
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insertion of CO2 to 2-methyl furoate promoted by a cobalt hypercrosslinked polymer catalyst to obtain a monomer of CO2-based biopolyesters†
Elizabeth
Rangel-Rangel
,
Beatriz
Fuerte-Díez
,
Marta
Iglesias
 and
Eva M.
Maya
*
and
Eva M.
Maya
*
Instituto de Ciencia de Materiales de Madrid (ICMM-CSIC), Sor Juana Inés de la Cruz, 3, Cantoblanco, Madrid 28049, Spain. E-mail: eva.maya@csic.es
First published on 18th September 2024
Abstract
2,5-Furan Dicarboxylic methyl Ester (FDME), a highly valued monomer for the synthesis of biobased polyesters, has been prepared through a new synthetic strategy that consists of the direct carboxylation of methyl furoate in two steps: the first one involves a solvent-free reaction using a moderate CO2 pressure (10 bar), a base (Cs2CO3) and a cobalt-based heterogeneous catalyst (HCP-Salphen-Co) for 6 h, which was prepared using mechanochemical polymerization. The second step consists of an acid esterification using standard conditions. The CO2-based FDME synthesized with this strategy was successfully reacted with a diol derived from vanillin, thus obtaining a CO2 and a completely bio-based polyester which exhibited high thermal stability with a starting degradation temperature of 250 °C and a glass transition temperature of 104 °C.
Sustainability spotlight2,5-Furfural Dimethyl Ester (FDME) is a highly valued bio-based monomer. Its production from 2-methyl furoate (MF), a bio-based molecule very abundant in peanuts, cocoa, and coffee, has been little explored. Only one work has been reported where FDME is obtained by alkoxycarbonylation of MF in CHCl3. At present, we offer a more sustainable procedure that takes advantage of using CO2 as a feedstock in a solvent-free reaction. Moreover, the new process uses a heterogeneous catalyst prepared by mechano-chemical synthesis which involves a very short reaction time, absence of solvents, and lower energy consumption than conventional synthesis. Thus, the process reported here allows us to advance toward obtaining a biobased monomer of great interest using sustainable protocols. |
Introduction
The incorporation of furan rings in polymers is having a great impact due to the excellent final properties it provides: high glass transition temperature, low melting and processing temperature and, excellent gas barrier properties which makes it have very up-and-coming applications such as high-performance fibers and as packaging materials.1,2 2,5-Furfural Dicarboxylic methyl Ester (FDME) is an interesting monomer widely used in the synthesis of furan-based polyesters which has been mainly prepared by oxidative-esterification of 5-hydroxymethylfurfural (HFM)3–6 or galactaric acid7,8 using different homogeneous metal-based catalysts. The synthesis of FDME can also be achieved from 2-methyl furoate (MF) which is also a bio-based molecule very abundant in peanuts, cocoa, and coffee and offers a wide range of applications, for example in the synthesis of fuels or fuels additives, as an intermediate for the synthesis of compounds with medical applications or in the synthesis of renewable hydrophilic polyesters.9–11However, as far as we have been able to investigate, only one work has been published where the FDME has been prepared from MF (Fig. 1 up). The procedure consists of a direct alkoxycarbonylation in a one-step reaction of three components, MF, methanol, and CHCl3, using a copper catalyst. The reaction was conducted at 120 °C for 12 h to obtain FDME with good yield (80%).12
 | ||
| Fig. 1 Reported and patented syntheses of FDME (2,5-Furan Dicarboxylic Methyl Ester) synthesis from MF (2-methyl furoate) (up); contributions of this work (down). | ||
The synthesis of FDME from MF and CO2 is an interesting alternative because besides obtaining this value monomer, takes advantage of using CO2 as feedstock. The conversion of CO2 into value chemicals, fuels, and polymers has become very important in the last decade since it can produce many that are traditionally obtained from petroleum derivatives. This implies, in addition to using this gas so that it is not emitted into the atmosphere, a reduction in the carbon footprint of the products obtained from it.13 Thus, obtaining a monomer from CO2 such as FDME that can be used to obtain a CO2-based polymer is especially relevant.14 The only procedure recognized so far where methyl furoate is reacted with CO2 is patented.15 The method involves dissolving methyl furoate and mixing it with a metal-supported catalyst (ZSM-5 type containing cobalt (Co), manganese (Mn), rhodium (Rh), or palladium (Pd)) in a sealed reactor, containing CO2 at 10–60 bar and a temperature between 140 and 250 °C for 6–10 hours (Fig. 1 up).
In this work we reported the synthesis of FDME by the insertion of CO2 to methyl furoate catalyzed by a hypercrosslinked polymeric catalyst (HCP-Salphen-Co) which was prepared in two steps, using a mechanochemical polymerization (Fig. 1 down). HCPs are excellent metal supports that have been used as heterogeneous catalysts in a wide variety of organic reactions.16 The most extensive synthesis of HCPs is carried out in solution, by heating the corresponding monomers for 24–72 h in a chlorinated solvent, using a crosslinker agent and FeCl3 or AlCl3 as catalysts. However, in 2019 the synthesis of an HCP using mechanochemical synthesis has been reported. This strategy, in addition to the fact that it is a solvent-free procedure, is much faster (5–60 min) which implies a more sustainable synthesis.17
Finally, as a validation test, a bio-based polyester has been synthesized combining the CO2-based FDME with a diol derived from vanillin, so a new polyester derived from CO2 containing furan rings and completely bio-based has been also reported (Fig. 1 down).
Experimental
Materials
All reagents were used as received. Methyl furoate (MF) (99.0%) and cesium carbonate (99.5%), were purchased from Acros Organics; dimethoxymethane (DMM) (98%) was supplied by Alfa Aesar; salicylaldehyde (99%) was purchased from Fisher Chemical; o-phenylenediamine (99%) was supplied by FEROSA; cobalt(II) acetate monohydrate (98%) was obtained from Strem Chemicals; titanium tetraisopropoxide was purchased from Merck-(97%); iron(III) chloride (98%) and carbon dioxide (99.9% of purity) were supplied by Praxair.Vanillin produced from lignin was provided by Borregaard (Norway) for this research.
Solvents were purchased from JT Baker.
N,N′-Phenylenebis(salicylideneimine)(salphen) was prepared following the procedure reported by the reaction of o-phenylenediamine and salicylaldehyde.181H-NMR spectrum of this reagent is provided in Fig. S1.†
2-(4-(Hydroxymethyl)-2-methoxyphenoxy)ethanol (Va-Diol) was prepared from vanillin following a two steps procedure reported in the literature.191H-NMR spectrum of this reagent is provided in Fig. S2.†
Synthesis of HCP-Salphen-Co
The salphen monomer (400 mg, 1.26 mmol) and iron(III) chloride (1 g, 6.1 mmol) was added to a zirconia vessel provided containing three zirconia balls. Then, dimethoxymethane (0.56 mL, 6.1 mmol) was added to this mixture and was ball-milled at 600 rpm for 55 min. Then, MeOH was added to the zirconia vessel and the product was filtered out. The solid was thoroughly washed with, NH4OH, diluted HCl (4 mL of HCl in 100 mL of water), and water. Finally, the solid was purified by Soxhlet extraction with MeOH for 24 h and then, it was dried under vacuum at 100 °C for another 24 h. The hypercrosslinked polymer was obtained as a brown powder (HCP-Salphen) (365 mg (93% yield).HCP-Salphen (250 mg) was heated at 80 °C for 24 h with Co(CH3COO)2·4H2O (97 mg, 0.4 mmol) in 15 mL of MeOH. After cooling down to r.t., the solid was collected by filtration, washed with methanol and the solid was dried under vacuum at 100 °C for 12 h. HCP-Salphen-Co was obtained as dark brown solid (270 mg).
Synthesis of FDME from methyl furoate and CO2
2-Methyl Furoate MF (630 mg, 5 mmol) was introduced in a high-pressure autoclave engineers' reactor with HCP-Salphen-Co (750 mg, 1% Co) and Cs2CO3 (1.630 g, 5 mmol) and 10 bar of CO2. This mixture was heated at 200 °C for 10 h. After this time the reactor was opened and 50 mL MeOH was added and HCP-Salen-Co was removed by filtration. Then, H2SO4 was added at room temperature until cesium sulfate precipitated (pH = 7) (1.540 g), which was also separated by filtration. The mixture was heated at 80 °C for 24 h and then, by adding water CO2-FDME was obtained as a beige solid with moderated yield (246 mg, 27%). The reactions were followed by TLC and proton nuclear magnetic resonance (1H-NMR).Synthesis of a biopolyester (PVaF) based on a diol derived from vanillin and CO2-FDME
In a 25 mL round-bottom flask equipped with a magnetic stirring bar were added 400 mg (2.1 mmol) of CO2-FDME, 1.4 g (7.06 mmol) of Va-Diol, 12 mg of glycerol (2.5% relative to the Va-Diol), 12 μL of Ti(OiPr)4 and 0.6 mL of toluene. The flask was coupled to a distillation bridge connected to a vacuum pump and the reaction system was purged removing the air by vacuum and filled with nitrogen gas. After five evacuation and filling cycles, the flask was gradually heated to 160 °C and after 5 min at this temperature 4 mL of N-methylpirrolidone (NMP) was added and the mixture was heated for 1 h, at 170 °C for 1 h, and finally at 180 °C for 45 min to carry out the transesterification reaction. After this time, the vacuum was then slowly applied to remove the excess alcohol but without reaching dryness. Then the temperature was increased to 200 °C. The reaction was maintained at this temperature for 2 h during which the solution acquired viscosity, having to add 0.5 mL of NMP on two occasions to prevent the mixture from stopping stirring. After this time, the reaction was stopped and the vacuum was then slowly applied to remove the excess of NMP. Then, the mixture was dissolved in 12 mL of a mixture of chloroform and trifluoroacetic acid (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). The solution was poured into 400 mL of methanol to obtain the polyester in the form of fine brown fibbers. After filtration and washing three times with methanol, the solid was dried under vacuum at 100 °C to yield 450 mg polyester (quantitative yield).
:1 v/v). The solution was poured into 400 mL of methanol to obtain the polyester in the form of fine brown fibbers. After filtration and washing three times with methanol, the solid was dried under vacuum at 100 °C to yield 450 mg polyester (quantitative yield).
Equipment and characterization techniques
The mechanochemical polymerization was done out on a planetary ball mill Retsch PM100 model with Fritsch 50 mL steel-cased zirconia grinding bowl provided with three zirconia grinding balls of 2 cm of diameter and Viton seal O-rings.FTIR-ATR spectra were recorded in a Bruker Vertex 70v in a spectral range of 400–4000 cm−1 with a resolution of 2 cm−1; 1H-NMR spectra were done with a BRUKER AVANCE III HD (Larmor frequencies of 400) using CDCl3 as solvent; X-ray Photoelectron Spectroscopy (XPS) data were recorded with a SPECS GmbH system equipped with a hemispherical energy analyzer PHOIBOS 150 9MCD. Samples were located first in the pre-chamber at rt and degassed before the analysis. A power of 200 W and voltage of 12 kV was applied with a non-monochromatic Mg X-ray source. Pass energies of 50 and 20 eV were used for acquiring both survey and high-resolution spectra, respectively. The thermal stability of the catalyst and bio-polyester was evaluated by thermogravimetric analysis (TGA) with a TQ-500 apparatus from TA Instruments. The experiments were carried out under an air or nitrogen atmosphere by a heating rate of 10 °C min−1, from rt to 850 °C using around of 5 mg of sample. The glass transition temperature, Tg, of the polyester, was obtained by differential scanning calorimetry (DSC) in a DSC Q-100 equipment from TA Instruments. The sample was encapsulated in a standard aluminum DSC pan and was heated at a scanning rate of 10 °C min−1 from room temperature to 350 °C in the first cycle to remove its thermal history. Then, the sample was cooled and the Tg was calculated from the inflection of the heat flow versus temperature curves in the second heat cycle. The porous properties were analyzed by nitrogen adsorption–desorption isotherms recorded in a Micromeritics ASAP 2020 M surface provided with a porosity analyzer at 77 K. The samples were degassed overnight at 120 °C. Specific surface areas were determined by BET theory; isotherm of CO2 uptake was obtained in a Micromeritics ASAP 2010 volumetric instrument, using ∼60 mg of powder sample outgassed at 423 K for 6 h using a turbo-molecular high vacuum pump. The sample was placed in a sample holder immersed in a liquid-recirculating thermostatic bath (Julabo FP40-HL) to control the adsorption temperature (273 K).
Isotherm of CO 2 uptake was recorded in a Micromeritics ASAP 2010 volumetric instrument, using 50 mg of powder sample which was placed in a sample holder immersed into a liquid recirculating thermostatic bath (Julabo FP40-HL) to control the adsorption temperature (273 K). Before the experiment, the sample was outgassed at 423 K for 6 h using a turbo-molecular high vacuum pump.
Results and discussion
Synthesis and characterization of HCP-Salphen-Co
The catalyst, HCP-Salphen-Co, was prepared in two steps (Fig. 2). In the first one, the salphen monomer was polymerized in 55 min by mechanochemical synthesis using FeCl3 as a catalyst and dimetoxymethane (DMM) as a crosslinker. The formation of an insoluble dark brown solid was the first evidence of the growth of the hypercrosslinked network, HCP-Salphen. Then, in a second step, the HCP-Salphen support was reacted with Co(OAc)2 in ethanol to anchor the corresponding cobalt cations within the polymeric network. | ||
| Fig. 2 Synthesis of catalyst HCP-Salphen-Co. | ||
The cobalt content was determined by inductively coupled plasma spectroscopy (ICP, Fig. S3†), resulting in 7% (1.2 mmol g−1).This percentage of cobalt indicates around two out of three salphen units feature anchored cobalt. The incorporation of cobalt to the network was also confirmed by XPS (Fig. 3a). The first peak at 783.85 eV is assigned to Co 2p3/2 and is attributed to the Co–N bond whereas the peak, at 799.51 eV is ascribed to Co 2p1/2. Since the binding energies of Co3+ and Co2+ are very close, the presence of satellite peaks is very significant in determining the oxidation states of cobalt. Thus, the observed satellites at 788.42 and 805.28 eV can be assigned to the presence of Co2+ ions.20,21
 | ||
| Fig. 3 (a) Co 2p XPS spectrum of HCP-Salphen-Co; (b) FT-IR of HCP-Salphen-Co and HCP-Salphen. | ||
The FT-IR spectrum (Fig. 3b) shows a peak at 1609 cm−1 accompanied by a neighboring peak at 1579 cm−1 due to the strong coordination between the imine groups and the metal cations. The bands at 1538 and 1435 cm−1 are assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations and the peaks at 1210 and 1161 cm−1 were attributed to the vibration of the C–N and C–O bonds, respectively. This spectrum was similar to the HCP-Salphen support which also shows the peak at 1600 cm−1 attributed to the free imine vibration bands (CN) but also is accompanied by another band at 1577 cm−1 that could be ascribed to the coordination of residual iron from the polymerization reaction to some imine bonds.
C stretching vibrations and the peaks at 1210 and 1161 cm−1 were attributed to the vibration of the C–N and C–O bonds, respectively. This spectrum was similar to the HCP-Salphen support which also shows the peak at 1600 cm−1 attributed to the free imine vibration bands (CN) but also is accompanied by another band at 1577 cm−1 that could be ascribed to the coordination of residual iron from the polymerization reaction to some imine bonds.
The incorporation of cobalt does not greatly decrease the thermal stability (Fig. S4†), but the degradation pattern changes a lot concerning the starting support since the degradation occurs in at least three stages while in the support HCP-Salphen, the degradation occurs in only one. The HCP-Salphen-Co shows an initial decomposition temperature of 270 °C.
The presence of metals generates a percentage of residue due to the formation of metallic oxides during the thermogravimetric analysis. Thus, the support, HCP-Salphen showed a 9.98% residue that corresponds to a remaining iron content of 6.98% coming from the synthesis. In the case of HCP-Salphen-Co the residue obtained by the TGA was 7.9%. Considering the amount of cobalt determined by ICP (7%), it can be observed that there are no iron residues in this catalyst, which shows that the cobalt ions have displaced the iron ions.
The porous properties were evaluated by N2 adsorption–desorption isotherms (Fig. 4a). HCP-Salphen exhibited some N2 uptake at low pressure, which is attributed to the presence of micropores. The N2 adsorption increased when the pressure was increased as is ascribed to porous materials showing a specific surface area of 94.7 m2 g−1. However, the incorporation of cobalt decreased drastically the specific surface area to 16.8 m2 g−1 which was attributed to the space occupied by the metal cation in the pores, as it has been reported in many metal-based HCPs.16 The CO2 uptake was measured by CO2 adsorption isotherm (Fig. 4e) indicating a CO2 uptake of 0.75 mmol g−1 which was a low value compared with other HCPs previously reported22,23 due to the absence of specific CO2-philic groups in this structure.
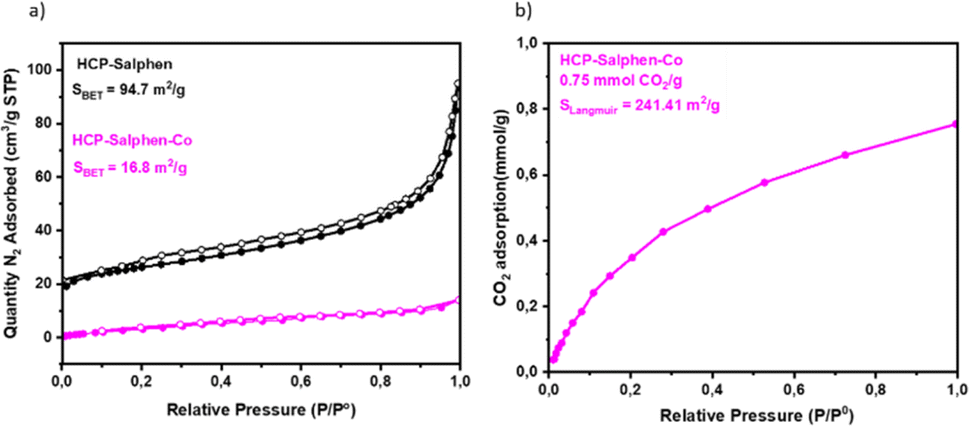 | ||
| Fig. 4 (a) N2 adsorption (solid circles)/desorption (open circles) of HCP-Salphen-Co and HCP-Salphen; (b) CO2 uptake of HCP-Salphen-Co. | ||
Synthesis of FDME from 2-methyl furoate and CO2
The strategy here presented consists of two steps, a carboxylation reaction of 2-methyl furoate (MF), and subsequent esterification, without the need to isolate any reaction intermediate (Table 1). The esterification is a known reaction that was carried out in the same way in all cases (see Experimental part for details).|
|
||||||
|---|---|---|---|---|---|---|
| Run | Cat. (% Co) | Base (mmol) | P (CO2) | T (°C) | t (h) | Yield (%) |
| a 2-Methyl Furoate, MF, (1 mmol), catalyst (0.18 mmol); selectivity of CO2-FMDE 100% in all cases. b MF (5 mmol), catalyst (0.9 mmol). | ||||||
| 1 | Co(OAc)2(1) | Cs2CO3(1) | 45 | 200 | 12 | <5 |
| 2 | 45 | 200 | 5 | <5 | ||
| 3 | 20 | 200 | 5 | 16 | ||
| 4 | 10 | 200 | 4 | 22 | ||
| 5 | Co(OAc)2(1.5) | Cs2CO3(1) | 10 | 200 | 4 | <5 |
| 6 | Co(OAc)2(2) | 10 | 200 | 4 | <5 | |
| 7 | HCP-Salphen-Co(1) | Cs2CO3 (1) | 10 | 200 | 4 | 12 |
| 8 | Cs2CO3(1) | 10 | 200 | 4.5 | 15 | |
| 9 | Cs2CO3(1) | 8 | 220 | 4 | 8 | |
| 10 | Cs2CO3(0.25) K2CO3(0.75) | 10 | 215 | 4 | <5 | |
| 11 | Cs2CO3 (0.5)/K2CO3(0.5) | 8 | 205 | 4 | <5 | |
| 12 | Cs2CO3 (1) | 10 | 200 | 6 | 27 | |
| 13 | Cs2CO3 (1) | 10 | 200 | 7 | 18 | |
| 14 | Cs2CO3(1.5) | 10 | 200 | 6 | <5 | |
| 15b | Cs2CO3 (5) | 10 | 200 | 10 | 27 | |
| 16 | — | Cs2CO3(1) | 10 | 200 | 6 | — |
| 17 | HCP-Salphen | Cs2CO3(1) | 10 | 200 | 6 | — |
To establish the reaction conditions of the first step, cobalt acetate was initially used as a catalyst (Table 1, entries 1–6). The ratio between methyl furoate (MF) and cesium carbonate was 1:1, the temperature was fixed at 200 °C and the CO2 pressure at 45 bar because they were conditions reported to promote C–H carboxylations.24 The reactions were done at different reaction times, obtaining CO2-FDME with low yields (entries 1 and 2). However, when the time was fixed to 5 hours and the reaction was carried out at lower pressure, 20 and 10 bars, achieving FDME with a yield of 16 and 22% (entries 3 and 4). The increase in the amount of catalyst was negative since very low yields were obtained again (entries 5 and 6). Despite the low yields of the reaction, it should be mentioned that no other by-products were detected, so the selectivity towards the desired product was 100%. No traces of MF were observed either, which has made us think that during this step undetectable degradation compounds are being formed, as it was previously observed by Banerjee et al. in the carboxylation of 2-furanoic acid.24
Thus, keeping the reaction conditions of entry 4, HCP-Salphen-Co was used as a catalyst obtaining CO2-FDME with a lower yield, of 12% (entry 7). To increase this yield, the reaction time, CO2 pressure, temperature and addition of other base were investigated (entries 7–14). The best yield achieved was 27%, using a ratio 1:1 between methyl furoate (MF) and cesium carbonate, 200 °C, 10 bars of CO2 pressure and 6 hours of reaction (entry 12). Scaling up the reaction by five times the amounts of reactants and catalyst led to a 27% yield (entry 15). The experiments in the absence of catalysts (entry 16) or using the starting support (entry 17) did not lead to the formation of any product.
To confirm the catalyst heterogeneity, the procedure was done 6 consecutive times (Fig. 5). HCP-Salphen-Co was separated after each reaction, washed with methanol, and dried under vacuum at 100 °C for 30 min to be used in new reactions. Performance decreases slightly in the first cycles but then recovers, reaching 32% of CO2-FDME yield in the last cycle.
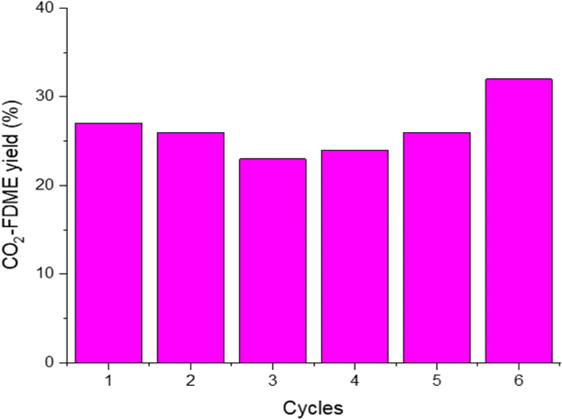 | ||
| Fig. 5 Recycling experiments of HCP-Salphen-Co of FDME from methyl furoate and CO2. | ||
The FT-IR spectrum and TGA (Fig. S5†) showed that the structure of the catalyst remains stable after the six runs. The FTIR shows the same characteristic bands that the catalyst before use. The TGA of the recycled catalyst showed the same thermal stability and residue (12.9%) indicating that there was no release of the metal during the six reactions, confirming its heterogeneous character.
The structure of the CO2-FMDE obtained in all experiments was confirmed by 1H-NMR. As an example, Fig. 6 shows the spectrum of reaction of entry 15. The spectrum shows two signals at 3.8 ppm and 7.3 that correspond to the protons of the methyl groups (a) and to the proton of the furan ring (b) respectively. The spectrum was identical to that of commercial FDME (Fig. S6†). As it can be observed, the CO2-FDME obtained showed high purity which allows us to use it directly to synthesize a polyester. The 13C-NMR (Fig. S7†) confirmed also the structure of the CO2-based monomer and the thermogravimetrical analysis showed a decomposition temperature of 124 °C (Fig. S8†).
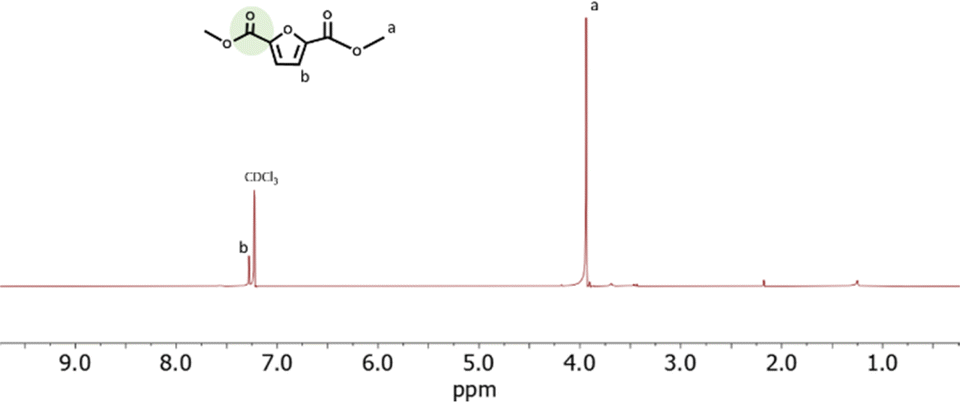 | ||
| Fig. 6 1H-NMR spectrum of CO2-FDME synthetized in this work. | ||
Based on the mechanisms of carboxylations of sp2 C–H bonds metal-catalyzed previously reported,25,26 a tentative mechanism for this reaction is proposed (Fig. S9†). The base (cesium carbonate) deprotonates the C–H bond of carbon 5 of methyl furoate (A) allowing the coordination of the catalyst. Then the CO2 insertion into the carbon–metal bond take places (B). Once the reaction is complete, methanol is added and the catalyst is removed. Then, after removing the catalyst, adding some drops of sulfuric acid, cesium sulfate and furan-2,5-dicarboxylic acid (FDCA) are generated (C). The cesium sulfate was removed by filtration and FDCA was heated with methanol to obtain FDME.
Finally, the procedure was evaluated in terms of sustainability fulfilling many principles of green chemistry.27 Thus, both reactants methyl furoate and CO2 are total and partially incorporated into the final product (Princ. 2 atom economy); the main waste product, cesium sulfate, is not dangerous to handle and is a useful inorganic salt used to prepare dense aqueous solutions for DNA purification and recovery (Princ. 3 less hazardous chemical synthesis); the synthesis of the catalyst was done using mechanochemical polymerization, without solvent, saving time and energy when compared with the conventional synthesis in solution (Princ. 5 safer solvent and Princ. 6 energy efficiency); a bio-based molecule, methyl furoate, very abundant in peanuts, cocoa, and coffee was used as starting material (Princ. 7 renewable feedstocks) and the synthesis of the target monomer was done through a catalytic process (Princ. 9 catalysis) and finally, this monomer has been reacted with a bio-based diol, derived from vanillin) to be applied in the synthesis of a bio-based polyester which is supposedly biodegradable (Princ. 10 design for degradation).
From the point of view of chemical efficiency of the process, the estimated E-factor (mass of waste/mass of product)28 is high, 167.65, taking into account the cesium sulfate and the methanol used in the esterification reaction as waste products. However, if methanol is recycled in the process (only traces of water would have to be removed), the fact would be corrected assuming a 14% of organic solvent loss in the solvent recovery process as it was proposed by Sun et al. in the synthesis of FMDE from of 5-hydroxymethylfurfural (HFM).29 Thus, the E-factor of the process will be 23.47, positioning within the segment of tolerable waste in the chemical industry (5–50)28 and also between the E-factors reported for the synthesis of FDME by oxidative-esterification of HMF (2.5–55.4).29,30
Synthesis of a CO2-based biopolyester (CO2-PVaF)
Vanillin is one of the bio-based compounds that can be obtained from lignin on an industrial large scale. Its structure is very interesting because has two reactive groups, an aldehyde and a phenolic hydroxyl group which means that it can be modified and used as a monomer to obtain a wide variety of polymeric materials with different applications, phenolic resins, polyurethanes, polycarbonates, polyester and polyimines, among others.31,32 Thus, the development of vanillin-based polymers is very attractive and appreciated.Vanillin has been previously used to obtain oligo- and polyesters with different properties depending on the other monomers with which it is combined (diols, diesters, diacids, etc.).33–37 However, as far as we know, only a series of polyesters from FDME and a diol prepared by the reaction of vanillin with guaiacol and 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) has been reported.38
Thus, the last part of this work deals with the synthesis and characterization of a new bio-based polyester. Vanillin was first converted into Va-diol (2-(4-(hydroxymethyl)-2-methoxyphenoxy) ethanol) following the procedure reported in the literature19 and then, it was reacted with the CO2-FDME obtained in this work (Fig. 7).
 | ||
| Fig. 7 Synthesis of Va-diol: (2-(4-(hydroxymethyl)-2-methoxyphenoxy) ethanol) from vanillin and polycondensation with CO2-FDME to obtain the CO2-based biopolyester (CO2-PVaF). | ||
The polyester was synthetized by adapting the procedure reported for furan-based polyester.39,40 Va-diol, CO2-FDME, titanium isopropoxide, glycerol, and toluene were gradually heated at 160 °C under N2 atmosphere. After 5 minutes of reaching 160 °C, NMP was added to favour stirring and the reaction was maintained at 160 °C for 1 h. The temperature was then raised to 170 °C, also maintained for 1 h, and then to 180 °C, kept at this temperature for 45 min to complete the transesterification reaction. After this time, a vacuum was applied to the system without drying it. The temperature was then elevated to 200 °C and allowed to react for 30 min to promote the polycondensation. The reaction was cooled to r.t. and the CO2-based biopolyester was removed from the flask by adding a mixture of chloroform and trifluoroacetic acid (2:1) which dissolved it. Then, this solution was added over methanol, and CO2-PVaF precipitates as fine brown fibers.
The novel polyester was initially characterized by FT-IR (Fig. S10†). The spectrum shows the characteristic band of vibration of the carboxylic esters groups at 1722 cm−1 and the bands attributed to the C–O stretch at 1139 cm−1. The band at 1503 cm−1 and the peaks between 1272 and 754 cm−1 were attributed to the C–O and C–C stretching bands of the vanillin-derived segment as it was previously observed for other polyesters prepared from vanillin-derived monomers.36
The 1H NMR (Fig. S11†) shows two singlets at 7.38 and 7.68 ppm attributed to the protons of the furan rings and a group of wide signals between 6.7 and 7.2 ppm assigned to the aromatic protons of the vanillin moiety. The spectrum also shows a group of signals between 4 and 5.30 ppm attributed to the aliphatic protons.
The thermal stability was evaluated by TGA (Fig. S12a†). The polyester degrades in two steps. The first one, with an initial degradation temperature of 250 °C and 9% of weight loss could be attributed to the degradation of the methoxy groups attached to the vanillin segment. The second step, with an initial degradation temperature of 370 °C is ascribed to the generalized degradation of the polymer. The glass transition temperature was determined by DSC (Fig. S12b†). The value obtained was 104 °C which was higher to those reported for the series of polyesters prepared from FDME and a diol containing vanillin, guaiacol and 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO) (82–95 °C).27
Conclusions
A new strategy to prepare 2,5-Furan Dicarboxylic Methyl Ester (FDME) using CO2 and 2-methyl furoate (MF) as feedstocks has been reported. The strategy consists of two steps: a direct solvent-free carboxylation over MF using a CO2 pressure of 10 bars, Cs2CO3 as a base, and a cobalt-based hypercrosslinked polymer catalyst (HCP-Salphen-Co) followed by an acid esterification using standard conditions. The HCP catalyst was prepared through mechanochemical polymerization, a more sustainable synthesis than the conventional one since it is faster (55 min vs. 24–72 h) and is a solvent-free polymerization.The process complies with at least six principles of green chemistry but it would be necessary to recycle the methanol used in the esterification stage in order to achieve a reasonable environmental factor of 23.47. FDME is a highly sought-after monomer due to the good properties it provides to the polymers to which it gives rise. For this reason, the new CO2-based FDME prepared in this work has been reacted with a diol prepared from vanillin to obtain the corresponding CO2-based polyester which showed good thermal stability with a starting degradation temperature of 250 °C and a glass transition temperature of 104 °C.
Data avalilability
The data from this research will be available upon request.Author contributions
E. Rangel-Rangel has carried out most of the experimental work and contributed to the conceptualization of the work; B. Fuerte-Díez has done some of the experimental work; M. Iglesias has participated in the corrections of the manuscript and E. M. Maya is responsible of the work (conceptualization, funding acquisition, supervision and writing – original draft).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project CATCO2NVERS funded by the European Union's Horizon 2020 research and innovation program under grant agreement no. 101000580. We thank the company BORREGAARD for providing bio-based vanillin to carry out this work.Notes and references
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- Y. Cui, C. Deng, L. Fan, Y. Qiu and L. Zhao, Green Chem., 2023, 25, 5836–5857 RSC.
- A. Salazar, A. Linke, R. Eckelt, A. Quade, U. Kragl and E. Mejía, ChemCatChem, 2020, 12, 3504–3511 CrossRef CAS.
- Y. Feng, W. Jia, G. Yan, X. Zeng, J. Sperry, B. Xu, Y. Sun, X. Tang, T. Lei and L. Lin, J. Catal., 2020, 381, 570–578 CrossRef CAS.
- X. Guo, Y. Si, Y. Huang, J. Zhang, X. Lyu, Y. Cheng and X. Li, Ind. Eng. Chem. Res., 2023, 62, 16309–16318 CrossRef CAS.
- A. Kumar, A. S. Chauhan, R. Bains and P. Das, Chem. Eng. J., 2024, 481, 148153 CrossRef CAS.
- G. Trapasso, M. Annatelli, D. Dalla Torre and F. Aricò, Green Chem., 2022, 24, 2766–2771 RSC.
- G. Trapasso, B. Chícharo, T. Gherardi, D. Redolfi-Bristol and F. Aricò, Catalysts, 2023, 13, 1114 CrossRef CAS.
- F. W. Gomes, R. C. Lima, C. R. Piombini, J. F. Sinfitele, F. G. De Souza, P. L. A. Coutinho and J. C. Pinto, Macromol. Symp., 2018, 381, 1800129 CrossRef.
- E. Forestier, C. Combeaud, N. Guigo, G. Corvec, C. Pradille, N. Sbirrazzuoli and N. Billon, Polymers, 2021, 13, 3295 CrossRef CAS.
- M. R. Miah, Y. Dong, J. Wang and J. Zhu, ACS Sustainable Chem. Eng., 2024, 12(8), 2927–2961 CrossRef CAS.
- W. Luo, K. Jiang, Y. Li, H. Jiang and B. Yin, Org. Lett., 2020, 22, 2093–2098 CrossRef CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- Y. Liu and X.-B. Lu, Macromolecules, 2023, 56, 1759–1777 CrossRef CAS.
- L. Lin, Z. Baozhong, L. Jun, C. Weina, Y. Qiang and Z. Kepin, CN115286598A, 2022.
- A. Valverde-Gonzalez, M. Iglesias and E. M. Maya, Chem. Mater., 2021, 33, 6616–6639 CrossRef CAS.
- J.-S. M. Lee, T. Kurihara and S. Horike, Chem. Mater., 2020, 32, 7694–7702 CrossRef CAS.
- Md. A. Asraf, C. I. Ezugwu, C. M. Zakaria and F. Verpoort, Photochem. Photobiol. Sci., 2019, 18, 2782–2791 CrossRef CAS.
- C. S. Lancefield, L. W. Teunissen, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2018, 20, 3214–3221 RSC.
- R. D. Bacelis-Martínez, G. Oskam, G. Rodriguez Gattorno and M. A. Ruiz-Gómez, Adv. Mater. Sci. Eng., 2017, 2017, 1–9 CrossRef.
- Md. A. Asraf, H. A. Younus, C. I. Ezugwu, A. Mehta and F. Verpoort, Catal. Sci. Technol., 2016, 6, 4271–4282 RSC.
- X. Liao, B. Pei, R. Ma, L. Kong, X. Gao, J. He, X. Luo and J. Lin, Catalysts, 2022, 12, 62 CrossRef CAS.
- B. Fuerte-Díez, E. Rangel-Rangel, M. Iglesias and E. M. Maya, J. CO2 Util., 2024, 80, 102679 CrossRef.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- M. Liu and C. Li, Angew. Chem., Int. Ed., 2016, 55, 10806–10810 CrossRef CAS.
- T. Saitou, Y. Jin, K. Isobe, T. Suga, J. Takaya and N. Iwasawa, Chem.–Asian J., 2020, 15, 1941–1944 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- K. Sun, S. Chen, Z. Li, G. Lu and C. Cai, Green Chem., 2019, 21, 1602–1608 RSC.
- M. Annatelli, J. E. Sánchez-Velandia, G. Mazzi, S. V. Pandeirada, D. Giannakoudakis, S. Rautiainen, A. Esposito, S. Thiyagarajan, A. Richel, K. S. Triantafyllidis, T. Robert, N. Guigo, A. F. Sousa, E. García-Verdugo and F. Aricò, Green Chem., 2024, 26, 8894–8941 RSC.
- H. Qiang, J. Wang, H. Liu and Y. Zhu, Polym. Chem., 2023, 14, 4255–4274 RSC.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Polym. Chem., 2015, 6, 6058–6066 RSC.
- C. Zhao, C. Huang, Q. Chen, I. D. V. Ingram, X. Zeng, T. Ren and H. Xie, Polymers, 2020, 12, 586 CrossRef CAS.
- W. Dong, H. Peng, R. Zhang, C. He, Y. Gu, Q. Yang, S. Chen, J. Lan and B. Yan, ACS Appl. Polym. Mater., 2023, 5, 10010–10020 CrossRef CAS.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- K. Lee and P. H. Lee, Tetrahedron Lett., 2008, 49, 4302–4305 CrossRef CAS.
- M. Jiang, Q. Liu, Q. Zhang, C. Ye and G. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1026–1036 CrossRef CAS.
- T. P. Kainulainen, J. A. Sirviö, J. Sethi, T. I. Hukka and J. P. Heiskanen, Macromolecules, 2018, 51, 1822–1829 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00426d |
| This journal is © The Royal Society of Chemistry 2024 |