
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese-mediated C(sp)–Si cross-electrophile coupling of alkynyl halides with chlorosilanes†
Quan Lin,
Liping Lin,
Jingjing Wang,
Daohong Yu *,
Zhengwang Chen* and
Zhong-Xia Wang*
*,
Zhengwang Chen* and
Zhong-Xia Wang*
Jiangxi Provincial Key Laboratory of Synthetic Pharmaceutical Chemistry, College of Chemistry and Materials Science, Gannan Normal University, Ganzhou 341000, P.R. China. E-mail: yudh@gnnu.edu.cn; chenzwang2021@163.com; zhongxiawang@ncu.edu.cn
First published on 3rd July 2025
Abstract
We present a manganese-mediated cross-electrophile coupling (XEC) that directly constructs C(sp)–Si bonds between alkynyl halides and chlorosilanes. By leveraging manganese as a reductant, this method enables a mild and operationally simple approach. Mechanistic studies reveal the in situ generation of key intermediates alkynylmanganese in amide media, which undergo SN2 substitution with chlorosilanes to afford diverse alkynylsilanes in high yields (up to 99%) with suppressed diyne byproducts. To our knowledge, this is the first discovery of alkynylmanganese species, derived from readily available alkynyl halides, for C(sp)–Si cross-electrophilic coupling. This work expands the scope of XEC in organosilicon chemistry and provides a robust alternative for C(sp)–Si bond formation.
Introduction
Cross-electrophile coupling (XEC) has emerged as a transformative strategy for C–C bond formation, holding considerable potential across diverse disciplines including pharmaceutical chemistry, materials science, and natural product synthesis.1 The XEC protocol achieves direct coupling of two electrophilic substrates via transition-metal catalysis under reductive conditions, exhibiting exceptional substrate compatibility while eliminating preformed organometallic reagents over conventional cross-coupling approaches.2 Beyond its well-established C–C coupling, XEC has recently demonstrated notable progress in the coupling of carbon electrophiles with commercial chlorosilanes as an indispensable strategy for constructing C–Si bonds, spanning C(sp3)–Si and C(sp2)–Si architectures, as evidenced by pioneering contributions from Shu and Oestreich, and others (Fig. 1a).3 Huang's recent report of a cobalt-catalyzed XEC reaction that couples pre-synthesized alkynyl sulfides with chlorosilanes to access C(sp)–Si alkynylsilanes represents a significant advancement (Fig. 1b, top).4 Nevertheless, the direct cross-coupling of readily available alkynyl halides with chlorosilanes to construct C(sp)–Si bonds has remained unreported.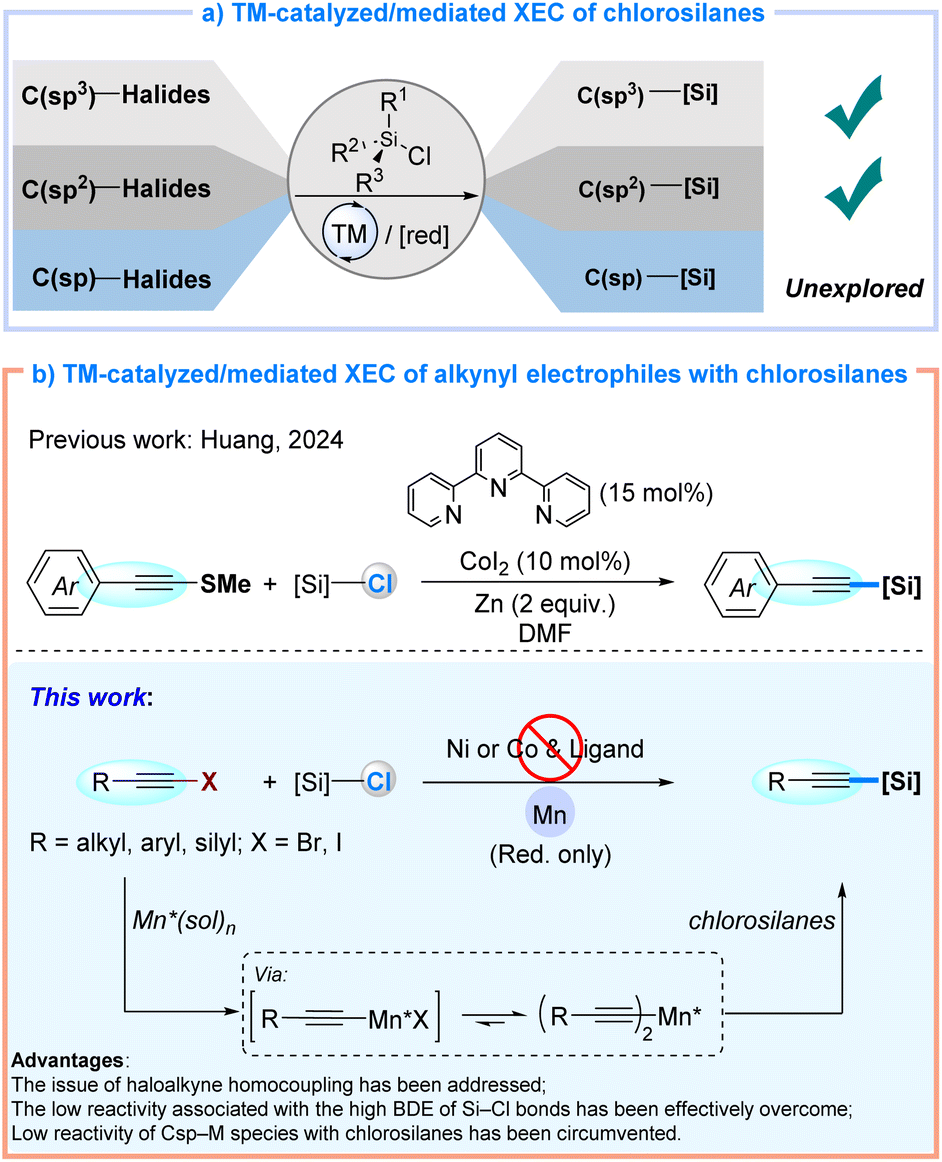 | ||
| Fig. 1 Cross-electrophile C–Si coupling reactions. | ||
