
Bending two-dimensional Cu(I)-based coordination networks to inverse electrocatalytic HER/CO2RR selectivity†
Kai
Zheng‡
 a,
Ding-Yi
Hu‡
a,
Xue-Wen
Zhang
a,
Xian-Xian
Xiao
a,
Zi-Jun
Liang
a,
Jun-Xi
Wu
a,
Duo-Yu
Lin
a,
Lin-Ling
Zhuo
a,
Heng
Yi
a,
Li
Gong
b,
Dong-Dong
Zhou
*a and
Jie-Peng
Zhang
*a
a,
Ding-Yi
Hu‡
a,
Xue-Wen
Zhang
a,
Xian-Xian
Xiao
a,
Zi-Jun
Liang
a,
Jun-Xi
Wu
a,
Duo-Yu
Lin
a,
Lin-Ling
Zhuo
a,
Heng
Yi
a,
Li
Gong
b,
Dong-Dong
Zhou
*a and
Jie-Peng
Zhang
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, GBRCE for Functional Molecular Engineering, IGCME, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhoudd3@mail.sysu.edu.cn; zhangjp7@mail.sysu.edu.cn
bInstrumental Analysis and Research Center, Sun Yat-Sen University, Guangzhou 510275, China
First published on 10th June 2024
Abstract
Two-dimensional (2D) coordination polymers have attracted great attention for catalysis because of their abundant exposed active sites. Here, we show that bending the local structure of 2D coordination polymers can inverse the electrocatalytic selectivity. A series of ultrathin nanosheets based on isoreticular/isostructural/isomeric 2D Cu(I) triazolate coordination polymers were successfully prepared. By introducing an amino group on the triazolate ligand, the shape of the 2D layer transforms from planar into wavy, which inverses the electrocatalytic selectivity from the HER (selectivity ∼ 80%) to the CO2RR (selectivity ∼ 76%, C2H4 up to 52%). Computational simulations showed that the wavy structure allows the amino groups to form attractive hydrogen-bonding interactions with the key reaction intermediates of the CO2RR for C2H4 and steric hindrance with the key reaction intermediates of the HER, giving lower and higher reaction energy barriers, respectively.
 Dong-Dong Zhou | Dr Dong-Dong Zhou was born in 1990. He received his BSc degree (2011) from South China Agricultural University and his PhD degree (2016) in inorganic chemistry under the supervision of Professor Jie-Peng Zhang from Sun Yat-Sen University. Then, he became an associate researcher in the Chen & Zhang Group at Sun Yat-Sen University. Since 2019, he has been an associate professor in the School of Chemistry at Sun Yat-Sen University. His current research interest focuses on the design and synthesis of porous coordination polymers or metal–organic frameworks, especially for their dynamic structural changes playing roles in applications of adsorption, separation, catalysis, etc. |
Introduction
The electrochemical CO2 reduction reaction (CO2RR) is considered a viable carbon-neutralization strategy.1–5 The hydrogen evolution reaction (HER) is the main competing reaction of the CO2RR. Therefore, CO2RR catalysts should show not only high product selectivity but also low HER selectivity.6–12 Besides the coordination geometry and/or electronic structure,13–16 the supramolecular microenvironment of the metal center also plays an important role in catalysis.17 Compared to traditional inorganic materials, coordination polymers (CPs) have defined and diverse structures with designability as well as tunable functionality, which facilitates the exploration of structure–activity relationships in catalysis. In this regard, coordination complexes could offer distinct advantages over traditional inorganic materials.18–22Compared with three-dimensional (3D) bulk materials, 2D nanomaterials with abundant exposed active sites have demonstrated exceptional catalytic performances.11,23,24 Ultrathin nanosheets of 2D coordination polymers (CPs) combining the advantages of CPs and 2D materials have attracted increasing attention for catalysis.25–29 However, the supramolecular microenvironment of 2D CPs has been scarcely considered for catalysis. Because coordination bonds are weaker than conventional covalent bonds, to maintain the 2D structure and/or facilitate exfoliating, these 2D CPs usually exhibit planar structures rather than more complex ones.
Recently, we reported high and tunable CO2RR performances of a series of isoreticular (nbo-a) 3D Cu(I)-based porous frameworks, i.e., [Cu(detz)] (MAF-2, Hdetz = 3,5-diethyl-1,2,4-triazole) and its analogs consisting of different ligand side groups (methyl, ethyl, and propyl).30 With even smaller ligand side groups, [Cu(tz)] (Htz = 1,2,4-triazole)31 and [Cu(atz)] (Hatz = 3-amino-1,2,4-triazole)32 can retain the coordination modes of MAF-2 analogs, i.e., three-coordinated Cu(I) ions and triazolate ligands, as well as planar Cu2(tz)2 units, but they adopt the 2D sql-a network topology (Fig. 1).33 If these 2D CPs can be exfoliated into ultrathin nanosheets, high CO2RR performances similar to those of 3D porous MAF-2 analogs may be achieved. More importantly, because the pores of the sql-a network are too small for amino groups, the 2D networks of [Cu(tz)] and [Cu(atz)] are planar and wavy in their crystal structures, respectively. In other words, the layer structure of [Cu(atz)] exhibits 3D characteristics of molecular complexes, enzymes, and metal–organic frameworks.
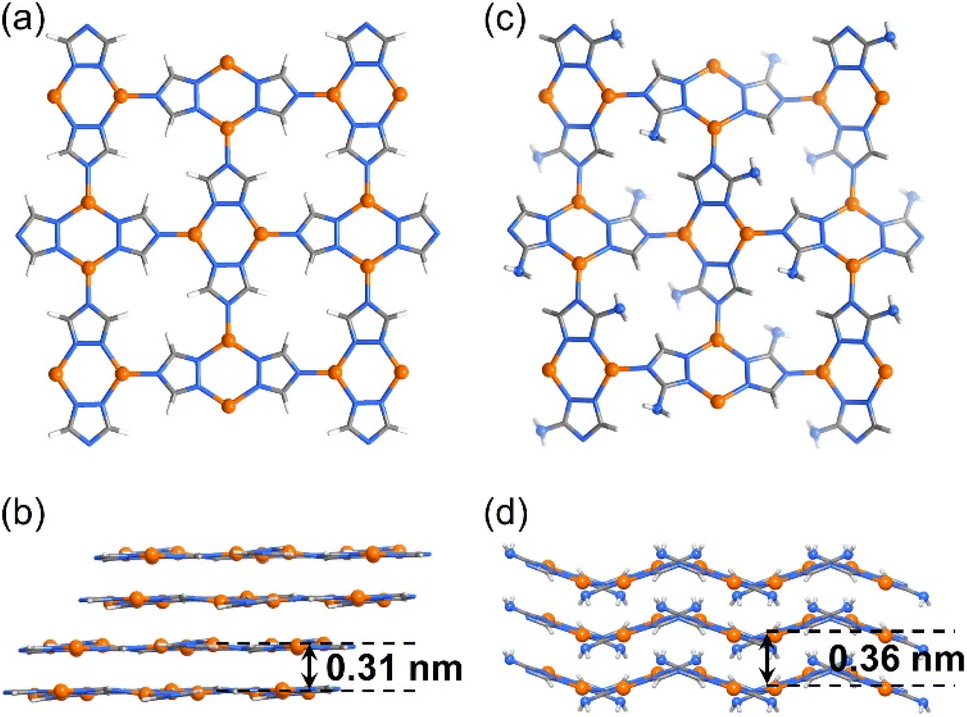 | ||
| Fig. 1 Crystal structures of 2D CPs. (a and b) [Cu(tz)] and (c and d) [Cu(atz)] viewed perpendicularly (a and c) and parallelly (b and d) to the coordination layers. | ||
In this work, we synthesize ultrathin nanosheets of not only isoreticular [Cu(tz)] and [Cu(atz)] but also isostructural/isomeric solid-solution framework structures containing different concentrations of amino groups and demonstrate that the bending of the layer structure is critical for enhancing the CO2RR and avoiding the HER.
Results and discussion
Syntheses and characterization
The solution reaction of [Cu(NH3)2]OH with Htz or Hatz at room temperature gave white microcrystalline samples of [Cu(tz)] or [Cu(atz)], respectively (Fig. S1 and S2†). SEM of [Cu(tz)] or [Cu(atz)] samples shows shale-like morphology, indicating that they could potentially be exfoliated into ultrathin nanosheets (Fig. S3†). Simple ultrasonic exfoliation treatments of microcrystalline [Cu(tz)] or [Cu(atz)] in methanol gave the corresponding nanosheets. Considering a common chemical formula of [Cu(tz)x%(atz)100−x%], the nanosheet samples prepared from [Cu(tz)] and [Cu(atz)] are denoted as (p-AxH100−x, x = 0, i.e., p-A0H100) and (w-AxH100−x, x = 100, i.e., w-A100H0) respectively (p/w for planar/wavy and A/H for amino/hydrogen).Transmission electron microscopy (TEM) and atomic force microscopy (AFM) showed ultrathin nanosheets with thicknesses of ca. 4.5 and 4.9 nm for p-A0H100 and w-A100H0, respectively (Fig. 2 and S4†). Considering that the thickness of each layer of [Cu(tz)] and [Cu(atz)] is 3.1 and 3.6 Å, respectively (Fig. 1), the ultrathin nanosheets contained ∼14 layers of the 2D coordination networks. X-ray photoelectron spectroscopy (XPS) showed characteristic peaks of Cu(I) at 914.8 eV, 932.6 eV and 952.6 eV, but no satellite peaks of Cu(II) appeared. Since the characteristic peaks of Cu(0) and Cu(I) cannot be distinguished, the 914 eV is further attributed to Cu(I) by the Cu LMM region (Fig. S5†).30,34 Powder X-ray diffraction (PXRD) of p-A0H100 and w-A100H0 showed only one or two of the strongest diffraction peaks of [Cu(tz)] or [Cu(atz)], without any typical inorganic species (Fig. S1 and S2†), respectively, indicating that p-A0H100/w-A100H0 should have a similar coordination structure to those of [Cu(tz)]/[Cu(atz)]. Moreover, these diffraction peaks are significantly broadened, being typical for ultrathin nanosheets.26,35,36 In CO2-saturated KHCO3 solution (0.1 M, electrolyte for the CO2RR), their PXRD patterns can remain unchanged for at least one week, indicating that they are suitable for the CO2RR (Fig. S1 and S2†).
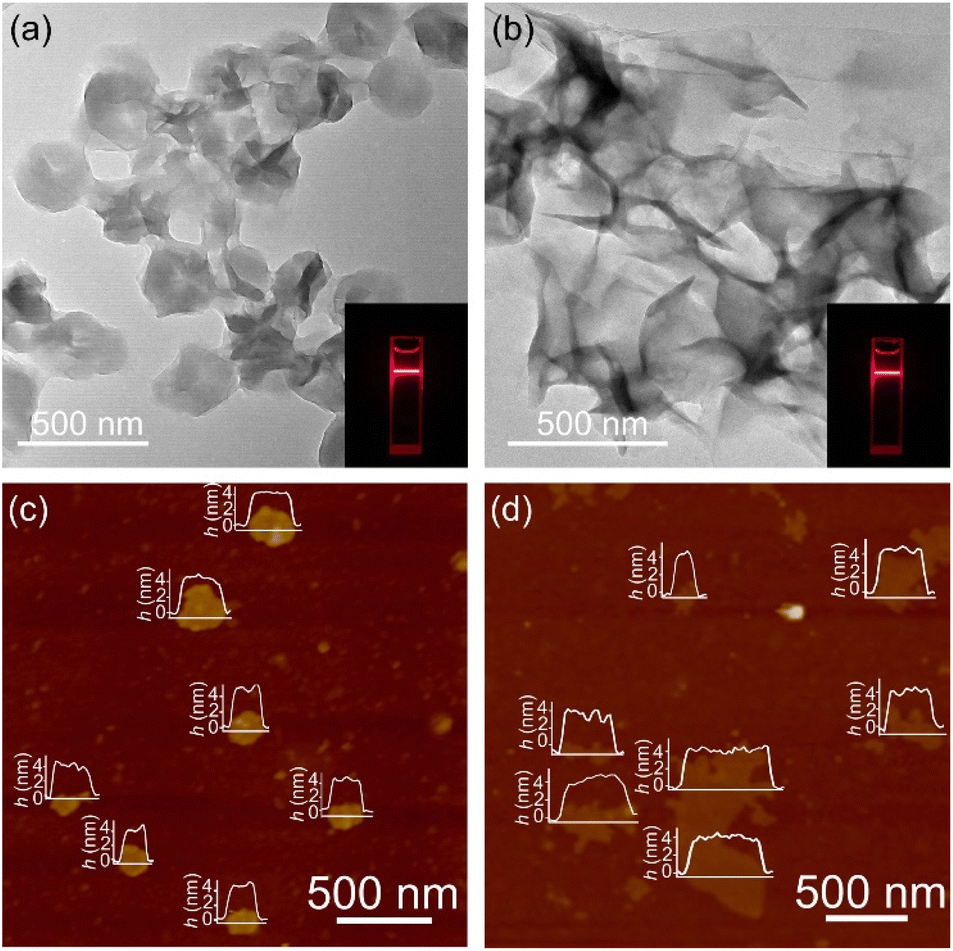 | ||
| Fig. 2 Morphology of ultrathin nanosheets. TEM images of (a) p-A0H100 and (b) w-A100H0 (insets: the Tyndall effect of a colloidal solution). AFM images of (c) p-A0H100 and (d) w-A100H0 (insets: the corresponding height profiles). | ||
Electrocatalytic CO2RR performances
The CO2RR performances of p-A0H100 and w-A100H0 (coated on a glassy carbon electrode) were investigated in 0.1 M KHCO3 solution (Fig. 3a and S6†). Under saturated CO2, their current densities were obviously higher than those in Ar, indicating catalytic activities for the CO2RR. Moreover, the current densities of w-A100H0 were much higher than those of p-A0H100, indicating that the wavy structure and/or the amino groups are beneficial for the CO2RR (Fig. 3a). Interestingly, p-A0H100 and w-A100H0 showed similar and high hydrophilicity (water contact angles < 20°) but very different CO2 affinities (Fig. 3a and S7†). The contact angles for CO2 gas bubbles and electrolytes of p-A0H100 and w-A100H0 were determined to be 135 ± 1° and 85 ± 1°, respectively. In other words, p-A0H100 is aerophobic and w-A100H0 is aerophilic.37,38 To the best of our knowledge, hydrophilic materials are always aerophobic and hydrophobic materials are generally aerophilic.39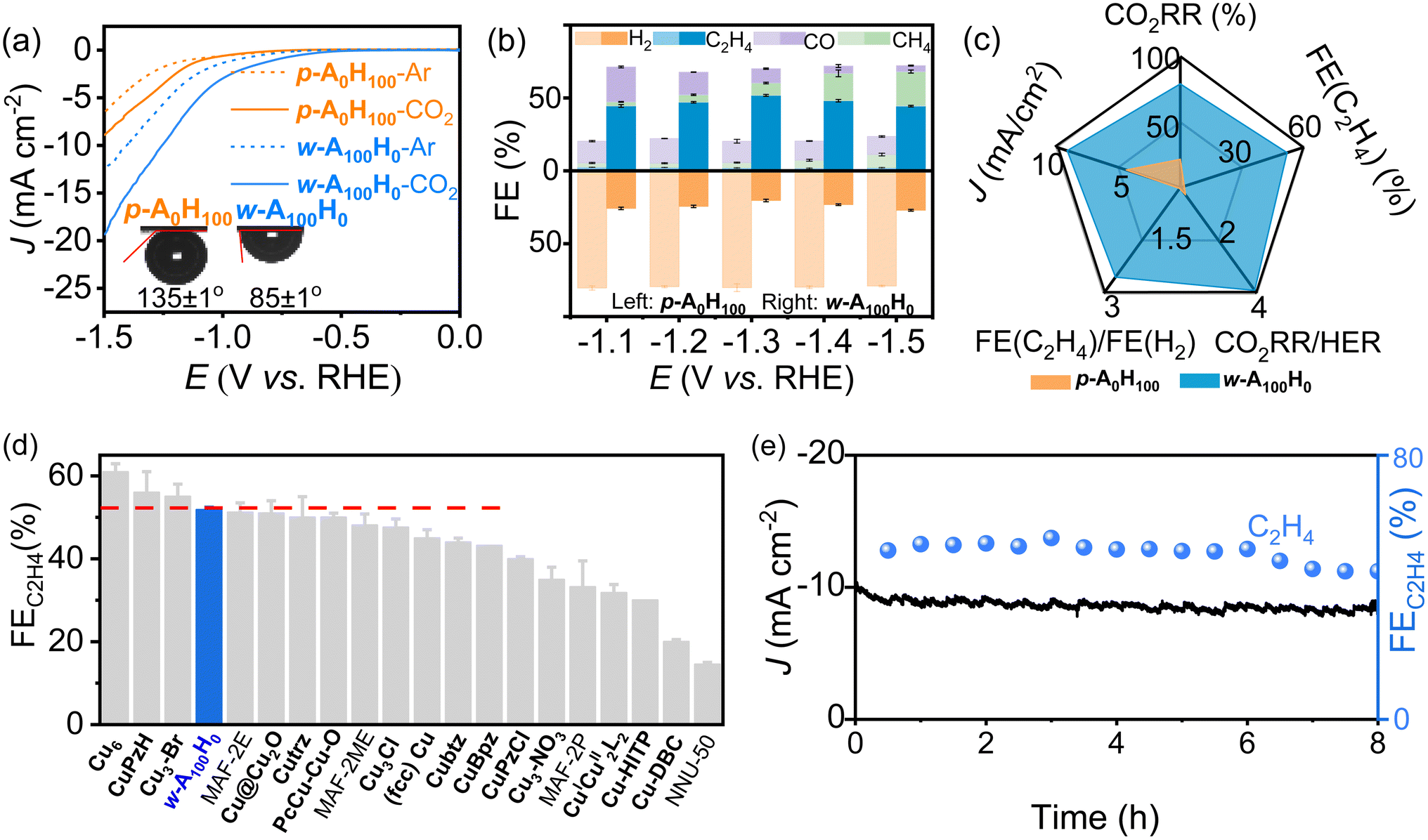 | ||
| Fig. 3 Electrocatalytic performances of p-A0H100 and w-A100H0. (a) Linear sweep voltammetry curves (Ar-saturated conditions were used for comparison). (b) Faradaic efficiency (FE) of gas products at different potentials. (c) Comparison of typical performances. (d) Comparison of FE (C2H4) of benchmark catalysts. (e) Chronoamperometry of w-A100H0 at −1.3 V. | ||
The different affinities toward H2O and CO2 can be explained by Grand Canonical Monte Carlo (GCMC) simulations and periodic density functional theory (PDFT) calculations (Fig. S8 and S9†). The results showed that H2O molecules evenly distribute on the surfaces of both p-A0H100 (C–H⋯O 3.53–3.62 Å) and w-A100H0 (N–H⋯O 3.07 Å and C–H⋯O 3.48–3.73 Å) with similar binding energies of −16.1 and −18.9 kJ mol−1, respectively. On the other hand, CO2 molecules interact very differently with p-A0H100 and w-A100H0. For w-A100H0, CO2 molecules concentrate in the groove sections of the wavy layer surface and simultaneously interact with Cu(I) and –NH2 groups (Cu⋯O 3.13 Å and N–H⋯O 3.18 Å) with a relatively strong binding energy of −24.7 kJ mol−1. For p-A0H100, CO2 evenly distributed on the flat layer surface (C–H⋯O 3.71–3.84 Å) with a weak binding energy of −12.2 kJ mol−1 (Fig. S8†).
Gas chromatography (GC) and 1H nuclear magnetic resonance (NMR) showed that most CO2RR products are gases (i.e., H2, CO, CH4 and C2H4) (Fig. 3b, S10–S15 and Tables S1–S4†). For p-A0H100, the H2 selectivity remained ∼80% over a wide potential range (−1.1 V to −1.5 V), and the total selectivity of CO2RR products was ∼20%. In contrast, w-A100H0 exhibited high CO2RR selectivity (∼76%) and low HER selectivity (∼24%) under the same conditions. Moreover, the C2H4 selectivity (44–52%) is significantly higher than those of other products, such as CO (4–24%) and CH4 (3–24%). It is noteworthy that the C2H4 selectivity of w-A100H0 can reach 51.8 ± 0.6% at −1.3 V, which is 26-fold higher than that of p-A0H100 (2.0 ± 0.2%) under the same conditions and 22-fold higher than the highest value of p-A0H100 (2.4% at −1.1 V) and comparable to the highest values of reported catalysts (Fig. 3c, d and Table S5†).40–45 Isotope labelling experiments confirmed that all the CO2RR products originated from CO2 rather than decomposition of the electrocatalyst (Fig. S16†).
Chronoamperometry tests at −1.3 V showed that the current density and C2H4 selectivity of w-A100H0 and p-A0H100 can remain unchanged for ca. 6 h (Fig. 3e and S17†). After chronoamperometry at −1.3 V for 8 h, PXRD showed that there was no obvious change before and after the CO2RR (Fig. S18†), which revealed that the coordination structure should be maintained; TEM showed that the morphology of the nanosheets was similar to that before catalysis and there was no occurrence of agglomeration or particles (Fig. S4†), which proved that there were no inorganic substances such as Cu or its derivatives present before and after the reaction; XPS combined with X-ray absorption near-edge structure (XANES) showed that the valence state of Cu did not change, and the N 1s region indicated that the intensity of Cu–N did not diminish after the CO2RR (Fig. S19 and S20†), suggesting that the Cu–N bonds were not broken.46,47 Therefore, the degraded catalysis performances after 6 h can be ascribed to the falling-off and/or poisoning effects rather than degradation of the catalysts (Fig. S21†), which have been widely observed in the literature.30,48,49
The operando electrochemical attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra of both p-A0H100 and w-A100H0 showed characteristic peaks (Fig. S22†) of *COOH, *CO, and *CHO (intermediates for CH4) and CO*–*CHO (intermediates for C2H4).43,47 In particular, comparing the ATR-FTIR spectra of p-A0H100 and w-A100H0 at −1.3 V for 800 s, the peaks of p-A0H100 are significantly weaker than those of w-A100H0 (Fig. S23†). Furthermore, the characteristic peaks of C2H4 intermediates in w-A100H0 are significantly stronger than those of CH4, being consistent with the experimental results.
To identify the roles of the amino group and layer bending, p-A0H100 and w-A100H0 were post-synthetically modified by ligand exchange with Htz or Hatz, respectively, i.e., change x in [Cu(tz)x%(atz)100−x%] (Fig. S24, S25 and Table S6†). PXRD patterns of the modified nanosheet products, i.e., p-A12H88, p-A25H75, w-A25H75 and w-A50H50, were similar to those of the parent nanosheets, and the characteristic PXRD peaks did not shift, indicating that the local structures of nanosheets changed little (Fig. S26†). TEM, AFM, and XPS further confirmed the retention of nanosheet morphologies and local coordination structures (Fig. S27–S29†). Note that p-A25H75 and w-A25H75 are supramolecular isomers, which have not been reported for ultrathin nanosheets. PDFT calculations showed that the planar structure of p-A0H100 can be retained when 25% of tz− was replaced by atz−, the wavy structure of w-A100H0 can be retained when 75% of atz− was replaced by tz−, and higher ligand exchanging ratios resulted in irregular layer deformation, possibly implying some unknown kinetic factors (Fig. S25 and 30†).50 These results indicated that the small pores of a planar sql-a network can only accommodate half an amino group, i.e., two pores for one amino group.33
Electrocatalysis tests clearly demonstrated that p-AxH100−x and w-AxH100−x preferred the CO2RR and HER, respectively (Fig. 4a, S31–S36 and Tables S7–S10†). For p-AxH100−x, when x increased from 0 to 25, the HER selectivity slightly decreased (<3.4%) and the CO2RR selectivity slightly increased (<3.7%), indicating that the concentration of amino groups in the planar structures plays a minor role in the selectivity. The isomers p-A25H75 and w-A25H75 offer a straightforward/reliable comparison of the roles of the amino group and the layer shape. Possessing the same chemical compositions and the same concentration of amino groups, w-A25H75 showed 5.3% higher CH4 selectivity, 20% higher C2H4 selectivity, and 25% lower HER selectivity, indicating the critical role of the wavy structure for CO2RR/HER selectivity. For w-AxH100−x, when x increased from 25 to 100, the CO and CH4 selectivities decreased slightly (<4.7%), whereas the C2H4 selectivity increased rapidly (>30%) and H2 selectivity decreased rapidly (>31%) (Fig. 4a). These results indicated that, when the layers are wavy, the amino groups also play important roles in improving not only CO2RR/HER selectivity but also C2H4 selectivity.
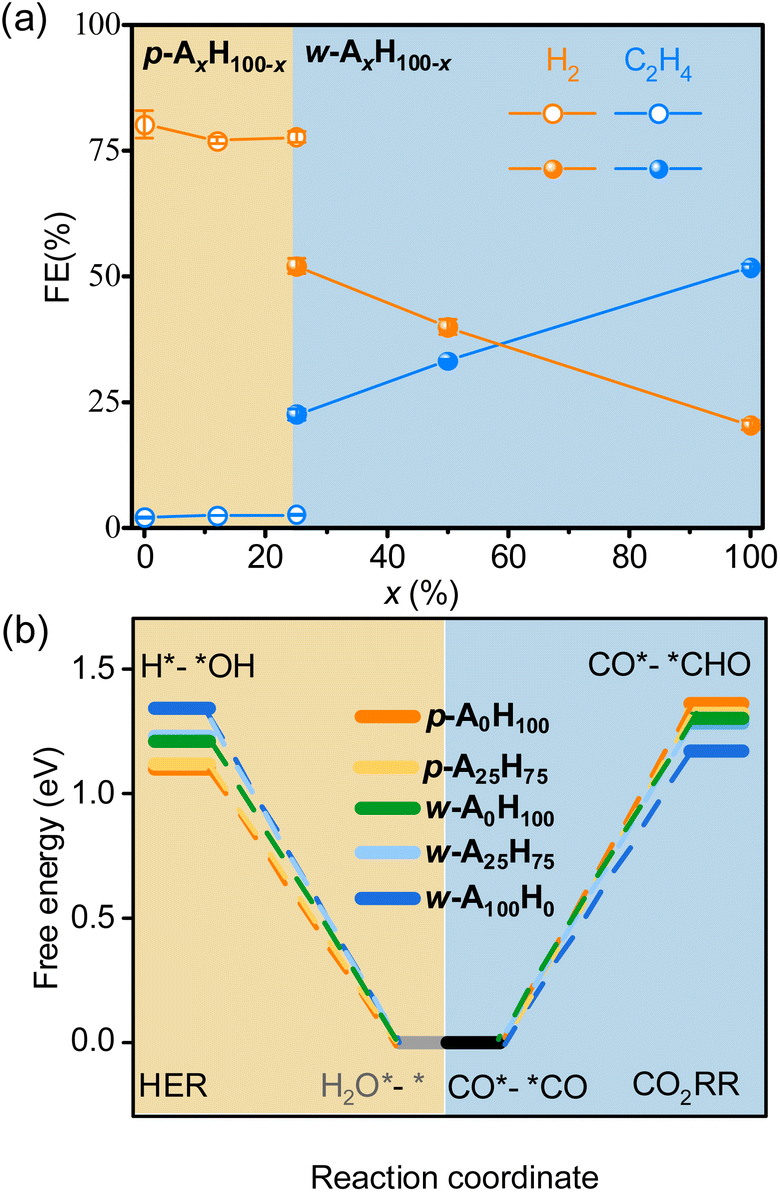 | ||
| Fig. 4 Selectivity and mechanisms for p-AxH100−x and w-AxH100−x. (a) CO2RR/HER selectivity at −1.3 V. (b) Comparison of ΔGmax of the CO2RR/HER. | ||
Mechanism investigations
