
A new sodium storage mechanism of TiO2 for sodium ion batteries
Dong
Yan
and
Likun
Pan
 *
*
Engineering Research Center for Nanophotonics & Advanced Instrument, Ministry of Education, Department of Physics, East China Normal University, Shanghai 200062, China. E-mail: lkpan@phy.ecnu.edu.cn; Fax: +86 21 62234321; Tel: +86 21 62234132
First published on 12th January 2016
Abstract
Sodium-ion batteries (SIBs) have attracted great interest for use as the next generation rechargeable batteries due to the abundant sodium natural resources and similar chemistry of lithium and sodium. TiO2 is an attractive candidate as an anode for SIBs due to its high safety, low cost, appropriate voltage platform and good structural stability during repeated charge–discharge processes. However, the sodium storage mechanism of TiO2 for SIBs remains unclear, which appears to be different from the working mechanism in lithium-ion batteries. This article highlights a recent report by Passerini's group, which successfully proposed a new sodium storage mechanism of TiO2 for SIBs.
 Dong Yan | Dong Yan received his Master's degree in 2014 at Henan University. Now he is pursuing his PhD under the supervision of Prof. Likun Pan in East China Normal University. His research interest lies in the synthesis of nanomaterials and their applications in lithium/sodium ion batteries. |
 Likun Pan | Prof. Likun Pan received his PhD in 2005 at Nanyang Technological University, Singapore. His research interests include the synthesis and properties of nanomaterials and their applications in lithium/sodium ion batteries, capacitive deionization, photocatalysis and solar cells. He has published more than 180 journal articles with over 4000 citations. |
In the last decades, lithium-ion batteries (LIBs) have become the promising important energy storage device for portable electronics due to their outstanding energy density, good rate capability and long cycle life.1,2 However, it is inevitable that lithium resources will be used up in the predictable future due to the increasing demand for large scale applications of LIBs.3,4 Instead, sodium-ion batteries (SIBs) have been highlighted as the most promising alternative to LIBs for next generation rechargeable batteries, due to abundant sodium resources, low cost and similar chemistry of lithium and sodium.3–5 Nevertheless, the radius of sodium ions (1.02 Å) is much larger than that of lithium ions (0.76 Å), making it difficult to find suitable electrode materials for SIBs.3,4,6,7 Besides, the sodium storage mechanism for SIBs may be different from that for LIBs.8 These issues have appealed to many researchers working for SIBs.
For an anode material, graphite has been commercialized for LIBs, but it just exhibits an extremely low capacity for SIBs in conventional carbonate based electrolytes.9 Partially graphitic materials and hard carbon can reversibly store sodium ions with high capacity, long-life cycling and satisfactory rate performance.10–12 However, the sodiation process of these materials occurs mainly around 0.01 V. If the cut-off potential is set around 0.01 V, sodium dendrite grows, and may lead to safety issues. Sn, Sb and P and their alloy based composites13–17 have also been regarded as significant anode materials due to their satisfactory theoretical capacity; however, the large volume change in these materials during repeated sodiation–desodiation reactions severely hinders their application in SIBs.18 Another important class of material is metal oxides (denoted as MOx; not including TiO2). In this section, the sodium storage mechanism is similar to that for LIBs, which can be summed up into a conversion reaction (eqn (1)) and a further alloying reaction (eqn (2)):18
| MOx + 2xNa+ + 2xe− ↔ xNa2O + M | (1) |
| M + yNa+ + ye− ↔ NayM | (2) |
(If the metal is an inactive element such as Fe, Co, Ni, Cu, Mn, and Mo, metal oxides only react with sodium ions through a conversion reaction (eqn (1)).8,18–21 These materials show satisfactory theoretical capacity; however, they have disadvantages such as poor cyclability and large hysteresis.18
Amongst various metal oxides, TiO2 is an attractive candidate as anode of LIBs and SIBs due to its high safety, low cost and excellent structural stability during repeated charge–discharge processes.22–33 In particular, the working voltage of TiO2 is located at lower values in SIBs than in LIBs, which can improve the energy density of the cells. In addition, the appropriate voltage plateau (above 0.2 V) for SIBs can reduce the growth of sodium dendrite, which is beneficial for improving safety. The reaction mechanism during the charge–discharge process is important to explain the electrochemical performance, and should significantly guide the exploration of electrode materials with superior electrochemical performance. Fig. 1 shows the schematic illustration of lithium ion insertion and extraction into anatase TiO2. As shown in Fig. 1, the interstitial sites (octahedral holes) in anatase structure can insert and extract lithium ions viaeqn (3).22
| Ti4+O2 + xLi+ + xe− ↔ Lix(Ti3+xTi4+1−x)O2 (x ≤ 1) | (3) |
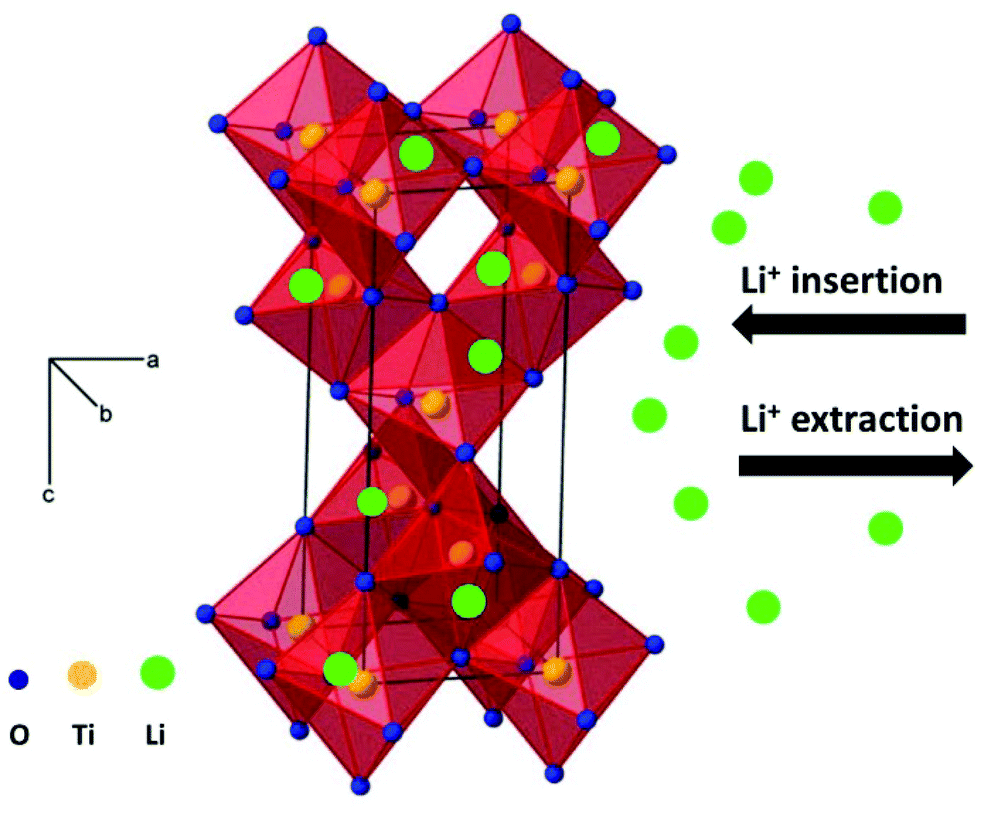 | ||
| Fig. 1 Schematic illustration of lithium ions insertion and extraction into anatase TiO2. | ||
Some early studies presented the sodium storage mechanism of TiO2 for SIBs being similar to the more frequently investigated one for LIBs. Kim et al.28 first claimed an insertion–extraction mechanism based on ex situ X-ray diffraction (XRD) and X-ray absorption spectroscopy (XANES) analysis of cycled electrodes, which showed the reduction of Ti4+ to Ti3+ upon sodium ion insertion and charge balancing caused by the sodium ion insertion. The reversible variation of the lattice parameter was calculated by the ex situ XRD analysis. Besides, no new peaks, especially for Ti and Na2O, appeared in the ex situ XRD analysis, indicating that no conversion reaction occurred. Similarly, Oh's, Yeo's and Jung's groups29–31 also obtained the same results based on an ex situ XRD or X-ray photoelectron spectroscopy (XPS) measurements. However, all these results presented appear debatable, because the above-mentioned characterization results does not support their points sufficiently. Other reaction mechanisms of TiO2 for SIBs may exist, and some objective factors may influence the experimental results. First some amorphous materials may exist during the repeated sodiation–desodiation process, which can not be detected by the ex situ XRD measurement. Some objective factors, especially the inappropriate sample preparation process, may influence the experimental results. Therefore, in situ technique and further analysis by other experimental measurements such as Raman spectroscopy, scanning electron microscopy (SEM) and energy dispersive X-ray (EDX) mapping should be conducted. Second, the variation of the chemical environment induced by the solid electrolyte interface (SEI) layer, electrolyte and other objective factors may contribute to the experimental results, which might be the reason for the groups to not find the presence of metallic Ti by XPS and XANES. Argon ion etching technology may be an effective method to remove the effect of the SEI layer as well as possible oxidation processes occurring during sample preparation. Therefore, a systematic study without the influence of the objective factors should be carried out to observe the variation in phase structure, morphology, chemical composition, and valence state of active materials during the charge–discharge process, and rigorously acquired experimental result should be obtained.
Recently, Passerini's group32 concluded that sodium ions appear to partially reduce the rather stable oxide and form metallic titanium (Ti), sodium oxide, and amorphous sodium titanate during the sodiation process, as revealed by in situ and ex situ XRD, ex situ XPS, ex situ SEM, EDX mapping, ex situ Raman spectroscopy and in situ gas chromatography and mass spectrometry (GC-MS). To follow structural changes in anatase TiO2 upon sodium ion insertion and extraction, they first used in situ and ex situ XRD measurement, which showed that almost all the anatase diffraction peaks disappeared with the exception of the (101) diffraction peak at the end of the first discharge process (sodiation process), revealing that the particles remained partially crystalline. The anatase diffraction peaks were not observed at the subsequent charged state (desodiation process), which implied that new amorphous materials may form after the desodiation process, replacing the previous crystal materials. For a further analysis of the structural change of anatase TiO2 during the initial sodiation process as explored by in situ and ex situ XRD, ex situ SEM analysis of electrodes at different charged–discharged states was carried out (Fig. 2). It was interesting that some new flower-like particles were formed during the first discharge (Fig. 2b). A cross-sectional SEM image of the discharged electrode was obtained (Fig. 2c) and flowers were found to exist at the surface of the electrode, and some spike and sheet like particles were formed within the coating layer, which indicated that the flower may start at the surface of the electrode, and then proceed throughout the electrode coating layer. However, the flowers disappeared during the subsequent first charging process (Fig. 2d). Besides, they did not reappear during the subsequent discharging process (Fig. 2e), indicating that their formation was related to some irreversible process occurring at the first sodiation process. Nonetheless, the abovementioned XRD and SEM measurements did not provide any information on the chemical composition of the electrode during the charge–discharge process. Therefore, to identify the elemental content of the flowers, a discharged electrode was investigated by EDX mapping analysis, which showed that the flower-like particles were sodium superoxide (NaO2). Indeed, the NaO2 particles must be amorphous because in situ and ex situ XRD analysis revealed no other crystalline phases. In the next step, the ex situ XPS analysis of cycled electrodes, which were treated by argon ion etching, was carried out to investigate the variation in the oxidation state of active materials at different charged–discharged states. Besides Ti3+ and Ti4+, the peaks for metallic Ti0 appeared in the fully discharged state. These results were in contrast to the study of Oh's and Jung's groups29,31 and suggested that sodium ion insertion into the anatase lattices was accompanied by a partial reduction of Ti4+ to Ti0. Furthermore, the electrodes were cycled to different cut-off potentials (Fig. 3) and then tested by XPS. The fitting results showed that the electrodes discharged to 0.3 V exhibited a Ti3+/Ti4+ ratio of 0.33, which should correspond to about 0.25 sodium per formula unit of TiO2 (Na0.25(TiO2), Fig. 3). When the electrodes were further discharged to 0.1 V, the ratio increased to around 2.23, corresponding to 0.69 sodium per TiO2 (Na0.69(TiO2), Fig. 3). In the subsequent charging process, the ratio decreased to about 0.35 (Na0.28(TiO2), Fig. 3), which indicated that about 0.41 sodium per TiO2 could be reversibly de-intercalated. The XPS results indicated that a new reversible sodium titanate (Nax(TiO2)) was formed at the different charged and discharged states, which was able to reversibly (de-)insert sodium ions. In a further attempt to explore the structural changes that occurred during the initial discharge process, ex situ Raman spectroscopy was performed. The Raman results indicated that the anatase TiO2 phase continuously disappeared upon the sodiation process. During the consecutive charging to 2.0 V, the anatase phase was not reformed. These results are in good agreement with those from the in situ and ex situ XRD as well as ex situ XPS experiments. Finally, in situ GC-MS was performed to investigate the evolution of gaseous species during the discharge process. The results showed that the steadily increasing oxygen (O2) evolution was remarkable over the whole discharge process, indicating the reduction of TiO2 to Ti, which further confirmed the abovementioned ex situ XPS results.
 | ||
| Fig. 2 Ex situ SEM study of cycled TiO2 electrodes at different discharged and charged states. (a) SEM image of pristine anatase TiO2 electrode. (b) SEM image of cycled discharged electrode at 0.1 V. (c) Cross sectional SEM image of an electrode discharged to 0.1 V. (d) SEM image of electrodes after the first charge up to 2.0 V and (e) after the second discharge down to 0.1 V.32 | ||
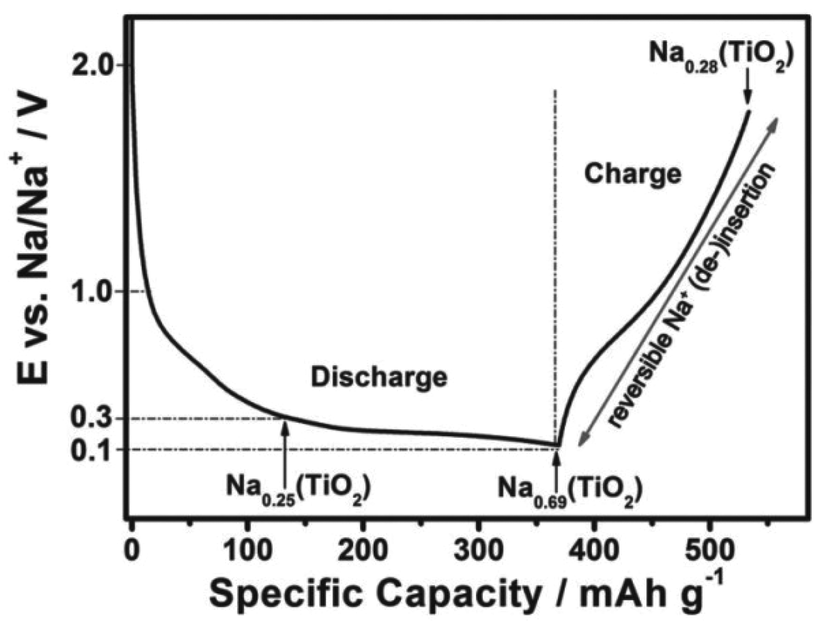 | ||
| Fig. 3 Scheme of the reaction mechanism of anatase TiO2. Charge–discharge curves of TiO2 electrode at the first cycle at 0.01 C in the voltage window of 0.1–2.0 V, including the proposed reaction mechanism and the sodiation degree of the newly formed Nax(TiO2) at 0.3, 0.1, and 2.0 V.32 | ||
In general, the systematic study proposes a new sodium storage mechanism of TiO2 including the initial formation of sodium titanate phase that decomposed into another sodium titanate phase, metallic Ti, NaO2 and O2. All these processes were irreversible, and the newly formed amorphous sodium titanate phase was able to reversibly (de-)insert about 0.41 sodium per TiO2, providing a reversible capacity of 140 mA h g−1. Besides, this study also opens up some challenges in developing TiO2 electrodes for SIBs. For example, first, many factors (e.g. phase structures, impurities, morphology and nanosizing effects) may influence the sodium storage mechanism of TiO2. It is necessary to carry out more systematic studies on the reaction mechanism of different TiO2. Second, TiO2 in this study exhibited an unsatisfied capacity, which only can reversibly insert about 0.41 sodium per TiO2. It is necessary to find effective methods to insert more sodium ions per TiO2: (i) TiO2 has intrinsically low electrical conductivity, and hence shows poor reversibility, which leads to its unsatisfied electrochemical performance. Doping and carbon coating may be the effective methods to improve its electrical conductivity, and thus enhance the reversibility. Very recently, Passerini's group reported improved electrochemical performance of carbon coated anatase TiO2 for SIBs33 with a specific capacity of 227 mA h g−1 at 0.1 C, corresponding to 0.72 sodium per TiO2. The study achieved some encouraging results, and further efforts for modifying TiO2 are still needed to meet its future practical application in SIBs. (ii) To improve the electrochemical performance of the TiO2 anode, another point to focus is further study of the sodium storage mechanism of the newly formed amorphous Nax(TiO2), which should give researchers some useful information and guidance to prepare TiO2 anodes with superior electrochemical performance.
Passerini's group has made exciting progress in developing a TiO2 anode for SIBs. They proposed a new sodium storage mechanism of TiO2 for SIBs and meticulously performed and analyzed this with a systematic study. The study addressed a key issue that puzzled researchers of TiO2 anodes for SIBs, provided strong evidence for understanding its sodium storage mechanism and made a significant impact on its future practical applications. This sodium storage mechanism of TiO2 should be suitable or at least valuable information for the other metal oxide anodes, and the systematic, effective and rigorous experimental analysis methods in this study also give directions to explore the sodium storage mechanism for other electrode materials for SIBs.
Acknowledgements
The authors acknowledge and thank Professor Stefano Passerini for critical reading of the manuscript and important input.Notes and references
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75 CrossRef CAS PubMed.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M. L. Doublet, D. Gonbeau, P. Novák, G. V. Tendeloo, R. Dominko and J. M. Tarascon, Science, 2015, 350, 1516 CrossRef CAS PubMed.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636 CrossRef CAS PubMed.
- M. S. Islam and C. A. J. Fisherb, Chem. Soc. Rev., 2014, 43, 185 RSC.
- T. Q. Chen, L. K. Pan, T. Lu, C. L. Fu, D. H. C. Chua and Z. Sun, J. Mater. Chem. A, 2014, 2, 1263 CAS.
- L. Liang, Y. Xu, X. Wang, C. Wang, M. Zhou, Q. Fu, M. Wu and Y. Lei, J. Power Sources, 2015, 294, 193 CrossRef CAS.
- K. He, F. Lin, Y. Zhu, X. Yu, J. Li, R. Lin, D. Nordlund, T. C. Weng, R. M. Richards, X. Q. Yang, M. M. Doeff, E. A. Stach, Y. Mo, H. L. Xin and D. Su, Nano Lett., 2015, 15, 5755 CrossRef CAS PubMed.
- M. M. Doeff, Y. Ma, S. J. Visco and L. C. D. Jonghe, J. Electrochem. Soc., 1993, 140, L169 CrossRef CAS.
- J. Ding, H. L. Wang, Z. Li, K. Cui, D. Karpuzov, X. H. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 941 CAS.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115 CrossRef CAS PubMed.
- J. T. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. X. Dou and L. M. Dai, Adv. Mater., 2015, 27, 2042 CrossRef CAS PubMed.
- L. Liang, Y. Xu, C. Wang, L. Wen, Y. Fang, Y. Mi, M. Zhou, H. Zhao and Y. Lei, Energy Environ. Sci., 2015, 8, 2954 CAS.
- J. Sun, H. W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980 CrossRef CAS PubMed.
- L. Wu, X. H. Hu, J. F. Qian, F. Pei, F. Y. Wu, R. Mao, X. P. Ai, H. X. Yang and Y. L. Cao, Energy Environ. Sci., 2014, 7, 323 CAS.
- J. F. Qian, X. Y. Wu, Y. L. Cao, X. P. Ai and H. X. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633 CrossRef CAS PubMed.
- J. C. Kim and D. W. Kim, Electrochem. Commun., 2014, 46, 124 CrossRef CAS.
- H. Y. Kang, Y. C. Liu, K. Z. Cao, Y. Zhao, L. F. Jiao, Y. L. Wang and H. T. Yuan, J. Mater. Chem. A, 2015, 3, 17899 CAS.
- X. J. Zhang, W. Qin, D. S. Li, D. Yan, B. W. Hu, Z. Sun and L. K. Pan, Chem. Commun., 2015, 51, 16413 RSC.
- Y. Xu, M. Zhou, X. Wang, C. Wang, L. Liang, F. Grote, M. Wu, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2015, 54, 8768 CrossRef CAS PubMed.
- M. M. Rahman, A. M. Glushenkov, T. Ramireddy and Y. Chen, Chem. Commun., 2014, 50, 5057 RSC.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364 CrossRef CAS PubMed.
- Y. Xu, M. Zhou, L. Wen, C. Wang, H. Zhao, Y. Mi, L. Liang, Q. Fu, M. Wu and Y. Lei, Chem. Mater., 2015, 27, 4274 CrossRef CAS.
- D. Yan, Y. Bai, C. Y. Yu and W. F. Zhang, J. Alloys Compd., 2014, 609, 86 CrossRef CAS.
- Y. Xu, E. M. Lotfabad, H. L. Wang, B. Farbod, Z. W. Xu, A. Kohandehghan and D. Mitlin, Chem. Commun., 2013, 49, 8973 RSC.
- L. Wu, D. Buchholz, D. Bresser, L. G. Chagas and S. Passerini, J. Power Sources, 2014, 251, 379 CrossRef CAS.
- D. Yan, C. Y. Yu, Y. Bai, W. F. Zhang, T. Q. Chen, B. W. Hu, Z. Sun and L. K. Pan, Chem. Commun., 2015, 51, 8261 RSC.
- K. T. Kim, G. Ali, K. Y. Chung, Y. K. Sun, K. Amine and S. T. Myung, Nano Lett., 2014, 14, 416 CrossRef CAS PubMed.
- S. M. Oh, J. Y. Hwang, C. S. Yoon, J. Lu, K. Amine, I. Belharouak and Y. K. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 11295 CAS.
- Y. Yeo, J. Jung, K. Park and Il.-D. Kim, Sci. Rep., 2015, 5, 13862 CrossRef PubMed.
- K. Jung, J. Seong, S. Kim, G. Leec and J. Lee, RSC Adv., 2015, 5, 106252 RSC.
- L. Wu, D. Bresser, D. Buchholz, G. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy Mater., 2015, 5, 1401142 Search PubMed.
- M. N. Tahir, B. Oschmann, D. Buchholz, X. Dou, I. Lieberwirth, M. Panthöfer, W. Tremel, R. Zentel and S. Passerini, Adv. Energy Mater. DOI:10.1002/aenm.201501489.
| This journal is © the Partner Organisations 2016 |