
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferential glycosylation does not modulate the conformational heterogeneity of a humanised IgGk NIST monoclonal antibody†
Fanny C.
Liu
 *a,
Jusung
Lee
a,
Thais
Pedrete
a,
Erin M.
Panczyk
b,
Stuart
Pengelley
c and
Christian
Bleiholder
*ad
*a,
Jusung
Lee
a,
Thais
Pedrete
a,
Erin M.
Panczyk
b,
Stuart
Pengelley
c and
Christian
Bleiholder
*ad
aDepartment of Chemistry and Biochemistry, Florida State University, 102 Varsity Way, Tallahassee, Florida 32306, USA. E-mail: cbleiholder@fsu.edu; fliu@fsu.edu
bBruker Daltonics, 40 Manning Road, Billerica, MA 01821, USA
cBruker Daltonics GmbH&Co, Fahrenheitstrasse 4, Bremen, 28359, Germany
dInstitute of Molecular Biophysics, Florida State University, 91 Chieftan Way, Tallahassee, Florida 32306, USA
First published on 3rd September 2024
Abstract
Investigating the structural heterogeneity of monoclonal antibodies is crucial to achieving optimal therapeutic outcomes. We show that tandem-trapped ion mobility spectrometry enables collision-induced unfolding measurements of subpopulations of a humanised IgGk NIST monoclonal antibody (NISTmAb). Our results indicate that differential glycosylation of NISTmAb does not modulate its conformational heterogeneity.
Monoclonal antibodies (mAbs) are the largest class of biotherapeutics, displaying a vast structural diversity that enables the recognition of various antigens.1 Biological activity and physicochemical stability are the two key aspects when rationally designing and engineering mAbs for therapeutic purposes. The biological activity of mAbs is determined by their specificity towards their respective antigens. The antibody-antigen binding follows a conformational selection mechanism where the binding-competent mAb conformation is selected from an ensemble of existing solution conformations.2 The physical and chemical stability3 of mAbs is just as important to characterise because denatured mAbs can self-assemble into multimeric aggregates that can be related to immunogenicity. These considerations underline the importance of characterising the conformational heterogeneity and the conformational-dependent stability of therapeutic mAbs.
However, it is challenging to investigate the structural heterogeneity of mAbs. Only four X-ray structures of intact mAb subclass IgG are available in the Protein Data Bank (PDB), highlighting the difficulties associated with crystallising intact mAbs. Further, crystal packing potentially distorts their three-dimensional structure.1 Moreover, studying glycoproteins such as mAbs by NMR spectroscopy remains technically challenging.4
Ion mobility/mass spectrometry (IM/MS) characterises proteins in the absence of bulk solvents by their mass-to-charge ratios and collision cross-sections.5 Mounting evidence suggests that these measurements largely retain native protein structures,5–7 because a rugged free-energy surface quenches protein unfolding by several orders of magnitude in the absence of a bulk solvent.8 In conjunction with collision-induced unfolding (CIU), IM/MS has been applied to probe structures and stabilities of multi-domain proteins, including mAbs.9–12 In combination with molecular dynamics (MD) simulations, IM/MS suggested that the broad peaks in IM/MS spectra of mAbs may be related to their hinge motions.13 Nevertheless, conventional IM/MS methods conduct ensemble-type measurements that characterise the entirety of the mAb conformational space and hence characterise their structures or stabilities only by their ensemble averages. Antibodies, however, are heterogeneous systems. Hence, conformationally resolved mAb characteristics could potentially be exploited to improve rational antibody design, engineering, or quality control.
Here, we present an approach for probing the structural heterogeneity and conformational-dependent stability of mAbs. We accomplish this by mobility-resolved collision-induced unfolding (mr-CIU) measurements enabled by tandem-trapped ion mobility spectrometry (Tandem-TIMS). Tandem-TIMS, developed in our laboratory jointly with Bruker Daltonics, comprises two adjacent TIMS devices linked by an interface region and is embedded in a QqTOF mass spectrometer (Fig. 1B).14–16 Tandem-TIMS is a versatile analytical method enabling non-ensemble mr-CIU measurements.17–19 Such mr-CIU workflows mobility-separate protein subpopulations and measure their collision cross-sections in TIMS-1, select a subpopulation by timing voltages at electrodes L1 and L2, collisionally activate the selected subpopulation between L3 and V1, and analyse the changes in their cross-sections in TIMS-2 (Fig. 1B). To test if mr-CIU allows probing the structural heterogeneity of mAbs, we used the recombinant humanised IgG monoclonal antibody NISTmAb (∼148 kDa) as prototype.20,21 At least six glycan sequences attached to Asn297 of NISTmAb were reported,22 and the crystal structure of the Fab domain (PDB ID: 5K8A) and the Fc domain (PDB ID: 5VGP) are known.23
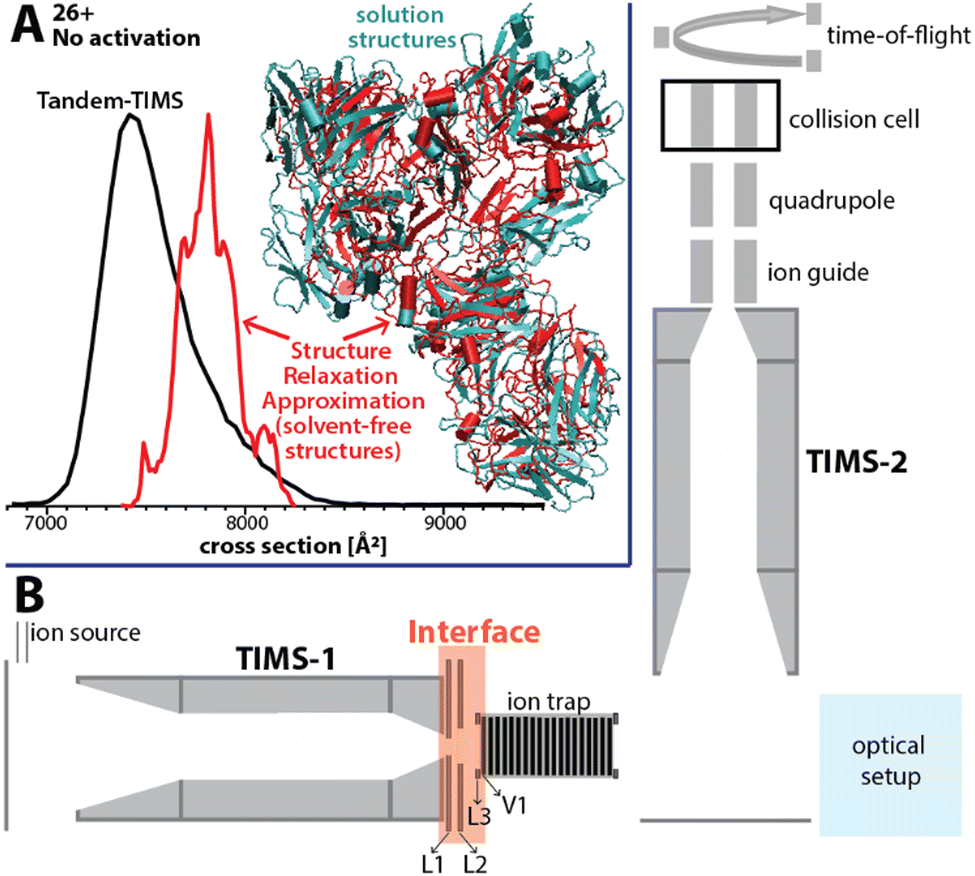 | ||
| Fig. 1 (A) The ion mobility spectrum computed by our computational Structure Relaxation Approximation for NISTmAb (red traces) agrees well with the experimental spectrum (black trace). The representative solvent-free NISTmAb structure computed by the SRA for charge state 26+ (red, cartoon) indicates strong retention of native contacts with respect to the representative structure obtained from explicit solvent molecular dynamics simulations (cyan, cartoon). (B) Schematics of the Tandem-TIMS instrument coupled to a QqTOF mass spectrometer. Tandem-TIMS comprises of two TIMS devices, an interface region with electrodes (L1, L2, L3, V1) capable of applying high electric fields to select and activate analyte ions. (See ref. 14–19 for details). | ||
First, we verified that Tandem-TIMS yields information on NISTmAb consistent with previously reported ensemble-type IM/MS measurements in terms of the nativeness of NISTmAb structures and its glycoforms.9,22,24 To retain native-like NISTmAb structures in Tandem-TIMS, we employed gentle conditions during electrospray ionisation (ESI) and minimised direct current (DC) and radio-frequency (RF) electric field strengths.25,26 In line with prior literature, NISTmAb charge states 23+ to 28+ dominate the Tandem-TIMS mass spectrum (Fig. S1A, ESI†).22 Fig. S2 and Table S1 (ESI†) show that the NISTmAb collision cross sections recorded by Tandem-TIMS agree to better than 1% with those recorded on an RF-confining drift tube ion mobility spectrometer.22 Next, we confirmed the ability of Tandem-TIMS to correctly identify the dominant intact NISTmAb glycoforms observed by Fourier-transform ion cyclotron resonance (FTICR) mass spectrometry. To this end, we removed solvent/salt adducts by collisional activation between TIMS-1 and TIMS-2 (200 V placed between electrodes L3 and V1) and applying 100 V between TIMS-2 and the quadrupole ion guide (see Fig. 1B). The deconvolved mass spectrum (Fig. S3, ESI†) reveals six well-resolved NISTmAb species that are consistent with the glycoforms (Table S2, ESI†) observed by FTICR in terms of their masses and relative abundances.12 Finally, we validated that ensemble-type CIU measurements with Tandem-TIMS are consistent with prior reports from other IM/MS instruments.22 Fig. S4 (ESI†) plots the cross-section distributions of charge state 26+ recorded upon varying the voltage between electrodes L3 and V1 (Fig. 1B) from 5 V to 200 V. The spectra reveal that an increased activation voltage induces the unfolding of NISTmAb in three main stages. First, NISTmAb compacts from ∼7420 Å2 to ∼7320 Å2 when the activation voltage is increased to 120 V. Subsequent activation to 180 V unfolds NISTmAb to ∼7660 Å2, which is followed by a significant unfolding transition to ∼8770 Å2 between 185 V – 200 V. The compaction at low activation voltages and the subsequent minor and significant unfolding stages are consistent with prior reports.22 Our findings thus demonstrate that ensemble-type CIU of NISTmAb charge state 26+ in Tandem-TIMS is comparable to that observed in conventional IM/MS instruments.
We used our structure relaxation approximation (SRA)27 to estimate NISTmAb structures for the native-like and collisionally-unfolded solvent-free experiments. Briefly, we carried out explicit-solvent molecular dynamics (MD) simulations starting from the proposed full-length NISTmAb structure20 and used the sampled structures as input for the SRA to estimate native-like NISTmAb structures for charge states 23+, 26+, and 27+ (Fig. 1A and Fig. S5 to S7, ESI†). The computed spectrum for charge state 26+ centres at ∼7800 Å2, which agrees with the experiment to within ∼5% (Fig. 1A). The corresponding structures proposed by the SRA (Fig. 1A, inset) suggest a fraction of native contacts Q of ∼0.75 (on a scale from 0 to 1.0 where 1.0 indicates identical structures; Fig. S6, ESI†) for all calculated charge states. Additionally, the SRA suggests that aspects of the solution-phase structural heterogeneity of NISTmAb are retained in the absence of bulk solvent (Fig. S11, ESI†). Thus, our data underscore that native IM/MS probes strongly native NISTmAb structures. Further, to identify the structural changes of NISTmAb upon CIU, we simulated the vibrational activation in the CIU experiments by successively increasing the molecular dynamics simulation temperature (Fig. S5 to S7, ESI†). This allowed us to correlate the experimentally observed increase in collision cross-section upon CIU to the loss of native contacts observed in the simulations. The data highlight that NISTmAb ions have lost essentially all native contacts after the first unfolding transition observed in Tandem-TIMS. The simulations further indicate that the major unfolding transition observed in Fig. S4 (ESI†) between 185 V and 200 V arises primarily from the unfolding of the Fab domain whereas binding between the heavy chains in the Fc-domain remains relatively intact.
Nevertheless, the biological activity and physicochemical properties of mAbs are dictated by their conformational heterogeneity1,3,28 because antigen recognition follows a conformational selection mechanism.2 Characterising the conformational diversity of mAbs thus appears imperative for optimising their biological activity and improving their clinical efficacy. However, conventional ensemble-averaged CIU measurements do not directly access this information.
By contrast, Tandem-TIMS partitions an ensemble of protein species into its constituent subpopulations and characterises their stability individually.14,17,19 Here, we carried out non-ensemble mr-CIU measurements with Tandem-TIMS to test if they enable the structural heterogeneity of NISTmAb and the differential stability of its subpopulations to be characterised.
The structural heterogeneity of NISTmAb is highlighted by the ensemble-type ion mobility spectrum featuring a single peak that is much broader than expected for a single conformation (Fig. 1A). We thus partitioned this ensemble into subpopulations by mobility-selecting NISTmAb ions eluting from TIMS-1. This is accomplished by timing the electric potential at electrodes L1 and L2 (Fig. 1B) with the mobility scan of TIMS-114 such that only the subpopulation of interest is transmitted through L1–L2 whereas all others are blocked.14 Subsequently, the cross-section of the selected subpopulation is measured in TIMS-2. Fig. 2A plots the ion mobility spectrum of the total ion population of charge state 26+ superimposed with the spectra recorded for four selected subpopulations. The data show that the cross-sections of the subpopulations (7260 Å2, 7370 Å2, 7625 Å2, and 7945 Å2) differ from that of the total ion population (∼7420 Å2). Fig. S8 (ESI†) shows the equivalent analysis for charge state 25+. Because the time for ions from selection to elution from TIMS-2 is roughly 50 ms to 100 ms, the data indicate that the broad NISTmAb peaks comprise non-interconverting species that are kinetically stable for at least 50 ms to 100 ms.
 | ||
| Fig. 2 Structural heterogeneity of NISTmAb probed by non-ensemble Tandem-TIMS measurements. (A) The ion mobility peak of charge state 26+ (black trace) is superimposed with four subpopulations (blue, red, orange, and light brown traces). The subpopulations correspond to distinct collision cross-sections and do not interconvert for at least ∼100 ms, which indicates that these are kinetically stable species. (B) The mass spectrum of each subpopulation shows six dominant glycoforms with the same m/z and similar relative abundance, indicating that the subpopulations comprise the same glycoforms. (C) The initial average mass difference between subpopulations 1 and 2 is ∼325 Da. The average m/z of subpopulation 1 at an activation voltage of 5 V is identical to that obtained at 80 V for subpopulation 2 (m/z ∼5880). (D) Despite having the same mass and, thus, the same number of attached solvent adducts, the subpopulations maintain the differences in their collision cross-sections of ∼10%. | ||
The most likely factors leading to kinetically stable, non-interconverting NISTmAb subpopulations are (1) variations in the NISTmAb glycoforms, (2) different numbers of solvent/salt adducts, and (3) conformational heterogeneity.
If glycoform variations were responsible for the observed NISTmAb subpopulations, then the mass spectrum of each subpopulation should show unique glycoform patterns. To test this, we plot the mass spectra of the four subpopulations of charge state 26+ in Fig. 2B. The spectra for all subpopulations exhibit the same patterns in terms of both the m/z of the glycoforms and their relative abundances, which indicates that the NISTmAb subpopulations are predominated by the same glycoforms in largely identical ratios. Fig. S8B (ESI†) shows the same observations for charge state 25+. Therefore, the presence of distinct NISTmAb subpopulations is unlikely due to glycoform variations. These results are consistent with prior literature indicating no impact of IgG1 glycosylation on its tertiary/quarternary structure.29
Next, we investigated if the subpopulations differ only in the number of solvent adducts. The initial average mass difference between the most compact (7260 Å2) and the most extended (7945 Å2) subpopulations for charge state 26+ is ∼325 Da (Fig. 2C). This mass difference corresponds to roughly 18 water molecules, which cannot physically account for a cross-section difference of ∼700 Å2. Further, this difference in collision cross-section is maintained even when their average m/z is comparable (Fig. 2D and Table S3, ESI†). Note that the equivalent observations are made for NISTmAb charge state 25+ (Fig. S8, ESI†).
Taken together, our data suggest that the kinetically stable, distinct NISTmAb subpopulations most likely reflect different NISTmAb conformations. To test this, we conducted mobility-resolved collision-induced unfolding (mr-CIU) experiments. Fig. S9 (ESI†) depicts the ion mobility spectra of two NISTmAb subpopulations for charge state 26+ that are collisionally activated by applying 5 V to 200 V between L3 and V1 and Fig. 3 plots the corresponding CIU fingerprints for the mobility-resolved subpopulations.
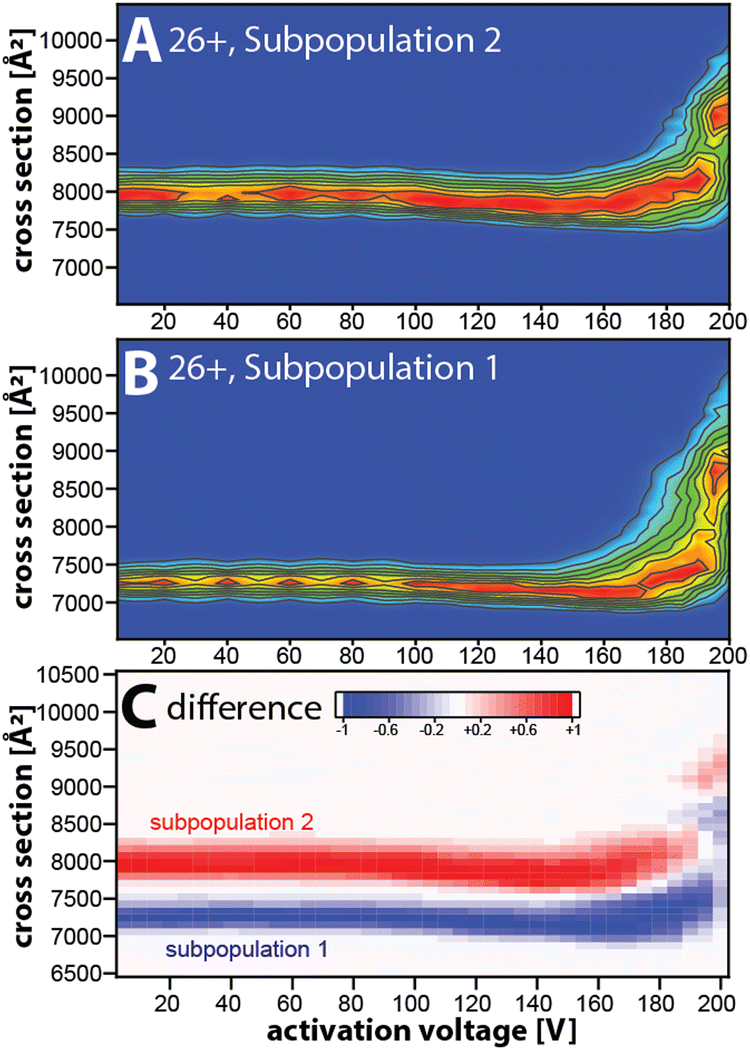 | ||
| Fig. 3 Conformational heterogeneity of NISTmAb probed by mobility-resolved CIU (mr-CIU) fingerprints obtained via non-ensemble Tandem-TIMS measurements. (A), (B) Mobility-resolved CIU fingerprints of NISTmAb 26+ subpopulations with initial cross-sections centred at ∼7260 Å2 and ∼7945 Å2, respectively. (C) Difference plot highlighting the differences in the CIU fingerprints of the NISTmAb subpopulations. | ||
Several observations in Fig. 3 deserve comment. First, the ion mobility spectra of the compact subpopulation do not show a kinetically stable feature with a cross-section of ∼7945 Å2 consistent with the extended subpopulation at any activation voltage. This implies that the unfolding pathway of the compact subpopulation does not proceed through conformational states associated with the more extended subpopulation. This observation is not expected if one assumes that the subpopulations were produced during or after ESI. Hence, our data suggest that the subpopulations originate from the NISTmAb solution-phase conformational heterogeneity. We underline that this notion aligns well with prior IM/MS studies.19,30 Second, CIU reveals differences in the unfolding characteristics of the NISTmAb subpopulations (Fig. 3C). For instance, the cross-sections for the subpopulations differ even at an activation voltage of 200 V where both populations have unfolded into structures with approximately 15–20% larger cross-sections than their initial, native-like conformations. Analogous observations are made for mobility-resolved CIU of charge state 25+ (Fig. S10, ESI†).
We summarise: (1) mobility-resolved CIU measurements enabled by Tandem-TIMS show that the broad peaks in the ion mobility spectra of NISTmAb consist of kinetically stable subpopulations that most likely originate from the solution-phase conformational heterogeneity of NISTmAb. (2) The mobility-resolved CIU fingerprints reveal the differences in the unfolding characteristic of the NISTmAb subpopulations.
With the rise of multispecific antibody therapeutics, there is a dire need for novel technologies that characterise heterogeneous and complex samples. Multispecific antibodies simultaneously target multiple antigens and, therefore, exhibit more complex biological activity and physicochemical stability than monovalent mAbs such as NISTmAb. Our data presented here highlights that non-ensemble CIU fingerprints enabled by mobility-resolved Tandem-TIMS measurements would be effective in characterising their structural and chemical complexity, thereby aiding in the rational design and engineering of complex biotherapeutics.
This work is supported by the National Science Foundation (CHE 2305173) and the National Institutes of Health (R01GM135682).
Data availability
The data supporting this article are found in the ESI.†Conflicts of interest
S. P. and E. M. P. are employees at Bruker Corporation, which sells timsTOF instruments.Notes and references
- M. L. Fernández-Quintero, G. Georges, J. M. Varga and K. R. Liedl, mAbs, 2021, 13, 1923122 CrossRef.
- M. L. Fernández-Quintero, J. Kraml, G. Georges and K. R. Liedl, mAbs, 2019, 11, 1077–1088 CrossRef.
- E. Garber and S. J. Demarest, Biochem. Biophys. Res. Commun., 2007, 355, 751–757 CrossRef CAS.
- L. Unione, A. Ardá, J. Jiménez-Barbero and O. Millet, Curr. Opin. Struct. Biol., 2021, 68, 9–17 CrossRef CAS PubMed.
- B. T. Ruotolo, K. Giles, I. Campuzano, A. M. Sandercock, R. H. Bateman and C. V. Robinson, Science, 2005, 310, 1658–1661 CrossRef CAS PubMed.
- T. Wyttenbach and M. T. Bowers, J. Phys. Chem. B, 2011, 115, 12266–12275 CrossRef CAS.
- S. L. Koeniger, S. I. Merenbloom, S. Sevugarajan and D. E. Clemmer, J. Am. Chem. Soc., 2006, 128, 11713–11719 CrossRef CAS.
- T. C. Cropley, F. C. Liu, M. Chai, M. F. Bush and C. Bleiholder, J. Am. Chem. Soc., 2024, 146, 11115–11125 CAS.
- Y. Tian, L. Han, A. C. Buckner and B. T. Ruotolo, Anal. Chem., 2015, 87, 11509–11515 CrossRef CAS.
- J. D. Eschweiler, R. M. Martini and B. T. Ruotolo, J. Am. Chem. Soc., 2017, 139, 534–540 CrossRef.
- E. J. Larson, D. S. Roberts, J. A. Melby, K. M. Buck, Y. Zhu, S. Zhou, L. Han, Q. Zhang and Y. Ge, Anal. Chem., 2021, 93, 10013–10021 CrossRef.
- I. D. G. Campuzano, J. H. Robinson, J. O. Hui, S. D.-H. Shi, C. Netirojjanakul, M. Nshanian, P. F. Egea, J. L. Lippens, D. Bagal, J. A. Loo and M. Bern, Anal. Chem., 2019, 91, 9472–9480 CrossRef PubMed.
- K. J. Pacholarz, M. Porrini, R. A. Garlish, R. J. Burnley, R. J. Taylor, A. J. Henry and P. E. Barran, Angew. Chem., Int. Ed., 2014, 53, 7765–7769 CrossRef.
- F. C. Liu, M. E. Ridgeway, M. A. Park and C. Bleiholder, Analyst, 2018, 143, 2249–2258 RSC.
- F. C. Liu, M. E. Ridgeway, M. A. Park and C. Bleiholder, Analyst, 2022, 147, 2317–2337 RSC.
- F. C. Liu, M. E. Ridgeway, J. S. R. V. Winfred, N. C. Polfer, J. Lee, A. Theisen, C. A. Wootton, M. A. Park and C. Bleiholder, Rapid Commun. Mass Spectrom., 2021, 35, e9192 CrossRef PubMed.
- F. C. Liu, T. C. Cropley, M. E. Ridgeway, M. A. Park and C. Bleiholder, Anal. Chem., 2020, 92, 4459–4467 CrossRef CAS.
- F. C. Liu, T. C. Cropley and C. Bleiholder, J. Am. Soc. Mass Spectrom., 2023, 34, 2247–2258 CrossRef CAS.
- T. C. Cropley, F. C. Liu, M. Chai, M. F. Bush and C. Bleiholder, J. Am. Chem. Soc., 2024, jacs.3c12892 CrossRef.
- C. Bergonzo and D. T. Gallagher, J. Res. Natl. Inst. Stand. Technol., 2021, 126, 126012 CrossRef CAS.
- M. Hilliard, W. R. Alley, C. A. McManus, Y. Q. Yu, S. Hallinan, J. Gebler and P. M. Rudd, mAbs, 2017, 9, 1349–1359 CrossRef CAS PubMed.
- I. D. G. Campuzano, C. Larriba, D. Bagal and P. D. Schnier, in ACS Symposium Series, ed. J. E. Schiel, D. L. Davis and O. V. Borisov, American Chemical Society, Washington, DC, 2015, vol. 1202, pp. 75–112 Search PubMed.
- D. T. Gallagher, C. V. Galvin and I. Karageorgos, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 524–529 CrossRef CAS.
- Y. Tian and B. T. Ruotolo, Int. J. Mass Spectrom., 2018, 425, 1–9 CrossRef CAS.
- F. C. Liu, S. R. Kirk and C. Bleiholder, Analyst, 2016, 141, 3722–3730 RSC.
- C. Bleiholder, F. C. Liu and M. Chai, Anal. Chem., 2020, 92, 16329–16333 CrossRef CAS.
- C. Bleiholder and F. C. Liu, J. Phys. Chem. B, 2019, 123, 2756–2769 CrossRef CAS PubMed.
- A. Niedziela-Majka, E. Kan, P. Weissburg, U. Mehra, S. Sellers and R. Sakowicz, SLAS Discovery, 2015, 20, 552–559 CrossRef CAS PubMed.
- G. P. Subedi and A. W. Barb, Structure, 2015, 23, 1573–1583 CrossRef CAS.
- S. J. Allen, R. M. Eaton and M. F. Bush, Anal. Chem., 2017, 89, 7527–7534 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Ion mobility and mass spectra; structure relaxation approximation calculations. See DOI: https://doi.org/10.1039/d4cc02125h |
| This journal is © The Royal Society of Chemistry 2024 |