Three fundamental challenges could persist in C(sp)–Si XEC reactions involving alkynyl halides and chlorosilanes. First, the high reactivity of alkynyl halides often results in undesired homocoupling and addition side reactions.5 Second, the low reactivity and elevated bond dissociation energy (BDE) of R3Si–Cl bonds (ca. 117 kcal mol−1),6 compared to R3C–Cl bonds (ca. 84 kcal mol−1),7 adversely affect the XEC reactivity of alkyne electrophiles. Third, the diminished nucleophilicity of commonly reported C(sp)–Ni/Co intermediates, relative to aryl or alkyl-Ni/Co species, further reduces their reactivity toward chlorosilanes.3a,8 Therefore, developing an efficient cross-electrophilic coupling protocol for the direct synthesis of alkynylsilanes from alkynyl halides is both timely and essential.
Herein, we report a manganese-mediated XEC reaction that enables direct C(sp)–Si bond formation between alkynyl halides and chlorosilanes. This method innovatively employs metal manganese as a reductant, circumventing requirements for sophisticated catalytic systems involving transition-metal catalysts and organic ligands. Notably, mechanistic investigations reveal the in situ generation of intermediate alkynylmanganese species from alkynyl halides as precursors in the presence of manganese metal and amide solvent, followed by SN2 nucleophilic substitution reactions with chlorosilanes.9 This protocol enables the synthesis of a variety of alkynylsilanes in high yields (up to 99%) under mild conditions, effectively suppressing the formation of diyne byproducts (Fig. 1b, bottom). As such, this discovery establishes an alternative synthetic toolkit for organosilicon chemistry.
Results and discussion
We initiated our studies by evaluating the coupling reaction between alkynyl bromide 1a and chlorosilane 2a (Table 1). Following extensive optimization, we identified the optimal combination of Mn (3 equiv.) in dimethylacetamide (DMA) (0.15 mol L−1), which produced the desired product 3a with an isolated yield of 80% within a reaction time of 2.5 hours (Table 1, entry 1). The addition of catalytic amounts of NiBr2(dtbbpy) or CoBr2/Terpy under these conditions notably reduced the yields, resulting in 23% and 3% of the desired alkynes, and 57% and 29% of diynes, respectively (Table 1, entries 2 and 3). The reaction did not proceed in the absence of Mn, nor when the organic reductant TDAE was used as a substitute (Table 1, entries 4 and 5). Alternative metal reductants, aside from iron, were capable of yielding the target product, albeit with lower yields; magnesium specifically may facilitate the formation of the alkynyl Grignard reagent (Table 1, entries 6–8). Amide solvents are essential for the success of this reaction, with DMA exhibiting particularly favorable properties (Table 1, entries 1 and 9–13). Under an air atmosphere, the reaction yielded only 22% of product 3a, alongside 29% of side product 3a′ (Table 1, entry 14). Furthermore, upon introducing D2O (1.0 equivalent) into the reaction conditions, the formation of 3a was inhibited, yielding 23% of deuteryne (Table 1, entry 15). These findings further support the presence of an organomanganese intermediate.2a,10 Additionally, we performed a gram-scale synthesis of 3a, achieving a yield of 64%, thereby underscoring the robustness of this protocol and its potential for synthetic applications (Table 1, entry 16).
|
||||
|---|---|---|---|---|
| Entry | Variation | Yield (%) | ||
| 3a | 3a′ | 3a′′ | ||
| a Standard conditions: 1a (0.15 mmol), 2a (0.45 mmol), Mn (0.45 mmol), DMA (1 mL), 25 °C, Argon, yields were determined by GC using dodecane as the internal standard.b Isolated yield.c Deuterated product. TDAE = tetrakis(dimethylamino)ethylene. Terpy = 2,2′:6′,2′′-terpyridine. w/ = with. w/o = without. | ||||
| 1 | None | 86 (80)b | 6 | 0 |
| 2 | w/ NiBr2(dtbbpy) (10 mol%) | Trace | 23 | 57 |
| 3 | w/ CoBr2 & Terpy (10 mol%) | 23 | 3 | 29 |
| 4 | w/o Mn | 0 | 0 | 0 |
| 5 | TDAE instead of Mn | 0 | 0 | 0 |
| 6 | Mg instead of Mn | 64 | 12 | 0 |
| 7 | Zn instead of Mn | 25 | 25 | 0 |
| 8 | Fe instead of Mn | 0 | 0 | 0 |
| 9 | DMF instead of DMA | 52 | 15 | 0 |
| 10 | THF instead of DMA | 0 | Trace | 0 |
| 11 | Toluene instead of DMA | 0 | 0 | 0 |
| 12 | DMSO instead of DMA | 0 | Trace | 0 |
| 13 | CH3CN instead of DMA | 0 | Trace | 0 |
| 14 | Under air | 22 | 29 | 0 |
| 15 | 1.0 equiv. D2O was added | 25 | 23c | 0 |
| 16 | 4 mmol of 1a (gram scale) | 64b | 8 | 0 |

Under optimized reaction conditions, we undertook an initial exploration of the scope of alkynyl bromides and representative alkynyl iodides in this manganese-mediated XEC utilizing TMSCl, VinylMe2SiCl, or VinylPhMeSiCl. The unactivated alkynyl halides exhibited significant reactivity with chlorosilanes, producing target products in yields ranging from 31% to 87%. Both silicon- and alkyl-substituted alkyne halides demonstrated compatibility within this reaction system (Fig. 2, products 3b–3f). Notably, tertiary alkyl-substituted alkynyl bromides (3f) exhibited significantly higher reactivity than secondary alkyl counterparts (3e), whereas primary alkyl-substituted alkynyl bromides (3c, 3d) required elevated amounts of chlorosilane and manganese, along with prolonged reaction times, to achieve complete conversion. We postulate that primary alkyl-substituted alkynyl bromides exhibit relatively low steric hindrance at the propargylic position may drive substrate isomerization to the allenic form, thereby inhibiting the formation of the alkynylmanganese species and enabling substrate recovery.11 Subsequently, we examined the scope of aromatic alkynyl halides (3g–3y). Remarkably, alkynylferrocene (3g) and alkynylbiphenyls (3h–3i) were found to be compatible with the standard conditions, yielding moderate results. The products 3j–3l indicate that aromatic alkynyl halides containing electron-donating groups (EDGs), such as methoxy and alkyl substituents on the benzene ring, are still able to yield the desired products in moderate to good yields. Similarly, the presence of electron-withdrawing groups (EWGs) such as –CO2Me (3m), –CO2Ph (3n), and –CF3 (3o) on the benzene ring produced comparably favorable outcomes. The reaction also exhibits excellent chemo-selectivity; we performed the XEC with alkynyl bromides containing various active functional groups, synthesizing products such as bromobenzene (3p), benzonitrile (3q), and benzaldehyde (3r) in yields ranging from 42% to 91%, while maintaining the integrity of their functional groups. Reactions involving aromatic alkynyl bromides with heteroaromatic rings proceeded smoothly, yielding alkynylsilanes (3s–3v) in yields of 48% to 95%. Furthermore, by increasing the amount of chlorosilanes, the bisilylation product 3w was obtained with a yield of 73%. In certain cases, silica-based drug analogs exhibited superior pharmacokinetic properties.12 We also explored the incorporation of alkynylsilane fragments into pharmaceutical and natural product scaffolds, including DL-menthol (3x), glycosides (3y), steroids (3z–3aa), icaridine (3ab), podophyllotoxin (3ac), ezetimibe (3ad), and ospemifene (3ae), achieving promising results. Additionally, when utilizing both unactivated and activated alkynyl iodides (1ea, 1ja, 1ka), as well as steroidal alkyne iodides (1za, 1aaa), corresponding silylation products were obtained with yields ranging from 49% to 99%.
 | ||
| Fig. 2 Scope of alkynyl halides. a Reaction conditions: 1 (0.15 mmol), 2 (0.45 mmol), Mn (0.45 mmol), DMA (1 mL), 25 °C, Argon, isolated yield. b Chlorosilane (0.75 mmol) and Mn (0.75 mmol) were used, 48 h. c 0.6 mmol 2 and 0.6 mmol Mn were used. | ||
We subsequently examined the scope of chlorosilanes by coupling them with alkynyl bromide 1k under standard conditions (Fig. 3). Depending on the varying chain lengths, steric properties, and functional groups of the chlorosilanes, we achieved yields ranging from good to excellent. For instance, TMSCl (3af), TESCl (3ag), chlorodimethyl(butyl)silane (3ah), and chlorodimethyl(isobutyl)silane (3ai) furnished the desired products in yields between 69% and 95%. Additionally, we assessed the compatibility of corresponding alkynyl halides under these conditions. Notably, alkynyl chloride (1kb) did not perform as anticipated, whereas alkynyl iodide (1ka) yielded product 3ab with a 90% yield. Chlorosilanes bearing sterically hindered or conjugated functional groups, such as aryl and alkenyl substituents (3aj–3al), resulted in target products with yields ranging from 60% to 78%. The incorporation of chloromethyl and chloropropyl functionalities onto the chlorosilanes was well tolerated, thus offering enhanced opportunities for late-stage modifications of the resultant products (3am–3an). Chlorohydrosilanes represent a fundamental class of materials in organosilicon chemistry.8c,13 Previous studies have shown that the bond dissociation energy of Si–H (ca. 77 kcal mol−1) is significantly lower than that of the Si–Cl bond.14 Products 3ao–3aq further illustrate that chlorohydrosilanes exhibit remarkable chemical selectivity under standard conditions. Moreover, the present method is also applicable to chlorogermanes, as demonstrated by examples 3ar and 3as.
 | ||
| Fig. 3 The scope of alkynyl chlorosilanes. Reaction conditions: 1k (0.15 mmol), 2 (0.45 mmol), Mn (0.45 mmol), DMA (1 mL), 25 °C, Argon, isolated yield. n.d. = not detected. | ||
The versatility of alkynylsilane functionality enables further structural variations (Fig. 4). The synthesized 3ao derivatives are readily accessible for cross-coupling reactions, exemplified by the formation of compound 5, which was obtained in 78% yield from the reaction of 3ao with methyl 4-vinylbenzoate and 1-(4-iodophenyl)ethan-1-one.15 In the nucleophilic addition of 4-methoxybenzenethiol to 3p, product 6 is preferentially formed, yielding 42% and demonstrating exceptional chemical selectivity.16 Additionally, alkynylgermane 3ar undergoes a Diels–Alder reaction with 2-pyrones to afford biphenyl compound 7 in a yield of 70%, accompanied by the cleavage of the trimethylgermyl group.17 Furthermore, a cobalt-catalyzed regioselective [3 + 2] annulation of 3v with ortho-formylphenylboronic acid produced cyclized product 8 in a remarkable yield of 95%.18 These compounds hold significant promise in biomedical applications and organic synthesis.5a,19
 | ||
| Fig. 4 Derivatization of alkynylsilanes/alkynylgermanes. Reaction conditions: a 1-(4-iodophenyl)ethan-1-one (0.15 mmol), 3ao (2 equiv.), Pd(OAc)2 (4 mol%), LiCl (4 equiv.), pyridine (2.4 equiv.), DMI, r.t. b 3p (0.15 mmol), 4-methoxybenzenethiol (1 equiv.), KOH (1 equiv.), DMSO, 120 °C. c 3ar (0.10 mmol), 2-pyrone (2 equiv.), ZnCl2 (10 mol%), DCE, 140 °C. d 3v (0.15 mmol), ortho-formylphenylboronic acid (1.5 equiv.), Co(acac)2 (10 mol%), DPPE (10 mol%), MeCN, 80 °C. | ||
To elucidate the mechanism underlying this process, several control experiments were conducted. Firstly, in the presence of Michael acceptors 4a–4f (3 equivalents), the reactions proceeded effectively, yielding the desired products with results ranging from 56% to 89%. Notably, no radical trapping product 9 was observed (see Scheme S1†). Furthermore, radical capture and inhibition experiments involving the addition of TEMPO and BHT to the silylation reaction demonstrated no significant effect on the formation of the desired product. In contrast, the introduction of 1,4-dinitrobenzene, a known single-electron transfer (SET) inhibitor, completely inhibited the reaction (Fig. 5a), indicating that the SET process may be involved in the oxidative addition of alkynyl bromide with active manganese (Mn*). Secondly, when the reaction was quenched with HCl (1 M, aqueous), no hydrogenation products were detected in the absence of chlorosilane, and nearly all of reactant 1a was remained (Fig. 5b, top). Substituting AcCl (2 equiv.) for chlorosilane yielded the hydrogenated product in 87% yield with Mn as the reductant, while the yield decreased to 6% with Mg, leaving a surplus of 28% of reactant 1a (Fig. 5b, bottom). The decomposition of chlorosilane in the reaction milieu Mn* facilitates the formation of the alkynylmanganese reagent.5b,20 Thirdly, to identify the type of alkynylmanganese reagent involved in this reaction, the 1ki solution generated from 1k and manganese activated with acyl chloride was analyzed by LC-HRMS (Fig. 5c, eqn (1)). The results corresponded with the di(alkynyl)manganese species (HRMS data: exact mass [M + H]+: 318.0447, found: 318.0445). This 1ki solution was subsequently reacted with TMSCl, resulting in the formation of 3ab with a yield of 17% (Fig. 5c, eqn (1)). Alternatively, 1ki was synthesized through a previously established transmetallation of an acetylene lithium reagent with MnBr2, followed by reaction with TMSCl, yielding 3ab with a yield of 30% (Fig. 5c, eqn (2)). To isolate or trap the di(alkynyl)manganese species 1ki, 1,10-phenanthroline (Phen) was added. Upon the addition of Phen to the 1ki solution formed via both pathways, an intermediate (Int-I) precipitated from the solution, with both exhibiting consistent paramagnetic NMR spectra. Unfortunately, the X-ray structure of Int-I was not identified as a di(alkynyl)manganese species; rather, it corresponded to (Phen)2MnCl2 (Fig. 5c). We speculate that the formation of MnX2 occurs concurrently with the generation of the di(alkynyl)manganese species or stems from the activation of manganese with acyl chloride or TMSCl. Furthermore, results from the control experiments show that after the coupling of 1k with TMSCl under standard conditions, the filtrate remains suitable for the synthesis of Int-I (X = Cl), indicating that a significant amount of MnCl2 is produced during the reaction.21
 | ||
| Fig. 5 Experiments designed to elucidate the reaction mechanism. | ||
Based on these experimental results and previous reports,3,4,9a,12 a reaction pathway is illustrated in Fig. 6b. Alkyne bromide 1k reacts with Mn* through an oxidative addition mechanism that involves a solvent-facilitated process, resulting in the formation of alkynylMn(II) bromide TS3. This is subsequently followed by transmetallation to yield 1ki, accompanied by the generation of MnX2.22 The nucleophilic substitution of 1ki with chlorosilanes produces the desired product, and the reaction pathway via TS3 with chlorosilanes is ruled out.23 Density Functional Theory (DFT) computations from 1k to TS3 provide support for this mechanistic proposal (Fig. 6a).21 Our calculations indicate a significant decrease in free energy of 71.6 kcal mol−1 as the Mn(DMA)5 complex24 binds to the bromine atom of 1k via a SET process, forming the highly stable intermediate IM1, which features a prominent six-membered ring with notable hydrogen bonding between the DMA and Br atoms. This finding underscores the indispensable role of amide solvents in facilitating this reaction (Table 1 and Fig. 6a). The reductive Mn(I) complex interacting with the electron-accepting compound 1k leads to the formation of a three-membered transition state TS1, accompanied by the dissociation of a DMA molecule, requiring a moderate energy barrier of 6.2 kcal mol−1. To achieve the transition state TS3, the synergistic process includes the dissociation of multiple DMA molecules and necessitates overcoming an additional energy barrier of 28.1 kcal mol−1, which represents the rate-determining step of the entire reaction. DFT computations suggest that if the synergistic process fails to facilitate the dissociation of DMA, a less stable transition state TS2 would form, requiring the overcoming of a higher energy barrier, thereby rendering this pathway unfavorable.
 | ||
| Fig. 6 DFT computations and proposed mechanism. | ||
Conclusions
In summary, we have developed a novel method that enables the direct construction of C(sp)–Si bonds from alkynyl halides and chlorosilanes. This study marks the first reported XEC reaction mediated by the in situ-generated alkynylmanganese species, which subsequently engage in SN2 reactions with electrophilic chlorosilanes. Density Functional Theory (DFT) computations highlight the pivotal role of amide solvents in facilitating the formation of these key alkynylmanganese intermediates. This protocol exhibits a broad substrate scope under mild reaction conditions, tolerating various aliphatic and aromatic alkynyl halides, as well as natural products and pharmaceutical-relevant molecules. The unique reactivity of alkynylsilanes offers diverse opportunities for late-stage derivatization and functionalization. Notably, our approach can also be extended to the XEC of alkynyl halides with chlorogermanes, and further explorations are currently being conducted in our laboratory. We expect to publish the progress in due course.Data availability
All the data have been presented in the manuscript and ESI.†Author contributions
QL, LL and JW performed the experiments. QL drafted the manuscript. DY, ZC and ZXW designed the project and revised the manuscript drafting. All the authors participated in the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work from the National Natural Science Foundation of China (No. 22222502, 22301043, 22462001) is gratefully acknowledged, and the Gannan Normal University Start-up Fund (No. BSJJ202306). This research was also supported by the Research Team Program of Gannan Normal University. We also thank Ye Xu from SINAP for assisting us in the growth of crystals.Notes and references
- For selected reviews and examples about recent efforts to cross-electrophile coupling, see: (a) W. L. Williams, N. E. Gutiérrez-Valencia and A. G. Doyle, J. Am. Chem. Soc., 2023, 145, 24175–24183 CrossRef CAS; (b) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (c) J. Liu, Y. Ye, J. L. Sessler and H. Gong, Acc. Chem. Res., 2020, 53, 1833–1845 CrossRef CAS; (d) Q. Pan, K. Wang, W. Xu, Y. Ai, Y. Ping, C. Liu, M. Wang, J. Zhang and W. Kong, J. Am. Chem. Soc., 2024, 146, 15453–15463 CrossRef CAS; (e) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS; (f) J. Duan, Y.-F. Du, X. Pang and X.-Z. Shu, Chem. Sci., 2019, 10, 8706–8712 RSC; (g) H. Meng, J.-S. Jia, P.-F. Yang, Y.-L. Li, Q. Yu and W. Shu, Chem. Sci., 2025, 16, 4442–4449 RSC; (h) D. Sun, G. Ma, X. Zhao, C. Lei and H. Gong, Chem. Sci., 2021, 12, 5253–5258 RSC; (i) Y. Ye, X. Qi, B. Xu, Y. Lin, H. Xiang, L. Zou, X.-Y. Ye and T. Xie, Chem. Sci., 2022, 13, 6959–6966 RSC; (j) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed; (k) A. Tortajada, M. Börjesson and R. Martin, Acc. Chem. Res., 2021, 54, 3941–3952 CrossRef CAS PubMed; (l) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (m) Z. Wu, H. Jiang and Y. Zhang, Chem. Sci., 2021, 12, 8531–8536 RSC; (n) L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450–5457 RSC; (o) E. Richmond and J. Moran, Synthesis, 2018, 50, 499–513 CrossRef CAS; (p) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- (a) T. Hiyama, M. Obayashi and A. Nakamura, Organometallics, 1982, 1, 1249–1251 CrossRef CAS; (b) S.-H. Kim and R. D. Rieke, Synth. Commun., 1998, 28, 1065–1072 CrossRef CAS; (c) S.-H. Kim and R. D. Rieke, J. Org. Chem., 1998, 63, 6766–6767 CrossRef CAS; (d) G. Cahiez, A. Martin and T. Delacroix, Tetrahedron Lett., 1999, 40, 6407–6410 CrossRef CAS; (e) G. Cahiez, C. Duplais and J. Buendia, Chem. Rev., 2009, 109, 1434–1476 CrossRef CAS; (f) G. Dagousset, C. François, T. León, R. Blanc, E. Sansiaume-Dagousset and P. Knochel, Synthesis, 2014, 46, 3133–3171 CrossRef CAS; (g) W. Yan, A. T. Poore, L. Yin, S. Carter, Y.-S. Ho, C. Wang, S. C. Yachuw, Y.-H. Cheng, J. A. Krause, M.-J. Cheng, S. Zhang, S. Tian and W. Liu, J. Am. Chem. Soc., 2024, 146, 15176–15185 CrossRef CAS PubMed; (h) D. Knyszek, J. Löffler, D. E. Anderson, E. Hevia and V. H. Gessner, J. Am. Chem. Soc., 2025, 147, 5417–5425 CrossRef CAS; (i) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS PubMed; (j) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC.
- (a) J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed; (b) P.-F. Su, K. Wang, X. Peng, X. Pang, P. Guo and X.-Z. Shu, Angew. Chem., Int. Ed., 2021, 60, 26571–26576 CrossRef CAS; (c) P. Guo, X. Pang, K. Wang, P.-F. Su, Q.-Q. Pan, G.-Y. Han, Q. Shen, Z.-Z. Zhao, W. Zhang and X.-Z. Shu, Org. Lett., 2022, 24, 1802–1806 CrossRef CAS PubMed; (d) X. Pang, P.-F. Su and X.-Z. Shu, Acc. Chem. Res., 2022, 55, 2491–2509 CrossRef CAS; (e) Y.-H. Yang, X. Pang and X.-Z. Shu, Synthesis, 2022, 55, 868–876 Search PubMed; (f) X. Pang and X.-Z. Shu, Chem.–Eur. J., 2023, 29, e202203362 CrossRef CAS PubMed; (g) G.-Y. Han, P.-F. Su, Q.-Q. Pan, X.-Y. Liu and X.-Z. Shu, Nat. Catal., 2024, 7, 12–20 CrossRef CAS; (h) C. Li, S. Yang and X. Zeng, ACS Catal., 2023, 13, 12062–12073 CrossRef CAS; (i) Y. Lin, L. Zou, R. Bai, X.-Y. Ye, T. Xie and Y. Ye, Org. Chem. Front., 2023, 10, 3052–3060 RSC; (j) J. Manigrasso, I. Chillón, V. Genna, P. Vidossich, S. Somarowthu, A. M. Pyle, M. De Vivo and M. Marcia, Nat. Commun., 2022, 13, 1 CrossRef CAS.
- D. Xing, J. Liu, D. Cai, B. Huang, H. Jiang and L. Huang, Nat. Commun., 2024, 15, 4502 CrossRef CAS.
- (a) W. Wu and H. Jiang, Acc. Chem. Res., 2014, 47, 2483–2504 CrossRef CAS; (b) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS; (c) R. Pan, C. Shi, D. Zhang, Y. Tian, S. Guo, H. Yao and A. Lin, Org. Lett., 2019, 21, 8915–8920 CrossRef CAS; (d) Y. Jiang, J. Pan, T. Yang, Y. Zhao and M. J. Koh, Chem, 2021, 7, 993–1005 CrossRef CAS.
- (a) J. Z. Dávalos and T. Baer, J. Phys. Chem. A, 2006, 110, 8572–8579 CrossRef; (b) L. Lu, J. C. Siu, Y. Lai and S. Lin, J. Am. Chem. Soc., 2020, 142, 21272–21278 CrossRef CAS; (c) H. Yoo, P. J. Carroll and D. H. Berry, J. Am. Chem. Soc., 2006, 128, 6038–6039 CrossRef CAS PubMed; (d) L. Qiu, Y. Liu, H. Chen, L. Song and W. Xie, Chem. Sci., 2025, 16, 9454–9461 RSC.
- (a) C.-L. Ji, X. Zhai, Q.-Y. Fang, C. Zhu, J. Han and J. Xie, Chem. Soc. Rev., 2023, 52, 6120–6138 RSC; (b) S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS; (c) T. Xiao-Jun and C. Qing-Yun, Chem. Sci., 2012, 3, 1694–1697 RSC.
- (a) J. Duan, Y. Wang, L. Qi, P. Guo, X. Pang and X.-Z. Shu, Org. Lett., 2021, 23, 7855–7859 CrossRef CAS PubMed; (b) L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 18587–18590 CrossRef CAS PubMed; (c) Z.-Z. Zhao, X. Pang, X.-X. Wei, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2022, 61, e202200215 CrossRef CAS.
- (a) L. Qi, X. Pang, K. Yin, Q.-Q. Pan, X.-X. Wei and X.-Z. Shu, Chin. Chem. Lett., 2022, 33, 5061–5064 CrossRef CAS; (b) V. Lanke and I. Marek, Chem. Sci., 2020, 11, 9378–9385 RSC.
- (a) A. Fürstner and H. Brunner, Tetrahedron Lett., 1996, 37, 7009–7012 CrossRef; (b) S.-H. Kim, M. V. Hanson and R. D. Rieke, Tetrahedron Lett., 1996, 37, 2197–2200 CrossRef CAS; (c) H. Kakiya, S. Nishimae, H. Shinokubo and K. Oshima, Tetrahedron, 2001, 57, 8807–8815 CrossRef CAS; (d) P. DeShong, D. R. Sidler, P. J. Rybczynski, G. A. Slough and A. L. Rheingold, J. Am. Chem. Soc., 1988, 110, 2575–2585 CrossRef CAS; (e) R. Takahashi, P. Gao, K. Kubota and H. Ito, Chem. Sci., 2023, 14, 499–505 RSC.
- (a) C. J. Elsevier, H. Kleijn, J. Boersma and P. Vermeer, Organometallics, 1986, 5, 716–720 CrossRef CAS; (b) A. J. Eberhart, H. J. Shrives, E. Álvarez, A. Carrër, Y. Zhang and D. J. Procter, Chem.–Eur. J., 2015, 21, 7428–7434 CrossRef CAS; (c) P. B. Changala, P. R. Franke, J. F. Stanton, G. B. Ellison and M. C. McCarthy, J. Am. Chem. Soc., 2024, 146, 1512–1521 CrossRef CAS.
- A. A. Toutov, K. N. Betz, D. P. Schuman, W.-B. Liu, A. Fedorov, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1668–1674 CrossRef CAS.
- (a) M.-M. Liu, Y. Xu and C. He, J. Am. Chem. Soc., 2023, 145, 11727–11734 CrossRef CAS; (b) Y. Ge, J. Ke and C. He, Acc. Chem. Res., 2025, 58, 375–398 CrossRef CAS; (c) Y. Wu, L. Zheng, Y. Wang and P. Wang, Chem, 2023, 9, 3461–3514 CrossRef CAS.
- (a) R. Tacke and U. Wannagat, Bioactive Organo-Silicon Compounds, Topics in Current Chemistry, Vol. 84, Springer, Berlin, Heidelberg, 1979 Search PubMed; (b) T. Liu, X.-R. Mao, S. Song, Z.-Y. Chen, Y. Wu, L.-P. Xu and P. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216878 CrossRef CAS.
- A. Hamze, O. Provot, M. Alami and J.-D. Brion, Org. Lett., 2006, 8, 931–934 CrossRef CAS PubMed.
- M. Patel, R. K. Saunthwal, D. K. Dhaked, P. V. Bharatam and A. K. Verma, Asian J. Org. Chem., 2015, 4, 894–898 CrossRef CAS.
- M.-M. Xu, X.-Y. You, Y.-Z. Zhang, Y. Lu, K. Tan, L. Yang and Q. Cai, J. Am. Chem. Soc., 2021, 143, 8993–9001 CrossRef CAS.
- M. Ueda, T. Ueno, Y. Suyama and I. Ryu, Tetrahedron Lett., 2017, 58, 2972–2974 CrossRef CAS.
- (a) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed; (b) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS; (c) J. Zhu, S. Chen and C. He, J. Am. Chem. Soc., 2021, 143, 5301–5307 CrossRef CAS PubMed.
- (a) K. Takai, T. Ueda, T. Hayashi and T. Moriwake, Tetrahedron Lett., 1996, 37, 7049–7052 CrossRef CAS; (b) S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS; (c) K. A. Johnson, S. Biswas and D. J. Weix, Chem.–Eur. J., 2016, 22, 7399–7402 CrossRef CAS; (d) M. Xü, S. Wu and G. Zou, Eur. J. Org Chem., 2024, 27, e202400625 CrossRef; (e) K. E. Poremba, N. T. Kadunce, N. Suzuki, A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2017, 139, 5684–5687 CrossRef CAS.
- See the ESI† for details.
- (a) M. Tamura and J. Kochi, J. Organomet. Chem., 1971, 29, 111–129 CrossRef CAS; (b) I. Suzuki, M. Yasuda and A. Baba, Chem. Commun., 2013, 49, 11620–11622 RSC.
- (a) R. West and L. C. Quass, J. Organomet. Chem., 1969, 18, 55–67 CrossRef CAS; (b) S. HariPrasad and G. Nagendrappa, Tetrahedron, 1993, 49, 3387–3396 CrossRef CAS; (c) M. Kunishima, D. Nakata, S. Tanaka, K. Hioki and S. Tani, Tetrahedron, 2000, 56, 9927–9935 CrossRef CAS.
- (a) G. A. Carriedo, D. Miguel and V. Riera, J. Organomet. Chem., 1988, 342, 373–380 CrossRef CAS; (b) J. Chai, H. Zhu, H. W. Roesky, Z. Yang, V. Jancik, R. Herbst-Irmer, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2004, 23, 5003–5006 CrossRef CAS; (c) D. Unseld, V. V. Krivykh, K. Heinze, F. Wild, G. Artus, H. Schmalle and H. Berke, Organometallics, 1999, 18, 1525–1541 CrossRef CAS; (d) V. V. Krivykh, I. L. Eremenko, D. Veghini, I. A. Petrunenko, D. L. Pountney, D. Unseld and H. Berke, J. Organomet. Chem., 1996, 511, 111–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectroscopic characterization and crystallographic data. CCDC 2423603. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03449c |
| This journal is © The Royal Society of Chemistry 2025 |