
J-aggregation induced emission enhancement of BODIPY dyes via H-bonding directed supramolecular polymerization: the importance of substituents at boron†
Yongjie
Zhang‡
 a,
Siyuan
Yuan‡
a,
Ping
Liu
a,
Lei
Jing
a,
Hongfei
Pan
a,
Xiang-Kui
Ren
*ab and
Zhijian
Chen
*ab
a,
Siyuan
Yuan‡
a,
Ping
Liu
a,
Lei
Jing
a,
Hongfei
Pan
a,
Xiang-Kui
Ren
*ab and
Zhijian
Chen
*ab
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072, China. E-mail: zjchen@tju.edu.cn; renxiangkui@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Chemical Engineering (Tianjin), Tianjin University, Tianjin, 300072, China
First published on 20th May 2021
Abstract
Two new boron-dipyrromethene (BODIPY) dyes 1b and 1c, bearing two uracil units at the 2,6-positions and solubilizing alkyne groups at boron atoms, were synthesized and characterized. The UV/Vis absorption and fluorescence spectroscopic studies indicated that in nonpolar solvents these BODIPY dyes supramolecularly polymerized into J-aggregates, which exhibited outstanding optical properties, such as narrowed absorption and emission bands with reduced fluorescence lifetime and increased quantum yields with respect to that for monomers. The mechanism of the polymerization of 1b and 1c was analysed by temperature- and concentration-dependent spectroscopy and studied with a nucleation–elongation model. Measurements of concentration-dependent 1H NMR and AFM demonstrated the H-bonding directed self-assembly of J-aggregates of dyes 1b and 1c, which led to the formation of one-dimensional nanowires of these dyes. Further molecular modelling studies and calculations based on exciton theory indicated that the bulky alkyne substituents at boron atoms effectively hindered the close contact between the π-faces of BODIPY chromophores, implying that the appropriate segregation of supramolecular polymer chains could be crucial for the aggregation-induced emission enhancement (AIEE) for this class of BODIPY dyes, as compared with the fluorescence quenching observed for the J-aggregates of BODIPY 1a bearing F atoms at boron in our previous report.
Introduction
J-aggregates of functional dyes have been investigated extensively in multidisciplinary research since their serendipitous discovery in the 1930s.1 The most attractive features of these dye aggregates are their remarkable optical properties caused by strong excitonic coupling of the dye units and their applications in important fields such as artificial light harvesting2 and sensitization in colour photography.3 To form J-aggregates, molecular packing with strong slippage of the chromophores is crucial and supramolecular principles have been adopted for rational control of the spatial arrangement of the dye molecules. For example, amphiphilically modified carbocyanine dyes have been reported to form highly ordered nanotubular or ribbon-like J-aggregates in early studies.4 More recently, Würthner et al. have developed a series of highly fluorescent J-aggregates based on perylene bisimide dyes through supramolecular design,5 in which the head-to-tail alignment of chromophores is directed by imide hydrogen-bonding interactions. In a later report, this supramolecular strategy was successfully applied for the construction of J-aggregates for amine-substituted naphthalene-diimide by Fernández and Ghosh.6 Nevertheless, only a very limited number of examples with intriguing properties resembling those of cyanine dye-based J-aggregates, i.e. the bathochromically shifted and sharpened narrower absorption band than that of the respective monomers and enhanced fluorescence with reduced lifetime, have been achieved so far.Boron-dipyrromethene (BODIPY) dyes are a class of versatile organic chromophores and have attracted research interest in multiple fields.7 The chemical structure of BODIPY dyes can be facilely modified, yielding products with improved optical and self-assembly properties.8 Accordingly, considerable efforts have been devoted towards constructing supramolecular materials, e.g. organogels,9 nanovesicles10 and liquid crystals,11 by using BODIPY as building blocks. However, while many BODIPY dyes exhibit high fluorescence quantum yields in dilute solutions, severe aggregation caused quenching (ACQ) of fluorescence may still take place,12 even in the case when typical 1D J-aggregates were formed, as observed for the uracil-functionalized BODIPY dye 1a (Scheme 1) in our previous report.13 Thus, the development of dye assemblies of this class of BODIPYs without fluorescence quenching or with aggregation induced emission enhancement (AIEE)14 is an appealing objective owing to their potential application in polarized luminescent materials15 or in other fields.16
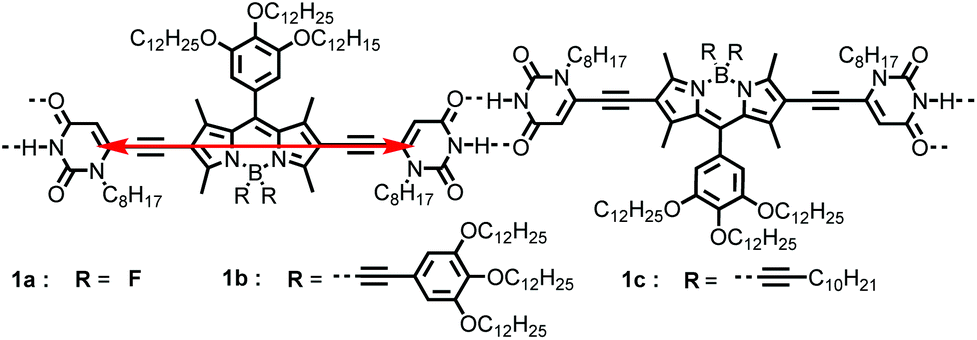 | ||
| Scheme 1 J-aggregation pattern of uracil-functionalized BODIPYs 1a, 1b, and 1c by H-bonding directed supramolecular polymerization along with the transition dipole moments (red arrow) of the dye molecules. | ||
In the present work, we report two newly synthesized BODIPY dyes 1b and 1c and their emission enhancement induced by J-aggregation via supramolecular polymerization directed by H-bonding interactions between the uracil groups at both sides (2,6-positions) of the BODIPY core. Meanwhile, the dyes 1b and 1c have solubilizing moieties containing multiple alkyl chains at the meso-position of the BODIPY core and the boron atom, which improves the solubility of the dyes in aliphatic solvents compared with the fluorine-unsubstituted analogue dye 1a. This feature is crucial for the supramolecular polymerization of 1b and 1c since it has been shown that the H-bonding interaction is much stronger in nonpolar aliphatic solvents, such as n-hexane, than in polar organic solvents.17 The optical and structural properties of the supramolecular J-aggregates based on BODIPYs 1b and 1c were investigated by spectroscopic and microscopic measurements as well as molecular modelling to reveal the self-assembly mechanism and elucidate the observed strong AIEE effect for these J-aggregates.
Results and discussion
As shown in Scheme 2, the synthesis of BODIPY dyes 1b and 1c started from BODIPY 3 with iodo substituents at the 2,6-positions.7b Dye 3 was reacted with the Grignard reagents 3,4,5-tridodecyloxyphenylacetylene and 1-dodecyne to provide boron-substituted chromophores 2b and 2c, respectively. Subsequently, the 6-ethynyl-1-n-octyluracil units were appended on the 2,6-positions of the BODIPY core by Sonogashira coupling to give BODIPY dyes 1b and 1c.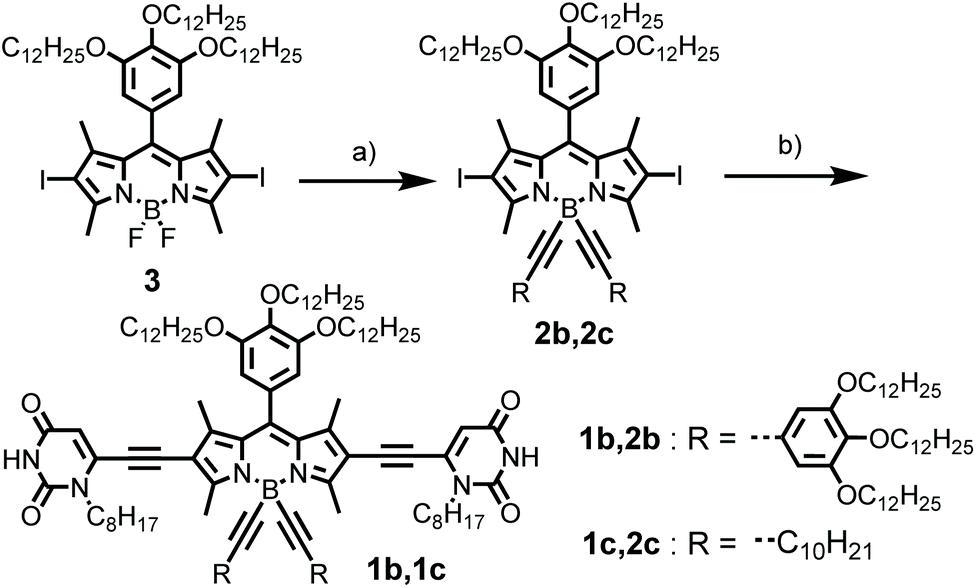 | ||
| Scheme 2 The synthetic route for BODIPY dyes 1b, 1c. Reagents and conditions: (a) 3,4,5-Tridodecyloxyphenylacetylene (1-dodecyne), C2H5BrMg, THF, 60 °C, 6 h, 64%; (b) 6-ethynyl-1-n-octyluracil, Pd(PPh3)4, CuI, TEA, 70 °C, 4 h, 74%. | ||
The UV/Vis absorption spectrum of 1b and 1c in CH2Cl2 (cT = 2.0 × 10−6 M) exhibits typical spectroscopic features for molecularly dissolved BODIPY dyes (Fig. 1a and Fig. S9†). For 1b, the absorption band in the range of 540–600 nm was assigned to the S0–S1 transition band (ε = 1.5 × 105 M−1 cm−1 at 557 nm). The fluorescence spectrum of 1b and 1c in CH2Cl2 displayed a mirror-image relationship to the absorption spectrum. The absorption and emission spectra of 1b and 1c in CH2Cl2 are highly comparable, indicating that the different substituents at boron atoms have only minor effects on the spectroscopic properties. In CH2Cl2, the fluorescence quantum yields of the monomers of dyes 1b and 1c were determined to be 0.42 and 0.39, respectively.
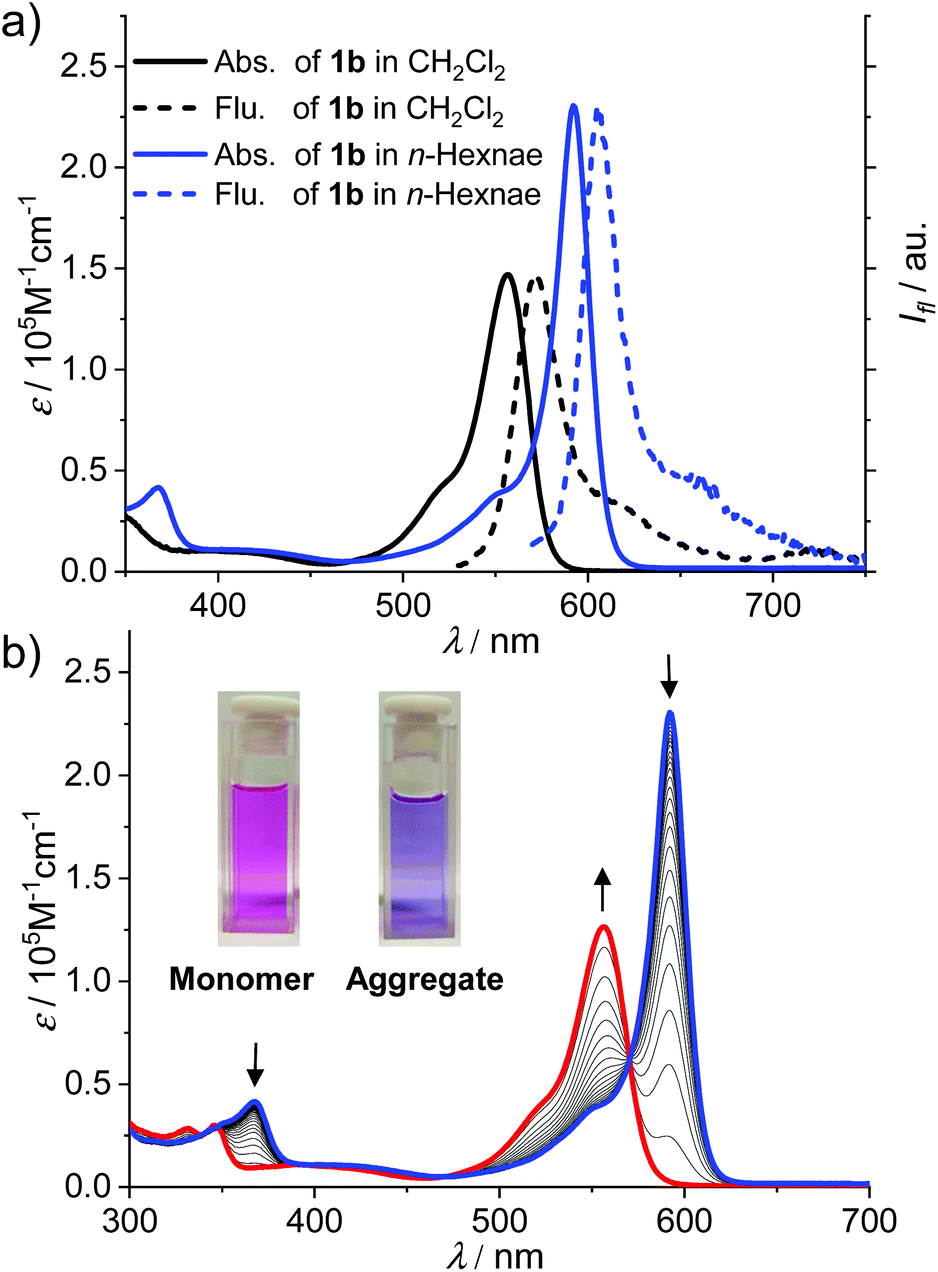 | ||
| Fig. 1 (a) UV/Vis absorption and fluorescence spectra (λEx = 350 nm) of dye 1b in CH2Cl2 (black, cT = 2.0 × 10−6 M) and n-hexane (blue, cT = 2.0 × 10−6 M). (b) Temperature-dependent UV/Vis absorption spectra of BODIPY dye 1b in n-hexane (cT = 2.0 × 10−6 M). The arrows indicate the spectra changing with the increase of the temperature from 277 to 333 K. Inset: Visual appearance of solutions of monomeric and aggregated dye 1b under daylight. | ||
For a solution of dye 1b in n-hexane at the same concentration as in CH2Cl2 (cT = 2.0 × 10−6 M), the spectral characteristics were obviously changed (Fig. 1a, blue lines). The maximum absorption wavelength (λmax) of BODIPY dye 1b in n-hexane was shifted bathochromically to 591 nm as compared with that in CH2Cl2 while the molar absorption coefficient at λmax was largely increased to 2.4 × 105 M−1 cm−1. Meanwhile, the band around 591 nm is obviously sharper than the S0–S1 band in DCM. Upon increasing the temperature of the solution in n-hexane (Fig. 1b), the absorption band of 1b at 591 nm was gradually decreased while a new band at 556 nm arose, which resembled the S0–S1 transition band of the molecularly dissolved 1b in CH2Cl2. In addition, an obvious colour change of the solution from purple (aggregate) to pink (monomer) was observed upon the increase in the temperature. All these observations pointed to the formation of J-aggregates of 1b in nonpolar n-hexane.
The J-aggregation of 1b in n-hexane was further confirmed by fluorescence spectroscopic measurements. As shown in Fig. 1a, the fluorescence spectrum displayed an approximately mirror-image relationship to the J-band with an emission maximum at 612 nm. Further temperature-dependent fluorescence spectra (Fig. 2a) indicated the gradual transition from the aggregated species to monomers upon heating. In addition, for the methylcyclohexane (MCH) solution of dye 1b as well as the n-hexane and MCH solutions of dye 1c, spectral characteristics of J-aggregates comparable with that for 1b in n-hexane were observed (Fig. S10–S15†).
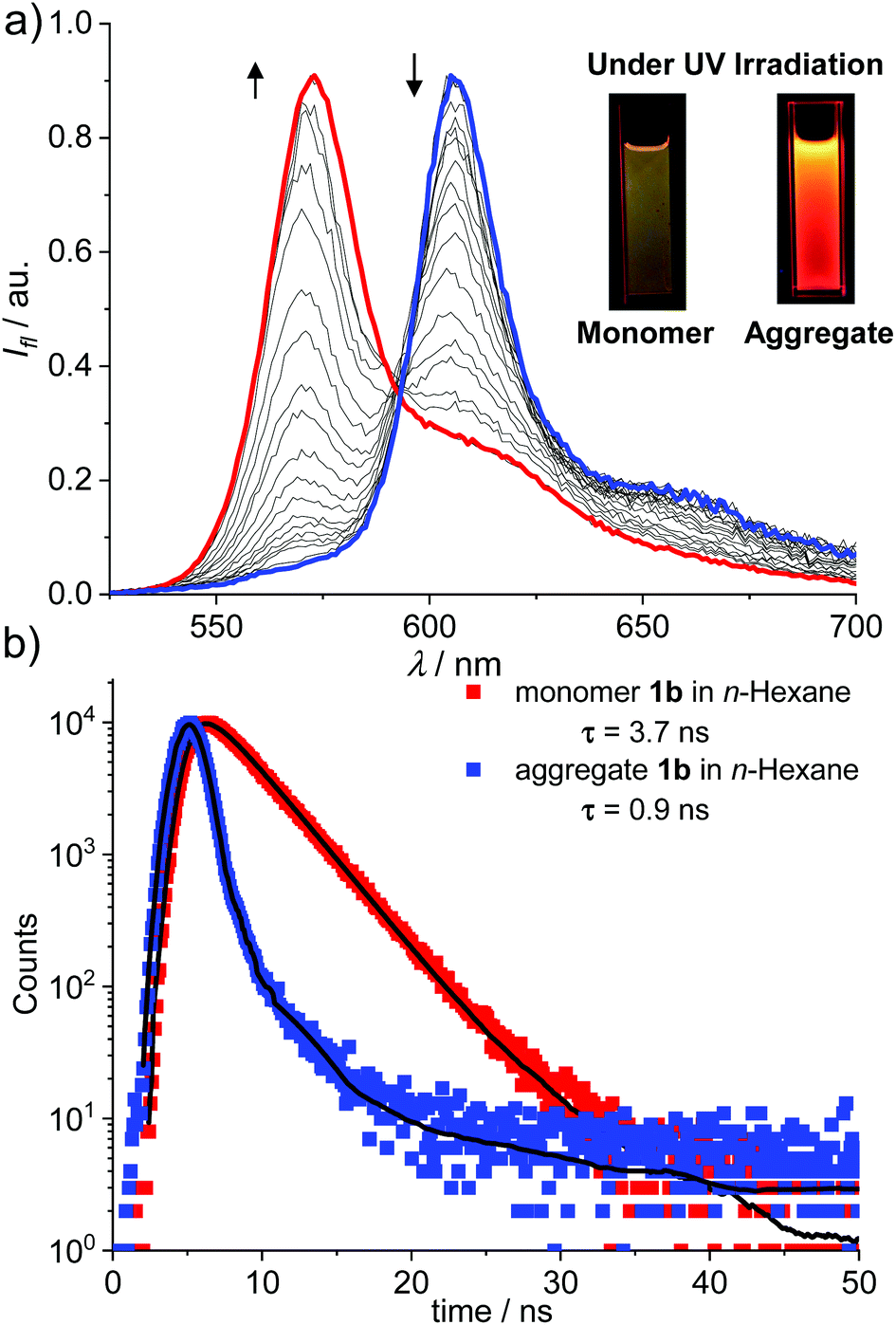 | ||
| Fig. 2 (a) Temperature-dependent fluorescence spectra of 1b in n-hexane (cT = 2.0 × 10−6 M). The arrows indicate the spectra change with the increase in the temperature from 277 to 333 K. Inset: Visual appearance of solutions of monomeric and aggregated dye 1b under a UV lamp (365 nm). (b) Time-resolved fluorescence decay for aggregates (cT = 1.0 × 10−5 M, λEx = 350 nm, λEm = 612 nm) and monomers (cT = 5.0 × 10−8 M, λEx = 350 nm, λEm = 572 nm) of 1b in n-hexane. | ||
More interestingly, for both 1b and 1c, much brighter fluorescence could be observed by the naked eye for the J-aggregates than that of monomers under irradiation of UV light, implying the occurrence of AIEE for these dyes (Fig. 2a, insets). Accordingly, more photophysical properties, including concentration-dependent fluorescence quantum yields (Fig. S16†) and fluorescence lifetimes (Fig. 2b and Fig. S17–S20†), were measured for dyes 1b and 1c in n-hexane as well as MCH and the selected results are presented in Table 1. Indeed, the J-aggregates of 1b and 1c exhibit much higher fluorescence quantum yields than the monomers. For example, the fluorescence quantum yields of the monomers of 1b in diluted n-hexane solutions (cT = 1.0 × 10−7 M) were measured as 0.27. With the increase in the concentration, the fluorescence quantum yields of 1b were gradually increased respectively to 0.62 (cT = 4.0 × 10−6 M). Meanwhile, the concentration-dependent fluorescence spectra (Fig. S16b†) of 1b exhibited bathochromically shifted emission bands that were characteristic of the formation of J-aggregates. Similar trends of the quantum yields at various concentrations were observed for these dyes in MCH. These results indicate explicitly the J-aggregation induced emission enhancement properties of these dyes.
| Dyes | Solvent | λ mon/nm | λ agg/nm | λ em,mon/nm | λ em,agg/nm | Φ mon | Φ agg | τ mon/ns | τ agg/ns |
|---|---|---|---|---|---|---|---|---|---|
| 1b | CH2Cl2 | 557 | N/A | 578 | N/A | 0.42 | N/A | 3.9 | N/A |
| 1c | CH2Cl2 | 555 | N/A | 568 | N/A | 0.39 | N/A | 4.5 | N/A |
| 1b | n-Hexane | 556 | 591 | 572 | 612 | 0.27 | 0.62 | 3.7 | 0.9 |
| 1c | n-Hexane | 555 | 590 | 570 | 604 | 0.19 | 0.51 | 3.4 | 2.4 |
| 1b | MCH | 559 | 588 | 578 | 608 | 0.39 | 0.58 | 3.0 | 2.1 |
| 1c | MCH | 558 | 589 | 570 | 603 | 0.23 | 0.48 | 3.2 | 2.5 |
For the J-aggregates of dyes 1b and 1c, the pronounced bathochromic shifts in the absorption and emission spectra with an obviously narrowed band shape indicated the strong excitonic coupling interactions between the aggregated chromophores. For 1b, distinct fluorescence lifetimes of 3.7 ns and 0.9 ns were measured for monomers and J-aggregates in n-hexane, respectively. The decreased fluorescence lifetime is indicative of enhanced radiative decay arising from the excitonic coherence between the aggregated molecules. To estimate the size of the coherent domain in J-aggregates, the radiative decay rate constants (kr) were calculated from kr = Φ/τ. Thus, kJr = 6.8 × 108 s−1 and kMr = 7.3 × 107 s−1 were evaluated for J-aggregates and monomers of 1b respectively. Furthermore, a coherent size18 of N ≈ 9 for J-aggregates of 1b can be estimated with N = kJr/kMr. Similarly, the coherent size in J-aggregates of 1c in n-hexane can be estimated to be ca. 4 dye molecules. These results confirmed that the AIEE property of J-aggregates of 1b and 1c is mainly caused by a superradiance effect. It is worth noting that the mechanism of J-aggregation induced emission enhancement observed for dyes 1b and 1c is obviously different from that of the other AIEgens such as tetraphenylethene and silole derivatives, for which a mechanism of restriction of intramolecular rotation (RIR) has been proposed to explain the enhancement of fluorescence.19
To give further insights into the mechanistic and structural aspects of the supramolecular polymerization of uracil-functionalized BODIPY dyes 1b and 1c, detailed analysis of the spectroscopic data as well as morphological studies of the J-aggregates were carried out. Based on the temperature-dependent UV/Vis spectroscopic data (Fig. 1b and Fig. S10–S12†), the fraction of aggregated molecules (αagg) versus temperature (T) was evaluated (for details, see the ESI†) and further fitted with the nucleation–elongation supramolecular polymerization model (Fig. 3a).20 The αagg–T plots exhibited satisfactory accordance with cooperative processes. For the aggregation process of 1b in n-hexane (cT = 2.0 × 10−6 M), the critical elongation temperature (Te) and molar enthalpy (ΔHe) were determined to be 330 K and −74.2 kJ mol−1 respectively (Table 2). For dye 1c, lower Te (318 K) and ΔHe (−64.5 kJ mol−1) were obtained at the same concentration, which indicates that 1b has a stronger tendency for aggregation. The dimensionless equilibrium constants (Ka) of the activation step at Te were determined to be 1.5 × 10−3 and 8.7 × 10−4 for 1b and 1c respectively, implying a higher degree of cooperativity for the aggregation process of 1b. As a comparison, the fitting results of the αagg–T plots in MCH (cT = 1.0 × 10−5 M) gave a similar trend to that in n-hexane, i.e., 1b possesses a higher Te and a smaller ΔHe than 1c (Table 2).
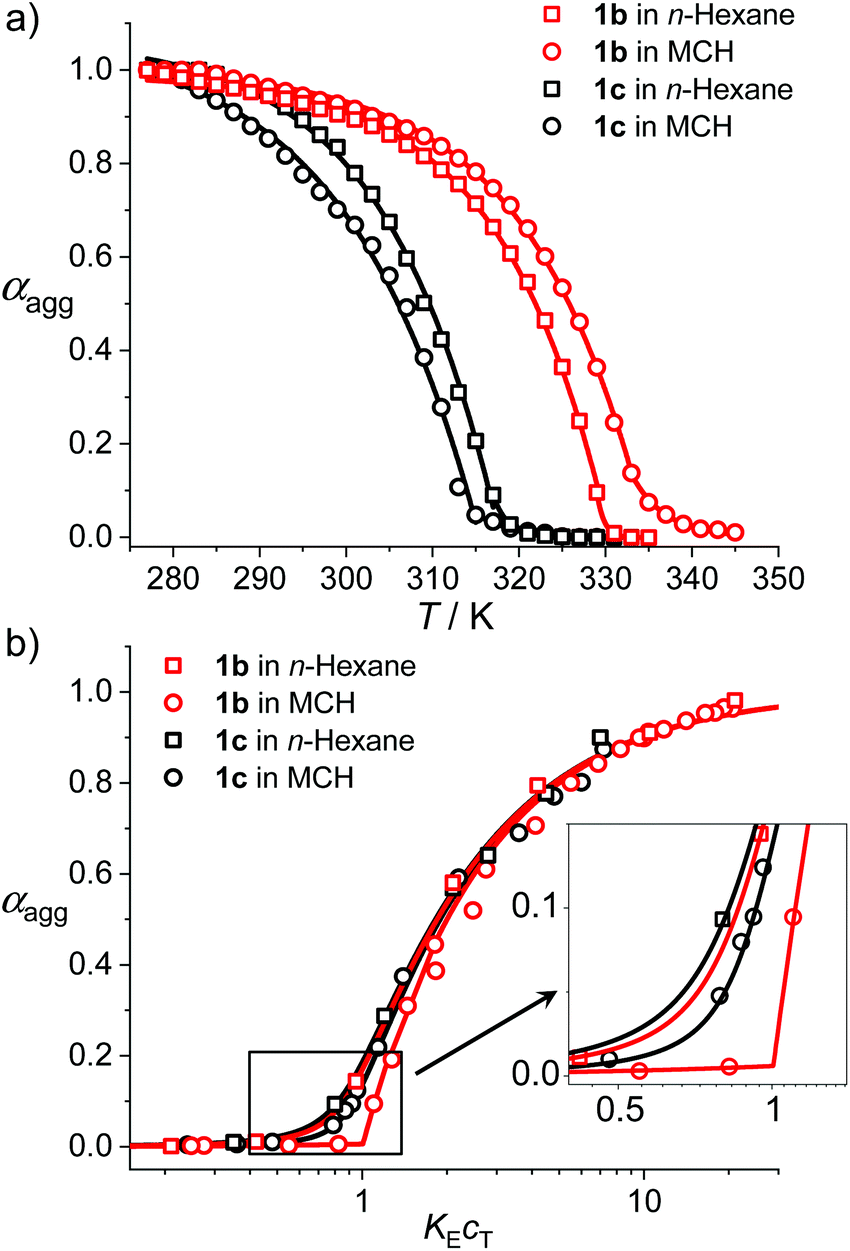 | ||
| Fig. 3 (a) Plot of the molar fraction of aggregated molecules of 1b and 1c in n-hexane (cT = 2.0 × 10−6 M) and MCH (cT = 1.0 × 10−5 M) as a function of the temperature and the fitting curves by applying the cooperative self-assembly model. (b) Plot of the molar fraction of aggregated molecules of 1b and 1c in n-hexane and MCH as a function of the dimensionless concentration KEcT and fitting curves by applying the Goldstein–Stryer model. | ||
| Dyes | T e/K | ΔHe/kJ mol−1 | K a | σ | s | K E /106 M−1 |
|---|---|---|---|---|---|---|
| a Measured in n-hexane. b Measured in MCH. c Data of 1b in n-hexane are measured at T = 313 K while data of 1b in MCH and 1c in both solvents are measured at T = 298 K. | ||||||
1b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
330 | −74.2 | 1.5 × 10−3 | 0.006 | 2 | 2.1 |
|
1bb |
334 | −69.7 | 3.2 × 10−4 | 0.003 | 3 | 0.28 |
|
1ca |
318 | −64.5 | 8.7 × 10−4 | 0.008 | 2 | 3.5 |
|
1cb |
315 | −54.4 | 8.9 × 10−5 | 0.003 | 2 | 0.12 |
Moreover, concentration-dependent UV/Vis absorption spectroscopic investigation was performed for BODIPY dyes 1b and 1c and spectral changes comparable with those in temperature-dependent studies were observed (Fig. S21 and S22†). By fitting the experimental data of αaggversus concentration cT with the Goldstein–Stryer model for nucleated supramolecular polymerization (Fig. 3),17b,21 parameters including nucleus size s, cooperativity factor σ and the elongation equilibrium constant KE were obtained (Table 2). These results corroborate the cooperative mechanism of the self-assembly processes of 1b and 1c. For 1b, the aggregates in n-hexane could not fully disaggregate to monomers at 298 K even upon diluting to a concentration of 1.0 × 10−7 M. Thus, the equilibrium constant of 1b in n-hexane was measured at 313 K and a KE of 2.1 × 106 M−1 was obtained. In MCH, the KE for the self-assembly of dye 1b is higher than that obtained for 1c, indicating that the aggregates of 1b have higher stability, which is consistent with the temperature-dependent spectroscopic studies. Moreover, the KE values of 1b and 1c in n-hexane are one order of magnitude higher than those in MCH, which is rational since the relatively less polar n-hexane is beneficial for the intermolecular H-bonding interaction.22
The H-bonding interactions between the 2,6-uracil groups of adjacent dye molecules were characterized by concentration-dependent 1H NMR spectroscopy (Fig. S23 and 24†) in CDCl3. For both dyes, significant concentration-dependency was observed for the signal of imide-H. As shown by 1b, the signal of imide-H displayed a constant downfield shift from 7.9 to 8.9 ppm while the dye concentration was gradually increased from of 9.8 × 10−5 M to 1.3 × 10−2 M, indicating the formation of intermolecular hydrogen bonds between uracil groups. According to a model developed by LaPlanche et al.,23 association constants of 410 M−1 and 450 M−1 in CDCl3 were obtained respectively for the H-bonding of dyes 1b and 1c. The nearly identical association constants for the two dyes suggested that the substituents at boron atoms have a minor effect on the H-bonding interactions. The H-bonding directed supramolecular polymerization often leads to the formation of 1D nanostructures, such as nanowires or nanofibers.24 Accordingly, the nanomorphology of the J-aggregates for BODIPY dyes 1b and 1c was characterized by atomic force microscopy (AFM). The results indicated that 1b self-assembled into nanowires with a width over 100 nm and a height of ca. 4 nm (Fig. 4). The J-aggregates of 1c exhibited a morphology similar to that of nanowires with a width of ca. 50 nm and a height of ca. 3 nm.
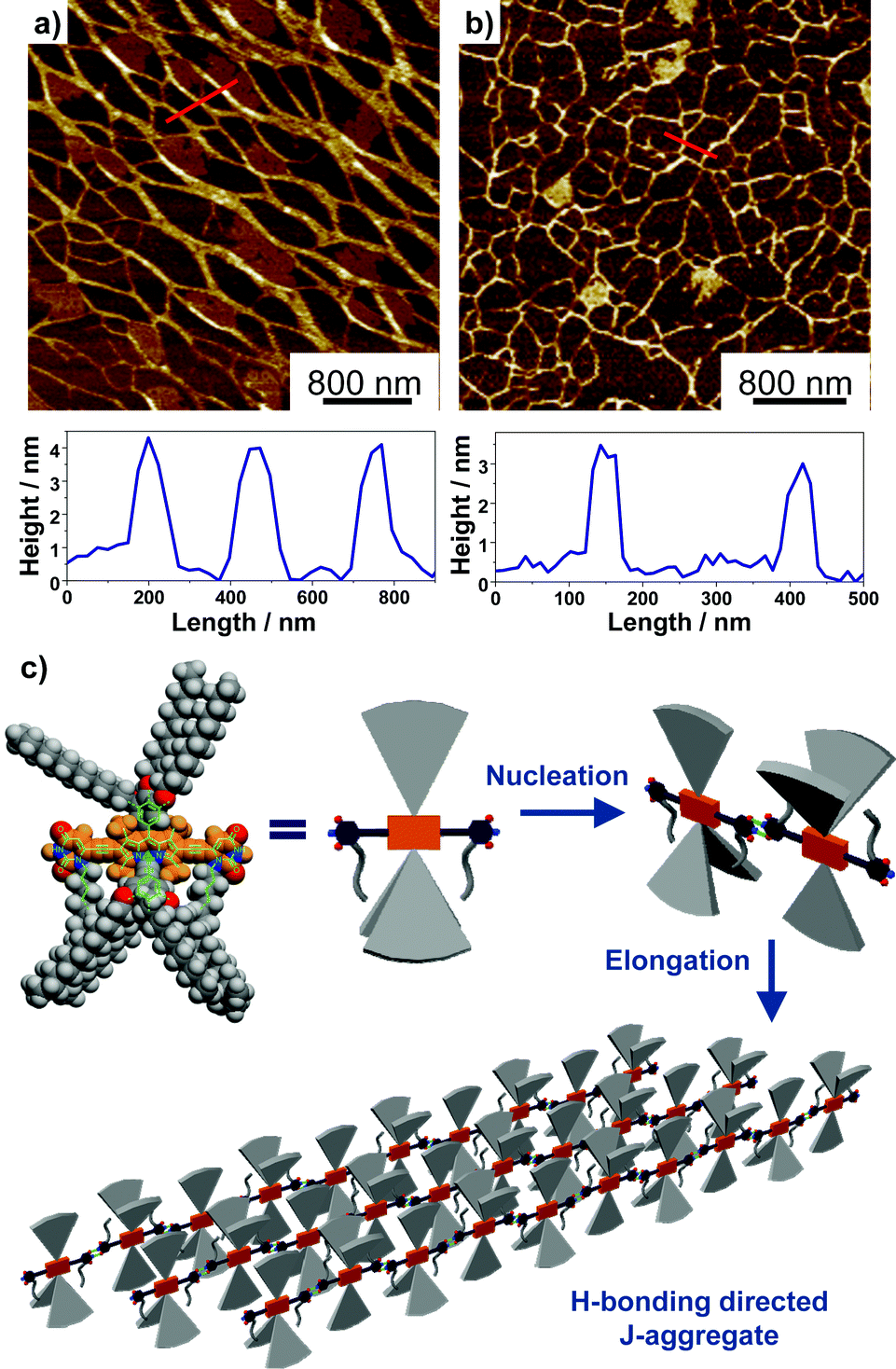 | ||
| Fig. 4 AFM images of J-aggregates of BODIPY dyes (a) 1b and (b) 1c by drop-casting n-hexane solution (cT = 1.0 × 10−5 M) on the mica surface and cross-section analysis along the red lines. (c) Schematic illustration of the formation of elongated 1D J-aggregates through the nucleation–elongation mechanism. | ||
Based on mechanistic analysis and the structural characterization for the J-aggregates of BODIPY 1b and 1c, a schematic illustration of the supramolecular polymerization process and molecular packing is proposed, as shown in Fig. 4c. Driven by the complementary intermolecular H-bonding interactions, the dimeric nucleus was first formed in the nucleation process and a subsequent elongation process gave the 1D H-bonding polymers in solution. Accordingly, highly “slipped” J-type arrangement of the transition dipole moments was obtained, as indicated by the large bathochromic shifts for these aggregates in absorption and emission spectroscopic measurements.
To shed more light on the observed J-aggregation induced emission enhancement for 1b and 1c, molecular modelling and calculations based on molecular exciton theory were performed to elucidate the molecular arrangement in the J-aggregates of these dyes. For the geometry-optimized tetrameric aggregates of 1a–c (Fig. 5 and Fig. S25†), it was observed that the π–π distances between the planes of dipyrromethene cores are significantly varied, confirming the difference in the molecular arrangement for the J-aggregates of dyes 1a–c. The chromophores of 1a exhibit close π–π stacking with a distance of about 0.46 nm, whereas for 1b and 1c, this distance increased to 0.88 and 0.75 nm, respectively. Meanwhile, the experimental bathochromic shift of the absorption band for 1a (−1469 cm−1) is obviously larger than that for 1b and 1c (−895 cm−1 and −957 cm−1), implying the stronger excitonic coupling interactions of 1a in the π–π-stacking direction since the center-to-center distance along the H-bonding direction of the chromophores should be nearly identical for these dyes (vide infra).
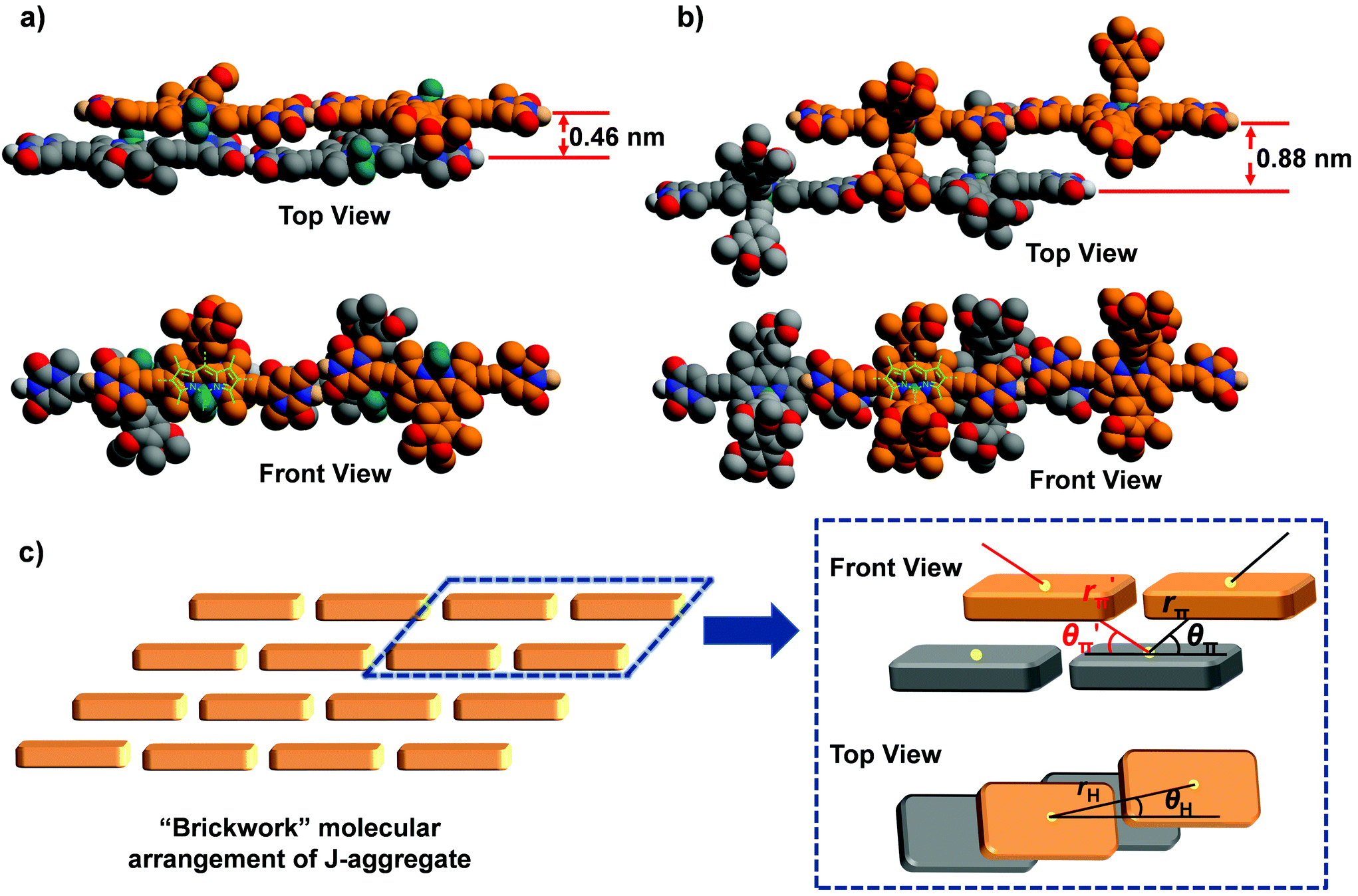 | ||
| Fig. 5 Geometrically optimized tetrameric aggregates for dyes (a) 1a and (b) 1b with the PM6-D3H4 method (for clarity, alkyls and hydrogen atoms are omitted and the molecules are shown in grey and orange colours). (c) Schematic representation of the “brickwork” model and the geometrical parameters that are considered for theoretical calculations with molecular exciton theory. | ||
Furthermore, the spectral shift of the absorption band upon J-aggregation was estimated by Kasha's molecular exciton theory25 based on the molecular modelling results. A “brickwork” model is proposed for the molecular arrangement in J-aggregates of 1a–c (Fig. 5c), in which only the nearest neighbour interactions were considered. Accordingly, one molecule had excitonic coupling interactions with two H-bonded neighbouring molecules and four π–π stacked ones. The geometrical parameters for the estimation of spectral shift in a “brickwork” model are presented in Fig. 5c, where the parameters rH and rπ/r′π refer to the center-to-center distances between two H-bonded or π–π stacked chromophores respectively while θH and θπ/θ′π refer to the angles defined by the direction of the transition dipole moment of one chromophore and the line connecting the centers of two neighbouring chromophores. These geometrical parameters for the J-aggregation of dyes 1a–c were obtained from the structure-optimized tetrameric aggregates and are listed in Table 3. As a result, the spectral shifts for J-aggregation of 1a–c are estimated as Δῠ ≤ −1371 cm−1, Δῠ ≤ −895 cm−1, and Δῠ ≤ −957 cm−1 respectively (for details, see the ESI†), which are consistent with the experimental data of these dyes obtained from UV/Vis spectroscopic measurements. In the π–π-stacking directions, the calculated Δῠ of 1b and 1c (−718 cm−1 and −822 cm−1) are much lower than that for 1a (−1276 cm−1), indicating that the bulky substituents on boron can cause considerable steric hindrance to suppress the excitonic interactions in the π–π stacking directions. Considering the ACQ and AIEE effects observed for the J-aggregates of 1a and 1b & 1c respectively, one can speculate that the segregation effect of bulky substituent groups is crucial to prevent the close π–π contact as well as ACQ of chromophores and lead to the J-aggregation-induced emission enhancement for dyes 1b and 1c.
| Dyes | μ eg/D | r H/Å | θ H/° | r π/Å | θ π/° | r′π/Å | θ′π/° | Δῠcal/cm−1 | Δῠex/cm−1 |
|---|---|---|---|---|---|---|---|---|---|
| BODIPY 1a | 7.9 | 23.6 | 6.0 | 8.1 | 37.3 | 17.3 | 15.5 | −1371 | −1469 |
| BODIPY 1b | 10.9 | 23.7 | 5.2 | 17.2 | 30.7 | 12.6 | 40.7 | −895 | −1065 |
| BODIPY 1c | 9.4 | 23.4 | 5.7 | 13.3 | 34.3 | 13.1 | 33.7 | −957 | −1069 |
Conclusions
In this work, two new uracil-functionalized BODIPY dyes 1b and 1c modified with bulky substituents on the boron atom were synthesized and characterized. Through supramolecular polymerization directed by intermolecular H-bonding interactions between the uracil groups, these dyes assembled into 1D J-aggregates with intense, bathochromically shifted absorption and fluorescence bands in nonpolar solvents n-hexane and MCH. Temperature- and concentration-dependent spectroscopic studies revealed the cooperative supramolecular polymerization of 1b and 1c with a nucleation–elongation mechanism. Moreover, the fluorescence quantum yields of J-aggregates of 1b and 1c were found to be significantly higher than that of the monomers, indicating extraordinary AIEE properties that could be ascribed to the excitonic coupling between aggregated molecules. Further structural characterization revealed that the dyes 1b and 1c supramolecularly polymerized into nanowires through intermolecular H-bonding interactions. Meanwhile, molecular modelling and calculations based on exciton theory indicated that the bulky substituent groups appended on boron atoms were able to largely hinder the close contact between the π-faces of BODIPY chromophores 1b and 1c. Accordingly, the combination of H-bonding interactions along the transition dipole moments and appropriate segregation of supramolecular polymer chains was demonstrated to be an effective strategy for the design of dye building blocks exhibiting both J-aggregation and AIEE properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 92056115 and 21875157).Notes and references
- (a) E. E. Jelley, Spectral absorption and fluorescence of dyes in the molecular state, Nature, 1936, 138, 1009 Search PubMed; (b) H. von Berlepsch, C. Böttcher and L. Dähne, Structure of J-aggregates of pseudoisocyanine dye in aqueous solution, J. Phys. Chem. B, 2000, 104, 8792 Search PubMed; (c) T. E. Kaiser, V. Stepanenko and F. Würthner, Fluorescent J-Aggregates of Core-Substituted Perylene Bisimides: Studies on Structure-Property Relationship, Nucleation-Elongation Mechanism, and Sergeants-and-Soldiers Principle, J. Am. Chem. Soc., 2009, 131, 6719 Search PubMed; (d) F. Würthner, T. E. Kaiser and C. R. Saha-Möeller, J-Aggregates: From Serendipitous Discovery to Supramolecular Engineering of Functional Dye Materials, Angew. Chem., Int. Ed., 2011, 50, 3376 Search PubMed.
- S. Kirstein and S. Dähne, J-aggregates of amphiphilic cyanine dyes: Self-organization of artificial light harvesting complexes, Int. J. Photoenergy, 2006, 2006, 1 Search PubMed.
- (a) M. T. Spitler, Dye Photo-Oxidation at Semiconductor Electrodes-A Corollary to Spectral Sensitization in Photography, J. Chem. Educ., 1983, 60, 330 Search PubMed; (b) C. Nasr, D. Liu, S. Hotchandani and P. V. Kamat, Dye-capped semiconductor nanoclusters. Excited state and photosensitization aspects of rhodamine 6G H-aggregates bound to SiO2 and SnO2 colloids, J. Phys. Chem., 1996, 100, 11054 Search PubMed.
- H. von Berlepsch, S. Kirstein, R. Hania, A. Pugzlys and C. Böttcher, Modification of the nanoscale structure of the J-aggregate of a sulfonate-substituted amphiphilic carbocyanine dye through incorporation of surface-active additives, J. Phys. Chem. B, 2007, 111, 1701 Search PubMed.
- (a) T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Supramolecular construction of fluorescent J-aggregates based on hydrogen-bonded perylene dyes, Angew. Chem., Int. Ed., 2007, 46, 5541 Search PubMed; (b) Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chiral J-Aggregates of Atropo-Enantiomeric Perylene Bisimides and Their Self-Sorting Behavior, Chem. – Eur. J., 2012, 18, 7060 Search PubMed; (c) F. Fennel, J. Gershberg, M. Stolte and F. Würthner, Fluorescence quantum yields of dye aggregates: a showcase example based on self-assembled perylene bisimide dimers, Phys. Chem. Chem. Phys., 2018, 20, 7612 Search PubMed.
- (a) H. Kar, D. W. Gehrig, N. K. Allampally, G. Fernández, F. Laquai and S. Ghosh, Cooperative supramolecular polymerization of an amine-substituted naphthalene-diimide and its impact on excited state photophysical properties, Chem. Sci., 2016, 7, 1115 Search PubMed; (b) H. Kar and S. Ghosh, J-aggregation of a sulfur-substituted naphthalenediimide (NDI) with remarkably bright fluorescence, Chem. Commun., 2016, 52, 8818 Search PubMed.
- (a) G. Fan, Y. Lin, L. Yang, F. Gao, Y. Zhao, Z. Qiao, Q. Zhao, Y. Fan, Z. Chen and H. Wang, Co-self-assembled nanoaggregates of BODIPY amphiphiles for dual colour imaging of live cells, Chem. Commun., 2015, 51, 12447 Search PubMed; (b) L. Yang, G. Fan, X. Ren, L. Zhao, J. Wang and Z. Chen, Aqueous self-assembly of a charged BODIPY amphiphile via nucleation-growth mechanism, Phys. Chem. Chem. Phys., 2015, 17, 9167 Search PubMed; (c) N. Boens, V. Leen and W. Dehaen, Fluorescent indicators based on BODIPY, Chem. Soc. Rev., 2012, 41, 1130 Search PubMed; (d) E. V. Antina, N. A. Bumagina, A. V'Yugin and A. V. Solomonov, Fluorescent indicators of metal ions based on dipyrromethene platform, Dyes Pigm., 2017, 136, 368 Search PubMed.
- (a) A. Loudet and K. Burgess, BODIPY dyes and their derivatives: Syntheses and spectroscopic properties, Chem. Rev., 2007, 107, 4891 Search PubMed; (b) L. Niu, Y. Guan, Y. Chen, L. Wu, C. Tung and Q. Yang, BODIPY-Based Ratiometric Fluorescent Sensor for Highly Selective Detection of Glutathione over Cysteine and Homocysteine, J. Am. Chem. Soc., 2012, 134, 18928 Search PubMed; (c) H. Lu, J. Mack, Y. Yang and Z. Shen, Structural modification strategies for the rational design of red/NIR region BODIPYs, Chem. Soc. Rev., 2014, 43, 4778 Search PubMed; (d) T. Kowada, H. Maeda and K. Kikuchi, BODIPY-based probes for the fluorescence imaging of biomolecules in living cells, Chem. Soc. Rev., 2015, 44, 4953 Search PubMed.
- F. Camerel, L. Bonardi, G. Ulrich, L. Charbonniere, B. Donnio, C. Bourgogne, D. Guillon, P. Retailleau and R. Ziessel, Self-assembly of fluorescent amphipathic borondipyrromethene scaffoldings in mesophases and organogels, Chem. Mater., 2006, 18, 5009 Search PubMed.
- A. Nagai, K. Kokado, J. Miyake and Y. Cyujo, Thermoresponsive Fluorescent Water-Soluble Copolymers Containing BODIPY Dye: Inhibition of H-Aggregation of the BODIPY Units in Their Copolymers by LCST, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 627 Search PubMed.
- (a) J.-H. Oliver, F. Camerel, G. Ulrich, J. Barbera and R. Ziessel, Luminescent Ionic Liquid Crystals from Self-Assembled BODIPY Disulfonate and Imidazolium Frameworks, Chem. – Eur. J., 2010, 16, 7134 Search PubMed; (b) M. Benstead, G. A. Rosser, A. Beeby, G. H. Mehl and R. W. Boyle, Mesogenic BODIPYs: an investigation of the correlation between liquid crystalline behaviour and fluorescence intensity, Photochem. Photobiol. Sci., 2011, 10, 992 Search PubMed.
- Y. Tokoro, A. Nagai and Y. Chujo, Nanoparticles via H-aggregation of amphiphilic BODIPY dyes, Tetrahedron Lett., 2010, 51, 3451 Search PubMed.
- Y. Zhang, P. Liu, H. Pan, H. Dai, X. K. Ren and Z. Chen, Alignment of supramolecular J-aggregates based on uracil-functionalized BODIPY dye for polarized photoluminescence, Chem. Commun., 2020, 56, 12069 Search PubMed.
- (a) S. Choi, J. Bouffard and Y. Kim, Aggregation-induced emission enhancement of a meso-trifluoromethyl BODIPY via J-aggregation, Chem. Sci., 2014, 5, 751 Search PubMed; (b) S. Kim, J. Bouffard and Y. Kim, Tailoring the Solid-State Fluorescence Emission of BODIPY Dyes by meso Substitution, Chem. – Eur. J., 2015, 21, 17459 Search PubMed; (c) Z. Liu, Z. Jiang, M. Yan and X. Wang, Recent Progress of BODIPY Dyes With Aggregation-Induced Emission, Front. Chem., 2019, 7, 712 Search PubMed.
- J. Koo, S. I. Lim, S. H. Lee, J. S. Kim, Y. T. Yu, C. R. Lee, D. Y. Kim and K. U. Jeong, Polarized Light Emission from Uniaxially Oriented and Polymer-Stabilized AIE Luminogen Thin Films, Macromolecules, 2019, 52, 1739 Search PubMed.
- (a) A. C. Benniston and G. Copley, Lighting the way ahead with boron dipyrromethene (Bodipy) dyes, Phys. Chem. Chem. Phys., 2009, 11, 4124 Search PubMed; (b) S. Lim, M. M. Haque, D. Su, D. Kim, J.-S. Lee, Y.-T. Chang and Y. K. Kim, Development of a BODIPY-based fluorescent probe for imaging pathological tau aggregates in live cells, Chem. Commun., 2017, 53, 1607 Search PubMed.
- (a) M. S. Cubberley and B. L. Iverson, H-1 NMR investigation of solvent effects in aromatic stacking interactions, J. Am. Chem. Soc., 2001, 123, 7560 Search PubMed; (b) Z. Chen, A. Lohr, C. R. Saha-Moeller and F. Würthner, Self-assembled pi-stacks of functional dyes in solution: structural and thermodynamic features, Chem. Soc. Rev., 2009, 38, 564 Search PubMed.
- H. Wang, T. E. Kaiser, S. Uernura and F. Würthner, Perylene bisimide J-aggregates with absorption maxima in the NIR, Chem. Commun., 2008, 10, 1181 Search PubMed.
- (a) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Pena-Cabrera and B. Z. Tang, Twisted Intramolecular Charge Transfer and Aggregation-Induced Emission of BODIPY Derivatives, J. Phys. Chem. C, 2009, 113, 15845 Search PubMed; (b) Y. Hong, J. W. Lam and B. Z. Tang, Aggregation-induced emission, Chem. Soc. Rev., 2011, 40, 5361 Search PubMed; (c) R. Hu, N. L. C. Leung and B. Z. Tang, AIE macromolecules: syntheses, structures and functionalities, Chem. Soc. Rev., 2014, 43, 4494 Search PubMed; (d) R. Hu, A. Qin and B. Z. Tang, AIE polymers: Synthesis and applications, Prog. Polym. Sci., 2020, 100, 101176 Search PubMed.
- (a) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Probing the solvent-assisted nucleation pathway in chemical self-assembly, Science, 2006, 313, 80 Search PubMed; (b) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, Insight into the mechanisms of cooperative self-assembly: The “sergeants-and-soldiers” principle of chiral and achiral C-3-symmetrical discotic triamides, J. Am. Chem. Soc., 2008, 130, 606 Search PubMed.
- R. F. Goldstein and L. Stryer, Cooperative Polymerization Reactions-Analytical Approximations, Numerical Examples, and Experimental Strategy, Biophys. J., 1986, 50, 583 Search PubMed.
- S. K. Yang and S. C. Zimmerman, Hydrogen Bonding Modules for Use in Supramolecular Polymers, Isr. J. Chem., 2013, 53, 511 Search PubMed.
- (a) L. A. LaPlanche, H. B. Thompson and M. T. Rogers, Chain AssociationEquilibria. A Nuclear Magnetic Resonance Study of the Hydrogen Bonding of N-Monosubstituted Amides, J. Phys. Chem., 1964, 69, 1482 Search PubMed; (b) W. S. Horne, C. D. Stout and M. R. Ghadiri, A heterocyclic peptide nanotube, J. Am. Chem. Soc., 2003, 125, 9372 Search PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, From supramolecular polymers to multi-component biomaterials, Chem. Soc. Rev., 2017, 46, 6621 Search PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, The exciton model in molecular spectroscopy, Pure Appl. Chem., 1965, 11, 371 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo00520k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2021 |