
Photoinduced oxidation of benzylic boronic esters to ketones/aldehydes via α-borylalkyl radicals†
Zhijun
Wang
,
Lanfeng
Wei
,
Jichang
Liu
 *,
Yu
Wei
* and
Liang
Xu
*
*,
Yu
Wei
* and
Liang
Xu
*
School of Chemistry and Chemical Engineering/State Key Laboratory Incubation Base for Green Processing of Chemical Engineering, Shihezi University, Shihezi, 832003, China. E-mail: xuliang4423@shzu.edu.cn
First published on 9th November 2022
Abstract
A method for direct oxidation of boronic esters to the carbonyl groups has been established herein. Under the irradiation of visible light and O2 atmosphere, ketones and aldehydes can be obtained in moderate to excellent yields. Using tetrabutylammonium tribromide (TBATB) as the catalyst, this procedure avoids using of metal-based (photo)catalysts. α-Borylalkyl radicals have been proposed as the key intermediates of this oxidation process.
Introduction
Organoboron species play significant roles in organic synthesis. Due to their versatile reactivity, they have been utilized to introduce diverse functionalities selectively, and thus build molecular complexity efficiently.1 A variety of transformations of C–B to C–C or C–heteroatom bonds2 have been intensively studied and widely applied, including Brown-type oxidation of organoboron reagents.3 In this context, oxidation of alkylboron species mostly affords alcohols with C–O single bonds. In comparison, the conversion from alkylborons to the carbonyl moieties with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds has been far less explored.
O double bonds has been far less explored.
A close inspection of the literature revealed several existing strategies. In 2016, Floreancig, Deiters and their coworkers disclosed that oxidation of α-O-substituted alkylboron with H2O2 would release aldehydes/ketones through the collapse of hemiacetal intermediates (Scheme 1a).4 In 2019, Partridge group reported a copper catalyzed method for the selective oxidation of benzylic boronic esters to benzoyl moieties, using air as the terminal oxidant (Scheme 1b).5 Later, Penhoat group realized continuous photocatalyzed aerobic oxidation of benzylic trifluoroborates to benzaldehydes, using UV irradiation and Ir-based photocatalyst to generate benzylic radical intermediates (Scheme 1c).6 In spite of these achievements,7 generally, from alkylborons to the carbonyl moieties, sequential oxidation processes are still required via the intermediate of alcohols.8 This situation leaves ample space to develop more step-economical pathways that directly connect alkylborons and the carbonyl groups in a single vessel.
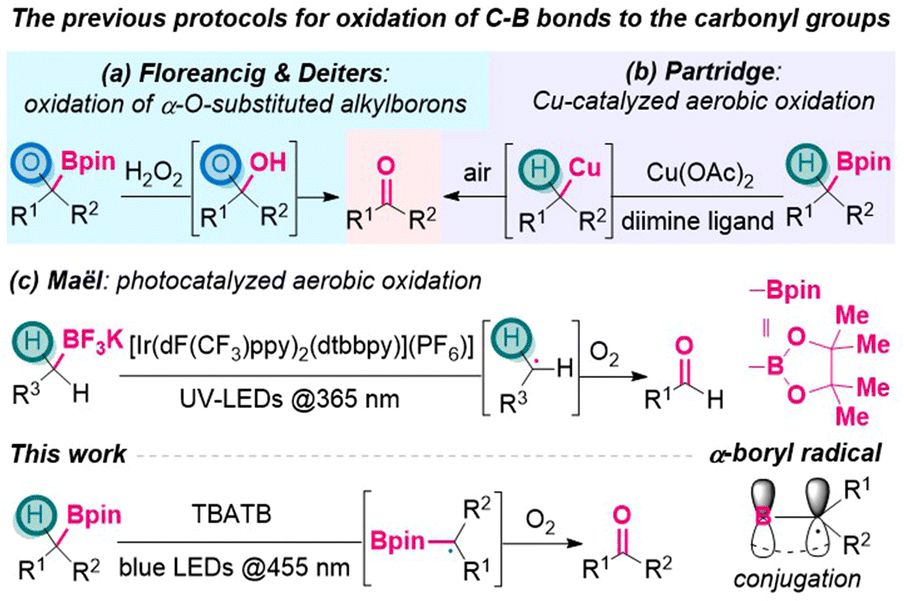 | ||
| Scheme 1 Construction of carbonyl groups from alkylboron species. | ||
On the other hand, the generation and application of α-borylalkyl radicals have been receiving increasing attention from the organic community,9 along with the prosperity of organoboron chemistry and radical reactions10 in the last decade. As depicted in Scheme 1, α-boryl radicals are usually stabilized by delocalization onto the empty p-orbital of the sp2-boron atom through conjugation effect,11 which renders their generation relatively easier and thus enables diverse downstream transformations,12 including the construction of α-heteroatom-substituted alkylborons.13 For example, in 1970, Brown group realized α-bromination of organoboranes via α-boryl radicals that were generated from hydrogen atom transfer (HAT) to bromine radicals.13d
Grounded on this, it was questioned whether α-borylalkyl radicals could be utilized to forge the carbonyl groups. To the best of our knowledge, no such processes had been described in the literature. As part of our efforts in the development of photocatalytic oxidation reactions,14 herein we disclose a metal-free visible-light induced protocol to oxidize boronic esters to ketones/aldehydes directly, using tetrabutylammonium tribromide (n-Bu4N+Br3−, TBATB) as the catalyst (Scheme 1).
Results and discussion
The investigation was initiated by exploring the reaction of (1-phenylethyl)boronic pinacol ester 1a in the presence of bromine radical donor TBATB, which was a shelf-stable and involatile solid, and also cheap and readily available.15 Fortunately, preliminary attempts led to the formation of the target product acetophenone 2a, under irradiation of visible-light and an O2 atmosphere.As indicated in Table 1, the optimal reaction conditions were established by systematically evaluating reaction parameters, including the dosages of TBATB, light sources, reaction time, and solvents. With tetrahydrofuran (THF) as the solvent, under an O2 atmosphere and irradiation of 10 W blue LEDs (455 nm), the HPLC yield of the target product 2a could reach 98% after 16 hours’ stirring, in the presence of 10 mol% of TBATB (Table 1, entry 1). The crucial roles of the above-mentioned parameters were then validated by control experiments. In the absence of TBATB, oxygen, or visible light, the signal of 2a could not be detected by HPLC analysis, indicating this transformation was a photoinduced TBATB-catalyzed aerobic process (entries 2–4). Furthermore, only trace 2a could be observed when the oxygen atmosphere was replaced by an air atmosphere (entry 6), implying a high concentration of oxygen was necessary to enable this transformation. Finally, the effect of different solvents was examined (entries 7–13). While alcoholic solvents, NMP and MeCN resulted in 67–93% yields of 2a, the application of DMSO and H2O was found detrimental, totally suppressing the desired transformation.
|
|
||
|---|---|---|
| Entry | Variations from the ‘standard’ conditions | Yieldb (%) |
| a Standard reaction conditions: 1a (0.20 mmol), TBATB (0.02 mmol, 10 mol%), O2 atmosphere, solvent (1.0 mL), rt = room temperature, 10 W 455 nm blue LEDs, 16 hours. b HPLC yield. c N.D. = not detected. | ||
| 1 | None | 98 |
| 2 | Without TBATB | N.D.c |
| 3 | Without O2 (under N2) | N.D. |
| 4 | Without LEDs | N.D. |
| 5 | White LEDs instead of blue LEDs | 69 |
| 6 | Air instead of O2 | Trace |
| 7 | EtOH instead of THF | 87 |
| 8 | n-PrOH instead of THF | 93 |
| 9 | i-PrOH instead of THF | 82 |
| 10 | NMP instead of THF | 67 |
| 11 | MeCN instead of THF | 85 |
| 12 | DMSO instead of THF | Trace |
| 13 | H2O instead of THF | Trace |
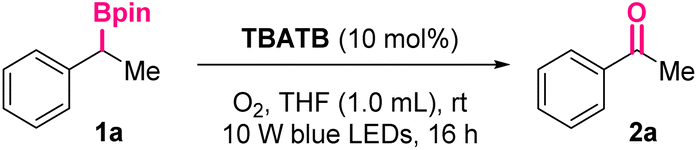
Under the above-mentioned optimal conditions, we investigated the substrate scope of this photoinduced oxidation reaction (Scheme 2). As shown, acetophenone 2a could be obtained in 94% isolated yield.
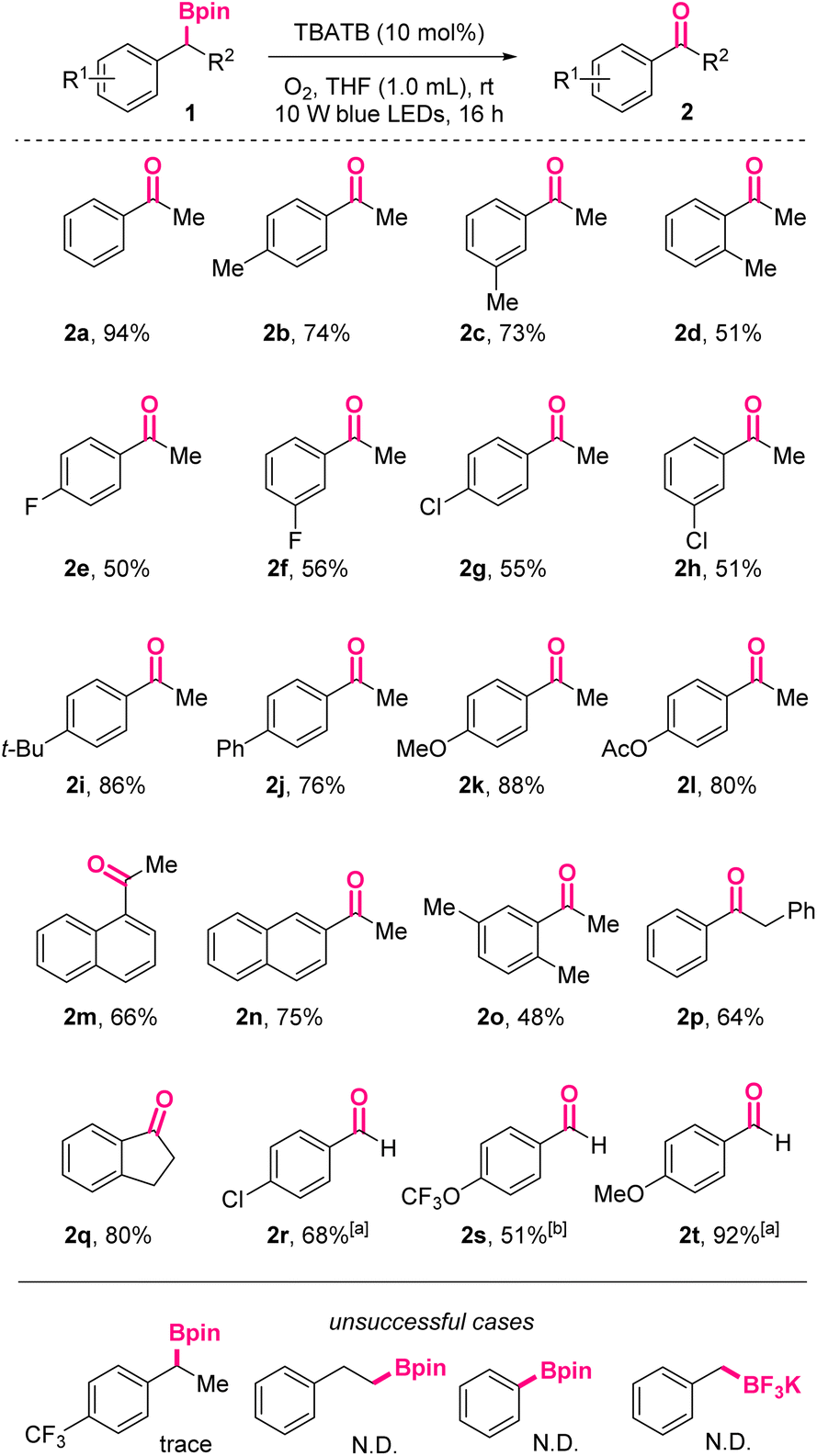 | ||
Scheme 2 Substrate scope for photoinduced oxidation of boronic esters to ketones/aldehydes. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NMP as solvent, TBATB (20 mol%). bNMP as solvent, TBATB (20 mol%), 30 hours. NMP as solvent, TBATB (20 mol%). bNMP as solvent, TBATB (20 mol%), 30 hours. | ||
Substrates with diverse substituents were well compatible with the conditions. Generally, the reactivity of benzylic boronic esters with electron-donating groups on the phenyl skeletons are higher than those with electron-withdrawing groups, no matter for the synthesis of ketones or aldehydes. For example, para-OMe-acetophenone 2k and para-OMe-benzaldehyde 2t were obtained in 88% and 92 yields, respectively, while their para-Cl counterparts were isolated in only 55% and 68% yields under the same conditions. When para-CF3-acetophenone was applied, only trace oxidized product was observed, further confirming the negative effect of electron-withdrawing groups. On the macro level, it was well-known electron-rich substrates were more susceptible to be oxidized. On the molecular level, this difference might be rationalized by the captodative or push–pull stabilization effect on the involved benzylic radical intermediates, which were substituted with electron-accepting functionalities like α-boryl. In addition, among ortho-, meta-, and para-methyl-substituted boronic esters, the ortho-substituted case resulted in the lowest yield (2d, 51% vs.2b, 74% vs.2c, 73%), indicating the negative influence of steric hindrance.
For the substrates with halogen atoms as substituents, the corresponding products 2e, 2f, 2g, 2h were obtained in moderate yields, leaving opportunities for further decoration of these products. Furthermore, naphthyl starting materials with more extended conjugated systems afforded 2m and 2n in 66% and 75% yields, respectively. When R2 was benzyl, 2p was obtained in 64% yield. Cyclic 1-indanone 2q could also be accessed via this protocol in 80% yield. When R2 was hydrogen atoms, the corresponding aromatic aldehydes would be exclusively obtained in moderate to excellent yields, without the formation of the over-oxidized carboxylic acids.
The preliminary attempts to oxidize inactivated aliphatic boryl species under these conditions reached no success. Also, trifluoroborates were found to be not suitable for this reaction system, probably due to the absence of vacant p-orbitals.
Subsequently, a series of control experiments were carried out to shed light on the reaction mechanism (Scheme 3). In the beginning, ethylbenzene 3 without substituents at the benzylic position was treated under the standard reaction conditions. Acetophenone 2a could not be detected by the HPLC analysis of the reaction mixture, thus ruling out the possibility of an initial deborylative protonation and subsequent benzylic oxidation pathway (Scheme 3a). Then, in the reaction was added water-18O (5.0 equiv.), which would not affect the overall yield of the oxidized product significantly (87%). However, mass spectrum analysis of the obtained product revealed the absence of 18O-labeled 2a. Taking the necessity of O2 for the conversion (Table 1, entries 3 and 6) into consideration, it was proposed the oxygen of 2a came from O2. Furthermore, when a typical radical scavenger, 2,2,6,6-tetramethylpiperidinooxy (TEMPO, 2.0 equiv.), was added, the formation of 2a was completely suppressed, indicating the reaction might go through a free radical process. Finally, after adding t-butyl alcohol (TBA, 2.0 equiv.), a hydroxyl radical scavenger, 2a was still generated in 58% yield, implying the hydroxyl radical might not be involved in the reaction mechanism.
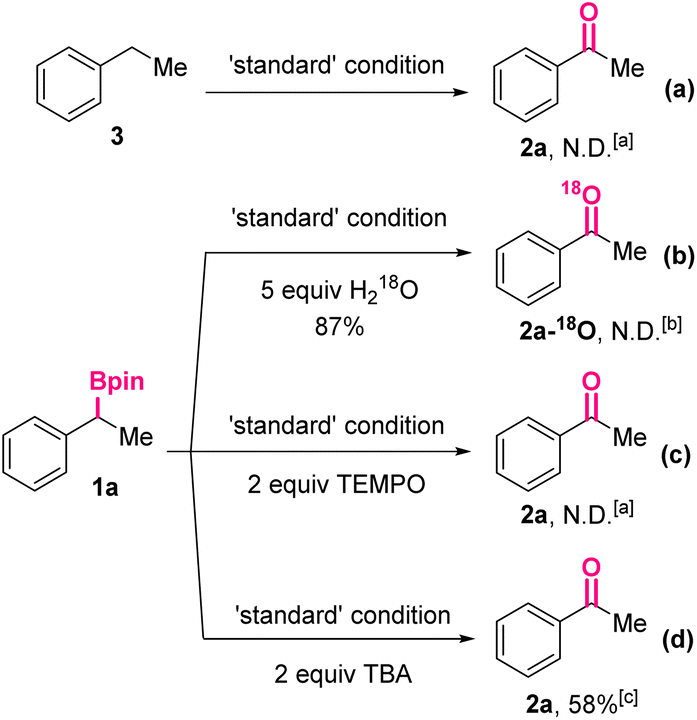 | ||
| Scheme 3 Control experiments. aNot detected by HPLC. bNot detected by HRMS. cHPLC yield. | ||
Furthermore, (1-bromoethyl)benzene 5 and para-tert-butylbenzyl bromide 6 were treated under the optimized conditions (Scheme 4). The corresponding carbonyl product 2a and 7 could be isolated in moderate yields, indicating the reactions using benzyl bromides as the starting materials might share similar intermediates with the reactions using benzyl boronic esters.
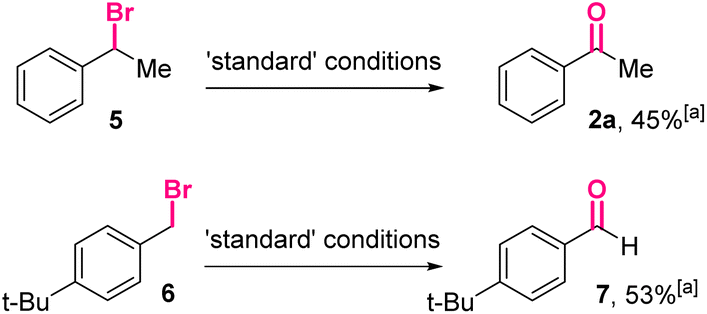 | ||
| Scheme 4 The oxidation of benzyl bromides. aIsolated yield. | ||
Accordingly, the possible reaction pathways have been depicted in Scheme 5. The reaction began with taking away the benzylic hydrogen atom from 2a by the bromine radical (Br˙), which originated from the homolytic cleavage of Br–Br bonds in TBATB under illumination.16 Under the oxygen atmosphere, the α-borylbenzyl radical IM1 would trap one O2 to generate peroxyl radical IM2. Then, IM2 and hydrogen bromide (HBr) underwent a hydrogen atom transfer (HAT) process to afford IM3 and regenerate the bromine radical (Br˙), thus completing the catalytic cycle. Finally, intramolecular elimination of HOBpin from IM3 occurred, leading to the product 2a. The above-mentioned process should also be applicable for the conversion of benzylic bromides, via the intermediates IMA and IMB.
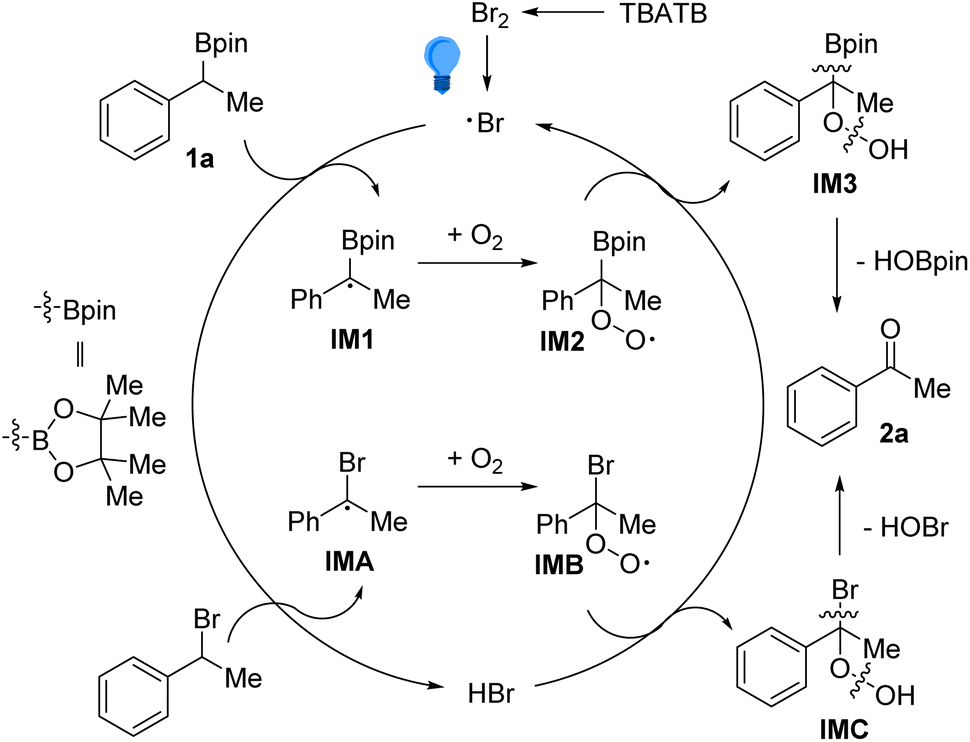 | ||
| Scheme 5 Proposed mechanisms. | ||
Conclusions
In conclusion, it has been shown that benzylic boronic esters can be converted to the corresponding ketones/aldehydes straightforwardly and chemoselectively. This reaction can be completed at room temperature under visible-light-induced conditions, in the absence of any metal-based (photo)catalysts, thus providing a complementary and environmentally friendly method that bridges alkylborons and the CO moieties directly. This study broadens the oxidation channels of boronic esters and the application of α-boryl radical intermediates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (21963010 and 22061036).References
- (a) J. W. B. Fyfe and A. J. B. Watson, Recent Developments in Organoboron Chemistry: Old Dogs, New Tricks, Chem, 2017, 3, 31–55 CrossRef CAS; (b) K. Yang and Q. Song, Tetracoordinate Boron Intermediates Enable Unconventional Transformations, Acc. Chem. Res., 2021, 54, 2298–2312 CrossRef CAS.
- A. M. Barcellos, M. Sacramento, G. P. da Costa, G. Perin, E. João Lenardão and D. Alves, Organoboron compounds as versatile reagents in the transition metal-catalyzed C–S, C–Se and C–Te bond formation, Coord. Chem. Rev., 2021, 442, 214012 CrossRef CAS.
- (a) L. Hao, G. Ding, D. A. Deming and Q. Zhang, Recent Advances in Green Synthesis of Functionalized Phenols from Aromatic Boronic Compounds, Eur. J. Org. Chem., 2019, 7307–7321 CrossRef CAS; (b) Y.-T. Xu, C.-Y. Li, X.-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Photoinduced hydroxylation of arylboronic acids with molecular oxygen under photocatalyst-free conditions, Green Chem., 2019, 21, 4971–4975 RSC; (c) W.-Z. Weng, H. Liang and B. Zhang, Visible-Light-Mediated Aerobic Oxidation of Organoboron Compounds Using in Situ Generated Hydrogen Peroxide, Org. Lett., 2018, 20, 4979–4983 CrossRef CAS PubMed; (d) J. J. Molloy, T. A. Clohessy, C. Irving, N. A. Anderson, G. C. Lloyd-Jones and A. J. B. Watson, Chemoselective oxidation of aryl organoboron systems enabled by boronic acid-selective phase transfer, Chem. Sci., 2017, 8, 1551–1559 RSC.
- R. D. Hanna, Y. Naro, A. Deiters and P. E. Floreancig, Alcohol, Aldehyde, and Ketone Liberation and Intracellular Cargo Release through Peroxide-Mediated α-Boryl Ether Fragmentation, J. Am. Chem. Soc., 2016, 138, 13353–13360 CrossRef CAS PubMed.
- J. D. Grayson and B. M. Partridge, Mild Cu-Catalyzed Oxidation of Benzylic Boronic Esters to Ketones, ACS Catal., 2019, 9, 4296–4301 CrossRef CAS.
- M. Roseau, N. Dhaouadi, C. Rolando, L. Chausset-Boissarie and M. Penhoat, Continuous photocatalyzed aerobic oxidation of benzylic organotrifluoroborates to benzaldehydes under Taylor flow conditions, J. Flow Chem., 2020, 10, 347–352 CrossRef CAS.
- W. Liu, P. Liu, L. Lv and C.-J. Li, Metal-Free and Redox-Neutral Conversion of Organotrifluoroborates into Radicals Enabled by Visible Light, Angew. Chem., Int. Ed., 2018, 57, 13499–13503 CrossRef CAS PubMed.
- C. Gerleve, M. Kischkewitz and A. Studer, Synthesis of α-Chiral Ketones and Chiral Alkanes Using Radical Polar Crossover Reactions of Vinyl Boron Ate Complexes, Angew. Chem., Int. Ed., 2018, 57, 2441–2444 CrossRef CAS.
- (a) A. Marotta, C. E. Adams and J. J. Molloy, The Impact of Boron Hybridisation on Photocatalytic Processes, Angew. Chem., Int. Ed., 2022, e202207067, DOI:10.1002/anie.202207067; (b) N. D. C. Tappin and P. Renaud, Radical Reactions of Boron-Ate Complexes Promoting a 1,2-Metallate Rearrangement, Chimia, 2020, 74, 33 CrossRef CAS PubMed; (c) N. Kumar, R. R. Reddy, N. Eghbarieh and A. Masarwa, α-Borylalkyl radicals: their distinctive reactivity in modern organic synthesis, Chem. Commun., 2020, 56, 13–25 RSC; (d) M. Kischkewitz, F. W. Friese and A. Studer, Radical-Induced 1,2-Migrations of Boron Ate Complexes, Adv. Synth. Catal., 2020, 362, 2077–2087 CrossRef.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, Radicals: Reactive Intermediates with Translational Potential, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- Q. Huang, J. Michalland and S. Z. Zard, Alternating Radical Stabilities: A Convergent Route to Terminal and Internal Boronates, Angew. Chem., Int. Ed., 2019, 58, 16936–16942 CrossRef CAS PubMed.
- (a) K. Jana and A. Studer, Allylboronic Esters as Acceptors in Radical Addition, Boron 1,2-Migration, and Trapping Cascades, Org. Lett., 2022, 24, 1100–1104 CrossRef CAS PubMed; (b) J. Michalland, N. Casaretto and S. Z. Zard, A Modular Access to 1,2- and 1,3-Disubstituted Cyclobutylboronic Esters by Consecutive Radical Additions, Angew. Chem., Int. Ed., 2022, 61, e202113333 CrossRef CAS; (c) S. Shi, F. Salahi, H. B. Vibbert, M. Rahman, S. A. Snyder and J. R. Norton, Generation of α-Boryl Radicals by H. Transfer and their Use in Cycloisomerizations, Angew. Chem., Int. Ed., 2021, 60, 22678–22682 CrossRef CAS; (d) L. Lewis-Borrell, M. Sneha, I. P. Clark, V. Fasano, A. Noble, V. K. Aggarwal and A. J. Orr-Ewing, Direct Observation of Reactive Intermediates by Time-Resolved Spectroscopy Unravels the Mechanism of a Radical-Induced 1,2-Metalate Rearrangement, J. Am. Chem. Soc., 2021, 143, 17191–17199 CrossRef CAS PubMed; (e) J. Michalland and S. Z. Zard, A Convergent, Stereoselective Route to Trisubstituted Alkenyl Boronates, Org. Lett., 2021, 23, 8018–8022 CrossRef CAS PubMed; (f) D. Wang, C. Mück-Lichtenfeld and A. Studer, Hydrogen Atom Transfer Induced Boron Retaining Coupling of Organoboronic Esters and Organolithium Reagents, J. Am. Chem. Soc., 2019, 141, 14126–14130 CrossRef CAS PubMed; (g) B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan and Z. Shi, An Olefinic 1,2-Boryl-Migration Enabled by Radical Addition: Construction of gem-Bis(boryl)alkanes, Angew. Chem., Int. Ed., 2019, 58, 9448–9452 CrossRef CAS PubMed.
- (a) T. Fang, J. Qiu, K. Yang and Q. Song, Photo-induced weak base-catalyzed synthesis of α-haloboronates from vinylboronates and polyfluoroalkyl halides, Org. Chem. Front., 2021, 8, 1991–1996 RSC; (b) L. Yang, D.-H. Tan, W.-X. Fan, X.-G. Liu, J.-Q. Wu, Z.-S. Huang, Q. Li and H. Wang, Photochemical Radical C–H Halogenation of Benzyl N-Methyliminodiacetyl (MIDA) Boronates: Synthesis of α-Functionalized Alkyl Boronates, Angew. Chem., Int. Ed., 2021, 60, 3454–3458 CrossRef CAS PubMed; (c) H. C. Brown, N. R. De Lue, Y. Yamamoto and K. Maruyama, Organoboranes. 22. Light-induced reaction of bromine with alkylboronate esters. A convenient synthesis of .alpha.-bromoalkylboronate esters, J. Org. Chem., 1977, 42, 3252–3254 CrossRef CAS; (d) H. C. Brown and C. F. Lane, Formation of alkyl bromides in the dark reaction of bromine with organoboranes. Evidence for an unusual pathway involving the prior bromination of the organoborane, J. Am. Chem. Soc., 1970, 92, 7212–7213 CrossRef CAS.
- (a) P. Dai and L. Xu, Visible-Light-Induced Benzylic C—H Oxygenation Reaction Using Tetrabutylammonium Tribromide as the Catalyst, Chin. J. Org. Chem., 2021, 41, 4690–4695 CrossRef; (b) L. Wei, Y. Wei, J. Zhang and L. Xu, Visible-light-mediated organoboron-catalysed metal-free dehydrogenation of N-heterocycles using molecular oxygen, Green Chem., 2021, 23, 4446–4450 RSC; (c) W. Zu, C. Day, L. Wei, X. Jia and L. Xu, Dual aminoquinolate diarylboron and nickel catalysed metallaphotoredox platform for carbon–oxygen bond construction, Chem. Commun., 2020, 56, 8273–8276 RSC; (d) L. Wei, J. Zhang and L. Xu, Visible-Light-Mediated Aminoquinolate Diarylboron-Catalyzed Metal-Free Hydroxylation of Organoboronic Acids under Air and Room Temperature, ACS Sustainable Chem. Eng., 2020, 8, 13894–13899 CrossRef CAS.
- M. Belal, S. Sarkar, R. Subramanian and A. T. Khan, Synthetic utility of biomimicking vanadium bromoperoxidase and n-tetrabutylammonium tribromide (TBATB) in organic synthesis, Org. Biomol. Chem., 2022, 20, 2562–2579 RSC.
- (a) A. Mardani, M. Heshami, Y. Shariati, F. Kazemi, M. Abdollahi Kakroudi and B. Kaboudin, A tunable synthesis of either benzaldehyde or benzoic acid through blue-violet LED irradiation using TBATB, J. Photochem. Photobiol., A, 2020, 389, 112220 CrossRef CAS; (b) A. Mardani, F. Kazemi and B. Kaboudin, Photo-tunable oxidation of toluene and its derivatives catalyzed by TBATB, J. Photochem. Photobiol., A, 2021, 414, 113301 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qo01457b |
| This journal is © the Partner Organisations 2023 |