
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative organocatalysed enantioselective coupling of indoles with aldehydes that forms quaternary carbon stereocentres†
Nomaan M.
Rezayee
 ,
Vibeke H.
Lauridsen
,
Line
Næsborg
,
Thanh V. Q.
Nguyen
,
Henriette N.
Tobiesen
and
Karl Anker
Jørgensen
*
,
Vibeke H.
Lauridsen
,
Line
Næsborg
,
Thanh V. Q.
Nguyen
,
Henriette N.
Tobiesen
and
Karl Anker
Jørgensen
*
Department of Chemistry, Aarhus University, DK-8000 Aarhus C, Denmark. E-mail: kaj@chem.au.dk
First published on 13th February 2019
Abstract
The first organocatalysed, metal-free cross-nucleophile coupling of indoles with α-branched aldehydes forming acyclic stereoselective quaternary carbon centres is presented. Applying an amino acid-derived catalyst with suitable organic oxidants affords the desired enantioenriched indole functionalised products with moderate to excellent yield and enantioselectivity. Two metal-free oxidative protocols employing either DDQ or a sequential approach that uses two organocatalysts to facilitate the use of O2 as the terminal oxidant are disclosed. These methods are compatible with various indoles ranging from electron-rich to -deficient substituents at the C-2, -5, -6, and -7-positions reacting with a series of different α-branched aldehydes.
Introduction
The oxidation of organic compounds serves as a powerful synthetic tool. Classically, transition-metal reagents have been employed to perform this transformation; however, redox-active organic compounds may also function in a similar capacity. While both classes are amenable to range from academic to industrial processes, organic oxidants may provide access to distinct reaction mechanisms leading to complementary reactivity and selectivity.1Quinones are an essential class of redox-active organic compounds found in diverse processes spanning from mitochondrial electron transport to the manufacture of industrial chemicals.1,2 Highlighting the versatility of quinones, three oxidation states are readily accessible, namely, the fully oxidised quinone, the one-electron reduced semi-quinone, and the two-electron reduced hydroquinone.1,2 As such, quinone oxidants may operate through diverse mechanisms via closed- or open-shell pathways.1,2 While quinone oxidants have been applied to typical oxidation reactions,3 investigations into their application for umpolung reactivity on enamine intermediates in organocatalysis remain unprecedented.4
A shift in the classical paradigm of reactivity for cross-nucleophile coupling of enolates was introduced by Kauffman et al. and Ito et al. using copper oxidants.5 This approach was later applied by Baran et al. for the α-functionalisation of mainly ketones and esters with indoles using a super-stoichiometric base and copper-oxidant (Scheme 1a).6 The method proceeded with diastereoselectivity in cases where chiral carbonyl compounds were employed (substrate control) and racemic for pro-chiral substrates. Underscoring the value of this methodology is the formation of cyclic quaternary carbon centres, however, only when employing either α-functionalised γ-butyrolactone or cyclohexenone scaffolds. Compounds bearing the indole motif are of great interest due to their privileged scaffold, since thousands of biologically active compounds contain the indole moiety.7
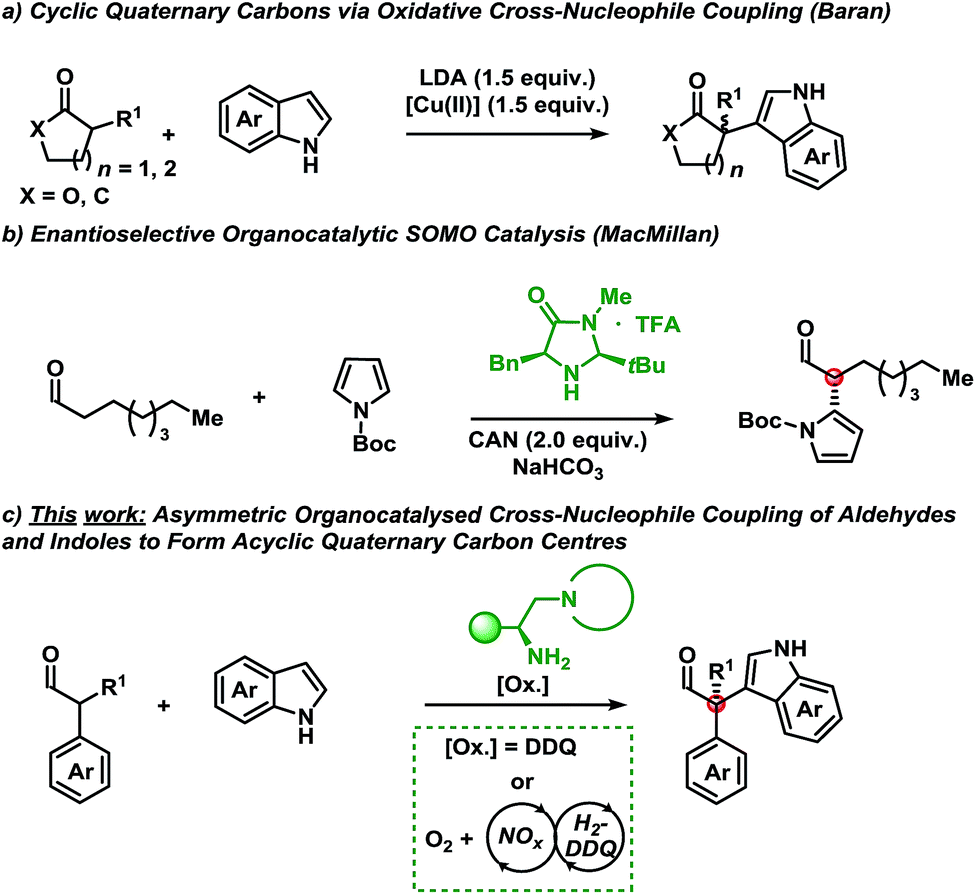 | ||
| Scheme 1 (a) Oxidative cross-coupling of indoles to aldehydes with LDA and Cu(II). (b) Enantioselective organocatalytic SOMO catalysis using CAN and 3a. (c) This work: organocatalytic asymmetric cross-nucleophile coupling of indoles with aldehydes applying organic oxidation systems. | ||
Owing to the congested nature of quaternary carbon stereocentres, asymmetric methodologies to construct these centres have been a long-standing challenge.8 As such, the majority of chemical compounds bearing an all carbon quaternary centre are derived from the chiral pool. This highlights the key challenges in the de novo synthesis of novel motifs. The advent of new methodologies to shift current synthetic paradigms may provide access to new points of diversity.
