
PAMAM–guanylthiourea conjugates mask furin's substrate binding site: mechanistic insights from molecular docking and molecular dynamics studies assist the design of potential furin inhibitors†
Chithra R.
Nair
and
K. G.
Sreejalekshmi
 *
*
Department of Chemistry, Indian Institute of Space Science and Technology, Valiamala Post, Thiruvananthapuram, 695 547, India. E-mail: sreeja@iist.ac.in
First published on 30th May 2023
Abstract
Furin, a subtilisin-like proprotein convertase human enzyme, is promising as a single therapeutic target for several viral manifestations. Hence, understanding the molecular mechanisms that are key to putative furin action would lead to the systemic development of specific and potent antivirals. To guide the rational design of potential furin inhibitors through a detailed mechanistic analysis of specific molecular interactions, we conducted in silico screening of an in-house library of a zero generation poly(amidoamine) dendrimer (G0 PAMAM) and its guanylthiourea derivatives (PAMAM–GTU). Molecular docking suggested substituent-guided tailored interactions and molecular dynamics simulation unveiled PAMAM–GTU's binding with furin's catalytic triad, whereas single arm GTU substitution influenced the interactions with Asp153, His194 and Ser368. The substituent effect on the binding free energy of dendrimer-furin interaction was assessed by MM/GBSA. One-step synthesis of PAMAM–GTU was achieved using a versatile thiocarbamoyl amidine transfer strategy. For the first time, a basic alumina column was optimized as an economically viable purification platform for PAMAM–GTUs, which were characterized by 1H- and 13C -NMR and HRMS (ESI) techniques.
Introduction
Furin is a ubiquitously expressed membrane endoprotease that catalyses proteolytic maturation of many proproteins with key roles in both physiological and pathological processes.1–7 Hence, furin has gained attention as a single therapeutic target for several diseases, where inhibiting furin activity is paramount to the prevention of pathogen invasion into human host cells. Proteins, peptides/peptidomimetics, small molecules and antibodies inhibit furin8,9 and offer an extra edge due to decreased viral resistance.6,10 Furin's specificity to the cleavage of the R-X-[R/K]-R↓ residue11 inspires the design of most peptide inhibitors to mimic this particular sequence.12 Asp and Glu moieties impart negative potential at furin's vast binding site13 and facilitate strong binding by inhibitors with multibasic sites. In the reported furin inhibitors, a guanidine unit was identified as a good H-bond donor facilitating their occupancy in different pockets of the catalytic site.14 Furthermore, replacing guanidine with less basic units could reduce the toxicity of the inhibitors with conserved furin inhibition activity. Interestingly, specific interaction with furin's catalytic pockets is determined by the strength of the electron density of the inhibitors as evident from the studies involving canavanine (oxyguanidine),15 citrulline16 and guanylhydrazone17 substituted guanidine inhibitors (Fig. 1). Inspired by this substitution effect imparted by guanidine, we designed a molecular fragment containing a guanylthiourea (GTU) unit to explore its interaction with furin's catalytic site. As a tether unit, the peptide-mimicking, biocompatible and non-immunogenic poly (amido amine) (PAMAM) dendrimer18 was selected owing to its vast implications as a carrier for biologically active species.19–22 Even though surface modified PAMAM dendrimers were studied as antiviral agents,23,24 molecular modelling studies of the PAMAM dendrimer with biological targets have received meagre attention, not to mention the gap in correlating the computational studies and experimental results.25 Similarly, very few reports have attempted the in silico screening of novel molecules for furin inhibition,26,27 specifically to understand the molecular level interactions at the catalytic site. Hence, we envisaged the design of a library of molecular conjugates comprising PAMAM and GTU units and explored their interactions within furin's catalytic pockets to decipher mechanistic insights and provide molecular level analysis of the resulting interactions. Although PAMAM is a widely explored class of dendrimer, to the best of our knowledge, the interaction of PAMAM and/or its conjugates with furin's binding site is not reported. | ||
| Fig. 1 Chemical structure of furin inhibitors containing (a) guanidine, (b) oxyguanidine, (c) guanylhydrazone and (d) urea units. (e) Structure of the guanylthiourea moiety (molecular fragment in the present study). | ||
To start with, we designed a small library which was primarily inspired by the similarity in chemical structures of G0 PAMAM GTU and m-guanidinomethyl-Phac-RVR-Amba,28 the strong furin binder (Fig. 2A and B). Here, the ethylenediamine (EDA) core (blue sphere) with four arms in the former resembled the dendritic architecture with P3 as the core (pink sphere) and P1, P2, P4 and P5 subunits as branches in the latter. Moreover, in both cases all the four arms terminated with basic residues. Hence, we hypothesized that the multibasic PAMAM GTU could effectively interact with furin's binding site. Anticipating success with the design strategy, we further aimed at a versatile, single-step synthesis of the dendrimer conjugates and an efficient purification, since the dendrimer conjugates are notorious when it comes to purification, both cost- and time-wise.
 | ||
| Fig. 2 Chemical structures of (A) m-guanidinomethyl-Phac-RVR-Amba and (B) G0 PAMAM GTU (R = alkyl/aryl) | ||
Results and discussion
Molecular docking studies
The designed 97-member G0 PAMAM–GTU library was composed of single arm substituted conjugates with aliphatic or aromatic substituents. The chemical structures are depicted in the ESI† (Fig. S1). Our choice of substituents was primarily based on their plausible interactions with furin's catalytic site coupled with the commercial availability of the reagents (to ensure synthetic viability). The designed library was initially subjected to XP molecular docking with furin (PDB ID: 5JXH) from Homo sapiens in complex with its native ligand, namely, meta-guanidinomethyl-Phac-RVR-Amba (G-Amba). The top scoring ligands, along with the major interacting residues in furin and the dock scores of furin-ligand complexes are summarized in Table 1.| Ligand name | Structure | Interacting residues | Dock score* |
|---|---|---|---|
| G0 3-ClPheGTU (1) |

|
Asp154, Asn192, Trp254, Asp264, Asn295, Glu299, Asp258, Pro256, Asp306 | −12.03 |
| G0 PAMAM |

|
Asp154, Glu236, Pro256, Glu257, Asp258, Asp306 | −11.9 |
| m-Guanidinomethyl-Phac-RVR-Amba (G-Amba) |

|
Asp154, Asn192, Val231, Glu236, Ser253, Pro256, Asp258, Asp264, Asp306, Tyr308 | −11.64 |
| G0 PheGTU (2) |
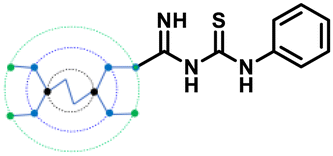
|
Asp154, Asn192, Glu236, Gly255, Asp258, Asp264, Glu299, Asp301, Asp306, Tyr308 | −10.5 |
| G0 2-OMePheGTU (3) |

|
Asp154, Asp191, Arg193, His194, Val231, Glu236, Gly255, Pro256, Asp258, Ser293, Asp306, Glu331 | −10.9 |
| G0 3,5-bis(CF3)PheGTU (4) |

|
Asp154, Asp191, Glu236, Pro256, Asp258, Asp264, Glu299, Asp306 | −10.8 |
| G0 amidinoPheGTU (5) |

|
Glu236, Asp264, Asp258, Gly255, Glu257, Ala292, Asn295, Glu299, Asp306 | −10.8 |

|
|||
As evident from the results, the 3-chlorophenyl derivative (G0 3-ClPheGTU) (1) was rewarded with the highest dock score (−12.03 kcal mol−1), which surpassed the score for the native ligand (−11.64 kcal mol−1). The 2D and 3D interactions of 1 with furin are depicted in Fig. 3A–C. The complex was mostly stabilized by H-bonds between the protonated primary amines of GTU and carbonyl oxygen of Asp residues of furin with a bond length in the range of 1.58 Å to 2.13 Å. Similar to G-Amba, three different arms in 1 occupied the S1, S2 and S4 pockets. Charged H-bonds between terminal primary amine at the unsubstituted arm and the carbonyl-oxygen of Trp254 and Asp306 and between the secondary amine and Pro256 assisted locating the arm deep into the S1 pocket. It is noteworthy that this is the site where the C-terminal Amba headgroup of G-Amba interacts with furin.28 The second arm of 1, in the S2 pocket, was stabilized by H-bonds with the terminal amine and Asp154 and Asn192, whereas in the case of G-Amba, an Arg side chain of P2 interacted with the S2 pocket. The GTU substituted arm of 1 occupied the S4 site with the phenyl ring projected outward to the solvent exposure region and Asp264 was bonded to amidine nitrogen through the H-bond and salt bridge, while it was the N-terminal guanidinomethyl group of G-Amba that interacted with the S4–S5 site. Interestingly, the third unsubstituted arm instead of occupying any of the catalytic pockets stabilized at the furin surface through H-bonds with Asn295 and Asp 258 and the salt bridge with Glu299. Nucleophilic attack by deprotonated Ser368 onto the carbonyl carbon of the substrate forms a tetrahedral intermediate with an oxyanion (‘O’ atom bearing negative charge) which in turn H-bonds to amides of Ser368 and Asn295 creating an oxyanion hole. Notably, the oxyanion hole formed between the amine of Ser 368 and the side chain amide of Asn295 additionally stabilizes the charged tetrahedral intermediate between Ser368 and the furin substrate.29,30 Here, unlike in G0 PAMAM, the H-bond between 1 and Asn295 enhanced the binding stability of the ligand with furin. Similar to G-Amba, other than H-bonds, the ligand was stabilized by electrostatic, van der Waals, hydrophobic and salt bridge interactions. Thus, the high dock score of 1 could be attributed to a greater number of stronger interactions with the furin residue.
 | ||
| Fig. 3 (A) 3D depiction of 1 at the catalytic cleft of furin. 1 is shown as an orange ball and stick model and G-Amba as a green ball and stick model. (B) 3D depiction and (C) 2D depiction of interaction of 1 with furin (purple arrows indicate H-bond, light blue residues and contour: polar attractions, light green residues and contour: hydrophobic interaction). | ||
A comparison of the docking poses of 1 and unsubstituted G0 PAMAM clearly indicated that GTU substitution influenced the nature of the interactions, type of residues and distribution of different arms of PAMAM into different pockets. In G0 PAMAM, the two arms connected to the same nitrogen atom of the EDA core were distributed in the S1 and S2 pockets, and the third arm in the S4 site whereas the fourth arm protruded out of the pocket. Noticeably, for 1, the two arms connected to the same nitrogen atom of the EDA core (one free and one substituted arm) occupied S2 and S4 pockets whereas one of the other arms connected to the second EDA tertiary nitrogen occupied the S1 site. The fourth unsubstituted arm was stabilized at the protein surface. Among the listed molecules in Table 1, G0 PheGTU (2) exhibited the same pattern where the GTU substituted arm occupied the S4 pocket. In the case of G0 2-OMePheGTU (3) and G0 3,5-bis(CF3)PheGTU (4) the arm was stabilized at the furin surface (Fig. S2A–D, ESI†). Notably, the OMe substituted phenyl ring interacted with His194, one among the residues in the catalytic triad, through π–π and π–cation interaction and a H-bond between Arg193 and the oxygen atom of the OMe group.
Inspired by the structure of G-Amba, we next made a fragment design modification by replacing the phenyl group of GTU with a benzimidamide unit to generate G0 amidinoPheGTU (5). Subsequent docking results reflected a change in the previously observed docking pose with the substituted arm occupying the S1 pocket, the core nitrogen appended free arm in the shared S4-S5 site, and the other two arms located along the protein surface (Fig. S2E, ESI†). From the literature, it was noticed that the non-interacting solvent exposed residue of the inhibitor also plays an important role in the furin inhibition activity, exemplified by the replacement of Tle with Leu, Val, or penicillamine at the solvent exposed P3 unit in the peptidomimetic furin inhibitor MI-1148, which resulted in its lower potency.31 In analogy, we anticipate that PAMAM GTU conjugates with single arm substitution that lacks direct interaction with the furin residues, which may play unique roles in their distribution within the catalytic cleft along with influencing the nature of the interactions.
The catalytic activity of furin is impeded by the inhibitors in two ways; by masking the catalytic cleft of the substrate binding site or by binding to the catalytic Asp153, His194 and Ser368 triad. Mostly, peptides or peptide mimetics follow the former pathway32 whereas a few small molecules follow the latter.33 Importantly, during molecular docking studies, we noticed that NH2 terminated G0 PAMAM GTU covered a larger surface area in the substrate binding site similar to G-Amba as evident from its orientation at the catalytic cleft of furin (Fig. 3A). Hence, we hypothesized that G0 PAMAM GTU could mask the catalytic site and subsequently may prevent the interaction of furin with certain viruses or bacteria substrates.
MD simulation analysis
In our structure-based furin inhibitor design approach, detailed understanding of the protein behaviour with respect to its flexibility and the conformational rearrangements during ligand binding being critical, we next decided to proceed with molecular dynamics (MD) studies. The results of 100 ns MD simulations, depicting interactions of furin with selected GTU derivatives are presented herein. Specifically, we were interested in identifying interactions with the catalytic triad (Ser 368, His 194 and Asp153), if any.Markedly, it was observed that all the selected PAMAM GTU derivatives exhibited conformation akin to the docking poses, especially in terms of their fitting in different pockets. Since 1 was awarded with the maximum dock score, we next explored the nature of this protein-ligand complex in more detail using MD. The 2D summary of the interaction analysis, including those interactions which occurred with a probability of over 30% during simulation, is presented in Fig. 4A. MD results also suggested that stabilization of the furin-1 complex occurred by the H-bond between the carbonyl oxygen of the Asp/Glu residue and primary amino groups/amide nitrogen of 1 as the acceptor–donor diad. Notably, all the four arms of 1 significantly interacted with the amino acid residues of furin.
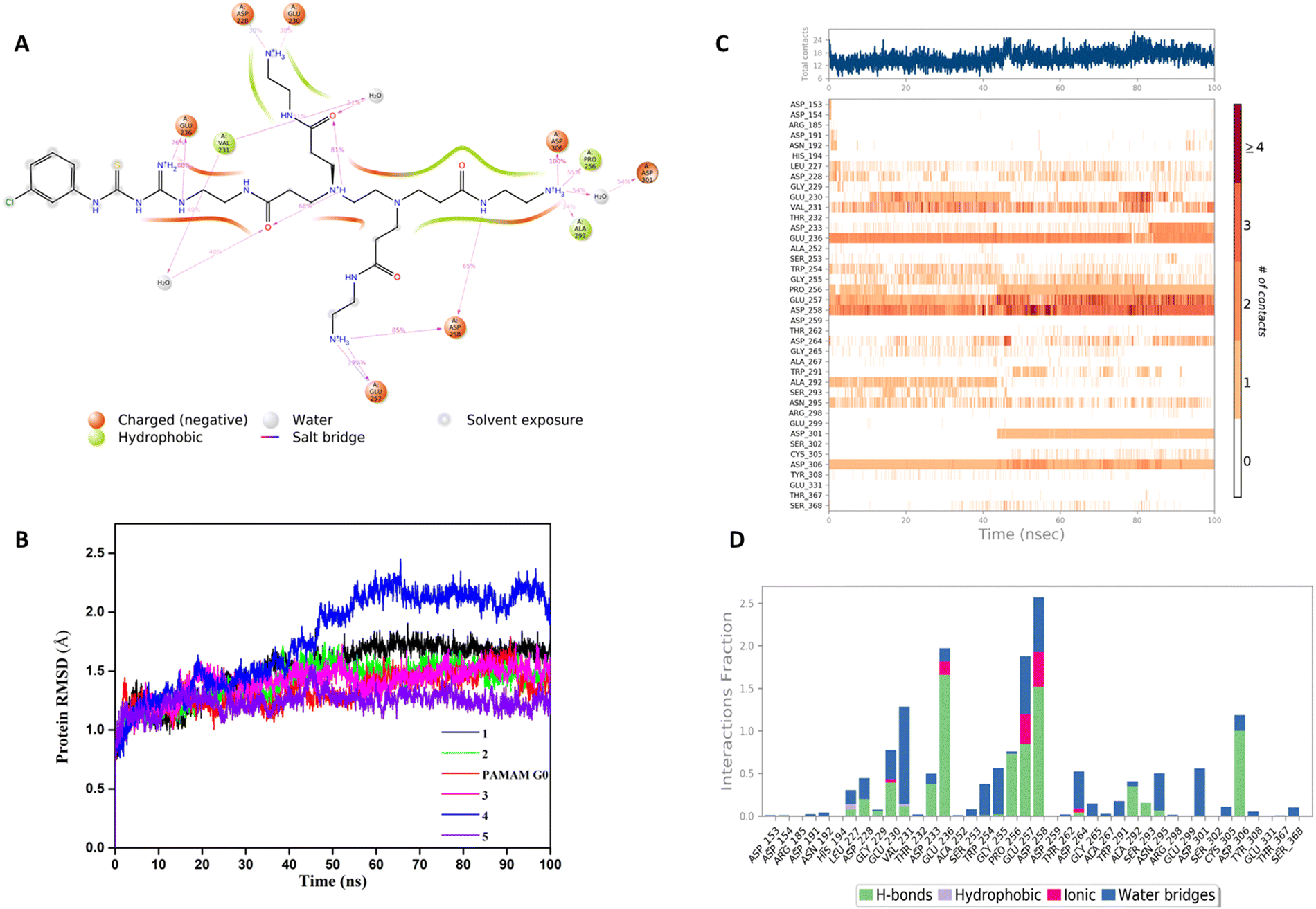 | ||
| Fig. 4 (A) 2D summary of interaction analysis results of 1 with furin. (B) Furin RMSD variation in the presence of different ligands. (C) Timeline representation of the interactions and contacts for furin and 1. (D) Interaction fraction summary of 1 in contact with furin. | ||
2D summary of the interaction analysis diagrams for G0 PAMAM, 2, 3, 4 and 5 are given in the ESI† (Fig. S3A–E). Some of the salient interactions observed were that of Asp153, one among the catalytic triad residues, which connected to the secondary amine of G0 PAMAM through a water mediated H-bond. One of the free arms of 3 formed a H-bond with Asp153 with an interaction probability of 99%. Whereas, 4 formed a H-bond with Asp153 with a 98% interaction probability and the amidine nitrogen of 5 exhibited a water-mediated H-bond with Asp153 and π-cation interaction with His194.
A root mean square deviation (RMSD) plot describes the time scale required to stabilize the protein structure upon its binding to a ligand. RMSD of furin Cα atoms in the presence of ligand molecules obtained from MD simulation is shown in Fig. 4B and the individual furin-ligand RMSD plots are shown in Fig. S4 (ESI†). RMSD of the G0 PAMAM-furin complex kept on fluctuating during the entire simulation time and could not get equilibrated even at 100 ns. However, in the presence of 1, furin RMSD fluctuated up to 55 ns and then stabilized around 1.75 Å. A similar pattern was observed for other selected PAMAM GTU-furin complexes which would imply that furin undergoes a conformational change upon ligand binding and the stability in the binding pocket is dictated by the chosen ligand. Among the different ligand-furin complexes studied, a comparatively stable RMSD plot was observed for the 5-furin complex and with 4 a significant fluctuation (>2Å) was observed.
The protein–ligand interaction timeline plot gives information about protein-ligand contact during the simulation along with the details of specific interacting amino acid residues (by means of H-bonds, water bridges and hydrophobic or polar interactions). The results indicate that a minimum of fifteen furin-ligand contacts (Fig. 4C) existed throughout the simulation for the selected ligands, suggesting a favourable interaction between the two. The interaction fraction summary of dendrimer-furin contacts was used to analyse the interface of the molecules with the catalytic triad of furin. Fig. 4D and Fig. S5A–E (ESI†) show that, in general, the molecules had more tendency to bind to Asp153 compared to His194 and Ser368. G0 PAMAM interacted with Asp153 with a fraction near to 1.5 through H-bonds, ionic interaction and water bridges. Also, hydrophobic interaction with His194 and a water bridge with Ser368 were noted. But for 2, a drastic reduction in the interaction fraction with Asp153 was evident, and with His194 and Ser368 it became still less. Furthermore, a reduction in interaction fraction was observed for 1, whereas surprisingly, 3 interacted more with Asp153 with a fraction more than 1.5, with H-bonds imparting a major contribution along with water bridge and ionic interactions. A similar trend was observed for 4. The interaction fraction of 5 with Asp153 was reduced and a significant hydrophobic interaction with His194 and a H-bond and water mediated H-bond with Ser368 was observed. Overall, MD simulation studies indicated that similar to the noncovalent 2,5-dideoxystreptamine derived furin inhibitors reported earlier,33 G0 PAMAM and the selected GTU conjugates interact with the furin catalytic triad. Apart from the interaction with the catalytic triad, binding of the furin inhibitor with the oxyanion hole32 and weak hydrophobic interaction26 can also influence the furin-inhibitor complex stability. From this point of view, we were interested to find out the interaction of PAMAM GTU conjugates with the oxyanion hole of furin. Interestingly, 1, 2 and 4 exhibited significant direct as well as water mediated H-bonding with Asn195, which suggested the plausible formation of a stable furin-PAMAM GTU complex where the interaction with the catalytic triad and the oxyanion hole would play a significant role in stability.
Binding free energy calculation of furin-PAMAM GTU complexes
The MM/GBSA method was used to find the effect of substituents on the binding free energy (ΔGbind) of dendrimers with furin. The contributions from various energy terms calculated using prime MM/GBSA are depicted in Table 2. The ΔGbind of G0 PAMAM for furin was −70.42 kcal mol−1, whereas 1 acquired a more negative value of −104.11 kcal mol−1. All the other selected PAMAM–GTU conjugates exhibited higher ΔGbind compared to G0 PAMAM, with 2, 3, 4 and 5 exhibiting values −88.38, −88.62, −75.11 and −109.90 kcal mol−1 respectively. These results suggest that single arm substitution of G0 can be a useful strategy in designing an energetically more favourable furin complex. Coulomb energy and electrostatic solvation energy nearly cancelled each other leaving van der Waals energy as a major contributor to the net binding free energy.| Complex | ΔGbind coulomba | ΔGbind covalent b | ΔGbind Hbondc | ΔGbind lipod | ΔGbind packinge | ΔGbind solv GBf | ΔGbind vdWg | ΔGbind totalh |
|---|---|---|---|---|---|---|---|---|
| a Coulomb energy. b Covalent binding energy. c Hydrogen bonding correction. d Lipophilic energy. e π–π packing correction. f Generalized Born electrostatic solvation energy. g van der Waals energy. h Total binding free energy. | ||||||||
| Furin-G0 PAMAM | −372.46 | 10.24 | −6.58 | −14.30 | 0.12 | 373.04 | −60.41 | −70.42 |
| Furin-1 | −405.60 | 19.40 | −7.29 | −21.13 | −0.16 | 393.10 | −81.98 | −104.11 |
| Furin-2 | −412.67 | 18.10 | −9.02 | −15.09 | 1.16 | 393.21 | −64.13 | −88.38 |
| Furin-3 | −352.77 | 9.21 | −6.84 | −17.25 | −1.35 | 349.42 | −70.33 | −88.62 |
| Furin-4 | −389.56 | 17.39 | −6.72 | −15.80 | −2.17 | 384.45 | −62.74 | −75.10 |
| Furin-5 | −491.947 | 5.12 | −10.37 | −15.74 | −0.91 | 465.94 | −61.70 | −109.89 |
Synthesis and characterization
With the encouraging results from in silico studies, we next proceeded with the validation of the synthetic feasibility of the designed PAMAM GTU derivatives. Our laboratories have established simple and versatile protocols to achieve the synthesis of several multifunctional systems.34–36 Here, pristine G0 PAMAM dendrimer was synthesised following the literature37 and regiospecific functionalization of G0 PAMAM was achieved using a thiocarbamoylamidine transfer (TCT) agent, 1-(N-arylthiocarbamoyl) amidino-3,5-dimethylpyrazole.38 A general procedure for the synthesis of G0 PAMAM GTU conjugates was developed by reacting equimolar proportions of G0 PAMAM with the corresponding TCT agent in MeOH at ambient temperature (Scheme 1). | ||
| Scheme 1 Single-step synthesis of PAMAM–GTU. | ||
Apart from the single arm substituted product, a two arm substituted GTU conjugate as well as unreacted G0 PAMAM were also present in the reaction mixture, which demanded its purification. Generally, the purification of dendrimers is a cumbersome and expensive affair mostly achieved either by dialysis or by chromatography using Sephadex as the stationary phase. Herein, a new cheap alternative method for the purification of G0 PAMAM conjugates is presented, wherein column chromatography using basic aluminium oxide as the stationary phase and 5% water in MeOH as the mobile phase was optimised to isolate pure PAMAM–GTU. MeOH removal under reduced pressure followed by lyophilisation afforded compounds 1 and 2 as white solids, which were highly hygroscopic. The complete structural assignment through 1H- and 13C-NMR and HRMS(ESI) data (Fig. S6–S12, ESI†) of the dendrimer conjugates attested to the success of our synthetic strategy in building a guanylthiourea platform on the PAMAM dendrimer and in establishing the utility of TCT for regiospecific modification of the amine terminus in PAMAM dendrimers. The molecular mass determined by HRMS(ESI) analysis was found to be 728.3927 and 694.4283 [M + H]+ for 1 and 2, respectively, as expected for single arm substituted G0 PAMAM GTU with the corresponding arylthiocarbamoylamidine unit. The 1H NMR spectra of 1 and 2 accounted for aromatic protons in the range of δ = 6.9 to 7.6 ppm and in the 13C NMR spectrum, along with the alkyl and aromatic carbon atoms, peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, CN and CO are present in the region δ = 160 to 175 ppm, which were absent in the DEPT-135 NMR spectrum. This confirms the successful transfer of a phenylthiocarbamoylamidine moiety to the PAMAM dendrimer.
S, CN and CO are present in the region δ = 160 to 175 ppm, which were absent in the DEPT-135 NMR spectrum. This confirms the successful transfer of a phenylthiocarbamoylamidine moiety to the PAMAM dendrimer.
The versatility of the synthetic scheme was established by reacting G0 PAMAM with 2-OMe/4-Me/4-nitro phenyl derivatives of TCT agents leading to the formation of 3, G0 4-MePheGTU (6) and G0 4-NO2PheGTU (7), respectively, as evident from the HRMS(ESI) spectra (Fig. S13–S15, ESI†). Thus, we achieved a simple and versatile protocol for the development of PAMAM–GTU complexes as potential furin inhibitors, which is much easier compared to the synthesis of peptide inhibitors.
Conclusion
In conclusion, by combining molecular docking studies with MD simulations, we demonstrated that rationally designed G0 PAMAM as well as its GTU derivatives could mask the binding site of furin. Besides, the molecules could bind to the catalytic triad – Asp153, His194 and Ser368 of furin, where the nature as well as the stability of the interactions could be modulated by judicious choice of the substituent on the PAMAM GTU arm. Furthermore, the molecules exhibit high binding free energy, with 1 and 5 with the most favourable energies in the studied molecules. The synthesis of the designed ligands was achieved in a single step reaction and purified by column chromatography using basic aluminium oxide as the stationary phase. Library expansion, in vitro evaluation of the library for furin inhibitors and the study of the effect of dendrimer generations on protein interactions will be performed in the future.Methods
Molecular docking
Molecular docking was performed on 2 Å X-ray crystal structure of proprotein convertase furin from Homo sapiens in complex with meta-guanidinomethyl-Phac-RVR-Amba (PDB ID: 5JXH) which was retrieved from the protein data bank (https://www.rcsb.org). We used different modules of software from Schrodinger 2020-4. The protein was prepared using protein preparation wizard39 by adding hydrogen, treating metal, deleting water molecules and assigning partial charges using the OPLS3e force field. Then protonation states were assigned and restrained. Furthermore, partial energy was minimized with 0.3 Å RMSD limit. The binding sites were defined after removing the native ligand and then a grid was generated using a grid box volume of 10 × 10 × 10 Å. The 2D structure of the ligands in the in-house library and reference molecules for comparison were drawn in 2D Sketcher of Maestro and a ligand library was created using R group enumeration. The Ligprep module of maestro was used to generate the 3D structure and the OPLS3e force field40 was used to generate the different conformations of each ligand. The Glide module was used to dock each ligand into the identified binding site of the grid. The lowest binding pose of each docking run was retained. The results were analysed using Glide XP visualizer.41MD simulation and binding free energy calculation
MD simulation was performed using Desmond. The protein–ligand complex was prepared using “System Builder” in Maestro. The protein–ligand complex was placed at the centre of an orthorhombic box with a buffer distance of 10 Å in order to create a hydration model using the TIP3P water model. Relaxation of the model system was performed before MD simulation. The sampling interval during the simulation was set to 20 ps and MD simulations were performed under the NPT ensemble for 100 ns with an initial temperature and pressure of 300 K and 1.01325 bar, respectively. The simulation interaction diagram tool was used to analyse the molecular dynamics simulation results. The prime MMGBSA tool in Maestro enabled the calculation of the binding free energy using the VSGB solvation model and OPLS3e force field.General chemistry procedure
All the commercially available reagents and solvents were of AR/spectroscopic grade obtained from Merck and used without further purification. Column chromatography was performed using basic aluminium oxide (activity I–II according to Brockmann) purchased from Finar Chemicals Limited, India. TLC was performed using silica gel 60 F254 (Merck) plates and visualization was done using iodine and a UV lamp of wavelength 254 nm. NMR spectra were recorded using a BRUKER AVANCE III HD 400 FTNMR spectrometer using D2O as a solvent and TMS as an internal standard. High resolution mass spectra (HRMS) were recorded using a Bruker microTOF-Q mass spectrometer (electrospray ionization (ESI). Infrared (IR) spectra were obtained using a PerkinElmer Spectrum 100 FT-IR spectrometer.Synthesis (general procedure)
Reaction of equimolar amounts of PAMAM G0 and a suitable TCT agent in MeOH at 50 °C for 12 h resulted in the formation of the corresponding PAMAM–GTU (Scheme 1). Methanol was evaporated under reduced pressure. Column chromatography using basic aluminium oxide as the stationary phase and 5% water in MeOH as the mobile phase resulted in the isolation of pure PAMAM–GTU. MeOH was removed under reduced pressure and the residue was lyophilized to get the desired pure products, which were further characterized.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
CRN acknowledges the University Grants Commission (UGC), Government of India for the Senior Research Fellowship. Authors thank Schrodinger for their generous support with the software (as part of Drug Discovery Hackathon 2020), Vikram Sarabhai Space Centre (VSSC) Thiruvananthapuram for recording NMR spectra and IIST for extending the Central Computing Facility for performing the screening.References
- Y. Cui, F. Jean, G. Thomas and J. L. Christian, BMP-4 Is proteolytically activated by furin and/or PC6 during vertebrate embryonic development, EMBO J., 1998, 17, 4735–4743 CrossRef CAS PubMed
.
- M. Vähätupa, Z. M. Cordova, H. Barker, S. Aittomäki, H. Uusitalo, T. A. H. Järvinen, M. Pesu and H. Uusitalo-Järvinen, Furin deficiency in myeloid cells leads to attenuated revascularization in a mouse-model of oxygen-induced retinopathy, Exp. Eye Res., 2018, 166, 160–167 CrossRef PubMed
- X. Yang, Q. Wang, Z. Gao, Z. Zhou, S. Peng, W. L. Chang, H. Y. Lin, W. Zhang and H. Wang, Proprotein convertase furin regulates apoptosis and proliferation of granulosa cells in the rat ovary, PLoS One, 2013, 8, 1–7 CrossRef
- Z. He, A. M. Khatib and J. W. M. Creemers, The proprotein convertase furin in cancer: more than an oncogene, Oncogene, 2022, 41, 1252–1262 CrossRef CAS PubMed
- P. Swärd, B. E. Rosengren, L. Jehpsson and M. K. Karlsson, Association between circulating furin levels, obesity and pro-inflammatory markers in children, Acta Paediatr., 2021, 110, 1863–1868 CrossRef PubMed
- G. Izaguirre, The Proteolytic regulation of virus cell entry by furin and other proprotein convertases, Viruses, 2019, 11, 837 CrossRef CAS PubMed
- B. A. Johnson, X. Xie, A. L. Bailey, B. Kalveram, K. G. Lokugamage, A. Muruato, J. Zou, X. Zhang, T. Juelich, J. K. Smith, L. Zhang, N. Bopp, C. Schindewolf, M. Vu, A. Vanderheiden, E. S. Winkler, D. Swetnam, J. A. Plante, P. Aguilar, K. S. Plante, V. Popov, B. Lee, S. C. Weaver, M. S. Suthar, A. L. Routh, P. Ren, Z. Ku, Z. An, K. Debbink, M. S. Diamond, P. Y. Shi, A. N. Freiberg and V. D. Menachery, Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis, Nature, 2021, 591, 293–299 CrossRef CAS PubMed
- F. Couture, F. DAnjou and R. Day, On the cutting edge of proprotein convertase pharmacology: from molecular concepts to clinical applications, Biomol. Concepts, 2011, 2, 421–438 CAS
- J. Zhu, J. Declercq, B. Roucourt, G. H. Ghassabeh, G. David, A. J. M. Vermorken, I. Lindberg, S. Muyldermans, J. W. M. Creemers, V. U. Brussel, U. A. Emirates, B. Raton and B. Raton, Generation and characterization of non-competitive furininhibiting nanobodies, Biochem. J., 2012, 448, 73–82 CrossRef CAS PubMed
- G. L. Becker, Y. Lu, K. Hardes, B. Strehlow, C. Levesque, K. Sandvig, U. Bakowsky, R. Day, W. Garten, K. Sandvig, U. Bakowsky, R. Day, W. Garten and T. Steinmetzer, Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases, J. Biol. Chem., 2012, 287, 21992–22003 CrossRef CAS PubMed
- S. S. Molloy, P. A. Bresnahan, S. H. Leppla, K. R. Klimpel and G. Thomas, Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen, J. Biol. Chem., 1992, 267, 16396–16402 CrossRef CAS PubMed
- S. A. Shiryaev, A. G. Remacle, B. I. Ratnikov, N. A. Nelson, A. Y. Savinov, G. Wei, M. Bottini, M. F. Rega, A. Parent, R. Desjardins, M. Fugere, R. Day, M. Sabet, M. Pellecchia, R. C. Liddington, J. W. Smith, T. Mustelin, D. G. Guiney, M. Lebl and A. Y. Strongin, Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens, J. Biol. Chem., 2007, 282, 20847–20853 CrossRef CAS PubMed
- R. J. Siezen, J. W. M. Creemers and W. J. M. V. A. N. D. E. Venz, Homology modelling of the catalytic domain of human furin a model for the eukaryotic subtilisin-like proprotein convertases, Eur. J. Biochem., 1994, 222, 255–266 CrossRef CAS PubMed
- E. E. A. Osman, A. Rehemtulla and N. Neamati, Why all the fury over furin?, J. Med. Chem., 2022, 65, 2747–2784 CrossRef CAS PubMed
- T. Van Lam Van, M. R. Heindl, C. Schlutt, E. Böttcher-Friebertshäuser, R. Bartenschlager, G. Klebe, H. Brandstetter, S. O. Dahms and T. Steinmetzer, The basicity makes the difference: improved canavanine-derived inhibitors of the proprotein convertase furin, ACS Med. Chem. Lett., 2021, 12, 426–432 CrossRef PubMed
- S. O. Dahms, K. Hardes, T. Steinmetzer and M. E. Than, X-ray structures of the proprotein convertase furin bound with substrate analogue inhibitors reveal substrate specificity determinants beyond the S4 pocket, Biochemistry, 2018, 57, 925–934 CrossRef CAS PubMed
- S. O. Dahms, T. Haider, G. Klebe, T. Steinmetzer and H. Brandstetter, OFF-state-specific inhibition of the proprotein convertase furin, ACS Chem. Biol., 2021, 16, 1692–1700 CrossRef CAS PubMed
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, A new class of polymers: starburst-dendritic macromolecules, Polym. J., 1985, 17, 117–132 CrossRef CAS
- J. Yang, Q. Zhang, H. Chang and Y. Cheng, Surface-engineered dendrimers in gene delivery, Chem. Rev., 2015, 115(11), 5274–5300 CrossRef CAS PubMed
- L. M. Kaminskas, V. M. McLeod, C. J. H. Porter and B. J. Boyd, Association of chemotherapeutic drugs with dendrimer nanocarriers: an assessment of the merits of covalent conjugation compared to noncovalent encapsulation, Mol. Pharmaceutics, 2012, 9, 355–373 CrossRef CAS PubMed
- H. Moorthy and T. Govindaraju, Dendrimer architectonics to treat cancer and neurodegenerative diseases with implications in theranostics and personalized medicine, ACS Appl. Bio Mater., 2021, 4, 1115–1139 CrossRef CAS PubMed
- S. Mignani, X. Shi, M. Zablocka and J. P. Majoral, Dendrimer-enabled therapeutic antisense delivery systems as innovation in medicine, Bioconjugate Chem., 2019, 30, 1938–1950 CrossRef CAS PubMed
- S. C. Günther, J. D. Maier, J. Vetter, N. Podvalnyy, N. Khanzhin, T. Hennet and S. Stertz, Antiviral potential of 3′-sialyllactose- and 6′-sialyllactose-conjugated dendritic polymers against human and avian influenza viruses, Sci. Rep., 2020, 10, 1–9 CrossRef PubMed
- M. Kandeel, A. Al-Taher, B. K. Park, H. J. Kwon and M. Al-Nazawi, A pilot study of the antiviral activity of anionic and cationic polyamidoamine dendrimers against the middle east respiratory syndrome coronavirus, J. Med. Virol., 2020, 92, 1665–1670 CrossRef CAS PubMed
- S. Ahmed, S. B. Vepuri, R. S. Kalhapure and T. Govender, Interactions of dendrimers with biological drug targets: reality or mystery-a gap in drug delivery and development research, Biomater. Sci., 2016, 4, 1032–1050 RSC
- C. Wu, M. Zheng, Y. Yang, X. Gu, K. Yang, M. Li, Y. Liu, Q. Zhang, P. Zhang, Y. Wang, Q. Wang, Y. Xu, Y. Zhou, Y. Zhang, L. Chen and H. Li, Furin: a potential therapeutic target for COVID-19, iScience, 2020, 23, 101642 CrossRef CAS PubMed
- B. Villoutreix, J. Creemers, Y. Léger, G. Siegfried, E. Decroly, S. Evrard and A. M. Khatib, Targeting furin activity through in silico and in vitro drug repurposing strategy for SARS-CoV-2 spike glycoprotein cleavage repression, Res. Sq., 2020, 1–16 Search PubMed
- S. O. Dahms, K. Hardes, K. L. Becker, T. Steinmetzer, H. Brandstetter and M. E. Than, X-ray structures of human furin in complex with competitive inhibitors, ACS Chem. Biol., 2014, 9, 1113–1118 CrossRef CAS PubMed
- S. O. Dahms, M. Arciniega, T. Steinmetzer, R. Huber and M. E. Than, Structure of the unliganded form of the proprotein convertase furin suggests activation by a substrate-induced mechanism, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11196–11201 CrossRef CAS PubMed
- T. Rungrotmongkol, P. Decha, P. Sompornpisut, M. Malaisree, P. Intharathep, N. Nunthaboot, T. Udommaneethanakit, O. Aruksakunwong and S. Hannongbua, combined qm/mm mechanistic study of the acylation process in furin complexed with the h5n1 avian influenza virus hemagglutinin's cleavage site, Struct., Funct., Genet., 2009, 76, 62–71 CrossRef CAS PubMed
- K. Hardes, G. L. Becker, Y. Lu, S. O. Dahms, S. Köhler, W. Beyer, K. Sandvig, H. Yamamoto, I. Lindberg, L. Walz, V. Vonmessling, M. E. Than, W. Garten and T. Steinmetzer, Novel Furin inhibitors with potent anti-infectious activity, ChemMedChem, 2015, 10, 1218–1231 CrossRef CAS PubMed
- S. Henrich, A. Cameron, G. P. Bourenkov, R. Kiefersauer, R. Huber, I. Lindberg, W. Bode and M. E. Than, The crystal structure of the proprotein processing proteinase furin explains its stringent specificity, Nat. Struct. Biol., 2003, 10, 520–526 CrossRef CAS PubMed
- S. O. Dahms, G. S. Jiao and M. E. Than, Structural studies revealed active site distortions of human furin by a small molecule inhibitor, ACS Chem. Biol., 2017, 12, 1211–1216 CrossRef CAS PubMed
- S. Titus and K. G. Sreejalekshmi, One-pot four-component synthesis of 4-hydrazinothiazoles: novel scaffolds for drug discovery, Tetrahedron Lett., 2014, 55, 5465–5467 CrossRef CAS
- S. Titus and K. G. Sreejalekshmi, Enriching biologically relevant chemical space around 2-aminothiazole template for anticancer drug development, Med. Chem. Res., 2018, 27, 23–36 CrossRef CAS
- R. Radhakrishnan and K. G. Sreejalekshmi, Computational design, synthesis, and structure property evaluation of 1,3-thiazole-based color-tunable multi-heterocyclic small organic fluorophores as multifunctional molecular materials, J. Org. Chem., 2018, 83, 3453–3466 CrossRef CAS PubMed
- D. A. Tomalia, B. Huang, D. R. Swanson, H. M. Brothers and J. W. Klimash, Structure control within poly(amidoamine) dendrimers: size, shape and regio-chemical mimicry of globular proteins, Tetrahedron, 2003, 59, 3799–3813 CrossRef CAS
- G. C. Jenardanan, M. Francis, S. Deepa and K. N. Rajasekharan, 1-(N-arylthiocarbamoyl)amidino-3,5-dimethyl pyrazoles - preparation and use in heterocycle synthesis, Synth. Commun., 1997, 27, 3457–3462 CrossRef CAS
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and Ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J. Comput. Aid. Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang, R. Abel, R. A. Friesner and E. D. Harder, OPLS3e: extending force field coverage for drug-like small molecules, J. Chem. Theory Comput., 2019, 15, 1863–1874 CrossRef CAS PubMed
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj00703k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |