
A natural food-grade supramolecular self-assembly system for creation of hierarchically structured hydrogels†
Xinke
Yu‡
a,
Jiyang
Cai‡
a,
Mengyue
Xu
ab,
Qing
Li
a,
Yunyi
Yang
a,
Zhili
Wan
 *ac and
Xiaoquan
Yang
a
*ac and
Xiaoquan
Yang
a
aLaboratory of Food Proteins and Colloids, School of Food Science and Engineering, Guangdong Province Key Laboratory for Green Processing of Natural Products and Product Safety, South China University of Technology, Guangzhou 510640, China. E-mail: zhiliwan@scut.edu.cn; Fax: +(086) 20-87114263
bLaboratory of Physics and Physical Chemistry of Foods, Wageningen University, Bornse Weilanden 9, 6708WG Wageningen, The Netherlands
cOverseas Expertise Introduction Center for Discipline Innovation of Food Nutrition and Human Health (111 Center), Guangzhou 510640, China
First published on 14th June 2024
Abstract
We develop a novel hierarchically structured hydrogel by the supramolecular self-assembly of all-natural food-grade building blocks, glycyrrhizic acid (GA) and carrageenan (CG). The co-assembled GA-CG hydrogel system displays an unusual structural transition with the appearance from opacity to translucence and then to opacity, as a function of the concentration of metal ions. The unique GA-CG supramolecular hydrogel system can serve as solid, edible, and responsive active cargo delivery platforms for food and biomedical applications.
Introduction
Supramolecular hydrogels emerge as ideal functional soft materials for versatile applications in a wide range of biological fields such as drug delivery, functional foods, tissue engineering, and biosensors, due to their facile fabrication process, excellent responsiveness to stimuli, and satisfactory rheological properties.1–5 In general, supramolecular hydrogels can be formed by self-assembly of low molecular weight gelators (LMWGs) through noncovalent interactions (e.g., hydrogen bonding, π–π stacking, etc.),6,7 or by self-assembly of hydrophilic and hydrophobic groups on the main or side chains of polymeric gelators (PGs) driven by electrostatic repulsion and hydrophobic interactions.8,9 In recent years, there is a growing interest in the development of multicomponent supramolecular hydrogels by combining LMWGs with PGs through co-assembly or self-sorting approaches.10–12 This innovative strategy is expected to form a new type of gel network and achieve multifunctional properties.13 Considering the safe and green applications in functional food and biomedical fields, it is highly desirable to develop supramolecular hydrogels by the use of naturally occurring, edible ingredients as building blocks that have high bioactivity, biocompatibility, and biodegradability.14–16Glycyrrhizic acid (GA) is a naturally occurring compound extracted from licorice root that possesses various biological activities, including anti-inflammatory, hepatoprotective, anticancer, and antiviral activities.17–19 From the point of view of chemical structure, GA is a chiral amphiphilic molecule consisting of a hydrophobic triterpenoid glycoside (18β-glycyrrhetinic acid) and a hydrophilic diglucuronic acid unit. The inherent amphiphilic and chiral nature of the GA molecules enables them to undergo anisotropic self-assembly to form supramolecular nanofibrils in water by hydrophobic interactions of the triterpene fragments and hydrogen bonds of the glucuronic acid fragments.20 The formed semi-flexible nanofibrils are right-handedly twisted with a width of about 2.5 nm and a period of 9 nm, and can further entangle to form supramolecular hydrogels with three-dimensional networks.21 Furthermore, the presence of numerous functional groups, rigid skeleton, and distinctive stacking behaviors within the GA molecules render them as ideal structural motifs to produce a broad range of GA derivatives, thereby promoting the development of multifunctional hydrogel materials.22–24 We have utilized the supramolecular self-assembly of GA to construct a variety of novel colloidal materials, such as robust gel emulsions for protection of nutrients,25,26 ultrastable thermo-responsive foams,27–29 and injectable hydrogels for wound healing.30 However, GA supramolecular hydrogels possess poor mechanical strength and limited ability to be shaped and can be easily destroyed during processing. Combining GA with biopolymers that have the capacity of self-assembly can endow GA supramolecular hydrogels with more functional properties and broaden application fields.
Carrageenan (CG), an algal sulfated linear polysaccharide with a galactan backbone, is an important class of food-grade biopolymers with superior gelling, thickening, and stabilizing properties and has been widely used in food, medical, cosmetic, and pharmaceutical applications.31,32 κ-CG, ι-CG, and λ-CG are three of the most common types of carrageenan, differing in the number and position of the sulfate groups: one for κ-CG repeat unit, two for ι-CG repeat unit, and three for λ-CG repeat unit.31,32 The formation of CG gels involves a conformational transition from random coils to well-ordered helices, and the helical structures are further associated by metal ion-mediated aggregation into supramolecular multi-filament structures.33,34 Therefore, as a unique naturally edible biopolymer with metal ion-induced supramolecular self-assembly, the combination of CG and GA may be able to create a new type of supramolecular hydrogels with satisfactory mechanical properties and multifunctionality by employing the multicomponent approach.
Herein, we explore the natural food-grade supramolecular self-assembly system of GA and CG with aim of creating novel hierarchically structured supramolecular hydrogels. The co-assembled GA-CG hydrogels showed an interesting structural transition and their appearance changes from opacity to translucence and then to opacity with the increasing concentration of metal ions. The assembly behavior and microstructure underlying this structural transition were systematically investigated. Finally, the delivery and controlled release properties of GA-CG hydrogel systems were evaluated. The obtained results are expected to have sustainable applications in those requiring high biosafety, such as functional foods and controlled delivery and release for biomedical use.
Results and discussion
In this study, we mixed glycyrrhizic acid (GA), carrageenan (CG), and metal ions (Na+ and K+) under stirring and heating using the one-pot method and then cooled the system to induce the formation of hydrogels (Fig. 1a). We observed that the GA-CG hydrogels underwent an interesting transition from opaque to translucent to opaque with increasing ion concentration (Fig. 1b and c), and the transparency of the hydrogel reached a maximum at the concentrations of 50 mM and 100 mM ions. All the samples were transparent solutions after heating (Fig. 1d), and differences in transparency appeared after cooling into hydrogels (Fig. 1e). In addition, this transparency transition was reversible, with no significant change in transparency during five heating–cooling cycles (Fig. 1f). Interestingly, for hydrogels of GA and other polysaccharides, such as guar gum (GG) and xanthan gum (XG), this transparency transition was not observed; instead, the turbidity consistently increased with increasing ion concentration (Fig. S1†). It is well known that salt ions can screen the electrostatic repulsion between polymer chains, resulting in tighter chain entanglement and twisted conformational changes, and thus a turbid appearance.33,35 The unusual transition phenomenon of GA-CG hydrogels suggests that a unique co-assembly behavior may occur in the supramolecular self-assembly system of GA and CG at different ion concentrations.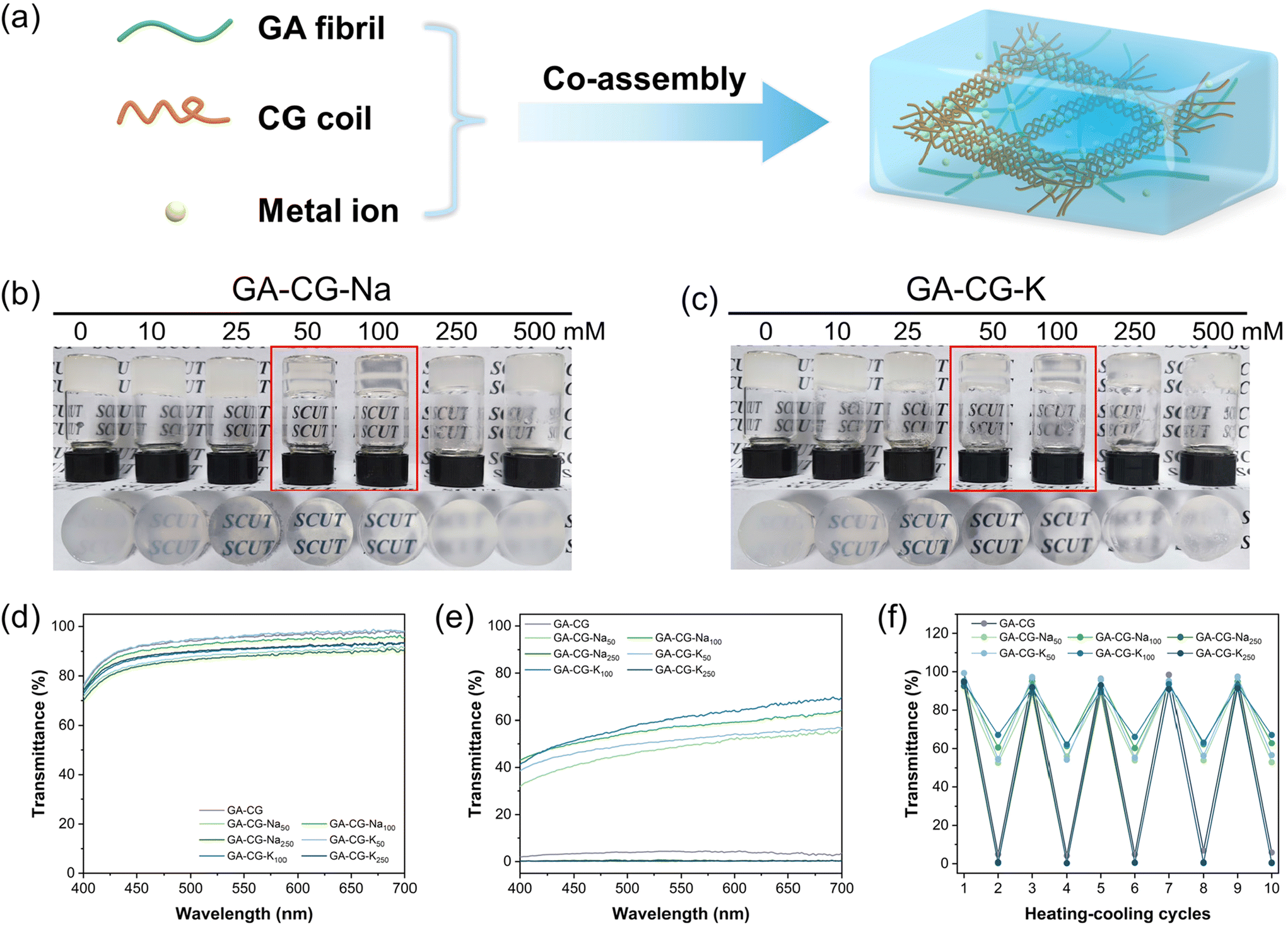 | ||
| Fig. 1 Schematic diagram of the fabrication process for GA-CG hydrogels (a). Digital photos of the gelation experiments formed from the GA-CG hydrogel with Na+ (b) and K+ (c). Transmittance spectra of GA-CG hydrogels after heating at 80 °C (d) and cooling to room temperature (e). Transmittance at 600 nm of GA-CG hydrogels during the five heating–cooling cycles (f). Pure water as blank control. Odd numbered cycles for heating, even numbered cycles for cooling. | ||
To further reveal this interesting phenomenon of transparency transition, the microstructure of the hydrogels was observed using Cryo-SEM. Compared to the GA-CG with homogeneous pores of around 3 μm diameters, upon addition of 50 mM salt ions, GA-CG-Na50 and GA-CG-K50 exhibited a hierarchical structure with large pores accompanying small ones. As the salt ions increased to 100 mM, the microstructure of the hydrogels showed larger pore size (Fig. 2a–e). However, when the salt ions reached 250 mM, the porous structure collapsed, displaying a collapsed network structure (Fig. S2c and d†). The AFM height images showed that the GA-CG formed fibrous structures with a few nanometers in width and micrometers in length, similar to that of the pure GA samples, indicating that during the co-assembly of GA and CG, the hydrogel network was dominated by GA molecules (Fig. 2f, Fig. S2e and f†). As a contrast, the GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100 showed aggregated fibrillar structure with larger width of 50–100 nm. This is attributed to the fact that in the existence of monovalent metal ions, the transition of supramolecular assembly behavior of CG governed the co-assembly process of GA and CG, and thus a hierarchical ordered double network structure was formed, where the larger scale of CG double helix motifs and aggregates wrapped GA nanofibrils within the network (Fig. 2g–j).33,34,36
 | ||
| Fig. 2 Cryo-SEM images of GA-CG (a), GA-CG-Na50 (b), GA-CG-Na100 (c), GA-CG-K50 (d), and GA-CG-K100 (e). AFM height images of GA-CG (f), GA-CG-Na50 (g), GA-CG-Na100 (h), GA-CG-K50 (i), and GA-CG-K100 (j). | ||
Furthermore, GA-CG hydrogel systems were further characterized by XRD, FTIR, XPS, and SAXS to reveal the molecular structural information during the transparency transition process. The XRD results showed that the GA-CG hybrid hydrogels retained the characteristic peaks of GA and CG without new peaks, and the peak intensities were increased compared with those of the individual components (Fig. 3a). This suggests that the hybrid hydrogels were formed mainly by orthogonal co-assembly between GA and CG, thus retaining the original properties of GA and CG.37 As shown in Fig. 3b, the GA-CG, GA-CG-Na100, and GA-CG-K100 retained the FTIR characteristic peaks of the GA (such as O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and CC stretching vibration) and CG (symmetry vibration of OSO at 1260 cm−1, O–C–O and –O–SO3− stretching vibrations at 928 cm−1 and 846 cm−1).29,36,38 However, the O–H and CC stretching vibration bands shifted to higher and lower wavenumbers, respectively, and the intensities of –CH3, CO, and C–O stretching vibration bands weakened. This suggests that the co-assembly between GA and CG mainly occurs through hydrogen bonding, hydrophobic interactions, and electrostatic interactions, while the introduction of metal ions does not induce significant changes in the molecular structures of GA-CG. It can be further confirmed in the XPS spectra, in which the intensities of the high-resolution S 2p and C 1s spectra decreased, the peak shapes became flatter, and no significant change in the positions of the binding energy of the groups was observed (Fig. 3c and Fig. S3b†). The one-dimensional SAXS intensity profiles of the GA-CG hydrogel systems in Fig. 3d showed different characteristics in the low q and intermediate q regions of the samples. In the low q region, the addition of metal ions in GA-CG hydrogel showed much higher I(q) values, suggesting that more intense aggregation occurred in their network structures (Fig. 3d).39 The intermediate q region, by converting the 1D SAXS intensity profiles into Kratky plots (Fig. 3e), had broader shoulder peaks for the GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100, resulting from the strong aggregation of the chains of CG.40 In the high q region, the scattering patterns all followed the Porod decay (slope of −4), implying that GA fibrils with sharp shape still exist in the complex aggregated structure of GA-CG.21 Besides, from the pair distance distribution function P(r) plots, the GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100 showed more pronounced skewed distributions (Fig. 3f), suggesting more long rod-like structures were developed in the GA-CG hydrogel systems with addition of metal ions.41Rg, viewed as the size of the aggregates formed during gelation, was further calculated from the P(r) plots (Fig. 3f). The Rg of GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100 were significantly larger than that of GA-CG, with an increase of ca. 2–8 folds, especially for GA-CG-K50 and GA-CG-K100, in agreement with the AFM results. Combined with the microstructure of GA-CG hydrogel systems, we can infer the reasons for the transparency transition of the GA-CG hydrogels. GA-CG nanostructures co-assemble into complex aggregates upon introducing certain concentrations of metal ions, which further form a hierarchically ordered spatial network structure with large pore size. This structure can enhance light transmittance and thus the transparency of the hydrogel. However, as the concentration of metal ions increases further, the strong co-assembly behavior of GA-CG leads to the collapse of the hydrogel network structure, resulting in a reduction in light transmittance and a turbid appearance of the GA-CG hydrogels.33,34,36,42
O, and CC stretching vibration) and CG (symmetry vibration of OSO at 1260 cm−1, O–C–O and –O–SO3− stretching vibrations at 928 cm−1 and 846 cm−1).29,36,38 However, the O–H and CC stretching vibration bands shifted to higher and lower wavenumbers, respectively, and the intensities of –CH3, CO, and C–O stretching vibration bands weakened. This suggests that the co-assembly between GA and CG mainly occurs through hydrogen bonding, hydrophobic interactions, and electrostatic interactions, while the introduction of metal ions does not induce significant changes in the molecular structures of GA-CG. It can be further confirmed in the XPS spectra, in which the intensities of the high-resolution S 2p and C 1s spectra decreased, the peak shapes became flatter, and no significant change in the positions of the binding energy of the groups was observed (Fig. 3c and Fig. S3b†). The one-dimensional SAXS intensity profiles of the GA-CG hydrogel systems in Fig. 3d showed different characteristics in the low q and intermediate q regions of the samples. In the low q region, the addition of metal ions in GA-CG hydrogel showed much higher I(q) values, suggesting that more intense aggregation occurred in their network structures (Fig. 3d).39 The intermediate q region, by converting the 1D SAXS intensity profiles into Kratky plots (Fig. 3e), had broader shoulder peaks for the GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100, resulting from the strong aggregation of the chains of CG.40 In the high q region, the scattering patterns all followed the Porod decay (slope of −4), implying that GA fibrils with sharp shape still exist in the complex aggregated structure of GA-CG.21 Besides, from the pair distance distribution function P(r) plots, the GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100 showed more pronounced skewed distributions (Fig. 3f), suggesting more long rod-like structures were developed in the GA-CG hydrogel systems with addition of metal ions.41Rg, viewed as the size of the aggregates formed during gelation, was further calculated from the P(r) plots (Fig. 3f). The Rg of GA-CG-Na50, GA-CG-Na100, GA-CG-K50, and GA-CG-K100 were significantly larger than that of GA-CG, with an increase of ca. 2–8 folds, especially for GA-CG-K50 and GA-CG-K100, in agreement with the AFM results. Combined with the microstructure of GA-CG hydrogel systems, we can infer the reasons for the transparency transition of the GA-CG hydrogels. GA-CG nanostructures co-assemble into complex aggregates upon introducing certain concentrations of metal ions, which further form a hierarchically ordered spatial network structure with large pore size. This structure can enhance light transmittance and thus the transparency of the hydrogel. However, as the concentration of metal ions increases further, the strong co-assembly behavior of GA-CG leads to the collapse of the hydrogel network structure, resulting in a reduction in light transmittance and a turbid appearance of the GA-CG hydrogels.33,34,36,42
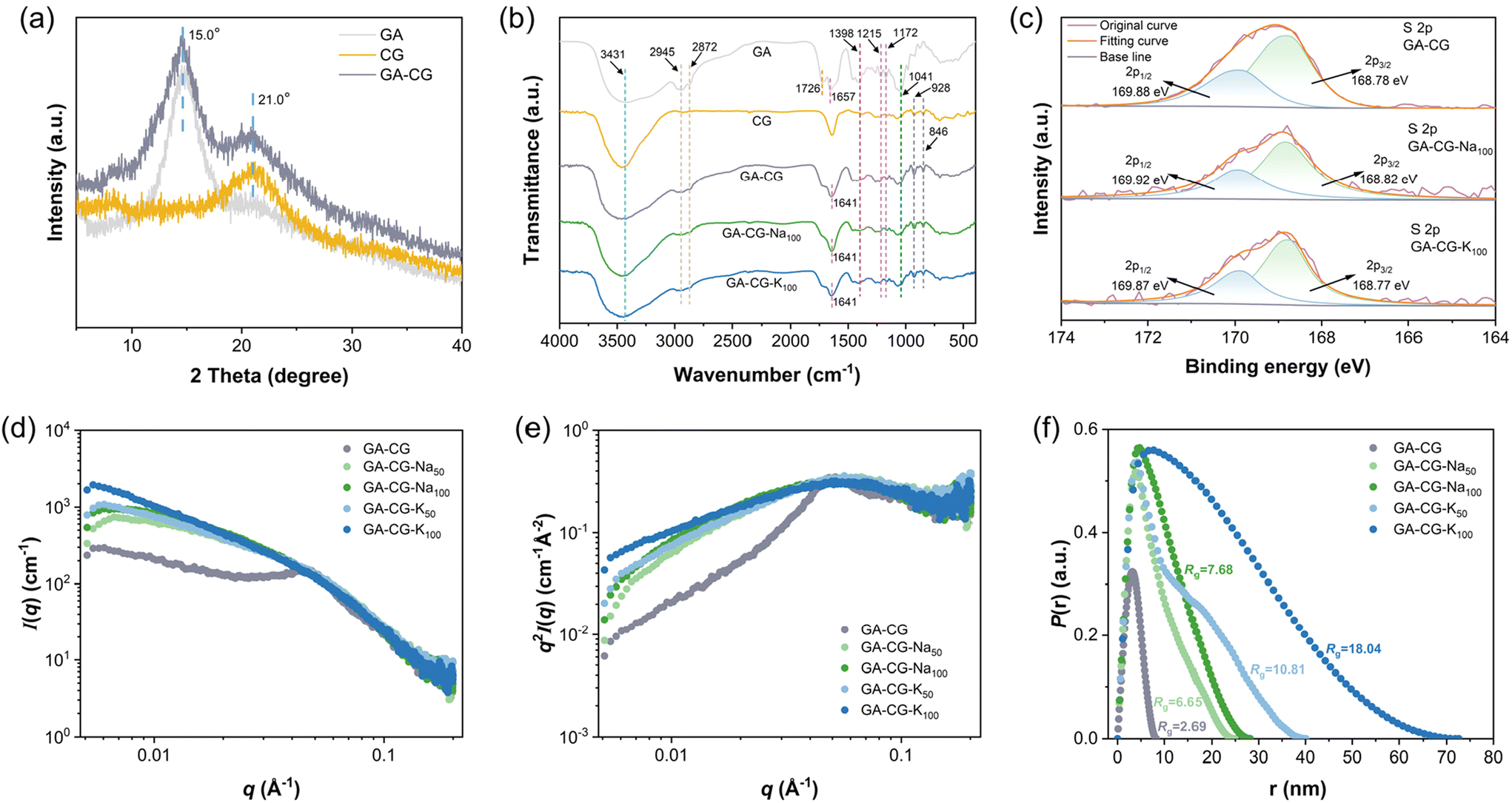 | ||
| Fig. 3 Characterization of interactions in GA-CG hydrogel systems. XRD (a), FTIR spectra (b), high-resolution XPS S 2p spectra (c), SAXS 1D intensity profile (d), Kratky plots (e), and pair distance distribution function P(r) plots (f) of GA-CG hydrogels. | ||
Dynamic oscillatory shear rheology is an important characterization tool to analyze the microstructural changes and mechanical response of hydrogel materials during deformation. From the frequency sweep data, G′ was significantly higher than G′′ for all samples within the LVR, and the G′ and G′′ hardly changed with frequency, indicating that all samples exhibited elastic solid-like properties (Fig. S5a and b†). The GA-CG hydrogel showed higher G′, G′′, and greater yield and flow stress increasing concentration of metal ions, and the effect of K+ was stronger than that of Na+, which was particularly pronounced in the compression test results (Fig. 5a, b, Fig. S5c, d, and Table S1†). The nonlinear rheological response behavior of the hydrogel samples was further analyzed in combination with the Lissajous–Bowditch curves. Similar to the GA, the elastic Lissajous curves of GA-CG showed a narrow ellipse shape at low strain amplitudes (less than 5%), suggesting a linear viscoelastic behavior. With increasing strain amplitudes, the elastic Lissajous curves of the GA-CG changed to an expanded ellipse (5–11% strain amplitudes) and further to a quadrilateral shape (11–500% strain amplitudes), indicating a rapid accumulation of viscous dissipation, which allowed the gel structure to undergo a final transition from elastic to viscous dominant behavior (Fig. 4e). These shifts of the elastic Lissajous curves occurred even more vigorously when the metal ions were introduced, where an elliptical-to-quadrilateral transition already happened at 5% strain amplitude (Fig. 4e and Fig. S5e†). Strain stiffening ratio (S factor) was calculated to provide more detailed insights into the elastic nonlinear behavior of the hydrogel samples (Fig. 4c). The S factors of all GA-CG hydrogel systems were larger than 0 throughout the entire process (0.1–1000% strain amplitudes), meaning the intracycle strain stiffening. Among them, the S factors of GA-CG hydrogels with added metal ions reached about 1.0 at 5% strain amplitude and eventually even about 4.0. The intracycle strain stiffening exhibited throughout the strain process can be attributed to the increased contact of the GA fibrils with the multilevel helical structure of the CG, where the fibrous network requires more shear stress to be further deformed and thus exhibits stronger rigidity.43 With a perfect elliptical shape, the viscous Lissajous curves of GA-CG showed a linear viscoelastic behavior at low strain amplitudes (less than 5%) (Fig. 4f). These specific shape distortions of the viscous Lissajous curves occurred more rapidly and violently when the metal ions were added. It is noteworthy that secondary loops appeared at 11–500% strain amplitudes, suggesting the presence of a significant nonlinear stress response (Fig. 4f and Fig. S5f†). The appearance of secondary loops is usually associated with microstructural rearrangements occurring on a shorter time scale than the deformation one.44Fig. 4d reveals the viscous nonlinear behavior of hydrogel samples in more detail by calculating the shear thickening ratio (T factor). The positive shear thickening ratios (T factors), revealing the viscous nonlinear behavior, of GA and GA-CG at the whole range of strain amplitudes (0.1–1000%) indicate the intracycle shear thickening. In contrast, the T factors of GA-CG with addition of metal ions changed from positive to negative values at 11% strain amplitude and subsequently displayed intracycle shear thinning behaviors. The intracycle shear thinning behavior is because there is not enough time for relaxation and the reformation of the physical interactions is delayed at high strain amplitudes.43 Overall, the metal ions induced the hierarchically co-assembled structure of GA-CG, endowing them with unique rheological properties, which may further influence their application performance.
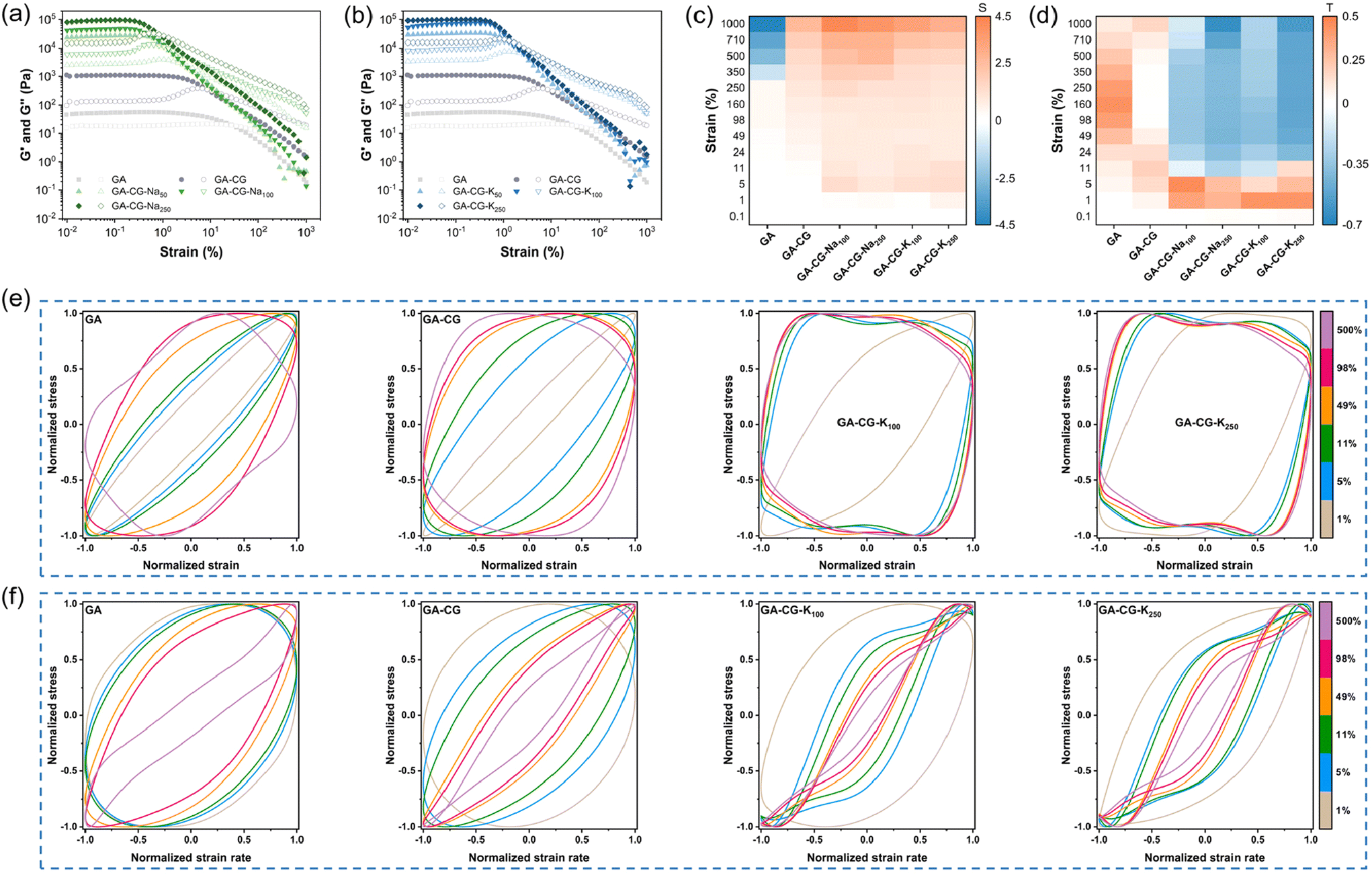 | ||
| Fig. 4 Strain sweeps of GA-CG-Nax (a) and GA-CG-Kx (b) samples. G′ and G′′ are shown as filled and open symbols, respectively. Strain stiffening ratio (S factor) (c) and Shear thickening ratio (T factor) (d) of hydrogel samples. Elastic (e) and viscous (f) Lissajous–Bowditch loops of hydrogel samples, acquired at the frequency of 6.28 rad per s and different strains of 1, 5, 11, 49, 98, and 500%. Stress, strain, and strain rate data are normalized with respect to their corresponding maximum values in the oscillation cycle. | ||
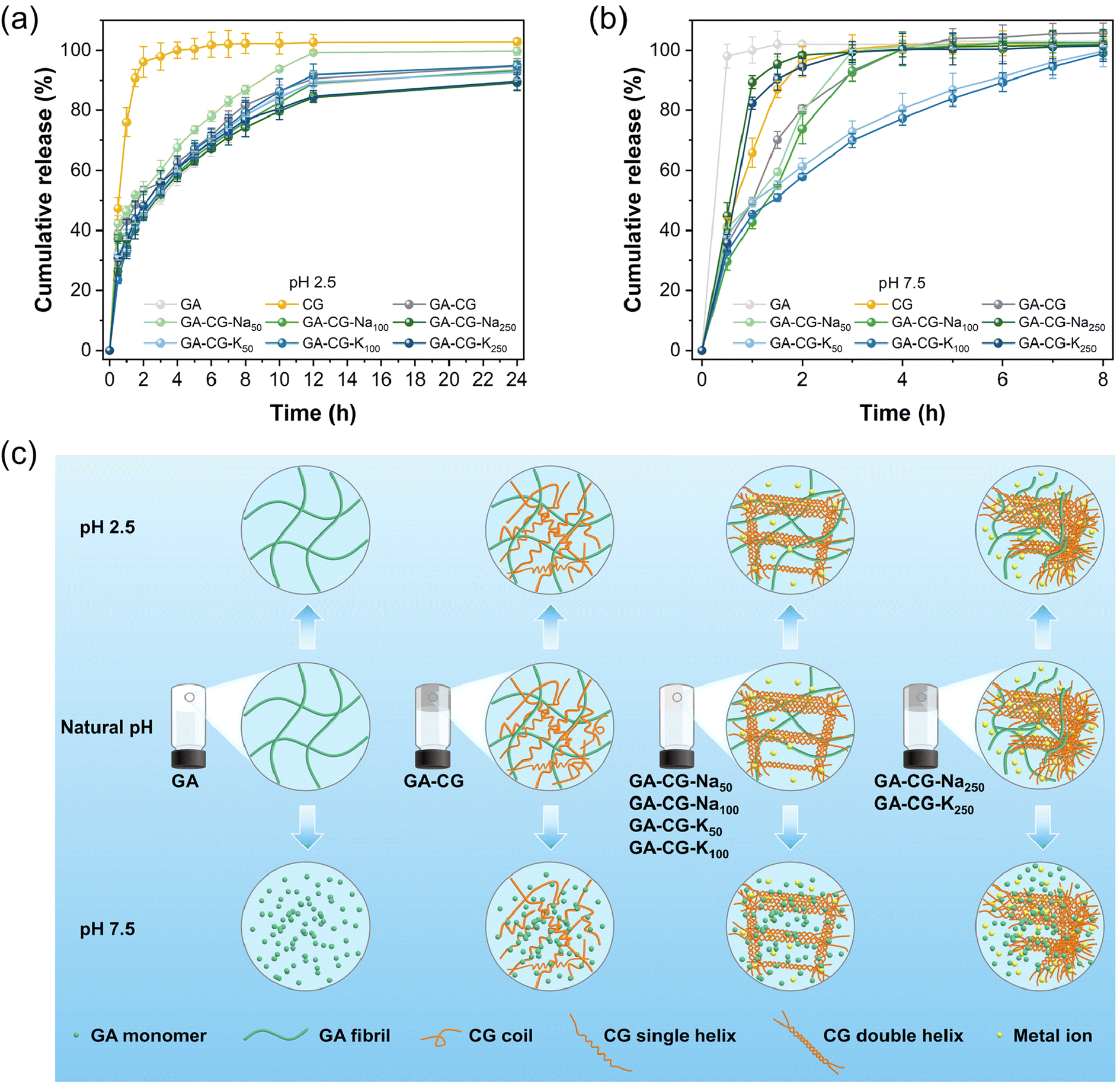 | ||
| Fig. 5 In vitro release profiles of a model nutrient (VB12) at 37 °C from hydrogel samples in simulated gastric fluid at pH 2.5 (a) and simulated intestinal fluid at pH 7.5 (b). Schematic illustration of structural changes of GA-CG hydrogels at different pH values (c). | ||
Supramolecular hydrogels have been widely used as the delivery platform to achieve spatial and temporal control over the release of drugs and nutrients. The controlled release properties of GA-CG hydrogels were thereby evaluated at different pHs. VB12 was selected as a model cargo. All the samples except CG showed a relatively slow-release rate at pH 2.5, where the release content reached about 60% after 4 h and was saturated around 12 h (Fig. 5a). The release profile was further fitted by the Ritger–Peppas model. The release profiles of all samples except CG at pH 2.5 were suitably fitted by the Ritger–Peppas model (R2 > 0.99), where the release exponent a was in the range of 0.3–0.4 indicating a dominated Fickian diffusion behavior.45 This also indicates that during the release process under acidic conditions, the GA-CG hydrogels exhibited minimal swelling and maintained an intact network structure (Fig. S6†). On the contrary, in the simulated intestinal fluid at pH 7.5, VB12 released rapidly at around 4 h for all hydrogel samples, except for GA-CG-K50 and GA-CG-K100 (Fig. 5b). It is attributed to the stronger assembly behavior between κ-CG and K+, resulting in the more complex and stable network structures and thus a slower release manner of VB12.33 The release profiles of GA-CG-K50 and GA-CG-K100 at pH 7.5 were well fitted by Ritger–Peppas model (R2 > 0.99) with a value in the range of 0.3–0.4, while the model fit for the rest of the hydrogel samples was bad (R2 < 0.99). This suggests that remarkable structural changes have occurred in the remaining hydrogel samples.45 The GA fibrillar network remains stable under acidic conditions, while at a pH levelled exceeding 7, GA exists as a monomer and cannot effectively co-assembled with CG into a complete network structure.46 By comparison, GA-CG-K50 and GA-CG-K100 contained ordered double helical assembled CG supramolecular structures, and thus displayed a sustainable release manner for VB12 (Fig. 5c). It is worth noting that the GA-CG-Na250 and GA-CG-K250 fail to retard the release of VB12, although the double helical assembled CG supramolecular structure is also present (Fig. 5c). This is mainly due to the high osmotic pressure generated by the exceedingly high ionic concentration as well as the excessively fast reaction kinetics, which prevent the formation of an ordered network structure. After 24 h, all hydrogel samples except GA-CG-K50 and GA-CG-K100 were dissolved in simulated intestinal fluid at pH 7.5, further confirming the structural stability of GA-CG-K50 and GA-CG-K100 (Fig. S6†). In addition to the pH-responsive controlled-release properties, the GA-CG hydrogels exhibited great protective effects on VB12. Compared to the control samples, the retention of VB12 in the GA-CG hydrogels was above 80% after both light and thermal degradation treatments (Fig. S7†).
Conclusions
In summary, a natural, edible supramolecular self-assembly system of GA and CG has been developed for fabrication of hierarchically structured hydrogels with remarkable characteristics and properties. An interesting phenomenon that the appearance of the GA-CG hydrogels transited from turbid to translucent and then back to turbid with the increase of metal ions, was observed. At 50–100 mM metal ions, the co-assembles of GA and CG nanostructures served as the building blocks to form a hierarchical ordered double network structure with larger pore size, which reduced the scattering of the light and exerted a translucent appearance. When the metal ions further increased to 250 mM, the huge osmotic pressure and ultra-fast reaction kinetics led to the collapse of gel network and thus the reduction in the transmittance of light. The hierarchically co-assembled structure of GA-CG hydrogels also endowed them with unique rheological properties. Moreover, the GA-CG hydrogels with different levels of transparency displayed highly tunable controlled-release behaviors of active cargoes, which can serve as an efficient and stimuli-responsive gastrointestinal-based delivery platforms. These findings are expected to provide a new perspective for the development of robust and multifunctional supramolecular hydrogels based on natural edible bio-building blocks (i.e., GA) in food and biomedical fields.Author contributions
Xinke Yu and Jiyang Cai contributed equally to this work. Xinke Yu: investigation, methodology, formal analysis, visualization, writing – original draft. Jiyang Cai: investigation, methodology, formal analysis, writing – original draft. Mengyue Xu: investigation, methodology, formal analysis. Qing Li: formal analysis. Yunyi Yang: formal analysis. Zhili Wan: conceptualization, methodology, supervision, project administration, funding acquisition, resources, formal analysis, writing – review & editing. Xiaoquan Yang: conceptualization, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the Outstanding Youth Project of Guangdong Natural Science Foundation (2024B1515020081), the National Natural Science Foundation of China (32172347), and the 111 Project (B17018).References
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Mater., 2019, 4, 463–478 CrossRef CAS.
- D. K. Smith, Soft Matter, 2024, 20, 10–70 RSC.
- D. J. Adams, J. Am. Chem. Soc., 2022, 144, 11047–11053 CrossRef CAS.
- L. Su, J. Mosquera, M. F. J. Mabesoone, S. M. C. Schoenmakers, C. Muller, M. E. J. Vleugels, S. Dhiman, S. Wijker, A. R. A. Palmans and E. W. Meijer, Science, 2022, 377, 213–218 CrossRef CAS.
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS.
- C. Zhao, X. Li, X. Han, Z. Li, S. Bian, W. Zeng, M. Ding, J. Liang, Q. Jiang, Z. Zhou, Y. Fan, X. Zhang and Y. Sun, Nat. Commun., 2024, 15, 1488 CrossRef CAS.
- A. M. Fuentes-Caparrós, F. de Paula Gómez-Franco, B. Dietrich, C. Wilson, C. Brasnett, A. Seddon and D. J. Adams, Nanoscale, 2019, 11, 3275–3280 RSC.
- M. Hu, Y. Wang, Z. Yan, G. Zhao, Y. Zhao, L. Xia, B. Cheng, Y. Di and X. Zhuang, J. Mater. Chem. A, 2021, 9, 14093–14100 RSC.
- K. Nakamura, R. Kubota, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2023, 14, 1696 CrossRef CAS.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- K. Zhi, H. Zhao, X. Yang, H. Zhang, J. Wang, J. Wang and J. M. Regenstein, Nanoscale, 2018, 10, 3639–3643 RSC.
- C. C. Piras, P. Slavik and D. K. Smith, Angew. Chem., Int. Ed., 2020, 59, 853–859 CrossRef CAS.
- Q. Li, X. Yu, S. Zhang, M. Xu, Y. Yang, Z. Wan and X. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 43633–43647 CrossRef CAS.
- M. N. Asl and H. Hosseinzadeh, Phytother. Res., 2008, 22, 709–724 CrossRef CAS.
- B. Schröfelbauer, J. Raffetseder, M. Hauner, A. Wolkerstorfer, W. Ernst and O. H. J. Szolar, Biochem. J., 2009, 421, 473–482 CrossRef.
- Z. Zhao, Y. Xiao, L. Xu, Y. Liu, G. Jiang, W. Wang, B. Li, T. Zhu, Q. Tan, L. Tang, H. Zhou, X. Huang and H. Shan, ACS Appl. Mater. Interfaces, 2021, 13, 20995–21006 CrossRef CAS.
- Q. Li, Z. Wan and X. Yang, Curr. Opin. Food Sci., 2022, 43, 107–113 CrossRef CAS.
- A. Saha, J. Adamcik, S. Bolisetty, S. Handschin and R. Mezzenga, Angew. Chem., Int. Ed., 2015, 54, 5408–5412 CrossRef CAS.
- X. Yu, Y. Meng, H. Zhang, J. Guo, S. Wang, H. Li, J. Hu and M.-H. Li, Nanoscale, 2021, 13, 15257–15266 RSC.
- L. Zhao, H. Zhang, Z. Guo, X. Yu, X. Jiao, M.-H. Li and J. Hu, ACS Appl. Mater. Interfaces, 2022, 14, 51394–51403 CrossRef CAS.
- W. Liu, Z. Li, Z. Wang, Z. Huang, C. Sun, S. Liu, Y. Jiang and H. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 7767–7776 CrossRef CAS.
- L. Ma, Z. Wan and X. Yang, J. Agric. Food Chem., 2017, 65, 9735–9743 CrossRef CAS.
- M. Xu, L. Ma, Q. Li, J. Wu, Z. Wan, T. Ngai and X. Yang, Food Funct., 2022, 13, 280–289 RSC.
- Z. Wan, Y. Sun, L. Ma, F. Zhou, J. Guo, S. Hu and X. Yang, Langmuir, 2018, 34, 3971–3980 CrossRef CAS.
- L. Ma, Q. Li, Z. Du, E. Su, X. Liu, Z. Wan and X. Yang, Adv. Mater. Interfaces, 2019, 6, 1900417 CrossRef.
- E. Su, Q. Li, M. Xu, Y. Yuan, Z. Wan, X. Yang and B. P. Binks, J. Colloid Interface Sci., 2021, 587, 797–809 CrossRef CAS.
- Q. Li, S. Zhang, R. Du, Y. Yang, Y. Liu, Z. Wan and X. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 17562–17576 CrossRef CAS.
- G. A. De Ruiter and B. Rudolph, Trends Food Sci. Technol., 1997, 8, 389–395 CrossRef CAS.
- J. Necas and L. Bartosikova, Vet. Med., 2013, 58, 187–205 CrossRef CAS.
- L. Schefer, J. Adamcik, M. Diener and R. Mezzenga, Nanoscale, 2015, 7, 16182–16188 RSC.
- M. Diener, J. Adamcik, A. Sánchez-Ferrer, F. Jaedig, L. Schefer and R. Mezzenga, Biomacromolecules, 2019, 20, 1731–1739 CrossRef CAS.
- Y. Ling and M. Lu, J. Polym. Res., 2009, 16, 29–37 CrossRef CAS.
- X. Yu, M. Xu, J. Cai, Q. Li, Y. Yang, Z. Wan and X. Yang, Giant, 2024, 17, 100240 CrossRef CAS.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS.
- M. Tang, Y. Lei, Y. Wang, D. Li and L. Wang, Food Hydrocolloids, 2021, 112, 106251 CrossRef CAS.
- C. Yu, K. Cui, H. Guo, Y. N. Ye, X. Li and J. P. Gong, Macromolecules, 2021, 54, 9927–9936 CrossRef CAS.
- L. C. Geonzon, K. Hashimoto, T. Oda, S. Matsukawa and K. Mayumi, Macromolecules, 2023, 56, 8676–8687 CrossRef CAS.
- D. I. Svergun and M. H. J. Koch, Rep. Prog. Phys., 2003, 66, 1735–1782 CrossRef CAS.
- R. Du, Y. Hu, R. Hübner, J.-O. Joswig, X. Fan, K. Schneider and A. Eychmüller, Sci. Adv., 2019, 5, eaaw4590 CrossRef.
- S. Tarashi, H. Nazockdast, A. Bandegi, S. Shafaghsorkh, G. Sodeifian and R. Foudazi, J. Rheol., 2023, 67, 15–33 CrossRef CAS.
- O. C. Duvarci, G. Yazar and J. L. Kokini, Trends Food Sci. Technol., 2017, 60, 2–11 CrossRef CAS.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Soft Matter, 2011, 7, 6222 RSC.
- K. Matsuoka, R. Miyajima, Y. Ishida, S. Karasawa and T. Yoshimura, Colloids Surf., A, 2016, 500, 112–117 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01410c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |