Biomedicine, ed. Ramanathan Nagarajan, ACS Symposium Series, vol. 1119, ch. 3, pp. 57–79, 2012 Search PubMed.
- S. Figueiredo, J. C. Cutrin and S. Rizzitelli, et al., MRI tracking of macrophages labeled with Glucan particles entrapping a water insoluble paramagnetic Gd-based agent, Mol. Imag. Biol., 2013, 15(3), 307–315 CrossRef PubMed.
- J. S. Yoo, R. K. Das, Z. Y. Jow and Y. T. Chang, In Vivo Detection of Macrophage Recruitment in Hind-Limb Ischemia Using a Targeted Near-660 Infrared Fluorophore, PLoS One, 2014, 9(7), e103721 Search PubMed.
- A. N. Pande, R. H. Kohler, E. Aikawa, R. Weissleder and F. A. Jaffer, Detection of macrophage activity in atherosclerosis in vivo using multichannel, high-resolution laser scanning fluorescence microscopy, J. Biomed. Opt., 2006, 11, 021009 CrossRef PubMed.
- A. Saxena, C. W. Kessinger and B. Thompson, et al., High-resolution optical mapping of inflammatory macrophages following endovascular arterial injury, Mol. Imag. Biol., 2013, 15, 282–289 CrossRef PubMed.
- C. Tu, X. Ma, P. Pantazis, S. M. Kauzlarich and A. Y. Louie, Paramagnetic, silicon quantum dots for magnetic resonance and two-photon imaging of macrophages, J. Am. Chem. Soc., 2010, 132, 2016–2023 CrossRef CAS PubMed.
- X. Chen, P. S. Conti and R. A. Moats, In vivo Near-Infrared Fluorescence Imaging of Integrin αVβ3 in Brain Tumor Xenograft, Cancer Res., 2004, 64, 8009–8014 CrossRef CAS PubMed.
- A. Becker, C. Hessenius and K. Licha, et al., Receptor-targeted optical imaging of tumor near infrared fluorescent ligands, Nat. Biotechnol., 2001, 19, 327–333 CrossRef CAS PubMed.
- X. Zhang, S. Bloch, W. Akers and S. Achilefu, Near-infrared molecular probes for in vivo imaging, Current Protocols in Cytometry, 2012, 60(12.27), 1–20 Search PubMed.
- M. Rudin and R. Weissleder, Molecular imaging in drug discovery and development, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS PubMed.
- D. D. Brand, K. A. Latham and E. F. Rosloniec, Collagen-induced arthritis, Nat. Protoc., 2007, 2, 1269–1275 CrossRef CAS PubMed.
- R. O. Williams, Collagen-induced arthritis as a model for rheumatoid arthritis, Methods Mol. Med., 2004, 98, 207–216 CAS.
- K. Phadke, J. Carroll and S. Nanda, Effects of various anti-inflammatory drugs on type II collagen-induced arthritis in rats, Clin. Exp. Immunol., 1982, 47(3), 579–586 CAS.
- M. Ziff, Pathways of mononuclear cell infiltration in rheumatoid synovitis, Rheumatol. Int., 1989, 9(3–5), 97–103 CAS.
- H. Yong, L. Yingxun, C. Zhongping, G. Ning and W. Jinke, Manufacture of IRDye800CW coupled Fe3O4 nanoparticles and their applications in cell labeling and in vivo imaging, J. Nanobiotechnol., 2010, 8, 25 CrossRef PubMed.
- E. Terreno, F. Uggeri and S. Aime, Image guided therapy: the advent of theranostic agents, J. Controlled Release, 2012, 161(2), 328–337 CrossRef CAS PubMed.
- E. Cittadino, M. Botta and L. Tei, et al., In Vivo Magnetic Resonance Imaging Detection of Paramagnetic Liposomes Loaded with Amphiphilic Gadolinium(iii) Complexes: Impact of Molecular Structure on Relaxivity and Excretion Efficiency, ChemPlusChem, 2013, 78, 712–722 CrossRef CAS PubMed.
- S. D. McKenna, G. Feger and C. Kelton, et al., Tumor necrosis factor (TNF)-soluble high-affinity receptor complex as a TNF antagonist, J. Pharmacol. Exp. Ther., 2007, 322(2), 822–828 CrossRef CAS PubMed.
- A. A. Garcia, P. Oden, U. Knipping, G. Ostroff and R. Druyor, Characterization of a β-Glucan Particle Using the Scanning Tunneling and Atomic Force Microscopes, in Synthetic Microstructures in Biological Research, ed. J. M. Schnur and M. Peckerar, Springer, New York, 131–144, ch. 4, 1992 Search PubMed.
- E. J. Sutton, T. D. Henning, B. J. Pichler, C. Bremer and H. E. Daldrup-Link, Cell tracking with optical imaging, Eur. J. Radiol., 2008, 18(10), 2021–2032 CrossRef PubMed.
- L. Marinova-Mutafchieva, C. Gabay, K. Funa and R. O. Williams, Remission of collagen-induced arthritis is associated with high levels of transforming growth factor-beta expression in the joint, Clin. Exp. Immunol., 2006, 146(2), 287–293 CrossRef CAS PubMed.
- P. P. Tak, T. J. Smeets and M. R. Daha, et al., Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity, Arthritis Rheum., 1997, 40(2), 217–225 CrossRef CAS PubMed.
- A. Hansch, O. Frey and D. Sauner, et al., In vivo imaging of experimental arthritis with nearinfrared fluorescence, Arthritis Rheum., 2004, 50(3), 961–967 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00720h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |