
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterogenization of molecular chalcoxides for electro-& photochemical H2 production†
Mark Gray a,
Yu-Fei Song*b and
Haralampos N. Miras*a
a,
Yu-Fei Song*b and
Haralampos N. Miras*a
aSchool of Chemistry, The University of Glasgow, Glasgow G12 8QQ, UK. E-mail: charalampos.moiras@glasgow.ac.uk
bState Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Centre for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: songyf@mail.buct.edu.cn
First published on 19th December 2024
Abstract
The endeavour to develop high-performance cost-effective catalytic systems with a minimal amount of active components and generation of hazardous waste is a significant and formidable step towards enhancing the hydrogen evolution reaction and the development of design principles with potential value in large scale applications. Here we investigate the heterogenization process of molecular chalcoxide catalysts and explore their electro- and photo-catalytic efficacy in driving the hydrogen evolution reaction (HER).
One of the primary dilemmas facing today's society is the impending energy crisis, which is closely associated with the escalating problem of global warming and the continuous increase in greenhouse gas emissions. It is apparent that a significant transformation in our political and economic decisions is imperative, given the growing dependence on renewable energy sources and the surge in research interest in materials science. Solar fuels offer a potential resolution to this problem, as they can be produced through the electro1 or photo2–4 driven hydrogen evolution reaction (HER). These reactions are complex and require a carefully designed catalytic system. Therefore, there has been a significant amount of research dedicated to the development of cost-efficient catalysts for the HER that do not require precious metals such as platinum. Ideally, hydrogen produced via water splitting enabled through renewable energy or through visible light would be utilised. Various catalysts made from readily available materials have been reported, primarily utilizing first row transition metals such as nickel and cobalt,5,6 as well as transition metal chalcogenides. Considering the latter, both theoretical and experimental studies have established a direct relationship between the catalytic activity towards the HER and the number of unsaturated sulphur atoms located at the edges of MoS2.7 In addition to the above observations, the well-defined structural features, solution processability, and modularity of molecular chalcogenides and chalcoxides, a subgroup of discrete molecular species comprised of transition metal oxysulfide/oxyselenides,8,9 have been the key factors driving the expansion of this family and the development of intriguing derivatives.3,10–12 To enhance various facets concerning the performance, functionality, modularity, stability, and cost-effectiveness of catalytic systems, it is essential to design suitable multicomponent systems that leverage the potentially cooperative interactions arising among the individual constituents. Some potential candidates for this include nanomaterials and graphitic carbon materials, such as graphitic carbon nitride (gCN), graphene, and carbon nanotubes (CNTs), all of which have shown promising cooperative effects for a range of catalytic species, however, the majority of the reported composite materials are based on the use of precious or rare metal nanoparticles.13–17 Specifically for our studies, we employed CNTs as the support component in our design approach due to their wide range of advantageous properties, such as mechanical strength, chemical stability, thermal resistance, and electrical conductivity.
