 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding and activation of small molecules by a quintuply bonded chromium dimer†
Jingmei
Shen‡
,
Glenn P. A.
Yap
and
Klaus H.
Theopold
*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: theopold@udel.edu
First published on 15th January 2014
Abstract
The quintuply bonded [HLiPrCr]2 reacts with various small molecules, revealing a pattern of two kinds of transformations. Unsaturated molecules that are neither polar nor oxidizing form binuclear [2+n] cycloaddition products retaining Cr–Cr quadruple bonds. In contrast, polar or oxidizing molecules effect the complete cleavage of the Cr–Cr bond.
Occasioned by the discovery of a dinuclear chromium complex featuring a sterically accessible quintuple metal–metal bond, we have begun to explore the reactivity of this novel functional group unique to transition metal chemistry. Recent studies indicate that M–M quintuple bonds have a remarkable reaction chemistry.1–16 Herein we describe the products of reactions between quintuply bonded [HLiPrCr]2 (1, where HLiPr = Ar–N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)–(H)CN–Ar, with Ar = 2,6-diisopropylphenyl)17 and various small molecules (Scheme 1). These reactions are of interest in their own right and make for fascinating comparisons with the reactivities of other binuclear metal complexes.
C(H)–(H)CN–Ar, with Ar = 2,6-diisopropylphenyl)17 and various small molecules (Scheme 1). These reactions are of interest in their own right and make for fascinating comparisons with the reactivities of other binuclear metal complexes.
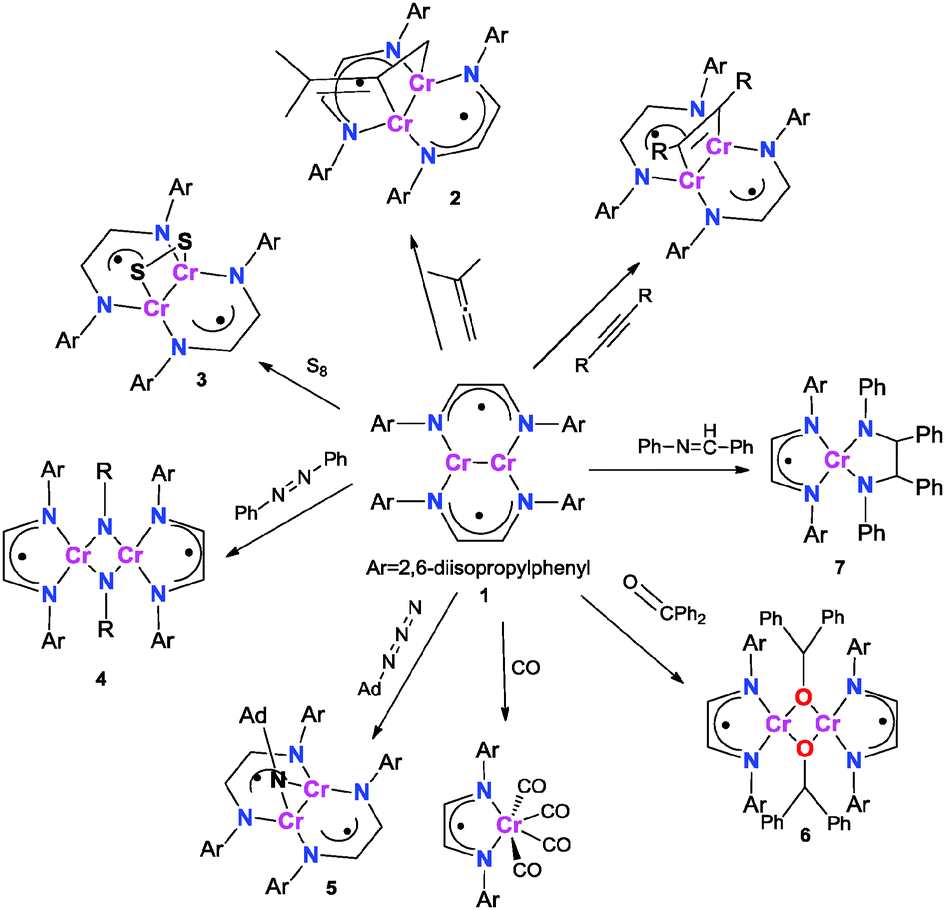 | ||
| Scheme 1 Reactions of 1 with alkyne, allene, sulfur, PhNNPh, AdN3, CO, benzophenone and benzylideneaniline. | ||
1 reacts rapidly with molecules containing multiple bonds. For example, we have previously described [2+2] cycloaddition reactions between 1 and alkynes.18 While the analogous reaction with ethylene is apparently reversible, 1 adds to the destabilized CC double bond of 1,1-dimethylallene, yielding another isolable [2+2] cycloaddition product, namely [HLiPrCr]2(μ-η1:η1-H2CCCMe2) (2, see Fig. 1). The terminal CC bond of the allene ligand has added across the two metal centers, forming a four-membered dimetallacycle. The C53–C54 distance of 1.466(5) Å and the Cr–Cr distance of 1.9462(8) Å are consistent with a two-electron reduction of allene and concomitant oxidation of the Cr–Cr center, which, however, retains the short Cr–Cr distance characteristic of a quadruple bond (see Table 1). The other CC bond of the allene remains essentially unperturbed (1.346(5) Å). The core of 2 adopts an almost planar geometry with a (C–C)–(Cr–Cr) twist angle of 24.3°, similar to the aforementioned alkyne adducts.18 The 1H NMR spectrum of 2 exhibited sharp resonances consistent with a diamagnetic ground state of the molecule.
| Cr–Cr | C–Cc | C–Nc | θ | δ | |
|---|---|---|---|---|---|
| a Twist angle (X–X)–(Cr–Cr) (X = C or S). b Dihedral angle between two ligand planes (see the ESI for details). c Average bond lengths in the α-diimine backbones. d Average. | |||||
| 1 | 1.8028(9) | 1.350(5) | 1.368(3) | N/A | N/A |
| 2 | 1.9462(8) | 1.337(5) | 1.380(4) | 24.3° | 151° |
| 3 | 1.9305(8) | 1.367(3) | 1.360(3) | 15.6° | 143° |
| 4 | 2.498(4) | 1.395(11) | 1.380(9) | N/A | N/A |
| 5 | 1.9575(11)d | 1.346(6) | 1.385(6) | N/A | 142°d |
| 6 | 3.1667(15) | 1.360(6) | 1.336(6) | N/A | N/A |
| 7 | N/A | 1.383(6) | 1.355(5) | N/A | N/A |
| 1-Butyne18 | 1.9248(7) | 1.352(4) | 1.370(4) | 23.7° | 146° |
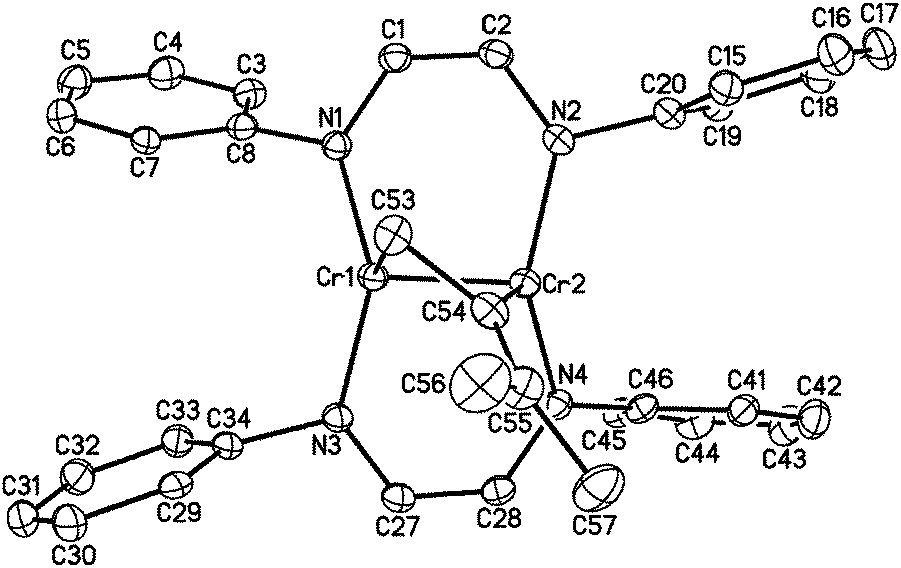 | ||
| Fig. 1 The molecular structure of 2 (30% probability level). Ligand i-Pr groups and H-atoms have been omitted for clarity. | ||
Oxygen atom sources, such as O2, N2O, and NO led to decomposition of 1 accompanied by loss of the diimine ligand. This motivated us to extend the exploration to less oxidizing chalcogens. Thus, treatment of an Et2O–toluene solution of 1 with elemental sulphur, at room temperature, caused the initially green solution to turn deep blue. A standard work-up of the reaction and recrystallization from diethyl ether yielded the simple binuclear adduct, [HLiPrCr]2(S2) (3) in modest yield (20%). The molecular structure of 3 is depicted in Fig. S1 (ESI†); it features a four-membered Cr2S2 ring. The “supershort” (Cr–Cr < 2.0 Å) Cr–Cr bond of 3 (1.9305(8) Å) is appreciably longer than that in 1 (1.8028(9) Å), indicating an oxidation from Cr(I) to Cr(II) and hence a bond order reduced to 4. The S–S bond length of 2.0513(10) Å approximates that of Kempe's disulfide analog (2.058(4) Å),2 which, however, features perpendicular coordination of the S22− unit and that of Cp2Cr2(μ-S)2(μ-η1-η1-S2) (2.028(2) Å).19 As is typical of the [2+2] cycloaddition products of 1, the Cr2S2 core is not perfectly planar. The (S–S)–(Cr–Cr) twist angle for the core is 15.6°, somewhat smaller than the analogous angles in the alkyne adducts and 2.
Table 1 contains selected bond lengths and angles for compounds 2–7. All the ‘cycloaddition’ products of 1 that maintain Cr–Cr bonds, i.e.2, 3, and 1-2-butyne, exhibit the twisted μ-η1:η1 bonding mode for the X2 ligands (X = C, S); this differs from the perpendicular (i.e. μ2-η2:η2) bonding motif more typically observed for complexes with metal–metal bonds, e.g. in Kempe's aminopyridinato dichromium complexes.2–4,20 At the same time, the dihedral angles (δ) between the α-diimine ligand planes are significantly larger than those of the aminopyridinato complexes (e.g. 107° for both the disulfide and the tolylacetylene adduct). In other words, the [L2Cr2] fragments of the α-diimine complexes are considerably flatter than those with aminopyridinato ligands. The near preservation of the planar geometry of 1 and the formation of unsaturated four-membered Cr2X2 rings as opposed to tetrahedrane-like structures is unlikely to be steric in origin. An electronic explanation may be rooted in the electronic flexibility afforded by the redox-active α-diimine ligands; this remains to be explored.
An isoelectronic – but less oxidizing – analog of O2 is azobenzene (PhNNPh). When one equivalent of the latter was added to a solution of (μ-η1:η1-HLiPr)2Cr2 (1) in diethyl ether, subsequent work-up and recrystallization produced red-brown crystals of dinuclear complex [HLiPrCr(μ-NPh)]2 (4) in 40% isolated yield. 4 is a dinuclear complex with bridging imido ligands (Fig. 2, top). This reaction may well go through an unstable [2+2] cycloaddition intermediate, which suffers oxidative addition, due to the high electronegativity of nitrogen. The molecular structure of 4 features four-coordinate chromium (ignoring the rather long Cr–C interactions) adopting pseudo-tetrahedral geometry, which is the preferred geometry of 4-coordinate Cr(III). The NN double bond has been severed completely (N⋯Navg = 2.695 Å). Similarly, the distance between the two chromium atoms in 4 is 2.498(4) Å, indicating the absence of any significant bonding interactions.
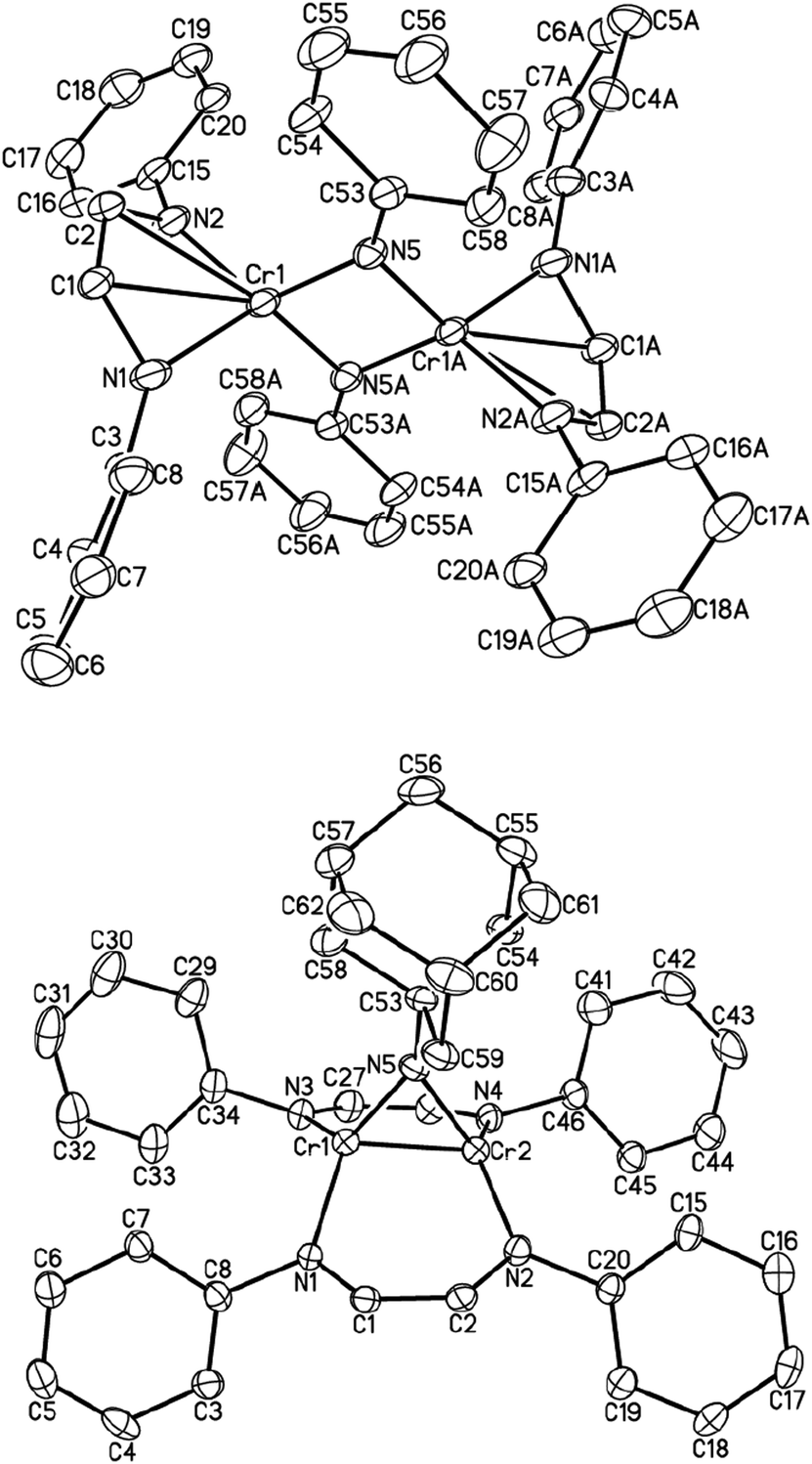 | ||
| Fig. 2 The molecular structure of 4 and 5 (both at 30% probability level). | ||
The average bond lengths of C–C, C–N bonds in the backbone of the α-diimine ligand are 1.395(11) and 1.380(9) Å, characteristic of a diimine radical anion; accordingly, chromium is in the formal oxidation state +III (S = 3/2). The effective magnetic moment of 4 at room temperature was 2.4(1) μB, consistent with antiferromagnetic coupling, both between the metal and its radical ligand as well as between the chromium atoms.
The reaction between (μ-η1:η1-HLiPr)2Cr2 (1) and sterically demanding Ad–N3 afforded another imido complex, namely [HLiPrCr]2(NAd) (5), as shown in Fig. 2 (bottom). Only one imido group has been added across the Cr–Cr bond. Once again, we suggest that a five-membered [2+3] cycloaddition product may be formed first, which rapidly extrudes N2. The bond distances and angles of 5 are comparable to those of other known bridging imido complexes of chromium.22–26 Similar to the geometries of the [2+2] cycloaddition products, the elongated Cr–Cr distance of 1.9575(11) Å is consistent with the two-electron oxidation of the Cr2 unit (to Cr(II)). 5 is also diamagnetic, presumably due to metal–metal quadruple bonding.
Finally, we were interested in studying the reactivity of 1 toward unsaturated molecules featuring X–Y bonds (X, Y = C, N, O). Exposure of a benzene solution of 1 to CO (1 atm) produced the dark blue carbonyl HLiPrCr(CO)4, as confirmed by 1H NMR spectroscopy.21 The reaction of 1 with benzophenone resulted in dinuclear [HLiPrCr(μ-OPh2)]2 (6). The structure of 6 (shown in Fig. S2, ESI†) reveals a benzophenone-bridged dimer with square planar Cr centers. The average carbon–oxygen bond length of the benzophenone is 1.355(5) Å, which is much longer than the 1.230(3) Å in benzophenone,27 suggesting some degree of reduction of the CO bonds. The average bond lengths of C–C, C–N bonds of the backbone of the α-diimine ligand are 1.360(6) and 1.336(6) Å, consistent with those of a monoanionic diimine ligand.21 These structural features suggest that 6 is a Cr(II) complex. Like [HLiPrCr(μ-Cl)]2,176 exhibited a simple isotropically shifted and broadened 1H NMR spectrum in C6D6, with chemical shifts at 96, 14.6, 3.2, 1.56, and −13.0 ppm. μeff(RT) of this complex was found to be 5.1(2) μB (3.6(1) μB per chromium), which is consistent with two antiferromagnetically coupled Cr(II) metal centers (S = 2) coordinated by ligand radicals (S = 1/2).
In contrast to 6, reductive coupling of CN double bonds was observed upon exposure of 1 to four equivalents of trans-benzylideneaniline. The reaction was found to form the coupling product, HLiPrCr(κ2-N2C26H22) (7). The crystal structure is shown in Fig. 3. 7 adopts tetrahedral coordination about chromium with the α-diimine apparently being in the singly reduced state (see Table 1). The room temperature effective magnetic moment of 7 was found to be 2.9(1) μB, consistent with a Cr(III) metal center (S = 3/2) strongly coupled to a ligand radical (S = 1/2).
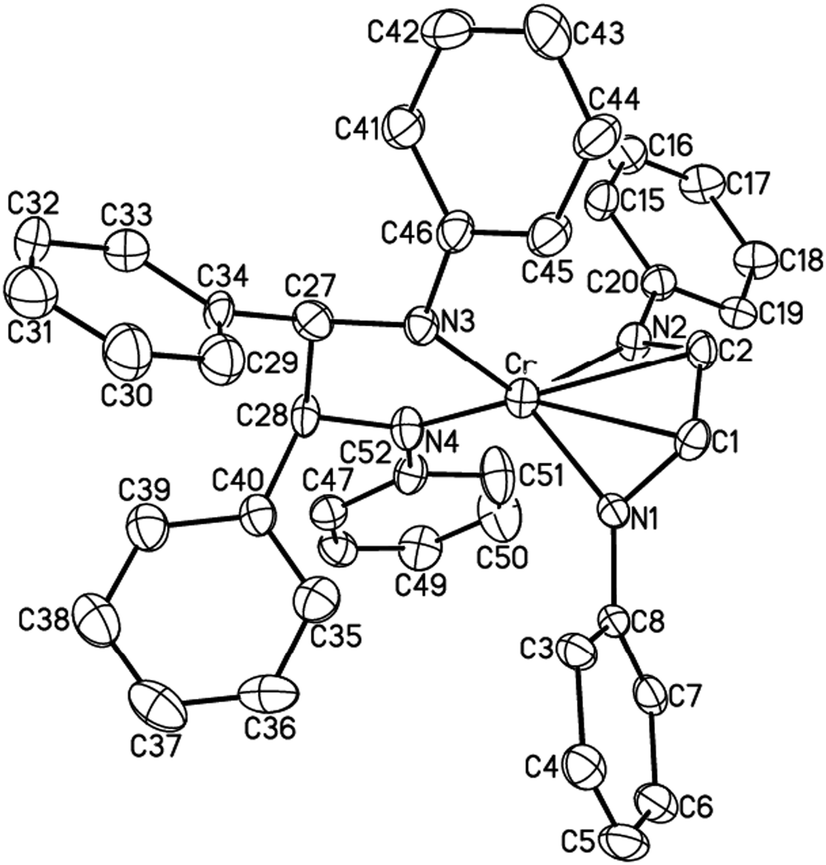 | ||
| Fig. 3 The molecular structure of 7 (30% probability level). | ||
In summary, reactivity studies on a quintuply bonded dichromium complex supported by α-diimine ligands have been extended to a variety of molecules. The products are varied and their structures differ from those established for quintuply bonded complexes supported by other ligands. A pervasive feature of 1 seems to be the formation of [2+n] cycloaddition products with nonpolar substrates. Polar, heteroatomic multiple bonds on the other hand effect complete cleavage of the metal–metal bond.
This work was supported by the NSF (CHE-0911081).
Notes and references
- A. Noor, T. Bauer, T. K. Todorova, B. Weber, L. Gagliardi and R. Kempe, Chem.–Eur. J., 2013, 19, 9825–9832 CrossRef CAS PubMed.
- E. S. Tamne, A. Noor, S. Qayyum, T. Bauer and R. Kempe, Inorg. Chem., 2012, 52, 329–336 CrossRef PubMed.
- C. Schwarzmaier, A. Noor, G. Glatz, M. Zabel, A. Y. Timoshkin, B. M. Cossairt, C. C. Cummins, R. Kempe and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 7283–7286 CrossRef CAS PubMed.
- A. Noor, E. S. Tamne, S. Qayyum, T. Bauer and R. Kempe, Chem.–Eur. J., 2011, 17, 6900–6903 CrossRef CAS PubMed.
- A. Noor and R. Kempe, Chem. Rec., 2010, 10, 413–416 CAS.
- F. R. Wagner, A. Noor and R. Kempe, Nat. Chem., 2009, 1, 529–536 CrossRef CAS PubMed.
- A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko and R. Kempe, Nat. Chem., 2009, 1, 322–325 CrossRef CAS.
- A. Noor, F. R. Wagner and R. Kempe, Angew. Chem., Int. Ed., 2008, 47, 7246–7249 CrossRef CAS PubMed.
- P.-F. Wu, S.-C. Liu, Y.-J. Shieh, T.-S. Kuo, G.-H. Lee, Y. Wang and Y.-C. Tsai, Chem. Commun., 2013, 49, 4391–4393 RSC.
- H.-G. Chen, H.-W. Hsueh, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., Int. Ed., 2013, 52, 10256–10260 CrossRef CAS PubMed.
- S.-C. Liu, W.-L. Ke, J.-S. K. Yu, T.-S. Kuo and Y.-C. Tsai, Angew. Chem., Int. Ed., 2012, 51, 6394–6397 CrossRef CAS PubMed.
- Y. L. Huang, D. Y. Lu, H. C. Yu, J. S. Yu, C. W. Hsu, T. S. Kuo, G. H. Lee, Y. Wang and Y. C. Tsai, Angew. Chem., Int. Ed., 2012, 51, 7781–7785 CrossRef CAS PubMed.
- Y.-C. Tsai, H.-Z. Chen, C.-C. Chang, J.-S. K. Yu, G.-H. Lee, Y. Wang and T.-S. Kuo, J. Am. Chem. Soc., 2009, 131, 12534–12535 CrossRef CAS PubMed.
- Y. C. Tsai, C. W. Hsu, J. S. Yu, G. H. Lee, Y. Wang and T. S. Kuo, Angew. Chem., Int. Ed., 2008, 47, 7250–7253 CrossRef CAS PubMed.
- Y.-C. Tsai, Y.-M. Lin, J.-S. K. Yu and J.-K. Hwang, J. Am. Chem. Soc., 2006, 128, 13980–13981 CrossRef CAS PubMed.
- C. Ni, B. D. Ellis, G. J. Long and P. P. Power, Chem. Commun., 2009, 2332–2334 RSC.
- K. A. Kreisel, G. P. Yap, O. Dmitrenko, C. R. Landis and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 14162–14163 CrossRef CAS PubMed.
- J. Shen, G. P. Yap, J. P. Werner and K. H. Theopold, Chem. Commun., 2011, 47, 12191–12193 RSC.
- L. Y. Goh and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1986, 1474–1475 RSC.
- M. J. Calhorda and R. Hoffmann, Organometallics, 1986, 5, 2181–2187 CrossRef CAS.
- K. A. Kreisel, G. P. Yap and K. H. Theopold, Inorg. Chem., 2008, 47, 5293–5303 CrossRef CAS PubMed.
- W. H. Monillas, G. P. A. Yap and K. H. Theopold, Inorg. Chim. Acta, 2011, 369, 103–119 CrossRef CAS PubMed.
- W. H. Monillas, G. P. Yap, L. A. MacAdams and K. H. Theopold, J. Am. Chem. Soc., 2007, 129, 8090–8091 CrossRef CAS PubMed.
- A. A. Danopoulos, D. M. Hankin, G. Wilkinson, S. M. Cafferkey, T. K. N. Sweet and M. B. Hursthouse, Polyhedron, 1997, 16, 3879–3892 CrossRef CAS.
- A. A. Danopoulos, G. Wilkinson, T. K. N. Sweet and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1995, 2111–2123 RSC.
- B. Moubaraki, K. S. Murray, P. J. Nichols, S. Thomson and B. O. West, Polyhedron, 1994, 13, 485–495 CrossRef CAS.
- E. B. Fleischer, N. Sung and S. Hawkinson, J. Phys. Chem., 1968, 72, 4311–4312 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparative and crystallographic data. CCDC 971178–971183. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc48746f |
| ‡ Current address: Department of Chemical and Biological Engineering, Northwestern University, USA. |
| This journal is © The Royal Society of Chemistry 2014 |