With these results serving as inspiration, we sought to develop a novel oxidative asymmetric organocatalytic concept for the oxidative coupling of α-branched aldehydes with indoles.9 This approach would provide access to acyclic scaffolds bearing a chiral quaternary carbon centre8 that would be otherwise unobtainable using traditional methods from the chiral pool.
MacMillan et al. have disclosed an example of the asymmetric coupling of a Boc-protected pyrrole with octanal using singly occupied molecular orbital (SOMO) catalysis (Scheme 1b).10 SOMO-activation strategies have mainly used stoichiometric metals as oxidants in combination with an organocatalyst.11
Disclosed herein is a metal-free, enantioselective protocol for the functionalisation of α-branched aldehydes with indoles at the C-3 position (Scheme 1c). The reactions proceed with moderate to excellent yields and enantioselectivities using an amino acid-derived primary amine catalyst and an organic oxidant. Generally, a broad spectrum of the indole coupling partner is well tolerated. Importantly, attempts at accessing these products using traditional single-electron oxidants proved challenging; indicating a potentially alternative mechanism to SOMO catalysis.
Results and discussion
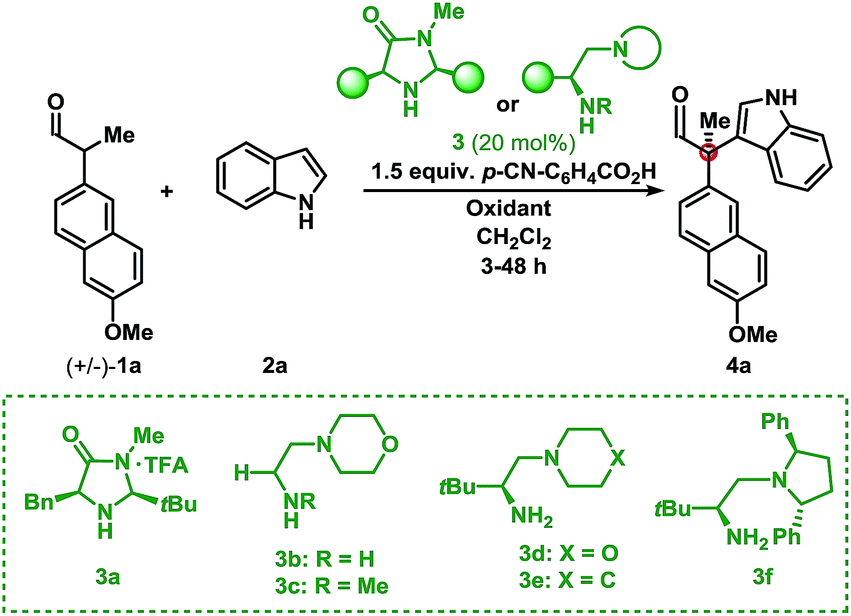
Initial investigations into organocatalysed cross-coupling were undertaken with racemic naproxen derivative 1a and indole 2a under standard SOMO-catalysis conditions.11 CAN, in combination with organocatalyst 3a in DME, led to complete consumption of 1a without any detection of the indole-coupling product 4a (Table 1, entries 1 and 2). Attempts to use milder oxidants also known for SOMO activation12 in combination with 3a (entry 3) and 3b (see ESI†) proved unfruitful, leading to a mixture of 1,2-addition products and undesired oxidation products (see Fig. 1).13 Finally, using the primary-amine catalyst 3b and the two-electron oxidant DDQ, the desired indole cross-coupled product 4a could be isolated. However, isolation of the desired α-functionalised aldehyde inadequately reflected the observed NMR yield. The remaining mass balance may be accounted for by the 1,2-addition of the indole to the aldehyde (Fig. 1). Use of pre-treated Iatrobeads remedied the undesired reactivity leading to a 38% yield of racemic 4a (entry 4). Interestingly, using the methylated analog of 3b, catalyst 3c did not provide the desired product, which suggests critical involvement of the N–H moiety of the primary amine catalyst to obtain the desired product (entry 5).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Oxidant | Temp. (°C) | Yield (%) | ee (%) |
| a Performed on a 0.10 mmol scale under Ar: 1.0 equiv. 1a, 5.0 equiv. 2a, 0.20 equiv. 3, 1.5 equiv. acid, 1.1 equiv. oxidant and 0.4 mL CH2Cl2. b 2.0 equiv. oxidant and 0.4 mL DME. c Acid omitted. d DDQ added in two portions. | |||||
| 1b | 3a | CAN | RT | 0 | — |
| 2b | 3a | CAN | −20 | 0 | — |
| 3b | 3a | Cu(OTf)2 | −20 | 0 | — |
| 4c | 3b | DDQ | RT | 38 | — |
| 5c | 3c | DDQ | RT | 0 | — |
| 6c | 3d | DDQ | RT | 41 | 66 |
| 7 | 3d | DDQ | RT | 70 | 70 |
| 8 | 3d | Fluoranil | RT | 31 | 63 |
| 9 | 3d | Chloranil | RT | 10 | 43 |
| 10 | 3e | DDQ | RT | 63 | 58 |
| 11 | 3f | DDQ | RT | 64 | 82 |
| 12d | 3f | DDQ | RT | 73 | 94 |
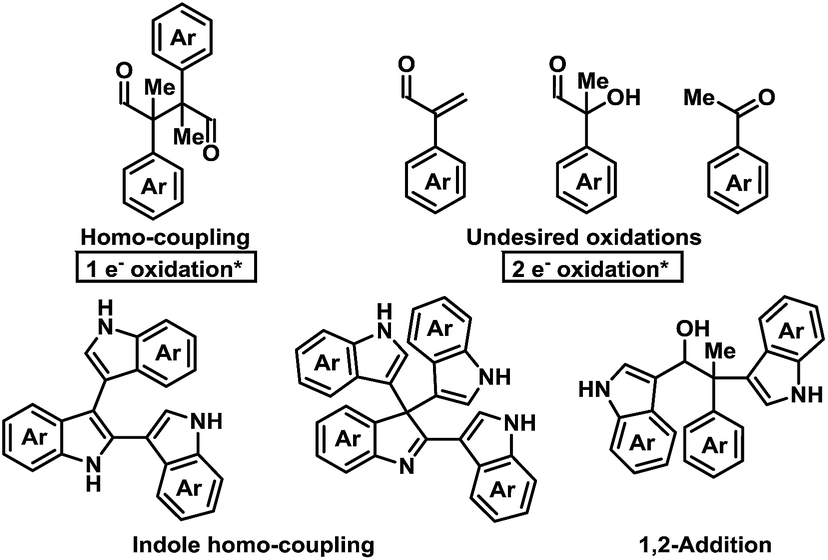 | ||
| Fig. 1 Potential byproducts during the screening for the oxidative coupling of indoles to aldehydes.* = Relative to aldehyde. | ||
As a proof-of-concept, chiral aminocatalyst 3d afforded the oxidative cross-coupling product 4a with 66% ee, albeit in a low 41% yield (Table 1, entry 6). The low yield can be attributed to poor product selectivity with 3d. Benzoic acid additives were investigated to improve the yield and enantioselectivity; however, the additive also affected the reaction outcome resulting in the formation of various byproducts (see ESI† and Fig. 1). While homo-coupling of the aldehyde was not observed, other α-functionalised products obtained from undesired oxidations were identified.14 The origin of these products was not investigated.
Substitution on the quinone oxidant was also investigated. Both fluoranil and chloranil were shown to be competent two-electron oxidants for this transformation, however in reduced yields compared to DDQ (Table 1, entries 8 and 9). The yields seem to be related to the first reduction potential of the oxidant (DDQ = 0.54 V, chloranil = 0.01 V, and fluoranil −0.04 V vs. SCE).14d,f Finally, other L-tert-leucine-derived organocatalysts were evaluated to perform this transformation. A survey of catalysts 3d, 3e, and 3f showed the same major enantiomer and improvements in yield and enantioselectivity (entries 7, 10, and 11). Applying catalyst 3f and portion-wise addition of DDQ15 further enhanced the yield and enantioselectivity of 4a to 73% and 94% ee (entry 12).
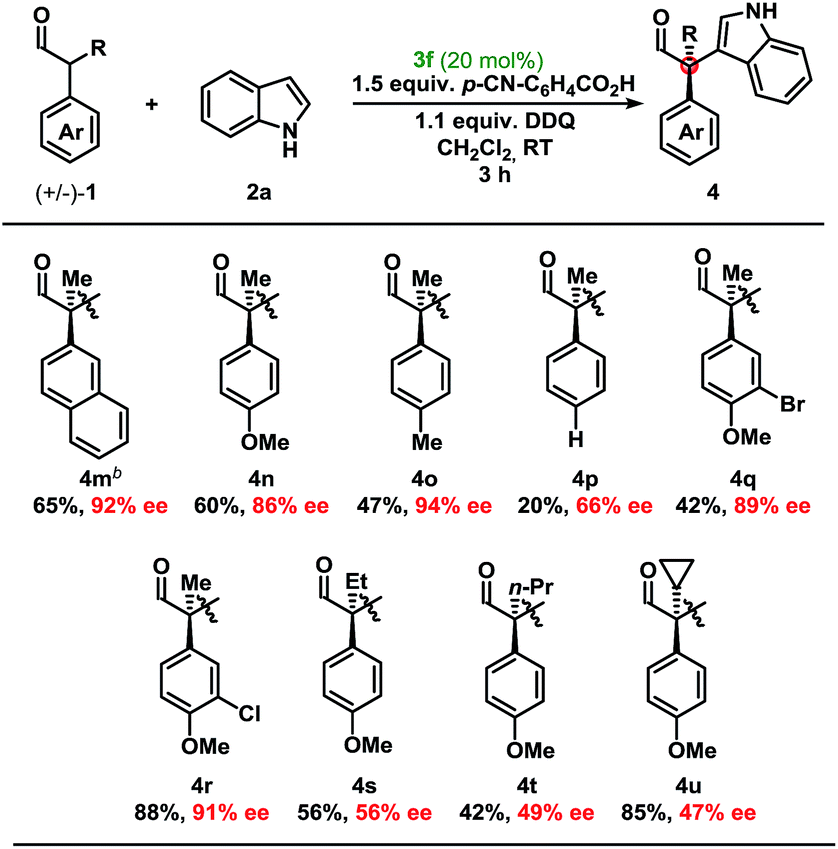
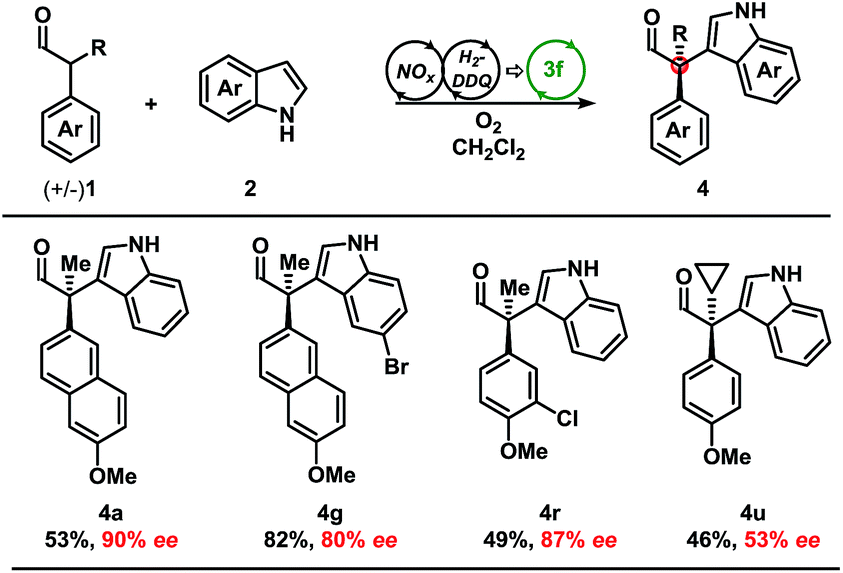
Development of a metal-free oxidative protocol with excellent enantioselectivity prompted us to evaluate the scope of the indole coupling partners (Table 2). The method proved general as indoles with electron-donating or electron-withdrawing groups were converted to the cross-coupled products.
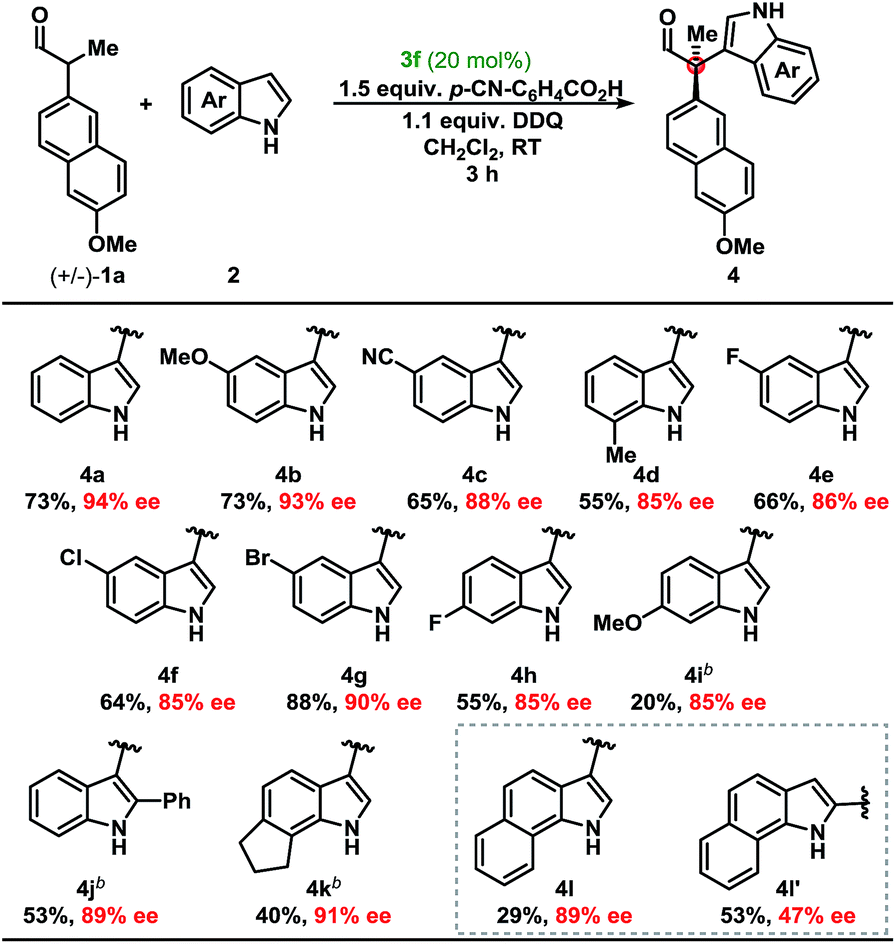
Indoles bearing halogens were well-tolerated leading to high enantioselectivity. Compounds 4f and 4g are of particular interest as they are amenable to subsequent palladium-catalysed cross-coupling reactions. Importantly, the absolute configuration of 4g was determined by X-ray crystallographic analysis and the remaining products were assigned by analogy. Further analysis demonstrated the scope of the indole coupling partner where functionalisation of the 7-, 6-, 2-, or even 6,7-positions of the indole were converted to the desired product with high enantioselectivity. It should also be noted that the presence of a C-2-phenyl substituent on the indole afforded 4j with high enantioselectivity. Notably, regioisomers were isolated in the case of 4l in a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio yielding 89% and 47% ee, respectively (vide infra).
:2 ratio yielding 89% and 47% ee, respectively (vide infra).
Moving from the electron-rich aldehyde 1a to the naphthyl-moiety, shown in product 4m, comparable yield and stereoselectivity were retained (Table 3). Transitioning from the extended π-system of the naphthyl-moiety to phenyl-derivatives 4n–p, minimal changes in enantioselectivities were observed except in the case of 4p. Taken in context with the results from 4n and 4o, a trend was observed where electron-rich aldehydes were more easily converted to the desired product.
Additional modifications to the anisole-bearing aldehyde moiety were made. We were pleased to find that halogens, specifically in the cases of 4q and 4r, were well tolerated, allowing for further synthetic elaborations upon formation of the chiral quaternary carbon centre. Alterations in the α-branched alkyl group showed a clear influence moving from Me- (4n, 86% ee) to Et- (4s, 56% ee) to n-Pr- (4t, 49% ee), and cyclopropyl- (4u, 47% ee) substitution. Although decreased enantioselectivities were observed with deviation from the methyl group, we were encouraged to find that this methodology could be extended to form highly congested quaternary carbon centres in up to 85% yield and with moderate enantioselectivity without optimisation of the aminocatalyst. It should be noted that non-α-branched aldehydes were not amenable to this methodology leading to decomposition.
Following the examination of the coupling partners, key empirical observations were used to reveal insights into the reaction mechanism.
First, stereoinduction using a chiral aminocatalyst implies that the catalyst is involved in the stereodetermining step (Scheme 2). Second, the stoichiometry of the oxidant relative to the indole suggests preferential oxidation of the enamine. This is consistent with the trend observed, where the more electron-rich aldehyde resulted in higher yields. Furthermore, delineations from previous methods under reaction conditions (acidic vs. basic), suggest that the deprotonation and subsequent oxidation mechanism are improbable under these conditions.6,16 Finally, the regioselectivity (C-3 vs. C-2) of the indole coupling partner, shown in 4l/4l′, indicates indole as a nucleophile. Extension of the fused aromatic in 2l allows for a decreased energetic penalty for the breaking of aromaticity in the phenyl core, thus revealing an electronically isolated pyrrole moiety that is also nucleophilic at the C-2 position.7b While a radical recombination mechanism cannot be dismissed, the stoichiometry of the aldehyde and indole (1.0 to 5.0 equiv.) vs. the oxidant (1.1 equiv.) deems the mechanism unlikely. In order to investigate the potential involvement of hydrogen-bonding of the NH-moiety of the indole with catalyst 3f in the enantioselectivity of the reaction, the oxidative coupling of N-methyl indole 2v with the aldehyde 1a was performed (Scheme 2a). Since the enantiomeric excess is maintained (compared to indole 2a) despite the lack of a hydrogen-donor in 2v, this indicates that 3f acts a steric-shielding catalyst. Based on this result and the absolute configuration of the chiral product obtained, we propose that the indole approaches from the bottom-face (Si-face) resulting in the absolute configuration as (R), due to steric-shielding of the Re-face as presented in Scheme 2b (for DFT-calculated models of the proposed intermediate, see ESI†). As a result we believe that 3f improves the enantioselectivity compared to 3d due the increased steric bulk of the diaryl pyrrolidine moiety.
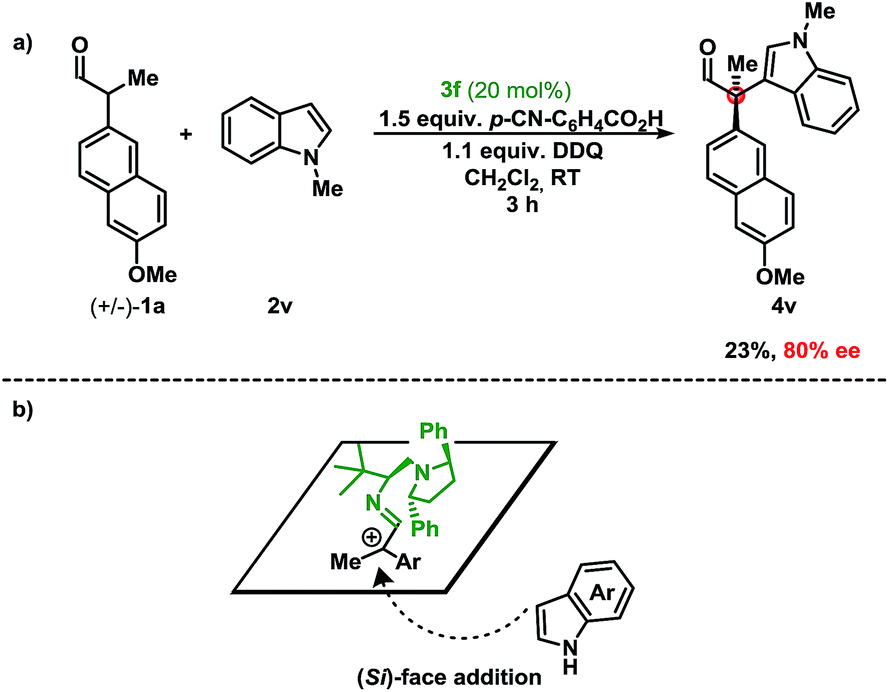 | ||
| Scheme 2 (a) Results for the oxidative coupling of N-methyl indole 2v with aldehyde 1a. (b) Stereochemical model for the approach of indole to the proposed intermediate. | ||
A more nuanced discussion regarding the oxidation of the enamine species using DDQ is warranted. Quinones are versatile oxidants and have access to three different oxidation states shown in Scheme 3a. While SOMO catalysis typically employs two equivalents of a single-electron oxidant, in the case of DDQ, one equivalent of quinone may facilitate the oxidation event. To probe whether DDQ is able to operate as a single-electron (SOMO catalysis) or two-electron oxidant under the present reaction conditions, we applied DDQ to a system proposed to operate via SOMO catalysis shown in Scheme 3b.
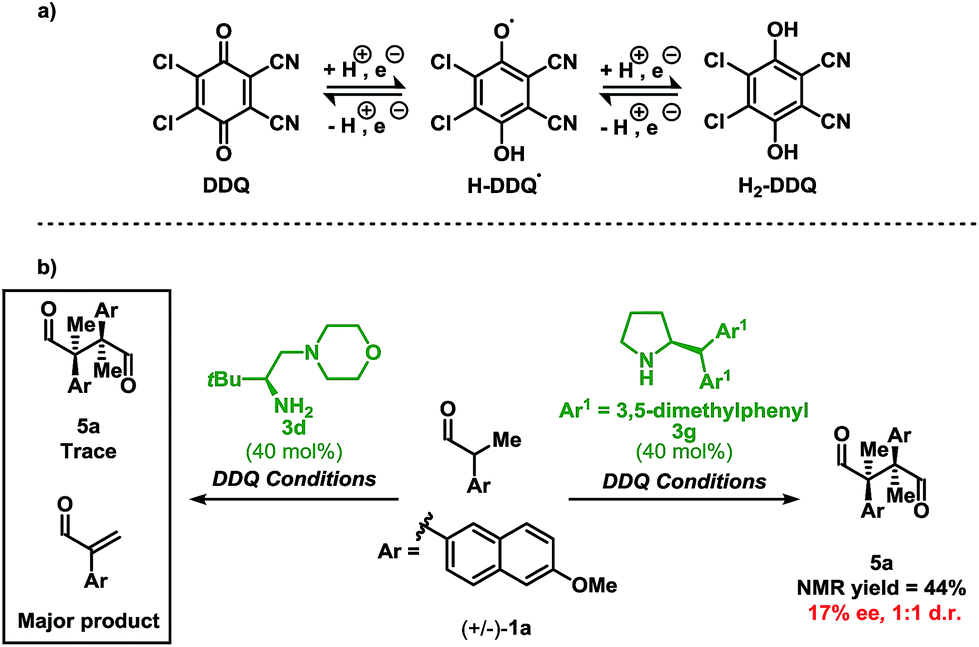 | ||
| Scheme 3 (a) Common oxidation states of DDQ. (b) Homo-coupling of 1a using a secondary amine catalyst (right) and a primary amine catalyst (left). DDQ conditions: 0.2 mmol 1a, 0.40 equiv. 3, 1.5 equiv. p-NO2-C6H4CO2H, 1.5 equiv. DDQ, and 0.4 mL CH2Cl2 at RT. | ||
When using DDQ combined with the secondary amine catalyst 3g, the homo-coupled product 5a was obtained in 44% yield (Scheme 3b). However, unlike Ag2CO3, which leads to 5a in 92% ee and 12:1 d.r.,13d only a 17% ee and a 1:1 d.r. was observed under the present reaction conditions. These results confirm that DDQ may operate as a one-electron oxidant when applying a secondary amine catalyst. Finally, highlighting a key distinction, replacing the secondary amine catalyst 3g with the primary amine catalyst 3d leads to only trace homo-coupled product 5a (see ESI†). The major product detected under these conditions was the α,β-unsaturated aldehyde which originates from a two-electron oxidation. Furthermore, other two-electron oxidation products, as shown in Fig. 1, were also detected.
Placing these results in context, a clear demarcation among primary amine and secondary amine catalysts can be distinguished. Shown in Scheme 4a is the direct comparison for the coupling of aldehyde 1n with indole 2a using primary amine catalyst 3b, and the secondary amine analog 3c. While the primary amine catalyst 3b furnished the coupled product in a 42% yield, application of the secondary amine catalyst 3c did not lead to the desired product.
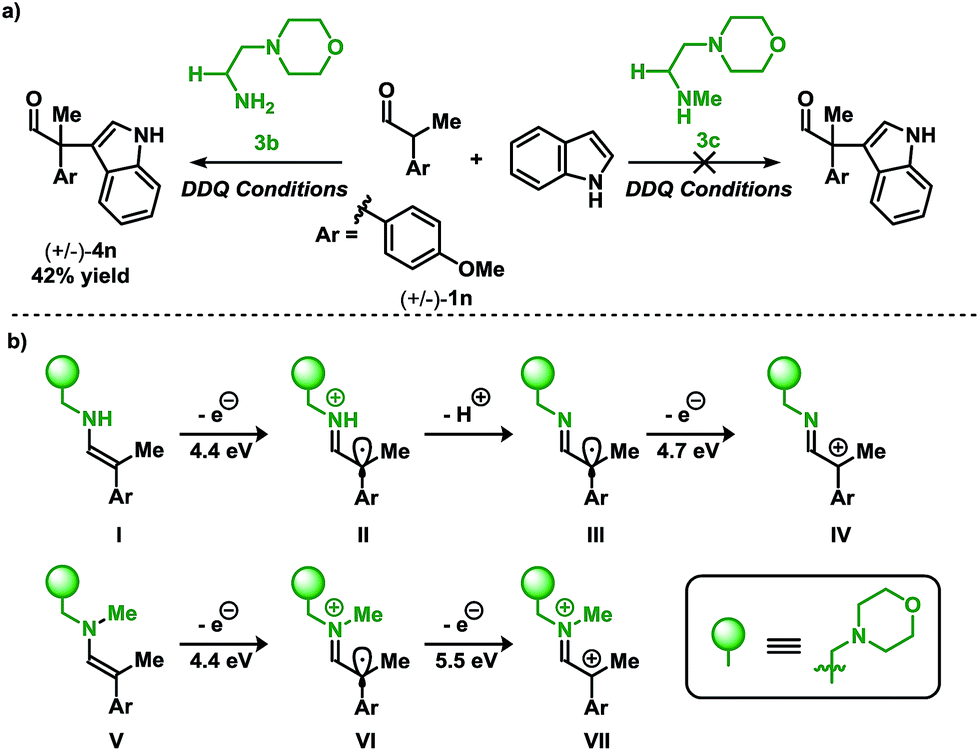 | ||
| Scheme 4 (a) Organocatalyzed coupling of indoles and aldehydes: primary vs. secondary amine catalysts (see Table 2 for conditions). (b) Calculated ionization potentials for putative intermediates.12 | ||
To gain insight into the divergence in reactivity, ionization potentials (IPs) of the enamines resulting from 3b and 3c were used to discern potential intermediates (Scheme 4b).13c In the case of primary amine catalyst 3b (enamine I) and secondary amine catalyst 3c (enamine V), the initial IP required to form the radical is 4.4 eV. These data imply that the radical intermediates (II and VI) may be accessible using either catalyst since the initial IP for both intermediates are degenerate. Thus, if the product results from SOMO catalysis, both catalysts would be sufficient to facilitate the transformation. However, the experimental results from Scheme 4a demonstrate that the desired coupling product (4n), for the reaction of aldehyde 1n with indole 2a, is only formed when employing the primary amine catalyst 3b, potentially implying an alternative reactive intermediate to the one operating in SOMO catalysis. Continued evaluation of IPs from the II and VI highlights a key energetic difference among the two radical cations. Oxidation of VI provides a dicationic intermediate with an IP of 5.5 eV. Unlike VI, II has the flexibility to vary in the protonation state which influences the IP. Deprotonation of the cationic II to III yields a neutral radical which may be oxidised to cationic IV requiring a significantly lower IP of 4.7 eV. These distinctions may contribute to the difference in reactivity between the primary- and secondary amine catalysts disclosed in Scheme 4a.
Furthermore, we have also attempted to apply typical single-electron probes such as radical clocks and traps which have proven inconclusive with no evidence of radical formation (see ESI†). Though not definitive, taken together these results indicate a preference for a two-electron oxidation prior to the nucleophilic attack when using DDQ in conjunction with a primary amine catalyst. A putative mechanism is illustrated in Scheme 5.
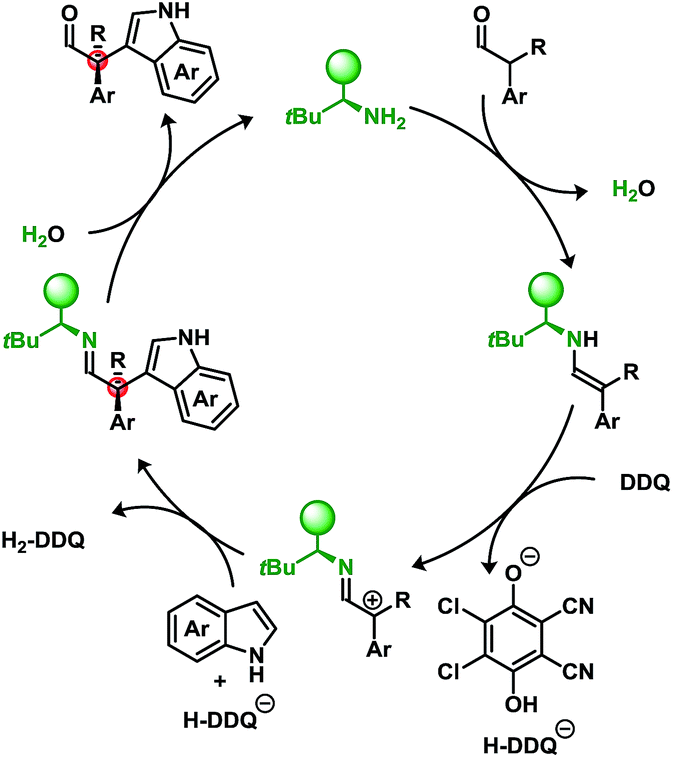 | ||
| Scheme 5 Plausible reaction mechanism. | ||
While DDQ may be operating as a two-electron oxidant, several oxidative manifolds may be accessible. Elegant studies by Mayr et al. have investigated the propensity to oxidise various nucleophiles through outer-sphere charge transfer or inner-sphere electron transfer via “C” or “O” attack of the quinone electrophile.14d,f Isolable products related to the latter case have recently been reported arising from the α-oxidation of ketones using a phosphoric acid catalyst and 1,4-benzoquinones.17
An underlying impetus for the use of DDQ in the developed methodology was to not only provide a metal-free route to these products but to also try to utilise O2 as the terminal oxidant. An NO/NO2-cycle can act as an intermediary catalyst to oxidise the resulting hydroquinone, H2-DDQ to DDQ, yielding H2O as the byproduct.18
Sub-stoichiometric quantities of t-butyl nitrite and organocatalyst 3f were employed with H2-DDQ to perform this cross-coupling. Due to incompatibility between NOx and the aldehyde, a sequential approach was undertaken where t-butyl nitrite and H2-DDQ were first combined under an O2 atmosphere to generate DDQ and then added to the reaction mixture. Table 4 demonstrates a proof-of-concept in applying O2 as the terminal oxidant.
| a Performed on a 0.10 mmol scale under O2: 0.20 equiv. tBuONO, 2.0 equiv. H2-DDQ in 0.4 mL CH2Cl2. After 1.5 h, the solvent was removed via evaporation and added portion-wise to a second vial containing 1.0 equiv. 1, 5.0 equiv. 2, 0.20 equiv. 3f, 1.5 equiv. acid, and 0.4 mL CH2Cl2 under Ar. Reactions were run for 3 h. |
|---|
|
The results in Table 4 demonstrate that the yields and stereoselectivities were generally comparable to those of their stoichiometric counterparts for the aerobic cross-coupling of indoles to aldehydes.
Conclusions
In summary, this article describes the first enantioselective oxidative cross-coupling of indoles with aldehydes using an amino acid-derived organocatalyst and an organic oxidant. Two different organic oxidative protocols have been disclosed using DDQ or O2 as the terminal oxidant to construct quaternary carbon centres bearing the indole moiety. Importantly, preliminary studies suggest that DDQ may operate as a two-electron oxidant prior to nucleophilic addition. This general method allows for the metal-free construction of acyclic quaternary carbon centres using α-branched aldehydes with indoles ranging from electron-rich to -deficient substitution patterns providing enantioselectivities up to 94% ee.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made possible by grants from Carlsberg Foundation's ‘Semper Ardens’ programme, FNU, Aarhus University and DNRF. H. N. T. thanks Novo Nordisk for a PhD grant. Thanks are expressed to Dr Gabriel J. Reyes-Rodríguez for performing the DFT calculations.Notes and references
- A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed.
- (a) D. Walker and J. D. Hiebert, Chem. Rev., 1967, 67, 153–195 CrossRef CAS PubMed; (b) E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC.
- (a) Y. Hayashi, T. Itoh and H. Ishikawa, Angew. Chem., Int. Ed., 2011, 50, 3920–3924 CrossRef CAS PubMed; (b) F. Huang, L. Xu and J. Xiao, Chin. J. Chem., 2012, 30, 2721–2725 CrossRef CAS; (c) Z. Meng, S. Sun, H. Yuan, H. Lou and L. Liu, Angew. Chem., Int. Ed., 2013, 53, 543–547 CrossRef PubMed; (d) G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645–2651 RSC; (e) Z. Xie, X. Zan, S. Sun, X. Pan and L. Liu, Org. Lett., 2016, 18, 3944–3947 CrossRef CAS PubMed.
- For a review on organocatalysis combined with oxidations: L. Zhu, D. Wang, Z. Jia, Q. Lin, M. Huang and S. Luo, ACS Catal., 2018, 8, 5466–5484 CrossRef CAS.
- (a) T. Kauffmann, G. Beissner, H. Berg, E. Köppelmann, J. Legler and M. Schönfelder, Angew. Chem., Int. Ed., 1968, 7, 540–541 CrossRef CAS; (b) Y. Ito, T. Konoike, T. Harada and T. Saegusa, J. Am. Chem. Soc., 1977, 99, 1487–1493 CrossRef CAS.
- P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2004, 126, 7450–7451 CrossRef CAS PubMed.
- (a) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (b) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC; (c) J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550–3567 RSC.
- For selected examples and reviews on the formation of quaternary stereocentres, see: (a) A. Gualandi, D. Petruzziello, E. Emer and P. G. Cozzi, Chem. Commun., 2012, 48, 3614–3616 RSC; (b) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS PubMed; (c) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (d) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed; (e) E. Picazo, S. M. Anthony, M. Giroud, A. Simon, M. A. Miller, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2018, 140, 7605–7610 CrossRef CAS PubMed; (f) A. E. Wendlandt, P. Vangal and E. N. Jacobsen, Nature, 2018, 556, 447–451 CrossRef CAS PubMed.
- For the formation of related products see: (a) M. A. Saputra, B. Nepal, N. S. Dange, P. Du, F. R. Fronczek, R. Kumar and R. Kartika, Angew. Chem., Int. Ed., 2018, 57, 15558–15562 CrossRef CAS PubMed; (b) M. Schlegel and C. Schneider, Chem. Commun., 2018, 54, 11124–11127 RSC.
- T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed.
- (a) J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640–11641 CrossRef CAS PubMed; (b) K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086–2087 CrossRef CAS PubMed.
- S. Rendler and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 5027–5029 CrossRef CAS PubMed.
- (a) M. Mečiarová, P. Tisovský and R. Šebesta, New J. Chem., 2016, 40, 4855–4864 RSC; (b) L. Næsborg, V. Corti, L. A. Leth, P. H. Poulsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 1606–1610 CrossRef PubMed; (c) L. A. Leth, L. Næsborg, G. J. Reyes-Rodríguez, H. N. Tobiesen, M. V. Iversen and K. A. Jørgensen, J. Am. Chem. Soc., 2018, 140, 12687–12690 CrossRef CAS PubMed; (d) L. Næsborg, L. A. Leth, G. J. Reyes-Rodríguez, T. A. Palazzo, V. Corti and K. A. Jørgensen, Chem.–Eur. J., 2018, 24, 14844–14848 CrossRef PubMed.
- (a) K. Kaneda, T. Itoh, N. Kii, K. Jitsukawa and S. Teranishi, J. Mol. Catal., 1982, 15, 349–365 CrossRef CAS; (b) J. H. P. Utley and G. G. Rozenberg, J. Appl. Electrochem., 2003, 33, 525–532 CrossRef CAS; (c) P. S. Baran and M. P. DeMartino, Angew. Chem., Int. Ed., 2006, 45, 7083–7086 CrossRef CAS PubMed; (d) X. Guo and H. Mayr, J. Am. Chem. Soc., 2013, 135, 12377–12387 CrossRef CAS PubMed; (e) Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2013, 53, 548–552 CrossRef PubMed; (f) X. Guo and H. Mayr, J. Am. Chem. Soc., 2014, 136, 11499–11512 CrossRef CAS PubMed; (g) S.-B. D. Sim, M. Wang and Y. Zhao, ACS Catal., 2015, 5, 3609–3612 CrossRef CAS.
- N. N. Karpyshev, O. D. Yakovleva, E. P. Talsi, K. P. Bryliakov, O. V. Tolstikova and A. G. Tolstikov, J. Mol. Catal. A: Chem., 2000, 157, 91–95 CrossRef CAS.
- (a) J. M. Richter, B. W. Whitefield, T. J. Maimone, D. W. Lin, M. P. Castroviejo and P. S. Baran, J. Am. Chem. Soc., 2007, 129, 12857–12869 CrossRef CAS PubMed; (b) P. S. Baran and M. Jessing, Heterocycles, 2011, 82, 1739–1745 CrossRef.
- G. A. Shevchenko, B. Oppelaar and B. List, Angew. Chem., Int. Ed., 2018, 57, 10756–10759 CrossRef CAS PubMed.
- (a) C. Qiu, L. Jin, Z. Huang, Z. Tang, A. Lei, Z. Shen, N. Sun, W. Mo, B. Hu and X. Hu, ChemCatChem, 2012, 4, 76–80 CrossRef CAS; (b) L. Bering, F. M. Paulussen and A. P. Antonchick, Org. Lett., 2018, 20, 1978–1981 CrossRef CAS PubMed; (c) C. Song, X. Dong, H. Yi, C.-W. Chiang and A. Lei, ACS Catal., 2018, 8, 2195–2199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1863137. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc00196d |
| This journal is © The Royal Society of Chemistry 2019 |