
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ monitoring of PISA morphologies†
Julia Y.
Rho
 *ab,
Georg M.
Scheutz
b,
Satu
Häkkinen
a,
John B.
Garrison
b,
Qiao
Song
a,
Jie
Yang
a,
Robert
Richardson
a,
Sébastien
Perrier
*acde and
Brent S.
Sumerlin
*b
*ab,
Georg M.
Scheutz
b,
Satu
Häkkinen
a,
John B.
Garrison
b,
Qiao
Song
a,
Jie
Yang
a,
Robert
Richardson
a,
Sébastien
Perrier
*acde and
Brent S.
Sumerlin
*b
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: Julia.Y.Rho@warwick.ac.uk; s.perrier@warwick.ac.uk
bGeorge & Josephine Butler Polymer Research Laboratory, Center for Macromolecular Science & Engineering, Department of Chemistry, University of Florida, Gainesville, Florida 32611, USA. E-mail: sumerlin@chem.ufl.edu
cFaculty of Pharmacy and Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
dAustralia Biological Physics, School of Physics and Astronomy, University of Manchester, Manchester M13 9PL, UK
eWarwick Medical School, University of Warwick, Coventry CV4 7AL, UK
First published on 1st July 2021
Abstract
Polymerization-induced self-assembly (PISA) is a facile method to obtain block copolymer aggregates with defined morphologies. However, the transitions between these morphologies have been difficult to monitor directly in real-time during the polymerization. Herein, we describe a straightforward and readily accessible in situ method to monitor the evolution of nanostructure via changes in internal hydrophobicity during the PISA process using a polymer-tethered pyrene fluorescent probe. We were able to correlate morphological transitions with changes of the pyrene emission and gain unprecedented insight into the evolution of core hydrophobicity during PISA.
Introduction
Amphiphilic block copolymers have been widely used to produce self-assembled nanostructures.1–4 In solution, these assemblies adopt not only spherical micellar morphologies but also higher-order structures, such as cylindrical micelles (worms) and vesicles to name a few.5–8 In particular, polymerization-induced self-assembly (PISA) has been used as a versatile method to obtain block copolymer particles in a highly tunable fashion at high concentrations.9,10 During PISA, a solvophilic precursor polymer is chain extended with a monomer that forms a solvophobic block. At a certain critical degree of polymerization (DP) of the solvophobic block, aggregation becomes energetically favorable and causes assembly of the polymer chains into multimolecular aggregates.11,12 Controlled radical polymerization techniques, such as reversible addition–fragmentation chain transfer (RAFT)13–15 or atom transfer radical polymerization (ATRP),5,16 have made the synthesis of well-defined block copolymers relatively straightforward, and for this reason, are widely adopted to conduct PISA.9,17–19Many approaches have focused on broadening the scope of PISA systems by introducing a wide range of polymer functionalities20–24 and exploring increasingly complex morphologies,25,26 such as framboidal shapes.27 Such nanoparticles have been utilized for a range of applications from coatings28 to nanomedicine.20,23,29–31
With these promising applications, a better understanding of the morphological progression from molecularly dissolved unimers to complex assembled structures could enable us to better predict, design, and direct these polymerizations. However, the sensitivity of the self-assemblies to fluctuations of temperature and concentration imposes major restrictions on the feasibility and reproducibility of ex situ morphology characterization. Both dry state transmission electron microscopy (TEM) and dynamic light scattering (DLS), which are commonly used to characterize PISA morphologies, are often conducted at room temperature, and require dilution or drying of the samples, which may alter assembly size and morphology. To circumvent the problems associated with ex situ methods, liquid-cell transmission electron microscopy32,33 and small-angle X-ray scattering34,35 have been adapted, using specialized setups and systems, for monitoring PISA in situ. However, the practicality and availability of these methods is a limitation in some cases.
Aggregation-dependent fluorescent probes, such as pyrene, have long been used to observe the onset of self-assembly in block copolymer micelles, i.e. the critical micelle concentration (CMC).36–40 When a ground state pyrene monomer (M) is excited (M*) at λmax ∼345 nm, three distinct monomeric emission bands are observed at I1 ∼375 nm, I3 ∼385 nm, and I5 ∼395 nm upon radiative decay. The I1/I3 ratio can be used to determine the changes in hydrophobicity in the local environment surrounding the pyrene – as the I1 emission band is sensitive to solvent polarity, and the I3 emission band is relatively insensitive. Upon micellization, the presence of pyrene embedded in the hydrophobic core will lead to a change in the I1/I3 ratio. When plotted over a range of concentrations, the CMC can be determined by the inflection point in the graph.41 If the excited pyrene (M*) is spatially proximal to another pyrene in its ground state (M), the pair can stack together to form a dimer (M·M*). This complex (M·M*) is referred to as an excimer and has an emission band around 475 nm (Iexi).42–44 The ratio of excimer to monomer has been used to monitor intra- and intermolecular interactions in proteins and membranes.45–47
There is considerable utility in employing aggregation-dependent fluorescent probes to interrogate supramolecular assembly and morphology. For example, Brédas and co-workers have elucidated how the intermolecular arrangements of organic π-conjugated molecules dictate electronic and optical properties.48 Importantly, π-conjugated systems often stack in columns and form face-to-face interactions (H-type aggregates) that have a low fluorescence quantum yield. Efforts have been made to enhance the emission of π-conjugated materials by modifying the π–π stacking arrangement via installation of bulky side groups.49 Utilizing supramolecular interactions to alter the arrangement of these systems, in particular, the adoption of a slipped stacking arrangement (J-type aggregates) can result in dramatic improvement in fluorescence quantum yield. Zhang, Wang, and co-workers reported that by introducing sterically demanding groups, bis(pyrene) derivatives could adopt a J-type aggregation which led to an almost 30-fold improvement in excimer emission.50 Importantly, the sterically bulky groups led these aggregates to adopt a dot-shaped (spherical) morphology with high excimer fluorescence, while the bis-pyrenes in the absence of sterically demanding groups formed sheet-like morphologies and adopted an H-type aggregation with much lower excimer emission. These examples highlight how changes in fluorescence excimer emission can be followed to provide insight into nanoscale transformations in supramolecular morphology.
In this article, we describe a straightforward and practical method to monitor changes in morphology and core hydrophobicity of the amphiphilic block copolymer polymer assemblies that form and evolve during the PISA process. Leveraging the aggregation-induced change in the emission spectrum of the fluorescent probe pyrene, we show that the ratio between the monomer (Imon at 375 nm) and excimer emission (Iexi at 475 nm) can be used to study morphological changes during PISA. At the same time, the I1/I3 ratio could also be used to monitor the hydrophobicity within the cores of nanoparticles prepared during PISA.51,52 Importantly, all this information can be obtained in situ, using a standard benchtop fluorometer.
Results and discussion
We synthesized a poly(N,N-dimethylacrylamide) (PDMA) macro-chain-transfer agent (macroCTA) with a carboxylic acid functionality on the Z group for the selective installation of the pyrene probe in proximity to the core-forming hydrophobic block of the polymer chain.25 Size-exclusion chromatography (SEC) indicated the resulting macroCTA had a number-average molar mass (Mn) of 7900 g mol−1 and dispersity (Đ) = 1.09 (Fig. S1†). To install the fluorescent pyrene probe at the growing chain end of the PDMA macroCTA, the amine-functionalized pyrene was coupled to the carboxylic acid on the Z-group of the macromolecular trithiocarbonate using standard peptide coupling conditions (see Fig. 1 and S1–16† for details).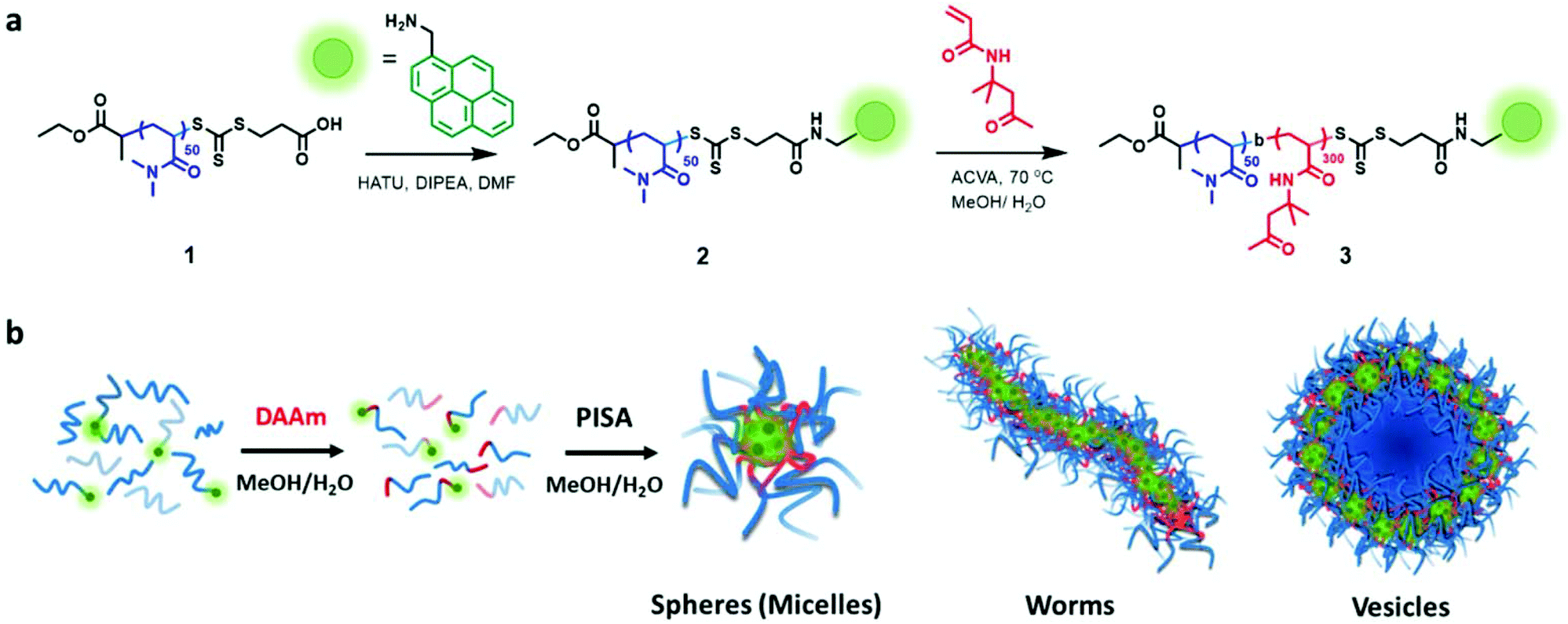 | ||
| Fig. 1 Synthetic outline of the synthesis of the macro-chain-transfer agent (macroCTA) and the polymerization-induced self-assembly (PISA) process. (a) Synthesis of PDMA macroCTA (1), pyrene-labeled PDMA macroCTA (2) and the chain extension polymerisation using diacetone acrylamide (DAAm) to yield the amphiphilic block copolymer poly(N,N-dimethylacrylamide)-block-poly(diacetone acrylamide) (PDMA-b-PDAAm) (3). (b) Schematic representation of the morphologies observed during PISA upon chain extension of the PDMA macroCTA with DAAm. | ||
Previously, Figg et al. showed that to access higher-order morphologies in aqueous DMA/diacetone acrylamide (DAAm)-based PISA systems, the incorporation of hydrophilic DMA monomer into the hydrophobic DAAm block was crucial to increase core hydration and chain mobility.53 As an alternative approach, we decided to alter the solvent polarity by using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of methanol and water to facilitate morphology transitions without the need for an additional co-monomer. Additionally, we added non-functionalized PDMA macroCTA (1) to the pyrene-functionalized PDMA macroCTA (2) to prevent fluorescent quenching and to minimize the contribution of the hydrophobic pyrene chain end in the assembly process. The ratio of 2 to 1 was maintained at 0.2 throughout the experiments. All the polymerizations were conducted at 70 °C under constant stirring using 4,4′-azobis(4-cyanovaleric acid) (ACVA) as a thermal radical initiator in a 1:1 (v/v) mixture of methanol and water.
:1 (v/v) mixture of methanol and water to facilitate morphology transitions without the need for an additional co-monomer. Additionally, we added non-functionalized PDMA macroCTA (1) to the pyrene-functionalized PDMA macroCTA (2) to prevent fluorescent quenching and to minimize the contribution of the hydrophobic pyrene chain end in the assembly process. The ratio of 2 to 1 was maintained at 0.2 throughout the experiments. All the polymerizations were conducted at 70 °C under constant stirring using 4,4′-azobis(4-cyanovaleric acid) (ACVA) as a thermal radical initiator in a 1:1 (v/v) mixture of methanol and water.
Initially, a range of DPs of the hydrophobic block was targeted to evaluate the scope of this PISA system (Table S1†). Each PISA reaction achieved full monomer conversion within 5 h, as confirmed by 1H NMR spectroscopy. Size-exclusion chromatography (SEC) of the chain extension products showed some residual uninitiated macroCTA. The deconvoluted SEC chromatogram revealed that the chain extension (PDMA-Pyr-b-PDMA) resulted in a blocking efficiency of 91%, see Fig. S16.† Despite this, we still observed a range of morphologies from spheres (DP = 100), worms/worm network (DP = 200) to vesicles (DP = 300), as confirmed by dry state TEM (Fig. 2b). Importantly, control experiments with 100% non-functionalized macroCTA showed similar morphologies at the same DPs (Table S1 and Fig. S18†), suggesting the pyrene residues play only a minimal role in governing self-assembly. It is worth noting that keeping the percentage of pyrene-functionalized macroCTA low (20%) was critical in ensuring that the pyrene aggregation did not affect the overall morphologies obtained during the PISA process. Increasing ratios of functionalized macroCTA led to different morphologies when compared to the control studies without pyrene-functionalized PDMA (2). Most notably, higher ratios of pyrene-functionalized macroCTA led to kinetically trapped spheres throughout the same polymerization.
 | ||
| Fig. 2 Size-exclusion chromatography (SEC) and transmission electron microscopy (TEM) characterization of polymers and assemblies formed via PISA. (a) SEC traces of PDMA macroCTA chain extended with DAAm to different targeted degrees of polymerization (DP) (50, 100, 200, and 300). (b) TEM images of spheres at a PDAAm DP of 100 (red, left), worms at PDAAm DP of 200 (orange, middle), and vesicles at a PDAAm DP of 300 (dark yellow, right). Scale bar is set to 500 nm. All PISA experiments for TEM were carried out to full conversion and crosslinked at 70 °C using a 10 wt% O-alkyl bishydroxylamine crosslinker relative to DAAm monomer to preserve the morphology for TEM analysis. | ||
After showing that we could obtain a range of morphologies with the chain extension of the PDMA macroCTA using only DAAm, we sought to study the emission behavior of the pyrene probes in morphological transitions during PISA. Using the same polymerization conditions, we targeted a final DAAm block with DP of 300, which generated vesicles in the previous experiments (Fig. 2b). Specifically, two polymerization sets under identical conditions, but one in sealed glass vials and one in the fluorometer, were conducted to correlate monomer conversion, nanoparticle morphology, and pyrene fluorescence during the PISA process. The polymerization set in glass vials was used to determine monomer conversion, molar mass, and morphology (Fig. 3a and Table S2†) by 1H NMR spectroscopy, SEC, and TEM, respectively. The polymerization in the fluorometer allowed monitoring of the pyrene emission throughout the reaction. Notably, we found that the emission ratio of pyrene excimer to monomer (Iexi/Imon), which is related to the stacking of the pyrene molecules, changed throughout the PISA process (Fig. 3b).
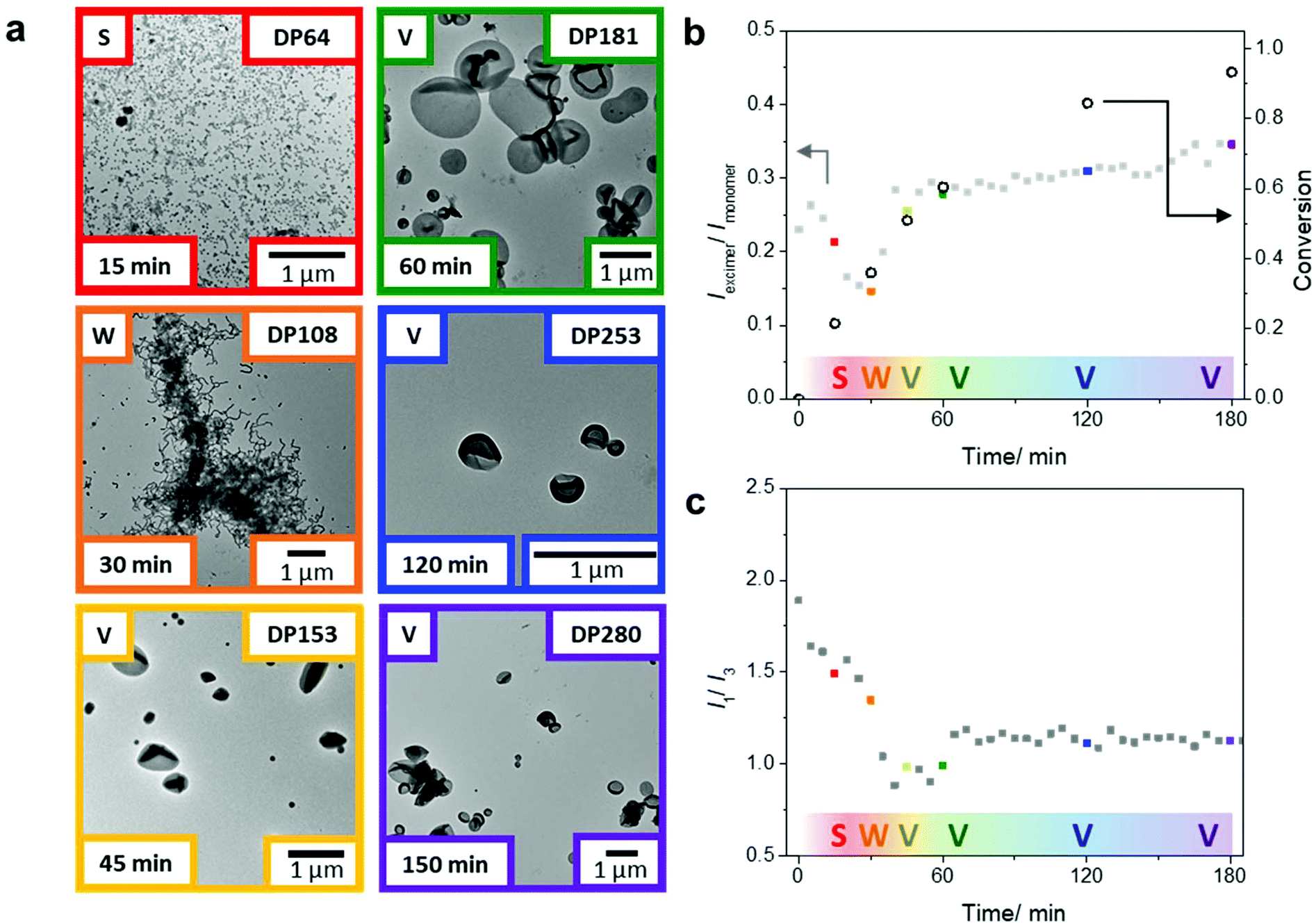 | ||
| Fig. 3 Correlation of polymerization kinetics (obtained by NMR spectroscopy), PISA morphology (obtained by ex situ TEM), and pyrene fluorescence (obtained by fluorescence spectroscopy). (a) TEM images of assemblies from polymerization aliquots at different conversions: spheres (21% red, top left), worms (36% orange, middle left), and vesicles (51% yellow, bottom left, 60% green, top right, 84% blue, middle right, and 93% purple, bottom right). Scale bars represent 1 µm. The ratio of (b) pyrene excimer to monomer emission (Iexi/Imon) and (c) pyrene monomer emission bands I1 to I3 (I1/I3) throughout the PISA process. | ||
Previously, Wang et al. reported that the arrangement of stacked fluorophores affected the fluorescence quantum yield of the pyrene excimer.50 We reasoned that during PISA, the pyrenes at the core of the self-assembly can undergo changes in their stacking orientation i.e., packing mode and packing efficiency. Therefore, the sensitivity of pyrene excimer emission to minor changes in fluorophore stacking orientation was used to monitor the structural rearrangements that accompany morphological transitions during PISA.43,50,54
After 15 min of the polymerization, we observed the aggregation of polymers into spherical micelles (Fig. 3a). As the self-assemblies transformed from spheres to worms after 30 min (DP = 108), we observed a decrease in the Iexi/Imon ratio. Throughout the PISA process, the transitions between different morphologies led to considerable structural rearrangements; we believe the fluctuations in the excimer ratio are due to changes in the packing efficiency and packing modes between the fluorophores during the reaction. The transitions during the classical PISA process (sphere-to-worm and worm-to-vesicles) are hypothesized to go through different mechanisms. The sphere to worm transition is often proposed to take place by collisions between self-assembled aggregates.55 In contrast, the worm to vesicle phase is hypothesized to proceed through a more continuous process. Blanazs et al. showed that the worm to vesicle transition proceeds via several intermediates: the branched network of worms leads to the partial fusion of worms, which then form nascent bilayers (as evidenced by the presence of ‘octopi’- and ‘jellyfish’-like intermediate structures) that eventually form pure vesicles.56 Nevertheless, both transitions require structural rearrangements in the polymer self-assembly.
Upon further polymerization of the DAAm block to DP = 153 at 45 min, vesicles were formed, which again led to a major change in the pyrene emission and, in this case, an increase in the Iexi/Imon ratio. These results suggest that transitions involving cylindrical structures are associated with changes in chain arrangements, which affect the fluorophore stacking and can be readily detected by changes in the Iexi/Imon ratio. Also, further increases of the PDAAm block size from this point onwards (DP = 181 at 60 min to DP = 280 at 180 min) showed only a small change in the Iexi/Imon ratio, consistent with no further significant change in morphology. It is worth noting that this gradual increase in the excimer ratio corresponds well with the fact that the hydrophobic block is continuing to grow, and in turn, the packing parameter, which affects the pyrene stacking arrangement, is still increasing throughout the polymerization.11
The pyrene monomer emission can also be used to gain information about the hydrophobicity in the local environment of the fluorescent probe. A decrease in the intensity ratio of the emission bands I1 to I3 is indicative of an increasingly hydrophobic local environment. From these studies, we can monitor the change in hydrophobicity near the growing chain end in the core of the assemblies throughout the polymerization. Upon chain extension of the PDMA macroCTA with DAAm, the pyrene on the active chain end experiences a progressively more hydrophobic environment, as shown in the decreasing I1/I3 ratio within the first 40 min of the polymerization (Fig. 3c). Interestingly, at 45 min a slight increase of the I1/I3 ratio was observed during the worm-to-vesicle transition, which suggests a slight increase in hydrophilicity that coincides with the formation of a bilayer vesicular structure where water is on the inside and outside of the bilayer. Notably, after the polymer chains have rearranged into vesicles the hydrophobicity (as indicated by the I1/I3 ratio) remains relatively constant as the pyrene moieties are buried in the center of the bilayer, presumably with very little solvation. These data suggest this non-invasive technique can monitor, in tandem, both changes in the self-assembly and hydration in the cores.
Conclusions
In summary, we demonstrated the use of pyrene as a fluorescent tag to monitor morphology and hydrophobicity during the PISA process. We were able to relate morphological transitions during the PISA process to changes in pyrene excimer-to-monomer ratio, which is dependent on the spatial proximity of pyrene molecules. Furthermore, the ratio of the I1 and I3 emission bands of the pyrene monomer provided unprecedented insight into the hydrophobicity at the growing chain end at the assembled core. In the future, we believe this straightforward and non-invasive in situ technique could be used to better understand and predict morphological progressions during the PISA process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Midlands Regional Cryo-EM Facility, hosted at the Warwick Advanced Bioimaging Research Technology Platform, for the use of the JEOL 2100Plus and University of Warwick Research Technology Platform for size exclusion chromatography (SEC) facilities. S. H. and R. R. acknowledge Lubrizol for financial support. J. R. would like to thank the Macro Group UK for their financial support for her travel to the US. J. R. This material is based on work supported by the National Science Foundation (DMR-1904631). This research was conducted with partial Government support under and awarded by DoD through the ARO (W911NF-17-1-0326).References
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- J. J. Shin, E. J. Kim, K. H. Ku, Y. J. Lee, C. J. Hawker and B. J. Kim, ACS Macro Lett., 2020, 9, 306–317 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- C. A. Machado, R. Tran, T. A. Jenkins, A. M. Pritzlaff, M. B. Sims, B. S. Sumerlin and D. A. Savin, Polym. Chem., 2019, 10, 6037–6046 RSC.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- D. Liu, J. He, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1660–1669 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef PubMed.
- G. Wang, M. Schmitt, Z. Wang, B. Lee, X. Pan, L. Fu, J. Yan, S. Li, G. Xie, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2016, 49, 8605–8615 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- X. Wang, C. A. Figg, X. Lv, Y. Yang, B. S. Sumerlin and Z. An, ACS Macro Lett., 2017, 6, 337–342 CrossRef CAS.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2013, 5, 350–355 RSC.
- M. Vakili, V. J. Cunningham, M. Trebbin and P. Theato, Macromol. Chem. Phys., 2019, 220, 1800370 CrossRef.
- A. Bagheri, C. Boyer and M. Lim, Macromol. Rapid Commun., 2019, 40, 1800510 CrossRef PubMed.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Angew. Chem., 2020, 59, 19136–19142 CrossRef CAS PubMed.
- B. Couturaud, P. G. Georgiou, S. Varlas, J. R. Jones, M. C. Arno, J. C. Foster and R. K. O'Reilly, Macromol. Rapid Commun., 2019, 40, 1800460 CrossRef PubMed.
- G. Mellot, P. Beaunier, J.-M. Guigner, L. Bouteiller, J. Rieger and F. Stoffelbach, Macromol. Rapid Commun., 2019, 40, 1800315 CrossRef PubMed.
- G. Mellot, J.-M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., 2019, 58, 3173–3177 CrossRef CAS PubMed.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- F. Ouhib, A. Dirani, A. Aqil, K. Glinel, B. Nysten, A. M. Jonas, C. Jerôme and C. Detrembleur, Polym. Chem., 2016, 7, 3998–4003 RSC.
- P. Gurnani, C. P. Bray, R. Richardson, A. E. R. Peltier and S. Perrier, Macromol. Rapid Commun., 2019, 40, 1800314 CrossRef PubMed.
- D. J. Rucco, B. E. Barnes, J. B. Garrison, B. S. Sumerlin and D. A. Savin, Biomacromolecules, 2020, 21, 5077–5085 CrossRef CAS PubMed.
- D. Le, F. Wagner, M. Takamiya, I. L. Hsiao, G. G. Alvaradejo, U. Strähle, C. Weiss and G. Delaittre, Chem. Commun., 2019, 55, 3741–3744 RSC.
- M. A. Touve, C. A. Figg, D. B. Wright, C. Park, J. Cantlon, B. S. Sumerlin and N. C. Gianneschi, ACS Cent. Sci., 2018, 4, 543–547 CrossRef CAS PubMed.
- G. M. Scheutz, M. A. Touve, A. S. Carlini, J. B. Garrison, K. Gnanasekaran, B. S. Sumerlin and N. C. Gianneschi, Matter, 2021, 4, 722–736 CrossRef.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141(34), 13664–13675 CrossRef CAS PubMed.
- S. Nishizawa, Y. Kato and N. Teramae, J. Am. Chem. Soc., 1999, 121, 9463–9464 CrossRef CAS.
- M. O. Guler, R. C. Claussen and S. I. Stupp, J. Mater. Chem., 2005, 15, 4507–4512 RSC.
- C. A. Parker and C. G. Hatchard, Nature, 1961, 190, 165–166 CrossRef CAS.
- J. B. Birks and L. G. Christophorou, Spectrochim. Acta, 1963, 19, 401–410 CrossRef.
- G. Basu Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248–254 CrossRef CAS PubMed.
- E. D. Goddard, N. J. Turro, P. L. Kuo and K. P. Ananthapadmanabhan, Langmuir, 1985, 1, 352–355 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- T. Förster and K. Kasper, Z. Elektrochem. Ber. Bunsenges. Phys. Chem., 1955, 59, 976–980 Search PubMed.
- T. Förster, Angew. Chem., Int. Ed. Engl., 1969, 8, 333–343 CrossRef.
- H.-J. Galla and E. Sackmann, Biochim. Biophys. Acta, Biomembr., 1974, 339, 103–115 CrossRef CAS.
- H.-J. Galla and W. Hartmann, Chem. Phys. Lipids, 1980, 27, 199–219 CrossRef CAS PubMed.
- P. Somerharju, Chem. Phys. Lipids, 2002, 116, 57–74 CrossRef CAS PubMed.
- J. Cornil, D. Beljonne, J.-P. Calbert and J.-L. Brédas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS.
- O. J. Dautel, G. Wantz, R. Almairac, D. Flot, L. Hirsch, J.-P. Lere-Porte, J.-P. Parneix, F. Serein-Spirau, L. Vignau and J. J. E. Moreau, J. Am. Chem. Soc., 2006, 128, 4892–4901 CrossRef CAS PubMed.
- L. Wang, W. Li, J. Lu, Y.-X. Zhao, G. Fan, J.-P. Zhang and H. Wang, J. Phys. Chem. C, 2013, 117, 26811–26820 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- D. Y. Chu and J. K. Thomas, Macromolecules, 1987, 20, 2133–2138 CrossRef CAS.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Macromolecules, 2017, 50, 935–943 CrossRef CAS.
- J. Duhamel, Langmuir, 2012, 28, 6527–6538 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00239b |
| This journal is © The Royal Society of Chemistry 2021 |