By harnessing the modularity and solution processability of discrete molecular species comprised of transition metal oxysulfide/oxyselenides (molecular chalcoxides) and their potential for heterogenization on carbon nanotubes (CNTs), such as by adsorption of these metal chalcoxides on the surface of appropriate nanomaterials, fruitful cooperative effects can be exploited (Fig. 1). Previous studies have demonstrated the use of nanotubes in engineering composite materials for diverse applications.18,19 However, the majority of the reported composite materials are based on the use of precious or rare metal nanoparticles by employing high cost and time-consuming processes.13–17 For example, there have been reports of Ru based catalysts immobilised on/in both single and multi-walled carbon nanotubes, with promising overall catalytic performance.20,21 The development of these catalytic systems relies on the precious metal Ru, posing a significant drawback due to the potential cost limitations in large-scale applications. There are a variety of examples demonstrating the synergistic effects of MoX (X = C, N, B, S) supported on nanotubes, as these materials benefit from their high conductivity and subsequent improvement in efficiency of the underlying charge transfer processes that are crucial in an efficient HER system.22–24
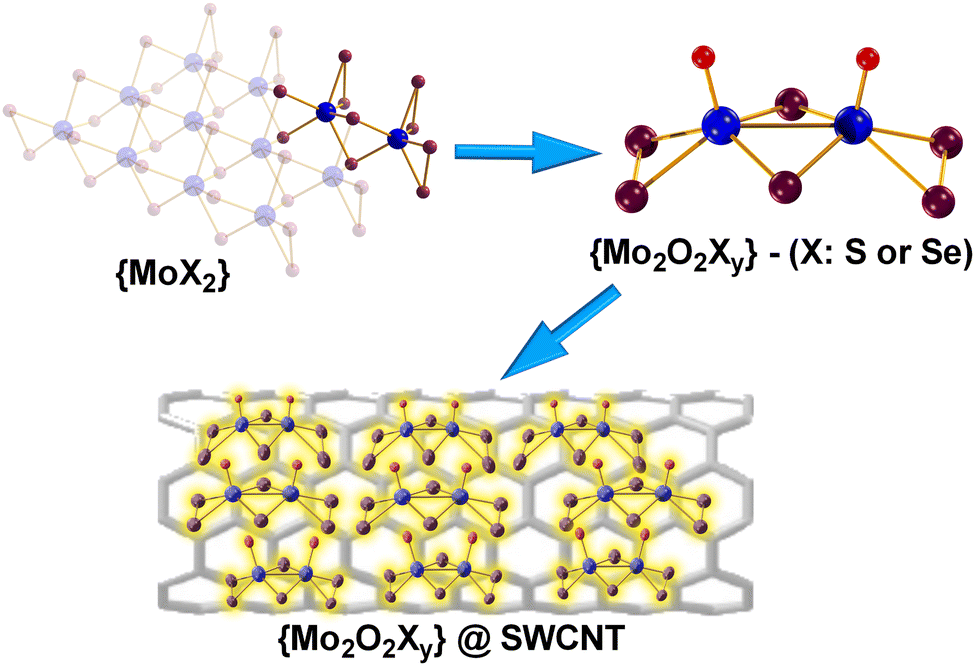 | ||
| Fig. 1 Ball-and-stick representation of: MoX2, [Mo2O2(μ-S)2(S2)(S4)]2− (1) and [Mo2O2(μ-Se)2(Se2)2]2−@SWCNT (2). Colour code: Mo, blue; X (S, Se), plum; O, red; counterions are omitted for clarity. | ||
Previous work conducted by our group has investigated a series of catalysts based on molecular transition metal chalcoxides, namely Mo2O2S62−, Mo2O2S82− (1), Mo2O2Se62− (2) and W2O2S62−.8,9 Considering the advantageous characteristics of CNTs mentioned above, it opens up possibilities for the design of catalytic composite materials and investigating the positive synergistic effects between the heterogenized molecular catalysts and the conductive CNT support. Consequently, this research endeavour is focused on exploring economically viable alternatives that could provide us with efficient catalytic systems for electrochemical and photochemical hydrogen evolution that is more durable, stable, and environmentally friendly.
The composite catalytic material's design entails the facile integration of the {Mo2} species by suspending the SWCNT in an acetone solution containing the respective dimers (1 or 2) at room temperature. To prevent potential oxidation, the reaction is carried out under a N2 atmosphere as an additional precaution due to the susceptibility of 2 to oxidation. The obtained composite materials 1@SWCNT and 2@SWCNT were subsequently characterised by FT-IR, Raman, EDS, SEM, XPS and TGA. Raman spectroscopy was used to confirm the adsorption of the catalytic species on the surface of the SWCNTs. Firstly, the study confirms the structural integrity of the SWCNTs during the heterogenization process. Additionally, the observed shifts of the relevant bands are assigned to surface processes influencing the C–C bond network and it is indicative of the adsorption of the molecular species on the CNT. Most importantly, the absence of characteristic Raman peaks of C–S (∼616, 660 cm−1) or C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼1770 cm−1) indicate the absence of both forms of bonding where it would be the case if we had coordinative interaction between the molecular catalyst and the CNT which is an additional indication that the molecular species are adsorbed on the surface of the CNT support without forming covalent bonds with it (see ESI†).
O (∼1770 cm−1) indicate the absence of both forms of bonding where it would be the case if we had coordinative interaction between the molecular catalyst and the CNT which is an additional indication that the molecular species are adsorbed on the surface of the CNT support without forming covalent bonds with it (see ESI†).
SEM measurements were recorded at various magnifications over multiple sites. Energy-dispersive X-ray measurement (EDX) elemental mapping (Fig. 2) showed that Mo, O, and S/Se elements were uniformly dispersed on the surface of the 1/2@SWCNT composite fibrous material. Detailed EDS analysis is provided in the ESI.†
 | ||
| Fig. 2 SEM image of composites 1@SWCNT and 2@SWCNT, with overlaid EDS (top), and elemental distribution from EDS (bottom). | ||
The TGA study revealed that the nanotubes retain their stability until a temperature of 450 °C. Beyond this point, the composite material decompose associated with weight losses of carbon, sulfur and selenium volatile by-products. Once the temperature reaches 695 °C, all volatile components are eliminated, resulting in the presence of molybdenum (in the form of MoO3). This enables the estimation of the catalyst loading by determining the moles of molybdenum present in the sample. The loading of the material was calculated to be ∼1% wt for 1, and 2% wt for 2 (based on EDX, TGA). Based on the calculated Mo content, this corresponds to a geometric catalytic loading on the working electrode of 0.05 μmol cm−2 and 0.082 μmol cm−2 respectively.
The catalytic performance of the composite materials towards HER was evaluated by drop casting the catalytic composites on a glassy carbon electrode (GCE, see ESI†). The scan rate for all linear sweep voltammetry (LSV) tests was 5 mV s−1, and the obtained polarisation curves were iR corrected to compensate for any potential loss arising from external resistance of the electrochemical system. Furthermore, a comparative study was conducted using mixtures of the same catalytic loading with carbon black, which were prepared from stock solutions of the respective dimers (Fig. 3). Moreover, the optimization of the heterogenization process for our studies on the catalytic dyes involved the assessment of various polar organic solvents. Our control experiments revealed that MeCN, specifically in the case of 2, provided satisfactory dispersity of the material and ensured the molecule's stability as well as the mechanical stability of the produced film on the electrode's surface. Additionally, both materials exhibited a faradaic efficiency of 100% (see ESI†). The results obtained from the comparative study indicated that the composite materials 1@SWCNT and 2@SWCNT exhibited a decrease of the associated onset potential for the HER within the range of 50–100 mV, in comparison to the physical mixture of the active dimer and carbon powder as shown in Fig. 3. More specifically, the inherent cooperative effect of the designed catalytic composite materials showed a 25.5% improvement for 1, and 24.3% for 2.8,9 That equates to a decrease in onset potential by 65 mV for 1@SWCNT, with η10 (the overpotential required to reach 10 mA cm−2) equal to 190 mV (vs. RHE, 25.5% improvement). In a similar manner, comparing the performance of 2@SWCNT against the physical mixture of 2 with carbon powder, cooperative effects influence the efficacy of the composite, a fact that is reflected to a decrease in onset potential by 90 mV and overpotential at current density of 10 mA cm−2 η10 = 280 mV (vs. RHE, 24.3% improvement). In order to evaluate the dynamics of the system, we conducted Tafel analysis revealing a slope of 58 mV dec−1 in the case of 1@SWCNT and 100 mV dec−1 in the case of 2@SWCNT, indicative of underlying Volmer–Heyrovsky processes.25 Furthermore, in order to assess the long-term stability of the materials, linear sweep voltammetry was used to compare η10 values after 1000 cycles (Fig. S8, ESI†), which revealed negligible reductions in activity, confirming the long-term stability of the composites. To confirm that the molecular species is indeed the true source of the observed catalytic activity, we performed cyclic voltammetry in organic media, which revealed no additional redox waves following the cycling process, indicating no structural or functional changes occur as a result of the prolonged catalytic cycling (Fig. S9, ESI†). Similar to the aforementioned improvements in bulk materials such as MoS2,22,23 this improvement can be attributed to the enhanced conductivity and efficient electron transfer process from the surface of the SWCNTs to the molecular chalcoxide catalytic species.26 Moreover, further increase of the applied overpotential in both cases results in a rapid rise of the cathodic current, which is indicative of accelerated HER activity. The analysis of the evolved gas using gas chromatography (Fig. S4, ESI†) confirmed further that the reaction occurring on the working electrode corresponds to the hydrogen evolution reaction (HER). The relevant turnover numbers expressed in amount of H2 produced per moles of catalytic composite found to be 859 and 57.2 for 1@SWCNT and 2@SWCNT respectively. These results unequivocally demonstrate the efficient evolution of hydrogen at full faradaic efficiency.
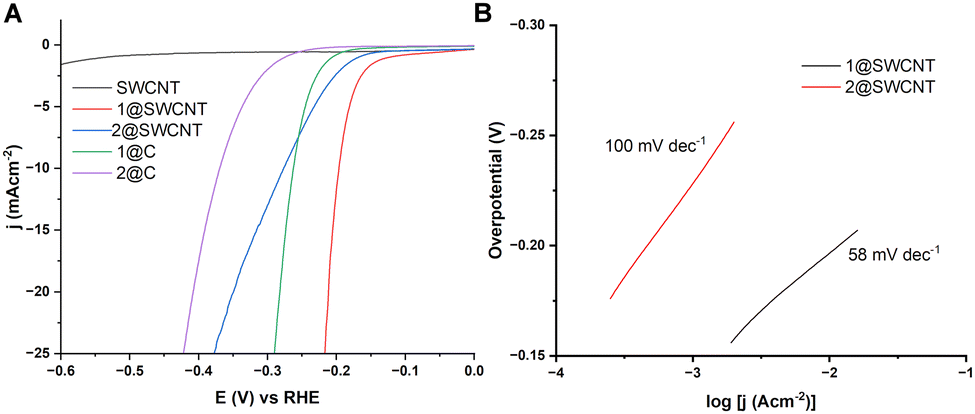 | ||
| Fig. 3 (A) Polarisation curves of the heterogenized molecular catalysts 1 and 2 on SWCNTs (red and blue) compared with the physical mixture of 1 and 2 with carbon powder (purple and green) in 1 M H2SO4. (B) Tafel slopes of the respective species. | ||
Subsequently, we explored the functionality of the heterogenized molecular catalysts for the production of H2 under photocatalytic conditions. More specifically, light driven HER activity was explored by combining individually the catalytic composites 1@SWCNT and 2@SWCNT in the presence of triethanolamine and Eosin Y as sacrificial electron donor and photosensitiser respectively, at neutral pH values (pH 7, see ESI† for details). Typical light driven experiments were performed under argon atmosphere at room temperature. Each experiment was carried out in triplicate and quantification of H2 was performed by gas chromatography of samples from the respective head spaces every 30 min of irradiation for a total duration of 3 hours. Gas chromatography revealed the potential of both catalytic composites in the photocatalytic production of H2 in the presence of a photosensitizer. Maximum activity was observed for both materials during the first 3 hours after which 1@SWCNT continued to produce H2 for the duration of the experiment in contrast to 2@SWCNT which produced minor additional amounts of H2 beyond that point (see Fig. 4). In either case, it becomes apparent that both materials can serve as functional photocatalysts for the HER, even at very low concentrations (0.011 mM for 1@SWCNT, 0.047 mM for 2@SWCNT, based on active catalyst loading), in neutral aqueous media. The maximum rate of H2 production found to be 0.027 μmol h−1 for 1@SWCNT and 0.080 μmol h−1 for 2@SWCNT.
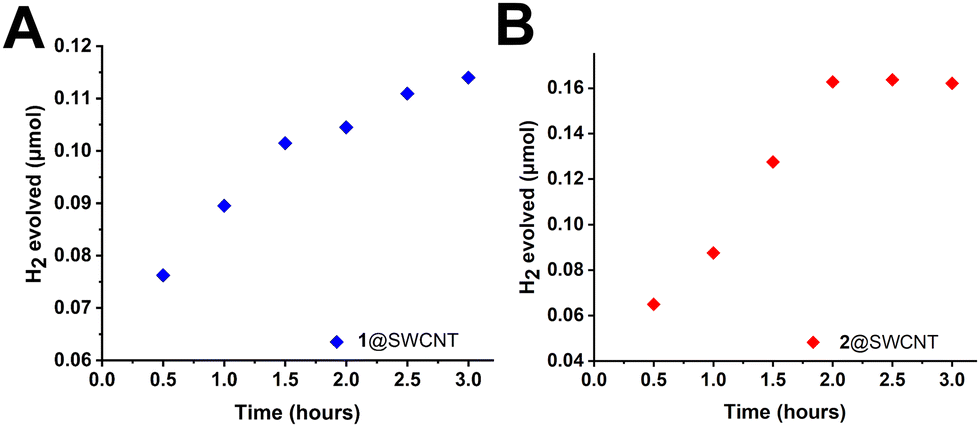 | ||
| Fig. 4 Photochemical hydrogen evolution for 1@SWCNT (A) and 2@SWCNT (B) in aqueous media at neutral pH. | ||
In conclusion, we discussed an accessible and facile design approach for the heterogenization of the [Mo2O2S2(S2)(S2)2]2− and [Mo2O2Se2(Se2)2]2− molecular catalysts on a SWCNT based catalytic system making it possible to identify effective cooperative effects in the electrochemical and photochemical production of H2. The considerable improvement of electrochemical activity at very low loadings of the generated composites (190 and 280 mV corresponding to 25.5 and 24.3% improvement in terms of overpotential at current densities of 10 mA cm−2 respectively) as well as in photochemical applications, paves the way for further exploration and development of next generation catalytic systems for H2 production. These results highlight a promising strategy for the design of economical, resilient, and eco-friendly electrocatalysts for hydrogen evolution reactions (HER). Investigating the modularity and processability of molecular species through the integration of transition metals or chalcogens within their molecular framework can provide valuable insights to refine catalyst design strategies and exploit potential cooperative and structural interactions. Most importantly, establishing a simple and cost-efficient synthetic approach for composite catalytic systems is vital for ensuring scalability in larger-scale applications.
This work was supported by the EPSRC grants (No. EP/J015156/1 and 2588464) and the University of Glasgow. H. N. M. thanks Dr Christopher Kelly for the XPS measurement.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. C. McGlynn, T. Dankwort, L. Kienle, N. A. G. Bandeira, J. P. Fraser, E. K. Gibson, I. Cascallana-Matías, K. Kamarás, M. D. Symes, H. N. Miras and A. Y. Ganin, Nat. Commun., 2019, 10, 4916 CrossRef PubMed.
- S. Repp, M. Remmers, A. S. J. Rein, D. Sorsche, D. Gao, M. Anjass, M. Mondeshki, L. M. Carrella, E. Rentschler and C. Streb, Nat. Commun., 2023, 14, 5563 CrossRef CAS PubMed.
- A. Abbas, E. Ostwald, J. Romer, A. Lenzer, M. Heiland, C. Streb, C. Kranz and A. Pannwitz, Chem. – Eur. J., 2023, 29, e202302284 CrossRef CAS PubMed.
- S. Batool, S. P. Nandan, S. N. Myakala, A. Rajagopal, J. S. Schubert, P. Ayala, S. Naghdi, H. Saito, J. Bernardi, C. Streb, A. Cherevan and D. Eder, ACS Catal., 2022, 12, 6641–6650 CrossRef CAS PubMed.
- Y. Aimoto, K. Koshiba, K. Yamauchi and K. Sakai, Chem. Commun., 2018, 54, 12820–12823 RSC.
- R. Takada, T. Nakazono, T. Nishimura, T. Shiga, M. Nihei, Y. Yamada and T. Wada, Sustainable Energy Fuels, 2023, 7, 3603–3608 RSC.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, J. Catal., 2000, 189, 129–146 CrossRef CAS.
- J. McAllister, N. A. G. Bandeira, J. C. McGlynn, A. Y. Ganin, Y.-F. Song, C. Bo and H. N. Miras, Nat. Commun., 2019, 10, 370 CrossRef CAS PubMed.
- A. Elliott, J. McAllister, L. Masaityte, M. Segado-Centellas, D. L. Long, A. Y. Ganin, Y. F. Song, C. Bo and H. N. Miras, Chem. Commun., 2022, 58, 6906–6909 RSC.
- X. L. Wang, C. Xue, N. Kong, Z. Wu, J. Zhang, X. Wang, R. Zhou, H. Lin, Y. Li, D. S. Li and T. Wu, Inorg. Chem., 2019, 58, 12415–12421 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248–253 CrossRef CAS PubMed.
- S. Batool, M. Langer, S. N. Myakala, M. Heiland, D. Eder, C. Streb and A. Cherevan, Adv. Mat., 2024, 36, 2305730 CrossRef CAS PubMed.
- M. A. Domínguez-Crespo, E. Ramírez-Meneses, A. Torres-Huerta, V. Garibay-Febles and K. Philippot, Int. J. Hydrogen Energy, 2012, 37, 4798–4811 CrossRef.
- S. A. Grigoriev, P. Millet and V. N. Fateev, J. Power Sources, 2008, 177, 281–285 CrossRef CAS.
- S. Zhou, X. Chen, P. Yu, F. Gao and L. Mao, Electrochem. Commun., 2018, 90, 91–95 CrossRef CAS.
- C. Huff, T. Dushatinski and T. M. Abdel-Fattah, Int. J. Hydrogen Energy, 2017, 42, 18985–18990 CrossRef CAS.
- C. Huff, J. M. Long, A. Heyman and T. M. Abdel-Fattah, ACS Appl. Energy Mater., 2018, 1, 4635–4640 CrossRef CAS.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. I. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS PubMed.
- R. K. Das, Y. Wang, S. V. Vasilyeva, E. Donoghue, I. Pucher, G. Kamenov, H. P. Cheng and A. G. Rinzler, ACS Nano, 2014, 8, 8447–8456 CrossRef CAS PubMed.
- M. A. Hoque, M. Gil-Sepulcre, A. de Aguirre, J. A. A. W. Elemans, D. Moonshiram, R. Matheu, Y. Shi, J. Benet-Buchholz, X. Sala, M. Malfois, E. Solano, J. Lim, A. Garzón-Manjón, C. Scheu, M. Lanza, F. Maseras, C. Gimbert-Suriñach and A. Llobet, Nat. Chem., 2020, 12, 1060–1066 CrossRef CAS PubMed.
- D. H. Kweon, M. S. Okyay, S. J. Kim, J. P. Jeon, H. J. Noh, N. Park, J. Mahmood and J. B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS PubMed.
- D. H. Youn, S. Han, J. Y. Kim, J. Y. Kim, H. Park, S. H. Choi and J. S. Lee, ACS Nano, 2014, 8, 5164–5173 CrossRef CAS PubMed.
- W. Tang, J. Jian, G. Chen, W. Bian, J. Yu, H. Wang, M. Zhou, D. Ding and H. Luo, Energy Mater. Adv., 2021, 140964 Search PubMed.
- Y. Huang, Y. Bao, T. Huang, C. Hu, H. Qiu and H. Liu, Molecules, 2023, 28, 192–203 CrossRef CAS PubMed.
- J. McAllister, N. A. G. Bandeira, J. C. McGlynn, A. Y. Ganin, Y.-F. Song, C. Bo and H. N. Miras, Nat. Commun., 2019, 10, 370 CrossRef CAS PubMed.
- J. W. Jordan, G. A. Lowe, R. L. McSweeney, C. T. Stoppiello, R. W. Lodge, S. T. Skowron, J. Biskupek, G. A. Rance, U. Kaiser, D. A. Walsh, G. N. Newton and A. N. Khlobystov, Adv. Mater., 2019, 31, 1904182 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental method, materials characterization, electrochemical and photocatalytic data. See DOI: https://doi.org/10.1039/d4cc05605a |
| This journal is © The Royal Society of Chemistry 2025 |