
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Materials discovery and design limits in MDABCO perovskites†
Samuel D.
Gale
 a,
Harry J.
Lloyd
ab,
Louise
Male
a,
Mark R.
Warren
b,
Lucy K.
Saunders
b,
Paul A.
Anderson
a and
Hamish H.-M.
Yeung
*a
a,
Harry J.
Lloyd
ab,
Louise
Male
a,
Mark R.
Warren
b,
Lucy K.
Saunders
b,
Paul A.
Anderson
a and
Hamish H.-M.
Yeung
*a
aSchool of Chemistry, University of Birmingham, B15 2TT, UK. E-mail: h.yeung@bham.ac.uk; Tel: +44 (0)121 414 8811
bDiamond Light Source Ltd., Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
First published on 17th August 2022
Abstract
We report the structures of three new materials in the MDABCO-based perovskite family. Analysis of the Goldschmidt tolerance factor and octahedral factor enable us to propose compositional limits for pseudo-cubic perovskite phase formation and suggest directions for further materials discovery in this family of new ferroelectrics.
1. Introduction
Discovery and development of new ferroelectric materials is important for the realisation of new memory storage technology, such as FE-RAM in flexible devices, soft robotics and wearable electronics. Perovskites, such as BaTiO3, have long been considered state-of-the-art in design of new FE materials owing to desirable properties and tunability of their composition.1 Perovskites adopt the general formula ABX3, where a larger A-site cation occupies the cavities within a pseudo-cubic corner-sharing network of BX6 octahedra. Substitutions on any of the three sites has been shown to result in remarkable changes in FE properties.The discovery of a new subset of hybrid organic–inorganic perovskites1e based on the A-site MDABCO dication (MDABCO = N-methyl-N′-diazabicyclo[2.2.2]octonium) has led to renewed interest in hybrid inorganic–organic FE materials, owing to their properties that are comparable to BaTiO3.2 Variation in the B and X sites has given rise to four MDABCO-based halide perovskites to date: [MDABCO]RbI3,2a [MDABCO]NH4I3,2b [MDABCO]KI3 (ref. 2c) and [MDABCO]NH4Br3.2b The ammonium iodide variant, in particular, has a large spontaneous polarization, high phase transition temperature and facile switching through eight possible polarisation directions. Factors thought to be important in driving spontaneous and switchable polarisation of MDABCO-based perovskites are the quasi-spherical nature of the A-site cation,3 orientation of the MDABCO dipole moment along the pseudo-cubic 〈111〉 direction, strain coupling and dipolar interactions.4 Hydrogen bonding, whilst it does not drive FE behaviour, has been shown to be important in determining the magnitude of polarisation and coercive field.5
Formation of new perovskites requires the assembly of the BX6 octahedra, which can empirically be predicted by the radius ratio or octahedral factor, OF = rB/rX , where rB and rX are the ionic radii of the B-site cation and X-site anion, respectively.6 Octahedra are expected to form in the range 0.41 ≤ OF ≤ 0.732. In addition, design rules typically consider the Goldschmidt tolerance factor (TF) as a necessary criterion for their formation:7
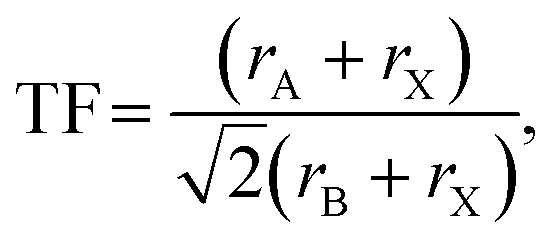
The discovery of new compositions is necessary to test how exactly each factor contributes to the ferroelectric performance, to refine our understanding of this important class of FE materials and suggest design rules for new materials. However, it is not yet clear where the compositional limits for MDABCO-based perovskite formation lie, knowledge of which would help to improve the success of future discovery efforts.
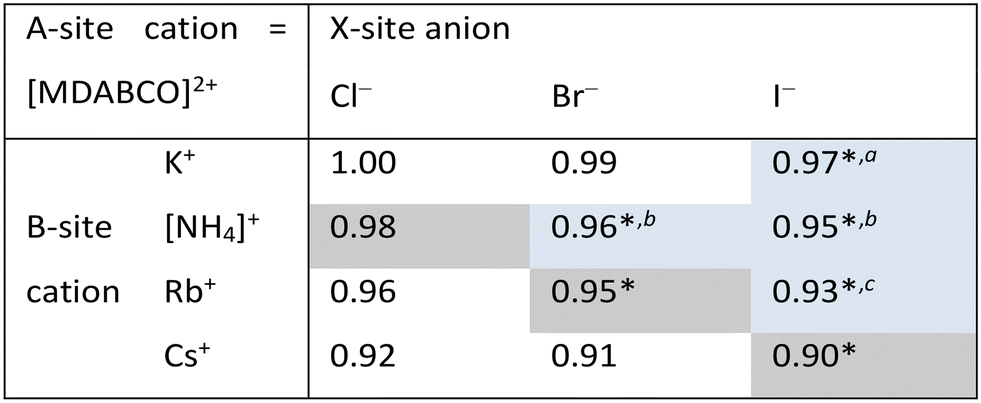
In this work, we report three new compositions in the [MDABCO]BX3 family, namely [MDABCO]CsI3, [MDABCO]RbBr3 and [MDABCO]NH4Cl3, discovered in the course of a systematic investigation of the different compositions afforded by combinations of K+, [NH4]+, Rb+ and Cs+ with Cl−, Br− and I−. We are thus able to define upper limits to the compositional range of pseudo-cubic MDABCO-based perovskites with respect to TF and OF, and suggest directions for the exploration of further compositions in future.
2. Experimental methods
Single crystals were synthesised at ambient temperature using protocols similar to previous reports and structures were determined at ambient temperature using single crystal X-ray diffraction either in-house or at beamline I19-2, Diamond Light Source. Full details of all synthesis and crystallography can be found in the ESI,† sections S1 and S2.3. Results and discussion
3.1 Pseudo-cubic perovskite [MDABCO]CsI3
Crystallisation at room temperature of a reaction mixture containing MDABCO, Cs+ and I− resulted in the formation of crystals with rhombohedral symmetry that solved in the polar space group R3. Owing to the threefold symmetry, the asymmetric unit consists of one third of an MDABCO molecule, one third of a Cs+ cation and one I− anion. The crystal structure overall has three ABX3 formula units per unit cell, which has the metric a = b = 9.8648(5) Å, c = 13.7921 Å, α = β = 90°, γ = 120° in the hexagonal setting. The cell volume, V = 1162.35(13) Å3; i.e., the volume per formula unit V′ = V/Z′ = 387 Å3.The structure consists of corner-sharing CsI6 octahedra with MDABCO dications occupying the large intra-framework A-site cavities (Fig. 1), isostructural with the previous compounds [MDABCO]RbI3 and [MDABCO]NH4I3. The N–N axes of the MDABCO molecules lie along the three-fold axes of the cell, parallel to the [111] direction with respect to the pseudo-cubic perovskite cell. Like the parent compounds, the MDABCO dication is involved in three weak hydrogen bonds to nearby framework I− anions via the N–H group (N–I distance 3.76(3) Å). The similarities between these features and those of existing ferroelectric MDABCO-based perovskites suggest that [MDABCO]CsI3 has the strong potential to exhibit ferroelectricity itself.
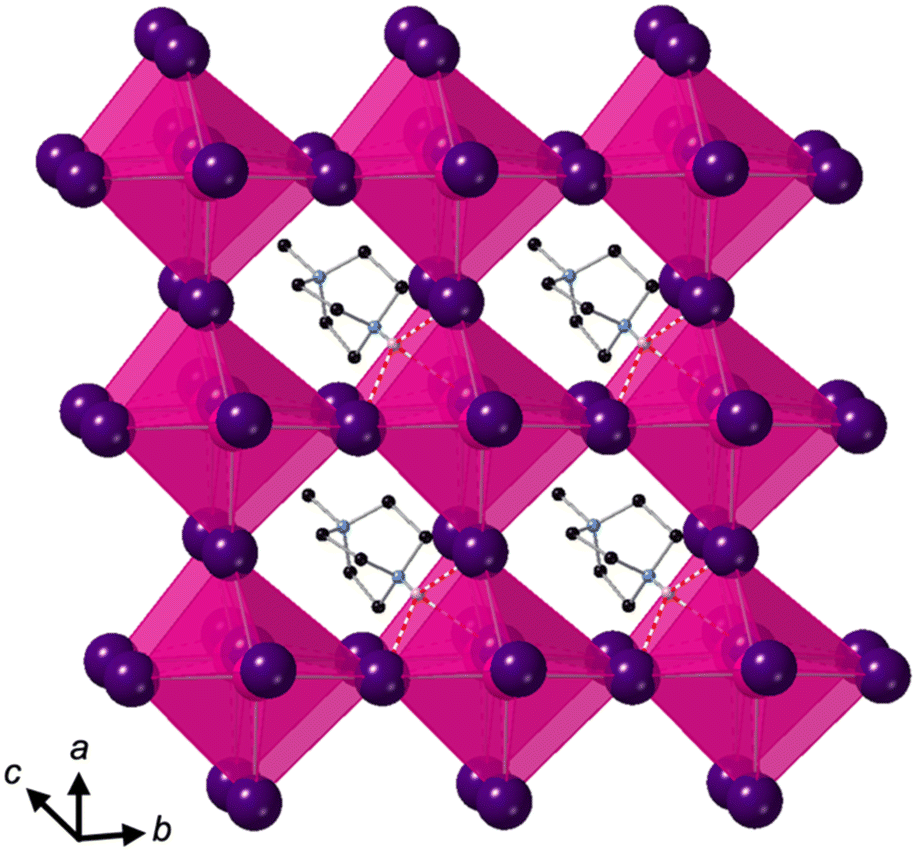 | ||
| Fig. 1 Four adjacent pseudo-cubic motifs of [MDABCO]CsI3, showing alignment of the MDABCO N–N axes along the unique axis (c). C, N, Cs and I are shown in black, blue, pink and purple, respectively. H atoms are omitted for clarity, apart from N–H atoms (pink) involved in hydrogen bonds, which are indicated by dashed red lines. | ||
The distorted CsI6 octahedra exhibit Cs–I distances within the range 3.682(3)–3.692(3) Å, which is slightly longer than the NH4+ and Rb+ analogues. The bond valence sum (BVS)8 for Cs is 1.54, which suggests that I− anions can approach the B-site more closely than would normally be expected for Cs+; this may be related to the polarizability of Cs and I, and the OF, 0.76, of [MDABCO]CsI3, which is larger than its previously reported analogues. We note also that although the structure refines well with full occupancy of all the atoms, we observe slightly improved fit statistics when the occupancy of the Cs+ cation is allowed to refine, converging to around 0.8.
This observation may be related to the above-average BVS and suggests that the structure contains randomly distributed substitutions of much lighter cations, such as H+, on the B-site and/or Cs+ vacancies co-existing with oxidation of the iodide sublattice, neither of which can easily be detected by X-ray diffraction.
The nearest neighbour I–Cs–I angles are within the range 78.89–102.89°, and the sum of deviations from the ideal octahedral angle,9σ = 81°, is within the range of existing MDABCO-based perovskites (σ = 70–92°). Although the CsI6 octahedra are highly distorted, they do not exhibit any tilting in the conventional sense. Application of ISODISTORT10 confirms that the [MDABCO]CsI3 structure can be assigned the Glazer notation a0a0a0.11
3.2 Pseudo-cubic perovskite [MDABCO]RbBr3
[MDABCO]RbBr3 forms at room temperature in the space group Pna21, which has spontaneous polarisation along the orthorhombic c-axis. There are four symmetry-related MDABCO dications per unit cell, which has the metric a = 14.1838(10) Å, b = 9.9322(6) Å, c = 9.6159(6) Å, α = β = γ = 90°. The cell volume, V = 1354.65(15) Å3; i.e., V′ = 339 Å3.The structure consists of a framework of distorted corner-sharing RbBr6 octahedra with MDABCO dications occupying the perovskite A-site within the pseudo-cubic intraframework cavities (Fig. 2). In contrast to existing MDABCO-based perovskites, the N–N axis of the MDABCO dications are oriented along alternating pseudo-cubic 〈110〉 directions, which are tilted with respect to the crystallographic c-axis. If the overall polarisation was found to be switchable it would, therefore, constitute “ferrielectricity”. Each MDABCO dication has a single short N–H⋯Br hydrogen bond (N–Br distance 3.159(8) Å) stabilising its interactions with the RbBr3 framework, rather than three weak hydrogen bonds as are found in other MDABCO-based perovskites.
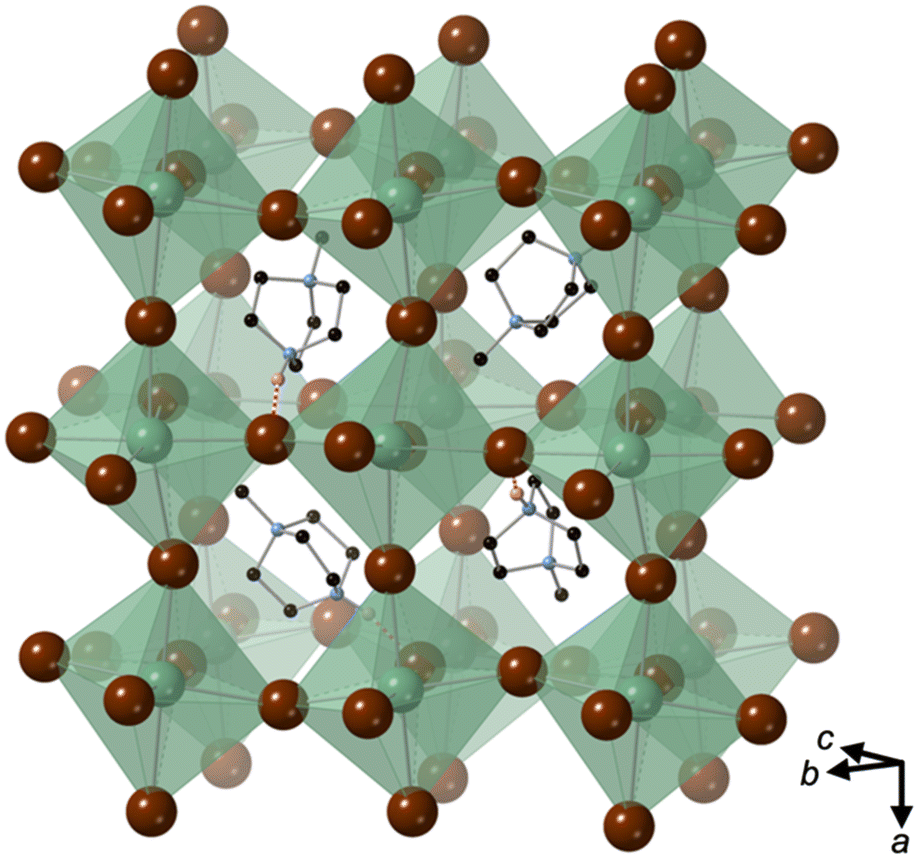 | ||
| Fig. 2 Extended unit cell of [MDABCO]RbBr3, showing the tilted orientations of four MDABCO dications within the pseudo-cubic framework of corner-sharing RbBr6 octahedra. C, N, Rb and Br are shown in black, blue, green and brown, respectively. H atoms are omitted for clarity, apart from N–H atoms (pink) involved in hydrogen bonds, which are indicated by dashed red lines. | ||
The distorted RbBr6 octahedra exhibit Rb–Br distances within the range 3.452(1)–3.654(1) Å. The BVS for Rb is 0.813, which indicates that the Rb+ cation is underbonded; it has the highest OF (vide infra) of the known pseudo-cubic perovskites. The average deviation from the mean Rb–Br distance is 18%, which is much larger than existing MDABCO-based perovskites (3–8%). However, nearest neighbour Br–Rb–Br angles are within the range 81.4–101.41°, such that the sum of deviations from the ideal octahedral angle, σ = 68°, is slightly lower than existing MDABCO-based perovskites. Application of ISODISTORT10 shows that the octahedral tilts of [MDABCO]RbBr3 can be assigned the Glazer notation a−a−c0.
3.3 Hexagonal-type perovskite [MDABCO]NH4Cl3
[MDABCO]NH4Cl3 forms at room temperature in the space group P21/n, which is centrosymmetric and, therefore, is not expected to exhibit FE properties at ambient temperature. The asymmetric unit consists of two MDABCO dications, six Cl− anions and two [NH4]+ cations. One of the [NH4]+ cations is disordered across an inversion centre with 50% occupancy on each of two sites. The other has an elongated anisotropic displacement ellipsoid, which is suggestive of similar disorder. These displacements coincide with the longest octahedral Cl–Cl axes, which suggests they occur in order to optimise ionic and/or hydrogen bonding interactions. The crystal structure overall has eight ABX3 formula units per unit cell, which has the metric a = 14.8238(4) Å, b = 10.1333(3) Å, c = 16.4441(4) Å, α = 90°, β = 90.577(2)°, γ = 90°. The cell volume, V = 2470.01(12) Å3; i.e., V′ = 309 Å3.The structure consists of face-sharing chains of NH4Cl6 octahedra aligned along the a-axis and the chains are arranged in a hexagonal close-packed array (Fig. 3) similar to CsNiCl3 and BaNiO3,12 and the analogous dabconium structure (dabconium = [C6H14N2]2+).13 MDABCO dications lie between the chains with their N–N axes oriented along a variety of directions within the bc-plane. There are two short hydrogen bond lengths between MDABCO N–H functionalities and Cl− anions of the chains, which have N–Cl distances of 2.998 Å and 3.005 Å.
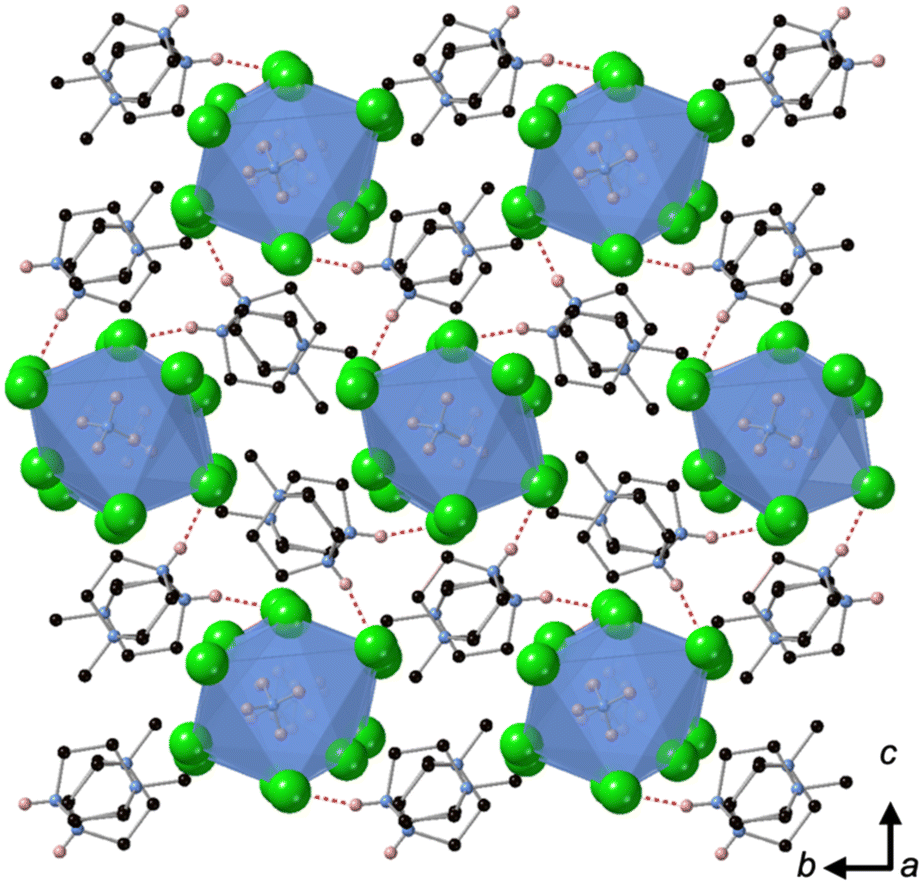 | ||
| Fig. 3 Crystal structure of [MDABCO]NH4Cl3 containing a hexagonal array of chains of face-sharing NH4Cl6 octahedra aligned along the a-axis, showing the orientations of MDABCO dications between them. C, N and Cl are shown in black, blue and green, respectively. H atoms are omitted for clarity, apart from ammonium and MDABCO N–H atoms (pink) involved in hydrogen bonds, which are indicated by dashed red lines. | ||
The distorted NH4Cl6 octahedra exhibit N–Cl distances in the range 3.154–4.25 Å; each [NH4]+ cation has one long N–Cl interaction and five shorter N–Cl interactions, which are indicative of dipole-assisted hydrogen bonding. The effective BVSs14 of the two [NH4]+ cations are 0.90 and 0.96, which indicates that the intermolecular interactions are satisfied in this arrangement with off-centred [NH4]+ cations. Nearest neighbour Cl–N–Cl angles are within the range 74.2° to 107.3°. However, the sum of deviations from the ideal octahedral angle, σ, are 71° and 83°, which is well within the range of existing MDABCO-based perovskites.
3.4 Phase behaviour
Analysis of the three structures reported herein alongside the existing MDABCO-based perovskites reveals certain compositional limits in their phase behaviour, which can be quantified by TF and OF ranges (Fig. 4). All three new structures have TF values that fall within the previously proposed range for pseudo-cubic perovskite formation (see sections S3 and S4 in the ESI†).2b However, in contrast to previous computational studies that assumed a pseudo-cubic perovskite for [MDABCO]NH4Cl3,15 we have found that it forms a hexagonal-type structure. Therefore, an upper TF limit can be defined between its TF, 0.98, and the highest TF of known pseudo-cubic structures, 0.97, which is observed for [MDABCO]KI3. This is somewhat reflected in the trends in the crystallographic volume per MDABCO dication, and a similar observation can be made regarding the lower limit of V′. Owing to the small size of the [NH4]+ and Cl− ions, [MDABCO]NH4Cl3 has a lower V′ (309 Å3) than the pseudo-cubic MDABCO halide perovskites (see section S5 in the ESI†). We can thus define a lower V′ limit for pseudo-cubic perovskite formation between 309 Å3 and 339 Å3, which is the value of V′ found for the smallest pseudo-cubic perovskite, [MDABCO]RbBr3.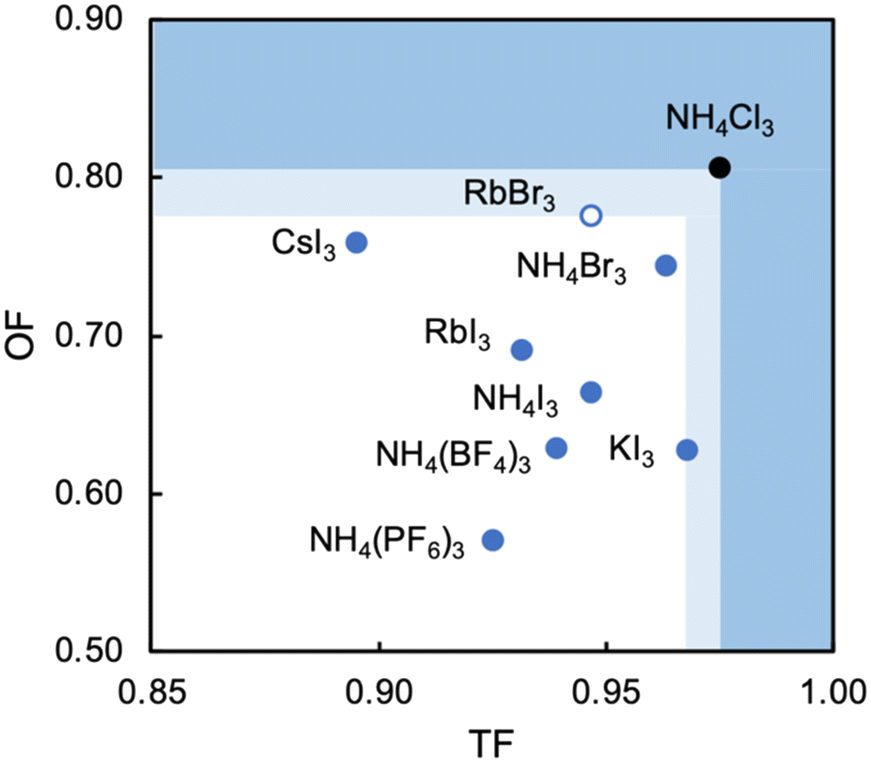 | ||
| Fig. 4 Phase behaviour of MDABCO-based perovskites as a function of tolerance factor (TF) and octahedral factor (OF), showing B- and X-site compositions. Blue circles are pseudo-cubic perovskites with 〈111〉 MDABCO orientations. The open circle is the pseudo-cubic perovskite with 〈110〉 MDABCO orientations. The black circle is the hexagonal-type structure, which defines upper limits of ranges for pseudo-cubic perovskite formation, 0.97 < TF < 0.98 and 0.78 < OF < 0.81, shown in light blue. Darker blue shaded regions lie above these limits. | ||
All the new structures reported herein have OF values above those of previously reported structures and, interestingly, all fall outside of the empirical range, 0.414–0.732, required for BX6 octahedron formation.6a However, both [MDABCO]RbBr3 and [MDABCO]CsI3 are found to form pseudo-cubic perovskites, owing to the high polarizability of the heavy ions involved. We can define an upper OF limit between the OF of [MDABCO]RbBr3, 0.78, and that of the hexagonal-type structure [MDABCO]NH4Cl3, 0.81. Our initial analysis of crystals formed from the reaction of MDABCO with Cs+ and Br− appears to confirm this trend. The high OF value of 0.85 should prohibit pseudo-cubic perovskite formation even though [MDABCO]CsBr3 falls within the TF range and, indeed, we observe a structure with similarities to other hexagonal-type halide perovskites,13 although we should note that refinement of this structure is ongoing.
Comparison of BVSs shows that structures close to the TF and OF limits deviate most from the ideal value of 1 for monovalent cations (see ESI section S6†), which may be related to their borderline status and other properties. [MDABCO]KI3 has exceptional mechanical properties,2c whilst [MDABCO]RbBr3 contains MDABCO dications in unusual 〈110〉 orientations.
4. Conclusions
In summary, we have described the new structures of three MDABCO-based materials in the ABX3 perovskite family. The cesium iodide variant adopts a polar R3 structure in common with its parent compounds and is expected to be ferroelectric. The low value of its TF, 0.90, extends the TF range of known MDABCO-based perovskites. The rubidium bromide variant contains MDABCO dications somewhat unusually oriented in 〈110〉 directions, tilted in alternating fashion such that the Pna21 structure is also polar. It has the highest OF, 0.78, of any known pseudo-cubic MDABCO-based perovskite. On the other hand, the ammonium chloride variant adopts a non-polar hexagonal-type structure, which allows us to define for the first time an upper TF limit of 0.97 < TF < 0.98 and an upper OF limit of 0.78 < OF < 0.81.Whilst the TF for [MDABCO]CsI3, 0.90, extends the range of TF in known MDABCO-based perovskites towards low TF values, the lower bounds for TF and OF remain undetermined. We expect that discovery of new MDABCO-based perovskites may be most successful using larger X-site anions to explore low TF and OF combinations, i.e., towards the bottom left region of Fig. 4. For example, [MDABCO]NH4(BF4)3 and [MDABCO]NH4(PF6)3 have recently been shown to adopt pseudo-cubic perovskite structures16 and our TF and OF calculations suggest that all K, Rb and Cs analogues with [BF4]− and [PF6]− on the X-site may also form, but to the best of our knowledge no other compositions have been reported.
The structures of the remaining halides shown in Table 1 currently remain undetermined, despite significant efforts in the lab. Our TF and OF analysis may explain why: Rb–Cl, Cs–Cl and Cs–Br have TF within the proposed perovskite formation range, and K–Cl and K–Br have OF within the expected range. However, none satisfy the criteria for both TF and OF together, which may explain why no chloride-based pseudo-cubic MDABCO perovskites have been reported to date. We may expect them to form alternative isomers, such as the hexagonal-type structure formed by [MDABCO]NH4Cl3 or different topologies altogether. Further work is required to synthesise them and determine their crystal structures.
Finally, we note that unusual 〈110〉 orientations of MDABCO in [MDABCO]RbBr3 run counter to our previous observation that 〈111〉 orientations are required for spontaneous polarisation.4 This suggests that new and interesting properties may arise at the TF and OF boundaries. Preliminary results of differential scanning calorimetry in our lab indicate the existence of high-temperature phase transitions in all three structures reported herein, and further investigation into structural features that affect phase transitions and dielectric properties of the materials is ongoing.
Author contributions
Conceptualization: HHMY, PAA. Formal analysis: SDG, HJL, HHMY. Funding acquisition: HHMY, HJL. Investigation: SDG, HJL, LM, MRW, LKS. Methodology: SDG, HJL, LM. Project administration: HHMY, PAA. Supervision: MRW, LKS, PAA and HHMY. Writing – original draft: HHMY. Writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Andrew Goodwin for useful discussions, Dominic Allen for the initial synthesis of [MDABCO]CsI3 powder, and Hanna Boström for determination of the Glazer tilt notations. We acknowledge the RSC for funding a Research Enablement Grant (E21-7701710937) and Diamond Light Source for beamtime on beamline I19-2 (CY26118-2). We thank Diamond Light Source and the University of Birmingham for studentship funding (HJL).Notes and references
- (a) W. J. Merz, Phys. Rev., 1953, 91, 513–517 CrossRef CAS; (b) L. G. Tejuca and J. L. G. Fierro, Perovskites and Applications of Perovskite-type Oxides, Marcel Dekker, New York, 1992 CrossRef; (c) D. B. Mitzi, J. Chem. Soc., Dalton Trans., 2001, 1–12 RSC; (d) R. E. Newnham and L. E. Cross, MRS Bull., 2005, 30, 845–848 CrossRef CAS; (e) W. Li, Z. Wang, F. Deschler, S. Gao, R. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 16099 CrossRef.
- (a) W.-Y. Zhang, Y.-Y. Tang, P.-F. Li, P.-P. Shi, W.-Q. Liao, D.-W. Fu, H.-Y. Ye, Y. Zhang and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 10897–10902 CrossRef CAS PubMed; (b) H.-Y. Ye, Y.-Y. Tang, P.-F. Li, W.-Q. Liao, J.-X. Gao, X.-N. Hua, H. Cai, P.-P. Shi, Y.-M. You and R.-G. Xiong, Science, 2018, 361, 151–155 CrossRef CAS PubMed; (c) L.-C. An, K. Li, Z.-G. Li, S. Zhu, Q. Li, Z.-Z. Zhang, L.-J. Ji, W. Li and X.-H. Bu, Small, 2021, 17, 2006021 CrossRef CAS PubMed.
- H.-Y. Zhang, Y.-Y. Tang, P.-P. Shi and R.-G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS PubMed.
- D. J. W. Allen, N. C. Bristowe, A. L. Goodwin and H. H.-M. Yeung, J. Mater. Chem. C, 2021, 9, 2706 RSC.
- H. S. Choi, S. Li, I.-H. Park, W. H. Liew, Z. Zhu, K. C. Kwon, L. Wang, I.-H. Oh, S. Zheng, C. Su, Q.-H. Xu, K. Yao, F. Pan and K. P. Loh, Nat. Commun., 2022, 13, 794 CrossRef CAS PubMed.
- (a) A. R. West, Solid State Chemistry and its Applications, Wiley, 2nd edn, 2014 Search PubMed; (b) Y. Cai, W. Xie, H. Ding, Y. Chen, K. Thirumal, L. H. Wong, N. Mathews, S. G. Mhaisalkar, M. Sherburne and M. Asta, Chem. Mater., 2017, 29, 7740–7749 CrossRef CAS.
- (a) V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477 CrossRef CAS; (b) G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2014, 5, 4712 RSC; (c) S. Burger, M. G. Ehrenreich and G. Kieslich, J. Mater. Chem. A, 2018, 6, 21785–21793 RSC.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- M. Buron-Le Cointe, J. Hébert, C. Baldé, N. Moisan, L. Toupet, P. Guionneau, J. F. Létard, E. Freysz, H. Cailleau and E. Collet, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 064114 CrossRef.
- (a) H. T. Stokes, D. M. Hatch and B. J. Campbell, ISODISTORT, ISOTROPY Software Suite, https://iso.byu.edu Search PubMed; (b) B. J. Campbell, H. T. Stokes, D. E. Tanner and D. M. Hatch, J. Appl. Crystallogr., 2006, 39, 607–614 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- Y. Takeda, F. Kanamaru, M. Shimada and M. Koizumi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 2464–2466 CrossRef.
- C. A. Bremner, M. Simpson and W. T. A. Harrison, J. Am. Chem. Soc., 2002, 124, 10960–10961 CrossRef CAS PubMed.
- L. Garciá-Rodríguez, A. Rute-Pérez, J. R. Piñero and C. González-Silgo, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 565–569 CrossRef PubMed.
- (a) T. W. Kasel, Z. Deng, A. M. Mroz, C. H. Hendon, K. T. Butler and P. Canepa, Chem. Sci., 2019, 10, 8187–8194 RSC; (b) H. Wang, H. Liu, Z. Zhang, Z. Liu, Z. Lv, T. Li, W. Ju, H. Li, X. Cai and H. Han, npj Comput. Mater., 2019, 5, 1–9 CrossRef CAS.
- (a) M.-J. Sun, C. Zheng, Y. Gao, A. Johnston, A. M. Najarian, P.-X. Wang, O. Voznyy, S. Hoogland and E. H. Sargent, Adv. Mater., 2021, 33, 2006368 CrossRef CAS PubMed; (b) H. S. Choi, S. Li, I.-H. Park, W. H. Liew, Z. Zhu, K. C. Kwon, L. Wang, I.-H. Oh, S. Zheng, C. Su, Q.-H. Xu, K. Yao, F. Pan and K. P. Loh, Nat. Commun., 2022, 13, 794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthesis procedures, tables of structure analysis and crystallographic information files (CCDC numbers 2181255–2181257). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00848c |
| This journal is © The Royal Society of Chemistry 2022 |