The tuneable CO2RR/HER performances can be elucidated using PDFT calculations, which showed that the rate-determining steps (RDSs) of the CO2RR (C2H4) and HER are CO*–*CO → CO*–*CHO and H2O*–* → H*–*OH (Fig. S37–S46†), respectively, similar to many literature studies.30,51–59 The ΔGmax values of the CO2RR/HER were calculated to be 1.36/1.10 and 1.17/1.34 eV for p-A0H100 and w-A100H0 (Fig. 4b and S37–S39†), meaning that the energy barrier of the CO2RR is higher than that of the HER for p-A0H100, while w-A100H0 showed an opposite trend, which is consistent with the experimental results. The ΔGmax values of the CO2RR/HER were 1.32/1.12 eV for p-A25H75, which were slightly lower/higher than those for p-A0H100 (Fig. 4b, S40 and S41†), indicating that increasing the concentration of the amino group in the planar structure has little effect on improving CO2RR/HER selectivity. For w-A25H75, the ΔGmax values of the CO2RR/HER were 1.28/1.23 eV, which were significantly lower/higher than those for p-A25H75, indicating that the wavy structure is critical for improving CO2RR/HER selectivity (Fig. 4b and S40 and S42†).To further illustrate the role of wavy structure in CO2RR/HER selectivity, a hypothetical w-A0H100 structure (fixed during simulation, otherwise relaxed to p-A0H100) was constructed to perform PDFT calculations, and the results showed that the ΔGmax values of the CO2RR/HER were calculated to be 1.30/1.21 eV, which were similar to those of p-A0H100 rather than w-A100H0 (Fig. S43 and S44†). This result indicates that a high CO2RR/HER selectivity requires not only the wavy structure but also the amino groups.
For the RDSs, the simulated intermediate structures showed that the amino groups interact weakly with the adsorbed CO and H2O molecules (Fig. 5 and S47†). In addition, the energies of the reactants (i.e., CO*–*CO and H2O*–*) are quite similar for all five simulated structures, indicating that the amino groups play trivial roles on the electronic structure of Cu(I), being consistent with the XPS results (Fig. S29†). Furthermore, the electrostatic potential (ESP) and the Mulliken population analysis of the five structures in both planar and wavy structures are similar, indicating that they possess similar coordination affinity (Fig. S48 and S49†).60,61 Therefore, the differences in ΔGmax values should mainly arise from the product structures (i.e., CO*–*CHO and H*–*OH). Similar to the literature results,55,56,62,63 the free energies of CO*–*CHO and H*–*OH were higher than those of CO*–*CO and H2O*–*, respectively. For CO*–*CHO, the amino groups can form attractive hydrogen-bonding interactions with the CHO fragments of w-A100H0 (N–H⋯O 3.02 Å, H⋯O 2.00 Å, ∠N–H⋯O 166.7°) and w-A25H75 (N–H⋯O 3.12 Å, H⋯O 2.13 Å, ∠N–H⋯O 161.5°), which decrease the system energies to give low CO2RR barriers (Fig. 5a and b). In contrast, for H*–*OH, the amino groups showed obvious steric repulsion with the adsorbed H atoms of w-A100H0 (H⋯H 2.05 Å) and w-A25H75 (H⋯H 2.08 Å), which increased the system energies to give high HER barriers (Fig. 5c and d). For planar structures p-A0H100 and p-A25H75, as well as amino-free structure w-A0H100, such attractive/repulsive effects cannot be formed. Therefore, the inversed CO2RR/HER selectivity from p-A0H100 to w-A100H0 can be explained by the cooperation of the wavy structure and amino groups.
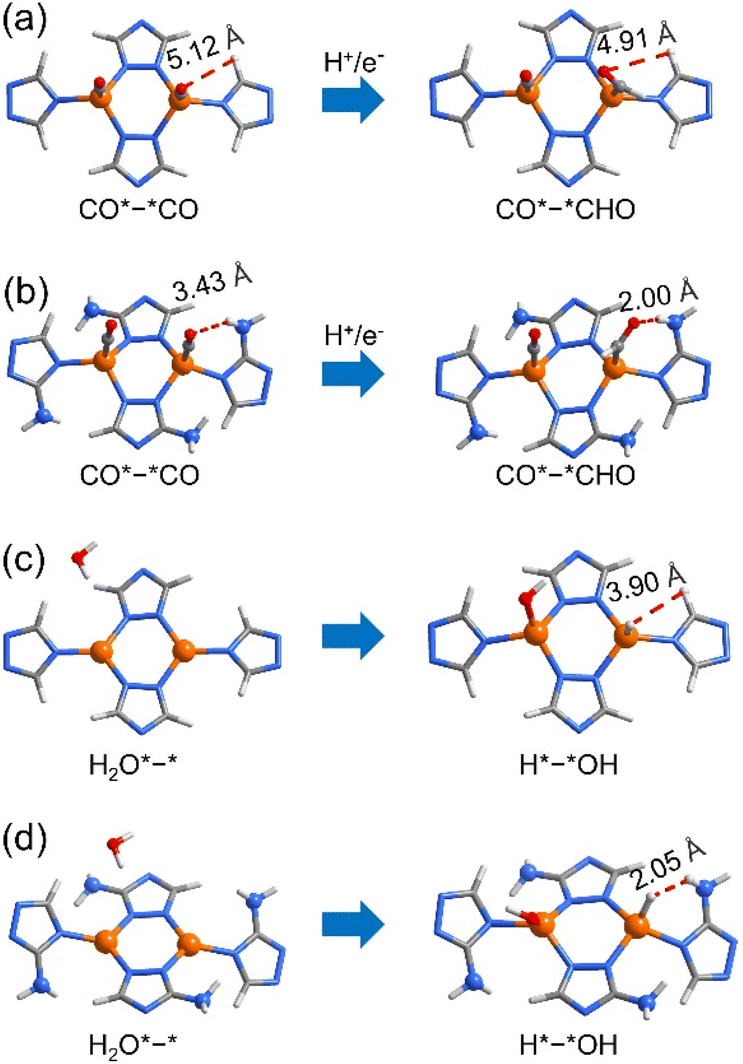 | ||
| Fig. 5 Key intermediates of the CO2RR/HER for p-A0H100 and w-A100H0. (a–d) Reactant/product structures of the RDSs of the CO2RR (a and b) and HER (c and d) for p-A0H100 (a and c) and w-A100H0 (b and d). | ||
Note that w-A100H0 has larger attractive/repulsive interactions for CO2RR/HER intermediates than w-A25H75, respectively, which can be attributed to the higher curvature degree of the layers (Fig. S50 and S51†). In other words, although experimental techniques such as TEM and AFM cannot distinguish the slightly different curvature degrees of the nanosheet samples, computational simulations indicated that the concentration of amino groups can fine tune the curvature degree and supramolecular microenvironment of the 2D coordination layers, as well as the CO2RR/HER selectivity.
Conclusions
In summary, a series of ultrathin nanosheets based on isoreticular/isostructural/isomeric 2D CPs have been synthesized. By systematically tuning the concentration of the amino group of the organic ligand, the ultrathin nanosheets adopt either planar or wavy structures, which show predominant electrocatalytic CO2RR or HER selectivity, respectively, and a high C2H4 selectivity of 52% is achieved. Computational simulations showed that amino groups not only play an important role in determining the shape of layer structures but also simultaneously promote the CO2RR and restrict the HER by interacting with the key reaction intermediates with attractive and repulsive interactions, respectively. These results demonstrate the delicate role of the supramolecular microenvironment for catalysis and the potential of 2D CPs for precise structural modulation and development of new catalysts. This work might also inspire future practical research to solve relatively practical issues in electrode preparation or cell devices.Data availability
The data supporting this article have been included as part of the ESI,† and more raw data are available from the corresponding authors upon reasonable request.Author contributions
D. D. Z. and J. P. Z. designed the research. K. Z. performed syntheses and measurements. D. Y. H. and X. W. Z. performed computational simulations. X. X. X., Z. J. L. and H. Y. performed XRD, FTIR and XPS analyses. J. X. W., D. Y. L. and L. L. Z. performed SEM and TEM analyses. L. G. performed AFM analyses. K. Z., D. D. Z. and J. P. Z. wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1500400) and the National Natural Science Foundation of China (22090061, 21821003, 22071272, 21975290 and 22231012).Notes and references
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS
.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 3506 CrossRef PubMed
- H. Shen, Z. Gu and G. Zheng, Sci. Bull., 2019, 64, 1805–1816 CrossRef CAS PubMed
- X.-D. Zhang, T. Liu, C. Liu, D.-S. Zheng, J.-M. Huang, Q.-W. Liu, W.-W. Yuan, Y. Yin, L.-R. Huang, M. Xu, Y. Li and Z.-Y. Gu, J. Am. Chem. Soc., 2023, 145, 2195–2206 CrossRef CAS PubMed
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed
- J. Feng, L. Wu, S. Liu, L. Xu, X. Song, L. Zhang, Q. Zhu, X. Kang, X. Sun and B. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS
- W. Wang, D. Chen, F. Li, X. Xiao and Q. Xu, Chem, 2024, 10, 86–133 CAS
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS
- A. R. Woldu, P. Talebi, A. G. Yohannes, J. Xu, X. D. Wu, S. Siahrostami, L. Hu and X. C. Huang, Angew. Chem., Int. Ed., 2023, 135, e202301621 CrossRef
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- D.-H. Nam, O. S. Bushuyev, J. Li, P. De Luna, A. Seifitokaldani, C.-T. Dinh, F. P. García de Arquer, Y. Wang, Z. Liang, A. H. Proppe, C. S. Tan, P. Todorović, O. Shekhah, C. M. Gabardo, J. W. Jo, J. Choi, M.-J. Choi, S.-W. Baek, J. Kim, D. Sinton, S. O. Kelley, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 11378–11386 CrossRef CAS PubMed
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS PubMed
- W. Zhang, S. Liu, Y. Yang, H. Qi, S. Xi, Y. Wei, J. Ding, Z.
J. Wang, Q. Li, B. Liu and Z. Chen, Angew. Chem., Int. Ed., 2023, 62, e202219241 CrossRef CAS PubMed
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech, E. Brunner, I. M. Weidinger, J. Zhang, A. V. Krasheninnikov, S. Kaskel, R. Dong and X. Feng, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719–729 CrossRef CAS
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef PubMed
- Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS PubMed
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC
- L. Cao and C. Wang, ACS Cent. Sci., 2020, 6, 2149–2158 CrossRef CAS PubMed
- Y. Quan, G. Lan, Y. Fan, W. Shi, E. You and W. Lin, J. Am. Chem. Soc., 2020, 142, 1746–1751 CrossRef CAS PubMed
- R.-J. Wei, P.-Y. You, H. Duan, M. Xie, R.-Q. Xia, X. Chen, X. Zhao, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2022, 144, 17487–17495 CrossRef CAS PubMed
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 1–10 Search PubMed
- J. Huang, Y. Li, R. K. Huang, C. T. He, L. Gong, Q. Hu, L. Wang, Y. T. Xu, X. Y. Tian, S. Y. Liu, Z. M. Ye, F. Wang, D. D. Zhou, W. X. Zhang and J. P. Zhang, Angew. Chem., Int. Ed., 2018, 130, 4722–4726 CrossRef
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu, D. D. Zhou and J. P. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed
- J.-P. Zhang, Y.-Y. Lin, X.-C. Huang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 5495–5506 CrossRef CAS PubMed
- D. Chen, Y.-J. Liu, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, CrystEngComm, 2011, 13, 3827 RSC
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- L. Cao, T. Wang and C. Wang, Chin. J. Chem., 2018, 36, 754–764 CrossRef CAS
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed
- G. Liu, W. S. Y. Wong, M. Kraft, J. W. Ager, D. Vollmer and R. Xu, Chem. Soc. Rev., 2021, 50, 10674–10699 RSC
- Z.-Z. Niu, F.-Y. Gao, X.-L. Zhang, P.-P. Yang, R. Liu, L.-P. Chi, Z.-Z. Wu, S. Qin, X. Yu and M.-R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed
- K. Lv, C. Teng, M. Shi, Y. Yuan, Y. Zhu, J. Wang, Z. Kong, X. Lu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1802339 CrossRef
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed
- J.-N. Lu, J. Liu, L. Zhang, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, J. Mater. Chem. A, 2021, 9, 23477–23484 RSC
- J. Wang, Y. Zhang, Y. Ma, J. Yin, Y. Wang and Z. Fan, ACS Mater. Lett., 2022, 4, 2058–2079 CrossRef CAS
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC
- H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Cent. Sci., 2022, 8, 1506–1517 CrossRef CAS PubMed
- R. Wang, J. Liu, L.-Z. Dong, J. Zhou, Q. Huang, Y.-R. Wang, J.-W. Shi and Y.-Q. Lan, CCS Chem., 2023, 5, 2237–2250 CrossRef CAS
- L. Zhang, X.-X. Li, Z.-L. Lang, Y. Liu, J. Liu, L. Yuan, W.-Y. Lu, Y.-S. Xia, L.-Z. Dong, D.-Q. Yuan and Y.-Q. Lan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS PubMed
- H.-L. Zhu, H.-Y. Chen, Y.-X. Han, Z.-H. Zhao, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2022, 144, 13319–13326 CrossRef CAS PubMed
- U. O. Nwabara, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, ChemSusChem, 2020, 13, 855–875 CrossRef CAS PubMed
- W. Gao, Y. Xu, H. Xiong, X. Chang, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202313798 CrossRef CAS PubMed
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 124, 12663–12666 CrossRef
- X. Qin, T. Vegge and H. A. Hansen, J. Am. Chem. Soc., 2023, 145, 1897–1905 CrossRef CAS PubMed
- X. Zou and J. Gu, Chin. J. Catal., 2023, 52, 14–31 CrossRef CAS
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed
- J. Jiao, Q. Yuan, M. Tan, X. Han, M. Gao, C. Zhang, X. Yang, Z. Shi, Y. Ma, H. Xiao, J. Zhang and T. Lu, Nat. Commun., 2023, 14, 6164 CrossRef CAS PubMed
- H. Liu, Q. Huang, W. An, Y. Wang, Y. Men and S. Liu, J. Energy Chem., 2021, 61, 507–516 CrossRef CAS
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 17, 2104445 CrossRef CAS PubMed
- Z. Q. C. Li, H. M. Sun, Y. j. Yang and C.-P. Li, Chin. J. Struct. Chem., 2022, 41, 2211084–2211099 Search PubMed
- J. D. Yi, D. H. Si, R. Xie, Q. Yin, M. D. Zhang, Q. Wu, G. L. Chai, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 17108–17114 CrossRef CAS
- J. Zhang, Y. Chen, F. Xu, Y. Zhang, J. Tian, Y. Guo, G. Chen, X. Wang, L. Yang, Q. Wu and Z. Hu, Small, 2023, 19, 2301577 CrossRef CAS
- X. Mao, W. Gong, Y. Fu, J. Li, X. Wang, A. P. O'Mullane, Y. Xiong and A. Du, J. Am. Chem. Soc., 2023, 145, 21442–21453 CrossRef CAS PubMed
- J. Sang, P. Wei, T. Liu, H. Lv, X. Ni, D. Gao, J. Zhang, H. Li, Y. Zang, F. Yang, Z. Liu, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 134, e202114238 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01982b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |