
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative study of the crystallization behavior within the Na2O–Y2O3–ZrO2–SiO2 system during heating and cooling
Christian
Thieme
 *a,
Marc
Dittmer
b,
Thomas
Höche
a,
Markus
Rampf
b and
Christian
Rüssel
a
*a,
Marc
Dittmer
b,
Thomas
Höche
a,
Markus
Rampf
b and
Christian
Rüssel
a
aFraunhofer Institute for Microstructure of Materials and Systems IMWS, Walter-Huelse-Str. 1, D-06120 Halle, Germany
bIvoclar Vivadent AG, Bendererstr. 2, LI-9494 Schaan, Principality of Liechtenstein. E-mail: christian.thieme@imws.fraunhofer.de; Web: https://www.imws.fraunhofer.de Fax: +49 345 5589 101; Tel: +49 345 5589 243
First published on 24th May 2022
Abstract
Phase formation in glasses from the system Na2O–Y2O3–SiO2 with different concentrations of ZrO2 were studied concerning phase formation and crystallization behavior. The compositions were near the stoichiometry of NaYSi2O6. A comparison of microstructure and phase content after instant crystallization during slow cooling and after subsequent thermal annealing is given. The phases as well as the microstructure differ depending on thermal history. During cooling, predominantly a phase with apatite structure (NaY9Si6O26) precipitates and the residual glassy phase is enriched in ZrO2. The composition of the glassy phase was quantified and an enrichment of ZrO2 of slightly above five mol% was measured. ZrO2 concentrations >5 mol% lead to the precipitation of Y2O3 enriched ZrO2. A fast cooling of the melt results in glass formation without instant crystallization. Subsequent heating at temperatures below 1000 °C leads to the precipitation of an unknown phase whereas at higher temperatures NaYSi2O6 crystallizes. Two-step heating in terms of nucleation and crystal growth were performed and small ZrO2-particles have been formed homogenously within the sample volume. Those particles do not trigger volume crystallization within the glasses and surface crystallization is the predominant crystallization mechanism.
1. Introduction
Glasses within the system Na2O–Y2O3–SiO2 are of great scientific interest because of their good sodium ion conductivity and their possible application as solid electrolyte.1,2 Especially sodium-rich glasses and glass-ceramics show the often desired high sodium conductivity.3 The glass formation range within the ternary Na2O–Y2O3–SiO2 system is evaluated by two different studies, as summarized in Fig. 1. In ref. 2, it was reported that the glass formation is limited by the maximum Y2O3-concentration of 9 mol%. In contradiction, ref. 4 reports a maximum Y2O3 concentration of around 15 mol%. The discrepancies cannot fully be explained by the synthesis temperatures or synthesis routes because in both studies, an almost identical experimental setup was used for the melting of the glasses. Independent from those investigations, it is known that other glass systems such as the Al2O3–Y2O3–SiO2 enable much higher Y2O3-concentrations.5 Hence, additions of Al2O3 should enlarge the glass formation range. Such glass-ceramics focus on the precipitation of yttrium aluminium garnet (YAG) crystals (mostly doped with Ce3+) for white light emission.6,7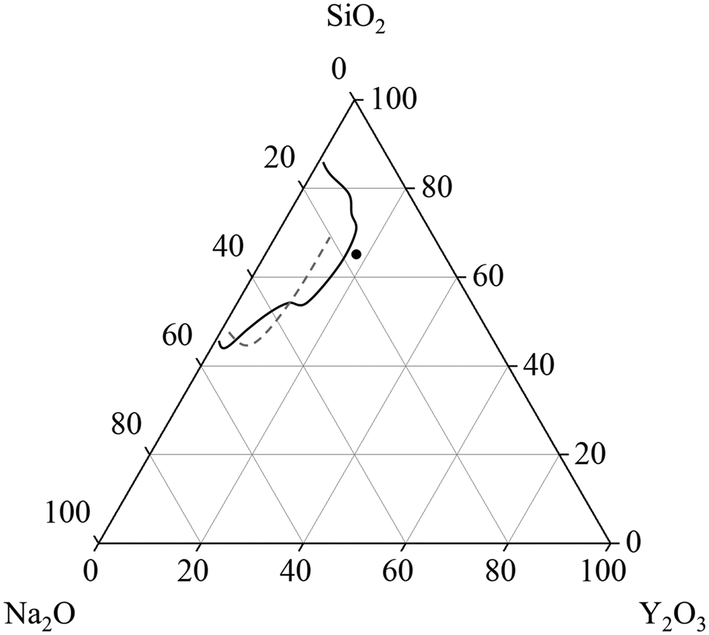 | ||
| Fig. 1 Glass formation range in units of [mol%] in the system Na2O–Y2O3–SiO2 reported in ref. 4 (solid line) and ref. 2 (dashed line). The dot depicts the starting composition for the investigations within this manuscript. | ||
However, the general crystallization behavior within the ternary system Na2O–Y2O3–SiO2 is scarcely described in literature.
The common technology to produce glass-ceramics is usually to quench a glass melt and to re-heat it to a temperature slightly above Tg in order to achieve nucleation and then in a second step to a higher temperature to allow crystal growth. Another strategy, however, is to slowly cool the glass and to achieve nucleation and crystal growth upon cooling. The phases formed during these different types of temperature–time schedules might be different and the formed microstructure, too. In this paper, glass-ceramics prepared by both concepts are reported on and compared with each other.
In order to prepare such compact materials, the crystallization behavior has to be studied with respect to phase formation. Furthermore, the effect of additives on the formation of volume crystals has to be investigated. One of the most popular oxidic nucleation agents is ZrO2, which is able to trigger volume crystallization in various glass systems.8 It should be noted that in some glass compositions, such as Li2O·2 SiO2 minor concentrations of ZrO2 (≤2 mol%) show the opposite effect, i.e. act as nucleation inhibitors.9,10
This work focuses on the glass formation in the range of high Y2O3 concentrations, near the border between glass formation and instant crystallization during cooling. The compositions within this manuscript are based on a stoichiometric disilicate with the composition NaYSi2O6. This stoichiometry is also depicted in Fig. 1 and serves as starting point for the investigations. Furthermore, the effect of different concentrations of ZrO2 on the glass stability and crystallization behavior is investigated.
2. Materials and methods
Samples with the compositions summarized in Table 1 were melted in Pt crucibles using an electric furnace with MoSi2 heating elements. The sample Zr0 possesses the same stoichiometry as crystalline NaYSi2O6. If ZrO2 is added, the molar ratios Na2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y2O3:SiO2 were kept constant. Reagent grade raw materials (Na2CO3, Y2O3, SiO2, ZrO2) were thoroughly mixed and afterwards heated to a temperature of 1650 to 1680 °C for several hours in order to obtain a homogenous melt. Afterwards, two different cooling regimes were used. In a first regime, samples were cooled from the melting temperature of 1650 or 1680 °C down to 800 °C with a rate of 1 K min−1. Afterwards, the furnace was switched off in order to allow a slow cooling of the sample down to room temperature. This procedure was used to achieve instant crystallization during cooling. Some of the compositions were re-melted at 1650 to 1680 °C and afterwards cast in a steel mould, which was then transferred into a muffle furnace pre-heated to a temperature in the range from 750 to 780 °C, which is slightly above the respective glass transition temperatures. Directly afterwards, the pre-heated furnace was switched off to allow cooling to room temperature with a rate of around 1 to 2 K min−1. After removal from the cold furnace, those samples are amorphous and were used for re-heating in order to achieve nucleation and crystallization.
:Y2O3:SiO2 were kept constant. Reagent grade raw materials (Na2CO3, Y2O3, SiO2, ZrO2) were thoroughly mixed and afterwards heated to a temperature of 1650 to 1680 °C for several hours in order to obtain a homogenous melt. Afterwards, two different cooling regimes were used. In a first regime, samples were cooled from the melting temperature of 1650 or 1680 °C down to 800 °C with a rate of 1 K min−1. Afterwards, the furnace was switched off in order to allow a slow cooling of the sample down to room temperature. This procedure was used to achieve instant crystallization during cooling. Some of the compositions were re-melted at 1650 to 1680 °C and afterwards cast in a steel mould, which was then transferred into a muffle furnace pre-heated to a temperature in the range from 750 to 780 °C, which is slightly above the respective glass transition temperatures. Directly afterwards, the pre-heated furnace was switched off to allow cooling to room temperature with a rate of around 1 to 2 K min−1. After removal from the cold furnace, those samples are amorphous and were used for re-heating in order to achieve nucleation and crystallization.
| Sample name | Na2O | Y2O3 | SiO2 | ZrO2 |
|---|---|---|---|---|
| Zr0 | 16.67 | 16.67 | 66.67 | — |
| Zr5 | 15.83 | 15.83 | 63.33 | 5 |
| Zr7 | 15.5 | 15.5 | 62 | 7 |
| Zr9 | 15.17 | 15.17 | 60.67 | 9 |
| Zr11 | 14.83 | 14.83 | 59.33 | 11 |
These heat treatments were performed in a muffle furnace using a heating rate of 5 K min−1 and a temperature accuracy of ±5 K. The supplied temperatures were in the range from 850 and 1150 °C kept for 5 h. Differential scanning calorimetry (DSC) was performed with a Linseis DSC Pt 1600. For this purpose, the glass samples were crushed using a steel mortar and subsequently sieved to a size fraction of 400 to 500 μm. This comparatively large grain size was chosen to predominantly observe volume effects. Afterwards, 60 mg of the crushed and sieved glass were given into a corundum crucible, which was heated with a rate of 10 K min−1.
The phase content was determined via X-ray diffraction (XRD) using a Bruker D8 Advance (θ/θ-geometry) and Ni-filtered Cu Kα radiation. Measurements were performed in a 2θ-range from 10 to 60°.
The microstructure and composition of the appearing phases was studied via scanning electron microscopy (SEM) in combination with elemental analyses (energy dispersive X-ray spectroscopy, EDXS). Therefore, a ZEISS Supra 55 VP in combination with a Bruker EDXS system were used. From elemental distribution maps, smaller regions of interest belonging to solely one phase were defined and afterwards quantified. This procedure was used to determine the composition of different crystalline or amorphous phases.
3. Results and discussion
3.1 Crystallization upon cooling
The stoichiometric sodium–yttrium–disilicate composition (composition Zr0 in Table 1) served as starting point for the investigations. From the glass formation ranges shown in Fig. 1, it can be seen that this composition is marginally outside the published glass formation range.4 However, the descriptions of the glass formation range vary in literature and the maximum concentration of Y2O3 in such glasses is not yet clear. During melting of the composition Zr0, a solid layer has formed at the top of the liquid. Very high melting temperatures of 1680 °C kept for several hours lead to the disappearance of this layer and hence enabled the preparation of clear glasses.If the melts are slowly cooled to room temperature, instant crystallization occurs. In order to investigate the crystallization and phase formation during slow cooling, the melts were cooled from 1680 to 800 °C using a rate of 1 K min−1. At 800 °C, the furnace was switched off and the samples were allowed to cool to room temperature with a rate of around 1 to 2 K min−1. Fig. 2 shows SEM micrographs of compositions with different ZrO2-concentrations after slow cooling. In sample Zr0, solely one crystalline phase is observed, whereas in the samples Zr7 and Zr9, a second phase precipitates. In Fig. 2, this latter phase is marked with arrows. In the case of sample Zr7, it precipitates in dendritic morphology with crystal sizes above 100 μm as known from other ZrO2-containing materials obtained via cooling of a melt, such as fused-cast refractories.11 In Zr9, a different morphology is observed. Dendrites were not found within this sample but spheres with diameters below around 20 μm. The sample Zr5 is not shown in Fig. 2 because it is similar to Zr0, i.e., only one phase appears. The compositions of the residual glassy phase and the crystalline phases are determined via EDXS on samples, where two crystalline phases are observed under the supplied conditions.
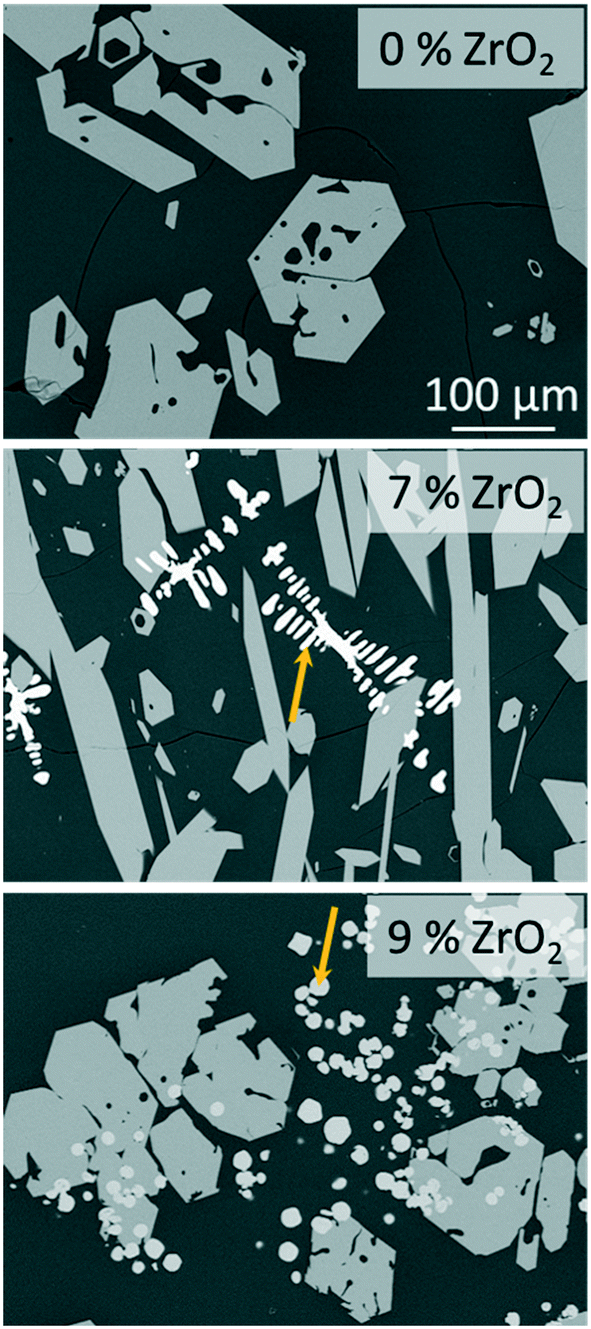 | ||
| Fig. 2 SEM micrographs (backscattered electrons) of samples Zr0, Zr7, and Zr9 after slow cooling to room temperature at identical magnifications. The arrows point at ZrO2 precipitates. | ||
The mol% compositions of the respective phases are summarized in Table 2. From those results, it can be seen that the phases have, within the limits of error of EDXS, the same composition which does not depend on the ZrO2-concentration. The glassy phase contains around 5.2 to 5.3 mol% ZrO2. The main crystalline phase is enriched in Na, Si, and Y. It also contains some ZrO2. This phase should be the NaY9Si6O26 with apatite structure and the space group P63/m (176).12
| Sample | Na2O | SiO2 | Y2O3 | ZrO2 | |
|---|---|---|---|---|---|
| Glassy phase | Zr7 | 22.8 | 67.5 | 4.4 | 5.3 |
| Zr9 | 22.0 | 68.5 | 4.2 | 5.3 | |
| Zr11 | 22.5 | 67.3 | 5.0 | 5.2 | |
| Main crystal phase | Zr7 | 5.5 | 56.8 | 33.7 | 3.9 |
| Zr9 | 5.2 | 57.4 | 33.5 | 3.9 | |
| Zr11 | 5.6 | 56.6 | 35.6 | 2.3 | |
| Secondary crystal phase | Zr7 | 0.1 | 2.3 | 15.9 | 81.7 |
| Zr9 | 0.0 | 1.9 | 15.1 | 82.9 | |
| Zr11 | 0.1 | 2.0 | 17.4 | 80.5 |
As reported in literature, the phase NaY9Si6O26 also appears if the stoichiometric compound NaYSi2O6 melts incongruently.13 The measured composition is not exactly equal to the stoichiometry of NaY9Si6O26, especially the Y-concentration is too low. However, also ZrO2 seems to be incorporated into the phase which changes the whole stoichiometry. It remains unclear, at which particular position, Zr4+ is incorporated. Reports in the literature about the formation of a solid solution, where quadrivalent ions are incorporated, also give no clear evidence on the incorporation.14
In this phase, Y3+ occurs at two different sites; one of them is only occupied by Y3+ which has a coordination number of 7 (in the following denoted as Y2-sites) and, while at the other site with the coordination number of 9, 75% are occupied by Y3+ (denoted as Y1-sites) the other 25% by Na+ (denoted as Na-sites). According to the EDXS analyses, the empirical composition of the crystalline phase is as follows: 3.1 Na, 15.8 Si, 18.8 Y, 1.1 Zr, and 61.2 O (at%), which correlates with a phase with a stoichiometry of around Na1.3Y8.0Si6.7Zr0.5O26. Since Zr4+ has a similar ionic radius as Y3+, (72 and 90 pm, respectively15), it should hence be assumed that Zr4+ occupies Y3+ sites, which of the two above described sites, however, remains unclear.
The basic composition in Kröger–Vink notation is as follows:
| Na(Na), 3 Y(Y1), 6 Y(Y2), 6 Si(Si), 26 O(O) |
NaY9Si6O26 has a very high melting temperature. Similar compositions are reported in ref. 13 to have melting points at around 1800 °C, or even higher. This also explains the different glass formation ranges reported in literature. At very high temperatures during synthesis (and a long melting time), residual crystalline phases should completely be dissolved and hence no longer act as seeds during subsequent cooling. This is observed, if the melts are casted and hence cooled rapidly.
The secondary phase which appears bright in the micrographs of Zr7 and Zr9 in Fig. 2 contains ZrO2 and Y2O3. From the high Y2O3-concentration it can be concluded that cubic ZrO2 should have formed. According to the literature, Y3+ occupies Zr4+ sites; the charge is balanced by O2− vacancies, which stabilizes the cubic phase down to room temperature. This is well-known from literature. As seen in the micrograph of sample Zr9, numerous ZrO2 crystals are embedded in the NaY9Si6O26 phase (see Fig. 2); hence the ZrO2 phase must have formed first and in a later stage, i.e., at lower temperatures, NaY9Si6O26 was crystallized. Nevertheless, there are also ZrO2 crystals surrounded by the glass phase and hence not obviously embedded in NaY9Si6O26 crystals. It might, however, be possible that the NaY9Si6O26 crystals have contact to the ZrO2(Y2O3)-phase outside the cut-plane visualized in the SEM-micrographs. There are also numerous ZrO2(Y2O3) crystals which do not have contact to the NaY9Si6O26 phase.
The results from quantification are supported by the respective XRD patterns displayed in Fig. 3. In the pattern of Zr5, solely the NaY9Si6O26 phase is identified, other phases are not detected within the limits of XRD. By contrast, in the patterns Zr7, Zr9, and Zr11, additionally XRD peaks attributable to cubic ZrO2 occur. The intensity of those peaks increases with increasing ZrO2 concentration. In the sample with 5 mol% ZrO2, cubic zirconia was not detected and hence ZrO2 is solely incorporated in the glass phase and the NaY9Si6O26 phase as previously described. ZrO2 concentrations of the ZrO2(Y2O3) and the NaY9Si6O26 crystals as well as of the glassy phase do not depend on the ZrO2 concentration of the respective samples. The ZrO2 concentration of the glass phase is always in the range from 5.2 to 5.3 mol% which seems to be the solubility limit applying a low cooling rate. This is also supported by the observation, that ZrO2 crystals are not observed in the sample Zr5.
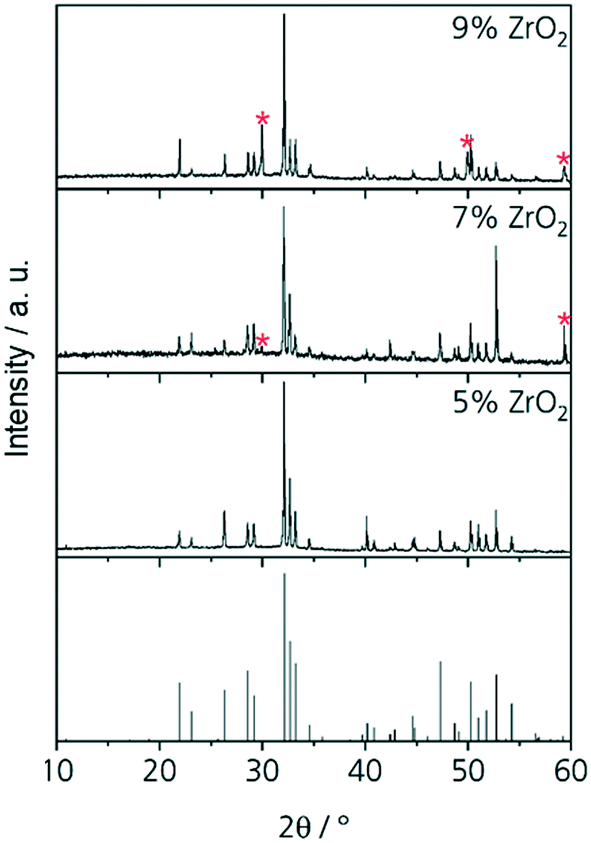 | ||
| Fig. 3 XRD patterns of Zr5, Zr7, Zr9 and Zr11 after slow cooling to room temperature. The theoretic pattern at the bottom belongs to NaY9Si6O26 (JCPDS no. 00-035-0404). The peaks of cubic zirconia (JCPDS no. 01-077-2112) are marked with red stars. | ||
If the slowly cooled samples, in which ZrO2 and NaY9Si6O26 have already been precipitated, are reheated to a temperature of 1100 °C for 10 h, it can be seen that further crystallization takes place within the sample volume (Fig. 4). In the micrograph (see Fig. 4) altogether four different phases occur. The NaY9Si6O26 and the ZrO2(Y2O3) crystals, a glassy phase and additionally a crystalline phase enriched in ZrO2 and Na2O. The latter phase grows preferably on the ZrO2(Y2O3) crystals, however, some of these crystals also grow on NaY9Si6O26. EDXS elemental mappings are displayed in Fig. 4. The composition of the new phase, which surrounds the ZrO2 crystals was quantified via EDXS. The quantification led to the conclusion that this crystal phase is Na2ZrSi2O7.
 | ||
| Fig. 4 EDXS elemental analysis of Zr7 after slow cooling to room temperature and subsequent reheating to 1100 °C for 10 h. | ||
3.2 Crystallization of the quenched glass upon heating
First heat treatments of the quenched glasses were performed on an amorphous sample with the composition Zr0 after casting on a metal mould. After heat treatments for 5 h at different temperatures in the range from 850 to 1150 °C, at least two crystalline phases can be detected by powder XRD. The respective diffractograms are displayed in Fig. 5. At 850 °C, crystallization can already be detected but the amorphous hump is also clearly visible. After thermal treatment at 900 and 950 °C, numerous sharp diffraction peaks occur; both diffractograms are almost identical. However, the crystalline phase or phases could not be identified. After crystallizing at 1000 °C, the formation of NaYSi2O6 crystals is detected. It should be noted that the composition of the sample Zr0 is the same as that of the crystal. Thermal treatment at temperatures in the range from 1050 to 1150 °C results in the sole crystallization of NaYSi2O6.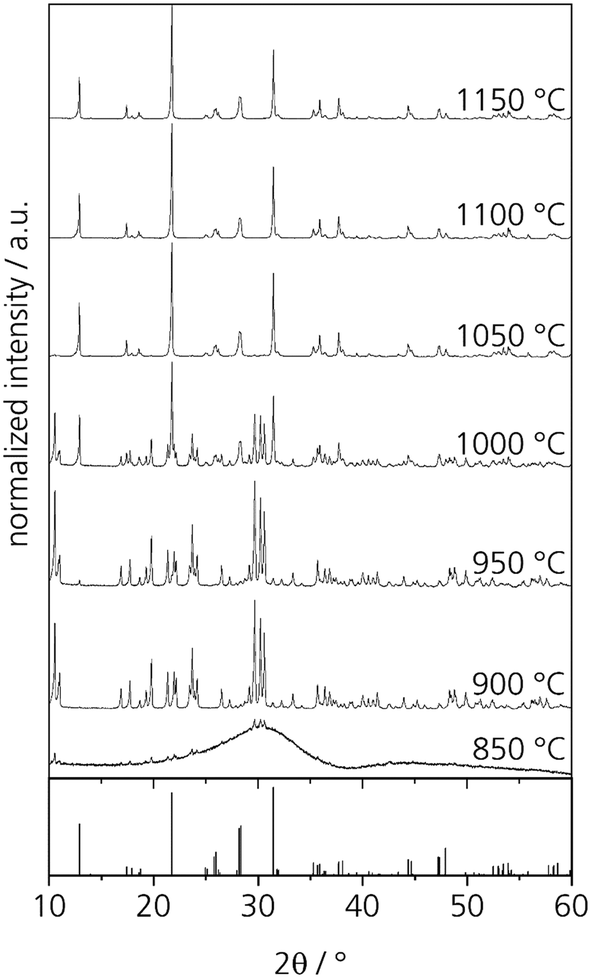 | ||
| Fig. 5 XRD patterns of glass Zr0 after heat treatments for 5 h at different temperatures. The theoretical peaks displayed at the bottom are attributed to NaYSi2O6 (calculated from ICSD 171255, ref. 16). | ||
In order to characterize the unknown phase, a glass specimen was heat treated at 850 °C for 5 h. This glass shows surface crystallization. However, the surface layer does not adhere to the bulk glass and flaked off from the glass easily. The crystalline layer was studied via EDXS and showed the same composition as the base glass. Hence, the unknown phase might be a different polymorph of NaYSi2O6.
From the cooling experiments, it can be concluded that long-term heat treatments below the melting point lead to a ZrO2 concentration of around 5.2 to 5.3 mol% in the glass matrix. Higher ZrO2 concentrations lead to the precipitation of cubic ZrO2-crystals containing high concentrations of Y2O3. Hence, in addition to the cooling experiments, the crystallization behavior was also studied upon heating of glass samples with the compositions Zr7 and Zr9.
In Fig. 6, the results from DSC of the stoichiometric glass Zr0 are illustrated together with Zr7 and Zr9. Glass Zr0 has a glass transition temperature of 761 °C, which increases to 799 and 794 °C for the glasses Zr7 and Zr9, respectively. The decrease of Tg from Zr7 to Zr9 might be due to the occurrence of phase separation. With increasing ZrO2 concentration, droplets enriched in ZrO2 can be formed, which consequently decrease the ZrO2 concentration in the residual glass phase. This results in a decrease of Tg. The formation of droplets with a size of around 5 nm enriched in ZrO2 has already been reported for the Li2O/Al2O3/SiO2 system.17 For all the three glasses, the same grain size fraction was used for DSC. Hence, also the exothermal crystallization peaks can be compared with each other. In the case of Zr0, a sharp peak with high intensity was measured. The onset is at 957 °C and the maximum is at 985 °C. An increase of the ZrO2 concentration leads to a shift of the crystallization peaks to higher temperatures. The onsets of the crystallization peaks are at 1013 and 1029 °C, while the maxima are at 1052 and 1058 °C for the glasses Zr7 and Zr9, respectively. The DSC curves of Zr7 and Zr9 are similar with respect to the shape of the crystallization peaks and the respective temperatures. However, there is a significant difference in comparison to the sample Zr0. The glass transition temperature shows a huge increase of 38 K between Zr0 and Zr7 and the crystallization temperatures increase by 56 K (onset) and 67 K (maximum), respectively. Also, the glass stability, illustrated by the difference between crystallization temperature (onset) and Tg, increases from Zr0 (197 K) to Zr7 (215 K) and Zr9 (226 K).
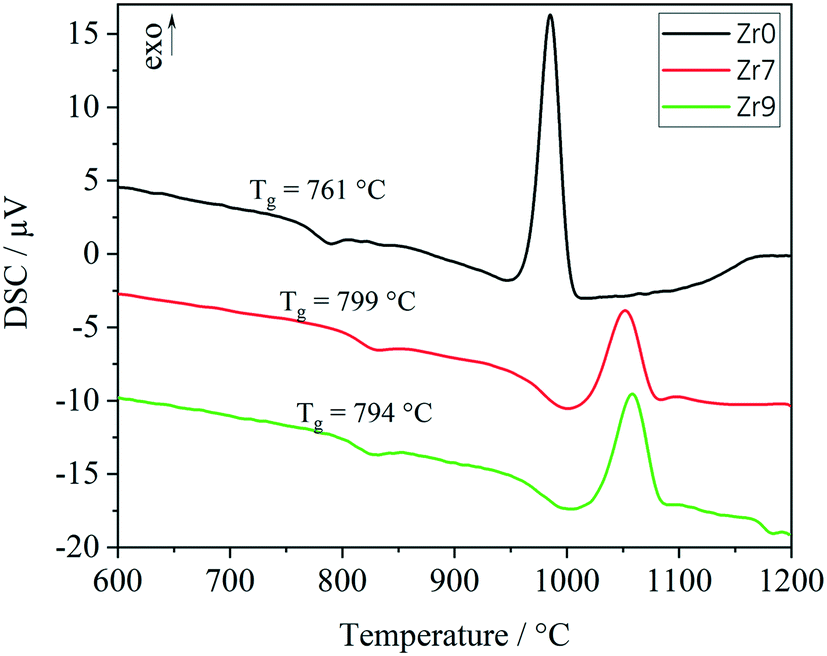 | ||
| Fig. 6 DSC curves of powder samples with a grain size of 400–500 μm. | ||
The exothermal crystallization peaks in Fig. 6 change their shapes from an intense and narrow signal (Zr0) to a broad and not as intense peak (Zr7 and Zr9), which should be due to a change of the crystallization mechanism from surface to volume crystallization or due to a decrease of the crystal growth velocity with increasing ZrO2 concentration.
For a more detailed study of the crystallization behavior, two step heat treatments of the samples with the compositions Zr0 and Zr7 were performed. The samples were first heated to a temperature in the range of 800 to 820 °C for 20 h in order to achieve nucleation and subsequently to 1000 or 1150 °C for 1 h to allow crystal growth. In the glass without ZrO2, nucleation solely occurred at the surface and hence crystallization starts from the surface (upper micrograph of Fig. 7), which leads to numerous cracks within the volume of the material. Volume crystallization was not observed. By contrast, in sample Zr7, volume crystals can be found after thermal treatment (lower micrograph of Fig. 7). However, the number density is not very high and hence, very huge crystals with diameters of more than 100 μm appear. It should be noted that the glass stability parameter of the Zr0 sample is somewhat lower than those of the samples Zr5 and Zr9. Nevertheless, the latter nucleates in the volume while the sample Zr0 does not. Hence ZrO2 acts as nucleation agent in the studied system despite of the higher glass stability parameter.
 | ||
| Fig. 7 SEM micrographs of glasses Zr0 and Zr7 after two-step heat treatments at 820 °C for 20 h and 1000 °C for 1 h. | ||
The elemental distribution of Zr7 after a two-step heat treatment is shown in Fig. 8. There, tiny Y2O3-enriched ZrO2 crystals can be seen, which are precipitated homogenously in the volume (see bright dots in the Y- and Zr-mappings). Those crystals are much smaller than the ZrO2 precipitations, which can be found after slow cooling.
 | ||
| Fig. 8 EDXS elemental mapping of sample Zr7 after a two-step heat treatment at 820 °C for 20 h followed by 1000 °C for 1 h. | ||
Increasing the ZrO2-conentration to 9 mol% (sample Zr9) does not improve the nucleation behavior of the glasses. In Fig. 9, SEM micrographs of sample Zr9 are shown after nucleation and crystallization. The number density of crystals is still very low and comparable to sample Zr7. The micrographs show a sample with the composition Zr9, heat treated at higher temperatures (820 °C for 20 h + 1150 °C for 1 h) and it can be seen that a large number of bright spots is enriched in between the larger crystals (see micrograph in the middle of Fig. 9). The right micrograph of Fig. 9 shows such a bright particle in more detail.
 | ||
| Fig. 9 SEM micrographs of sample Zr9 after a two-step heat treatment at 820 °C for 20 h and 1150 °C for 1 h. The dashed areas in the left micrograph mark large crystals and the framed area in the middle micrograph was used for an EDXS elemental analysis displayed in Fig. 10. | ||
Fig. 10 shows an EDXS elemental analysis, where it is seen that the bright spots are ZrO2 crystals enriched in Y2O3. Zirconia crystals are found all over the sample. Quantifications within Fig. 10 lead to the following conclusions concerning the appearing crystalline phases and their growth:
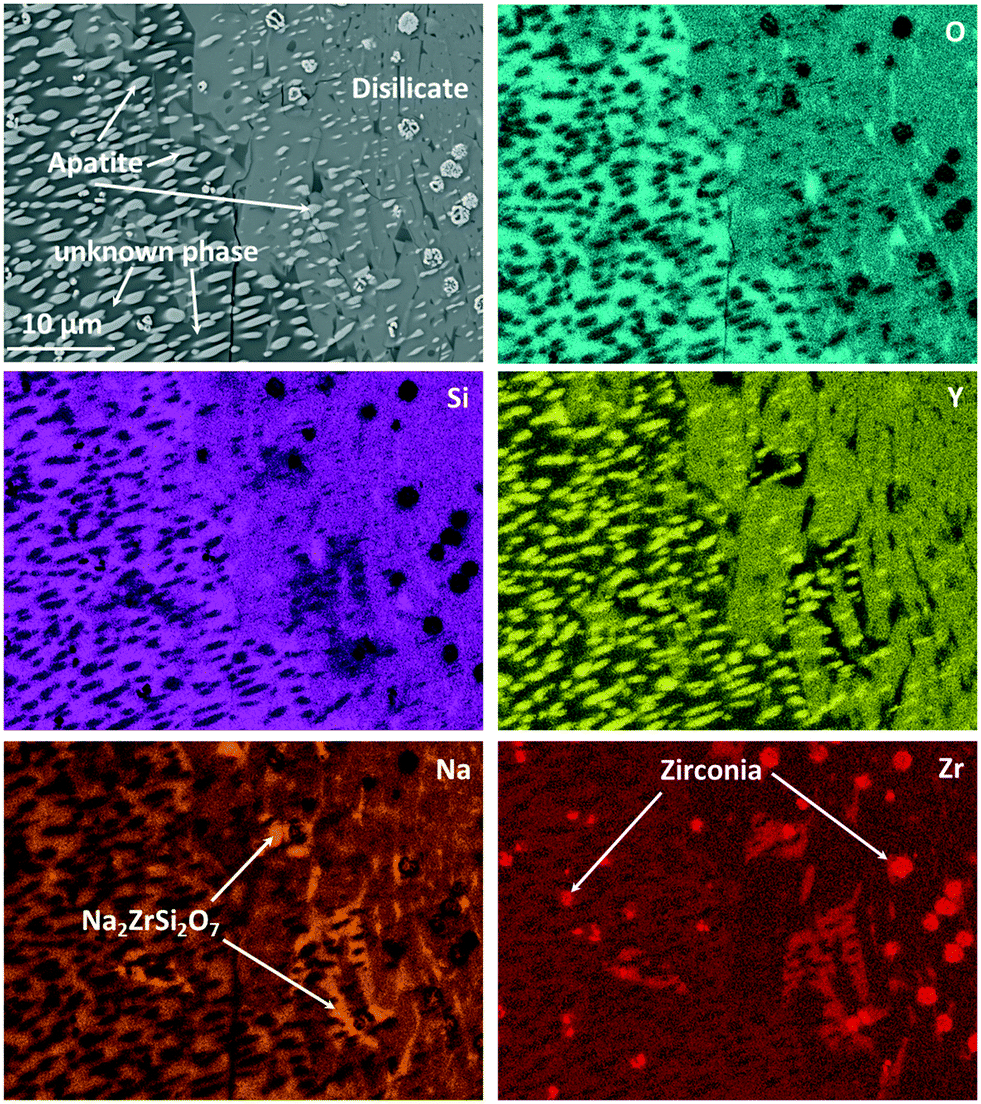 | ||
| Fig. 10 EDXS elemental mapping of Zr9 after a two-step heat treatment at 820 °C for 20 h + 1150 °C for 1 h. The mapping was recorded in the area of Fig. 9 marked with “EDXS”. | ||
The main phase in Fig. 10, which is visible in the left half of the micrograph, is apatite, NaY9Si6O26. This phase is embedded in another crystalline phase (see below), which, however, could not clearly be identified and in the following is denoted as “unknown phase”. The apatite crystals have an elongated morphology and surprisingly are all oriented with their long axis, probably the crystallographic (0001)-axis in the same direction. This is assumed because apatite crystals of different composition, (Ca5(PO4)3F), however, of the same space group show needle to rod-like shape when crystallized from homogeneous glass and the elongated direction is that of the crystallographic (0001)-axis.18,19 There are two possibilities to explain this: the first is that the apatite crystal grows up on a precursor crystal, i.e., the particular apatite crystal has a definite orientation relation to the precursor crystal. The second possible reason is that the apatite phase shows a dendritic growth and the bright crystalline spots in Fig. 10 are attributed to one dendrite which is cut in such a manner that the main branch of the dendrite is not in the cut plane. It should be noted that dendritic growth of calcium apatite has already been reported in ref. 20 and 21. The major crystalline phase in the right half of Fig. 10 is NaYSi2O6. However, this phase is only the main phase within the displayed micrograph. An overview of the microstructure is given in Fig. 9, where it is also seen that the NaYSi2O6 phase only appears in between the large crystals and is hence only a minor phase. In the volume fraction, where this phase is detected, the ZrO2-crystals are larger. An explanation might be that during heating, the ZrO2 crystals precipitate inside the glass. Some of the crystals lead to the formation of volume crystals. The apatite crystals as well as the unknown phase seem to grow at first, which leads to an enrichment of ZrO2 in the residual material. There, the zirconia crystals can grow to a larger size. In a later stage, the NaYSi2O6 phase forms and the residual glassy phase also crystallizes. There, Na2O is enriched and Na2ZrSi2O7 is additionally formed.
This was also proven by XRD. However, different from the composition without zirconia, which forms solely the NaYSi2O6 phase at high temperatures (see Fig. 5), in the composition with 9 mol% ZrO2 the unknown phase also precipitates in a large amount. This phase is marked in the micrograph in Fig. 10. The “unknown phase” is seen on the left side. Its composition was measured to be around: 61.7 O, 8.6 Na, 22.8 Si, 4.4 Y, 2.4 Zr (in at%) which is attributed to an empirical formula Na0.84Y0.44Zr0.24Si2.21O6. The phase is assumed to be a polymorph of NaYSi2O6 in which some Y3+ has been substituted by Zr4+.
Conclusions
Quenching of melts in the system Na2O/Y2O3/SiO2 with ZrO2 additions and subsequent thermal treatment resulted in phase compositions and microstructures fairly different from those obtained during slow cooling of melts with the same composition. Slow cooling results in the formation of NaY9Si6O26 crystals with apatite structure and for ZrO2-concentrations >5 mol% also of yttrium stabilized cubic zirconia. During reheating of the slow cooled glasses, zirconia crystals serve as starting points for the formation of Na2ZrSi2O7. If the material is prepared via melt quench technique, and if ZrO2 is given in concentrations above 5 mol%, homogenously distributed small ZrO2 crystals are precipitated. Furthermore, glasses are obtained from which NaYSi2O6 precipitates at high temperatures. At crystallization temperatures below around 1000 °C, a phase forms, which could not be identified, but is assumed to be a polymorph of NaYSi2O6 in which a part of Y3+ is substituted by Zr4+. It also precipitates at higher temperatures in the case of a sample with a ZrO2 concentration of 9 mol%. For a more detailed characterization of the crystallization processes within this glass system, the unknown phase has to be characterized in future experiments.Conflicts of interest
There are no conflicts of interest to declare.References
- J. Schilm, D. Wagner, C. Heubner, U. Langklotz, C. W. Lee, H. S. Kang, J. W. Park and M. Kusnezoff, J. Eur. Ceram. Soc., 2021, 41, 4876–4883 CrossRef CAS.
- M. Alexander and B. Riley, Solid State Ionics, 1986, 18–19, 478–482 CrossRef CAS.
- T. Okura, K. Kawada, N. Yoshida, H. Monma and K. Yamashita, Solid State Ionics, 2012, 225, 367–370 CrossRef CAS.
- P. W. Angel and R. E. Hann, J. Am. Ceram. Soc., 1992, 75, 3278–3282 CrossRef CAS.
- M. J. Hyatt and D. E. Day, J. Am. Ceram. Soc., 1987, 70, C-283–C-287 CrossRef.
- S. Alahraché, M. Deschamps, J. Lambert, M. R. Suchomel, D. De Sousa Meneses, G. Matzen, D. Massiot, E. Véron and M. Allix, J. Phys. Chem. C, 2011, 115, 20499–20506 CrossRef.
- A. Keshavarzi, W. Wisniewski and C. Rüssel, CrystEngComm, 2012, 14, 6904 RSC.
- W. Höland, V. Rheinberger and M. Schweiger, Philos. Trans. R. Soc., A, 2003, 361, 575–589 CrossRef.
- K. Thieme and C. Rüssel, J. Mater. Sci., 2015, 50, 1488–1499 CrossRef CAS.
- H. R. Fernandes, D. U. Tulyaganov and J. M. F. Ferreira, J. Mater. Sci., 2013, 48, 765–773 CrossRef CAS.
- W. Wisniewski, C. Thieme and C. Rüssel, J. Eur. Ceram. Soc., 2019, 39, 2186–2198 CrossRef CAS.
- R. P. Gunawardane, R. A. Howie and F. P. Glasser, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1564–1566 CrossRef.
- F. C. Lee, J. Marr and F. P. Glasser, Ceram. Int., 1981, 7, 43–47 CrossRef CAS.
- J. R. Tolchard, P. R. Slater and M. S. Islam, Adv. Funct. Mater., 2007, 17, 2564–2571 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- D. M. Többens, V. Kahlenberg and R. Kaindl, Inorg. Chem., 2005, 44, 9554–9560 CrossRef PubMed.
- T. Höche, M. Mäder, S. Bhattacharyya, G. S. Henderson, T. Gemming, R. Wurth and C. Rüssel, CrystEngComm, 2011, 13, 2550–2556 RSC.
- C. Moisescu, C. Jana and C. Rüssel, J. Non-Cryst. Solids, 1999, 248, 169–175 CrossRef CAS.
- W. Wisniewski, R. de Kloe and C. Rüssel, CrystEngComm, 2015, 17, 2969–2973 RSC.
- J. J. Shyu and J. M. Wu, J. Mater. Sci., 1994, 29, 3167–3171 CrossRef CAS.
- J.-J. Shyu and J.-M. Wu, J. Am. Ceram. Soc., 1991, 74, 1532–1540 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |