
Light-mediated olefin coordination polymerization and photoswitches
Mingyuan
Li
a,
Ruibin
Wang
 a,
Moris S.
Eisen
ab and
Sehoon
Park
*ac
a,
Moris S.
Eisen
ab and
Sehoon
Park
*ac
aDepartment of Chemistry, Guangdong Technion Israel Institute of Technology, Shantou 515063, China. E-mail: sehoon.park@gtiit.edu.cn
bSchulich Faculty of Chemistry, Technion-Israel Institute of Technology, Technion City, 32000 Haifa, Israel
cTechnion-Israel Institute of Technology, Technion City, 32000 Haifa, Israel
First published on 15th June 2020
Abstract
Polyolefins are important polymeric materials in the plastics industry. While classical Ziegler–Natta catalysts are still superior in global polyolefin production, it is challenging to control their polymerization behavior due to intrinsic heterogeneity. A significant number of molecular olefin polymerization catalysts based on transition metals are capable of harnessing polymerization behavior thanks to their finely-tunable ligand sets, where ligand variation acts as electronic and/or steric factors towards the active metal center. Light has been utilized as a green and specific stimulus in the context of coordination olefin polymerization to mainly alter the electronic state of active catalytic sites, which enables photo-controlled chain propagation. A handful of examples of polymerization catalytic systems that have precise controllability under photoirradiation have been developed to provide a broad range of polyolefins in terms of polymeric (micro)structure. In this Review, we wish to outline photo-responsive, transition metal-based coordination polymerization catalysts ranging from homogeneous to heterogeneous, and monometallic to bimetallic regimes. Specific but varied roles of light depending on the employed catalysts for olefin polymerization are presented with a strong emphasis on the photo-altered properties of the active species.
 Mingyuan Li | Mingyuan Li was born in China in 1992. He received his PhD degree (2019) in materials science from Donghua University (Professor Zhengguo Cai). In August 2019, he worked as a postdoctoral fellow with Professor Moris S. Eisen at Guangdong Technion-Israel Institute of Technology (GTIIT). His research interests are the synthesis and application of polyolefin materials catalyzed by transition metal complexes. |
 Ruibin Wang | RuiBin Wang was born in China in 1986. He received his Master's degree (2011) in Medicinal Chemistry from Sun Yat-sen University (Professor Gui Lu). From February 2012 to July 2016, he worked at the Chinese Academy of Sciences as a research fellow. Then, he moved to Guangdong Shunde Industrial Design Institute to work as a research fellow (2016–2020). In early 2020, he started to pursue his PhD degree under the tutelage of Prof. Sehoon Park at Guangdong Technion Israel Institute of Technology (GTIIT). His research is focused on synthetic and mechanistic organometallic chemistry, particularly involving catalysis. |
 Moris S. Eisen | Moris S. Eisen emigrated to Israel in 1977 from Colombia. He received his Ph.D. from the Hebrew University under the supervision of Prof. Jochanan Blum. In 1990, as a Weizmann Postdoctoral Fellow, he joined Prof. Tobin J. Marks’ group at Northwestern University. In 1992, as an Alon Fellow, he joined the Department of Chemistry at the Technion – Israel Institute of Technology (since 2007, Schulich Faculty of Chemistry). Since 2003, he has been a full professor and incumbent of the Samuel O. Friedlander Academic Chair in Chemistry. |
 Sehoon Park | Sehoon Park was born in Seoul (Korea). He received his Ph.D. (2008) from Tokyo Institute of Technology (Prof. Kohtaro Osakada). He then moved to the University of North Carolina at Chapel Hill to work as a postdoctoral fellow with Prof. Maurice Brookhart (2009–2012). From April 2013 to February 2019, he worked at the Institute for Basic Science (Korea) as a senior research fellow, mentored by Prof. Sukbok Chang. In early 2019, he started his independent research as an assistant professor at Guangdong Technion Israel Institute of Technology (GTIIT). His research interests are synthetic and mechanistic organometallic chemistry. |
1. Introduction
Synthetic polymeric materials are important commodity chemicals in our society, which have been greatly advancing in terms of the polymer's physical and chemical properties to provide people with higher quality and efficiency in life.1–3 These synthetic polymers are prepared from their monomers largely via either step-growth or chain-growth polymerization. A representative example of the former working mode is the condensation reaction of monomers that have reactive multi-functions, which proceeds in a stepwise manner through dimers and oligomers as key intermediates.4–6 While this method produces a variety of highly useful thermoplastic polymers such as polyesters and polyamides, the high conversion of monomers is crucial to achieving high molecular weight polymers and the reaction itself is difficult to control due to the nature of the step-growth mechanism.7–9 In contrast, chain-growth polymerization requires initiators to generate the active species, which causes polymer chain growth in a controlled manner, being able to give highly modulated polymers. Among them, polyolefins are chain-growth polymers that are widely utilized in modern life. In fact, more than 178 million tons of polyolefins were produced in a single year in 2015, which corresponded to >50% of the global production of synthetic polymers in the same year.10,11 Such largescale polyolefin production is likely due to low-cost olefin monomers and them having far less toxicity with broad end-applications of polyolefin products relative to other types of commodity polymers.12–14Ziegler–Natta catalysts are one of the most practical and powerful catalytic systems for the production of commercialized polyethylene [e.g. high density polyethylene (HDPE), linear low density polyethylene (LLDPE)]. However, these catalytic systems are extremely difficult to study and understand in terms of polymerization mechanism due to their heterogeneous nature.15–18 In this regard, a number of well-defined homogeneous catalytic systems based on transition metals have been developed in the last few decades. For example, Kaminsky communicated the first homogeneous catalyst system composed of group 4 metal-based metallocene complexes and methylaluminoxane (MAO) as a cocatalyst for the coordination polymerization of ethylene and α-olefins leading to HDPE and LLDPE in 1980.19–22 In 1995, Brookhart reported Pd and Ni-based complexes with α-diimine ligands as olefin polymerization catalysts to furnish highly branched polyethylene referred to as low density polyethylene (LDPE) under relatively mild conditions, when compared to LDPE production via a classical radical process that requires far harsher conditions.23–27 Following Brookhart's α-diimine polymerization catalysts, a number of research groups successfully developed well-tailored [N–O] and/or P-based ligand systems of late transition metals (e.g. bis-iminopyridine, salicylaldiminate, phosphine sulfonate, etc.) for the (co)polymerization of olefins.28–30 Molecular olefin polymerization catalysts with electronically and sterically modular ligand systems enable the control of the polymerization profile, including the molecular weight and dispersity, degree of branching, and tacticity of polyolefins.31–33 In general, the electrophilic nature of the active metal center stabilized by a non-coordinating counter anion is the key to accomplishing high polymerization catalytic activity, while the electron-donating substituents of the ligands contribute towards suppressed chain transfer to give rise to high molecular weight polymers.34–40 The steric bulkiness of ligands is well known to often block an axial coordination site of active metal centers and slow chain-transfer processes, which yields high molecular weight polyolefins with narrow dispersity and enables living polymerization to synthesize various block copolymers.41–49 Physical reaction parameters, including temperature and ethylene pressure under certain ligand systems, have also been shown to strongly influence catalyst productivity, polymer molecular weight and microstructure.50–53
Given the well-established relationship between electronic/steric variations in ligand and olefin polymerization behavior, a handful of research groups have been interested in utilizing light as an external stimulus and as a tool for the controlled synthesis of polyolefins.54–58 There have been remarkable reports on photo-switchable catalysts for non-coordination–insertion polymerization, including ring-opening metathesis polymerization (ROMP),59,60 ring-opening polymerization (ROP),61–65 and atom-transfer radical polymerization (ATRP),66–71 in which photosensitive organo(metallic) catalysts and/or photo-initiators can be reversibly activated to modulate polymerization pathways. The Inagaki and Akita group published a review on visible-light promoted bimetallic catalysis for olefin oligomerization via a coordination–insertion mechanism.72 Bielawski published a seminal review on switchable polymerization catalysts, including a few examples of photo-responsive polymerization catalytic systems.73 Similarly, Hecht reviewed a series of photoswitchable polymerization catalysts.74 Despite the remarkable advances in olefin coordination–insertion polymerization,23–33 a review regarding light-mediated olefin coordination polymerization has not been published thus far.
Since light is specific, tunable, and green, light-mediated olefin coordination polymerization (2010–2019) is a promising strategy for the construction of finely-tuned polyolefin architectures. In this regard, we wish to outline in this review the photoresponsive olefin polymerization catalysts ranging from monometallic to bimetallic, and homogeneous to heterogeneous systems. The light-driven electronic and steric alterations in the employed catalytic systems and their effects on polymerization behavior are discussed (Scheme 1).75 This review covers the literature up to March of 2020.
 | ||
| Scheme 1 Catalytic systems for light-mediated olefin coordination polymerization and photoswitches. | ||
2. Monometallic catalytic systems
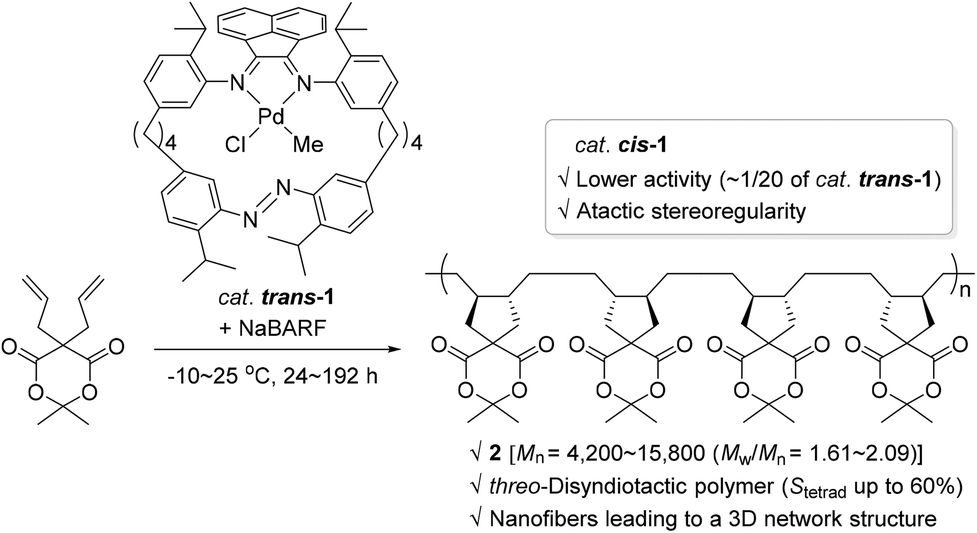
Photochromic moieties on the ligand systems of monometallic olefin polymerization catalysts affect polymerization behavior upon light irradiation generally via either of the following modes: (i) a photo-absorbing ligand plays the role of an electronic factor to bring about alteration in the electronic state of the metal center; or (ii) a photo-sensitizing ligand induces reversible changes in the steric environment surrounding an active metal site. The former working mode involving photo-induced electronic alteration at the active metal center has been well studied, whereas the catalytic system accompanied by significant steric changes in the light has rarely been covered (vide infra).In 2010, Aida and coworkers synthesized a Pd(II) complex 1 with a cyclic α-diimine ligand containing a photo-isomerizable azobenzene moiety, and applied complex 1 [trans-1 or 80% of cis-1 (azobenzene group geometry)] as a catalyst in the known cyclopolymerization76–78 of isopropylidene diallylmalonate in an attempt to control its polymerization activity by utilizing light as a switchable external stimulus (Scheme 2).79 The cyclopolymerization smoothly proceeded in the presence of catalytic amounts of trans-1 and sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBARF) as a cocatalyst at −10∼25 °C to give the corresponding polymer that has a trans-1,2-disubstituted cyclopentane repeating unit (2). The molecular weights, Mn, of the obtained cyclic polymer 2 increased up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 with a molecular dispersity (Mw/Mn) of 1.65 in 192 h. Notably, the nuclear magnetic resonance (NMR) spectroscopic analysis revealed that the tacticity due to relative configurations between the cyclopentane units in 2 prepared viatrans-1 catalysis, is rich in threo-disyndiotactic tetrad up to 60%. This was in stark contrast with the stereooutcome obtained using catalyst cis-1, where the syndiotacticity was lower. Another interesting comparison is the highly differing catalytic activities: trans-1 showed ca. 20 times higher diallylmalonate cyclopolymerization activity relative to cis-1 [kobsd (trans-1) = 2.50 × 10−5 s−1vs. kobsd (cis-1) = 0.13 × 10−5 s−1 under the assumption of pseudo-first-order kinetics]. These contrasting stereochemical and kinetic observations indicate that although the azobenzene group with a trans or cis geometry is embedded far from the Pd center, it counterintuitively and largely influences the catalytic behavior of the Pd species. The obtained polymeric materials rich in syndiotactic sequence formed barb-shaped polymers, which self-assembled into unusual nanofibers to eventually bring about a 3D network structure essential for physical gelation.
800 with a molecular dispersity (Mw/Mn) of 1.65 in 192 h. Notably, the nuclear magnetic resonance (NMR) spectroscopic analysis revealed that the tacticity due to relative configurations between the cyclopentane units in 2 prepared viatrans-1 catalysis, is rich in threo-disyndiotactic tetrad up to 60%. This was in stark contrast with the stereooutcome obtained using catalyst cis-1, where the syndiotacticity was lower. Another interesting comparison is the highly differing catalytic activities: trans-1 showed ca. 20 times higher diallylmalonate cyclopolymerization activity relative to cis-1 [kobsd (trans-1) = 2.50 × 10−5 s−1vs. kobsd (cis-1) = 0.13 × 10−5 s−1 under the assumption of pseudo-first-order kinetics]. These contrasting stereochemical and kinetic observations indicate that although the azobenzene group with a trans or cis geometry is embedded far from the Pd center, it counterintuitively and largely influences the catalytic behavior of the Pd species. The obtained polymeric materials rich in syndiotactic sequence formed barb-shaped polymers, which self-assembled into unusual nanofibers to eventually bring about a 3D network structure essential for physical gelation.
 | ||
| Scheme 2 Cyclopolymerization of isopropylidene diallylmalonate catalyzed by trans-1 or cis-1. | ||
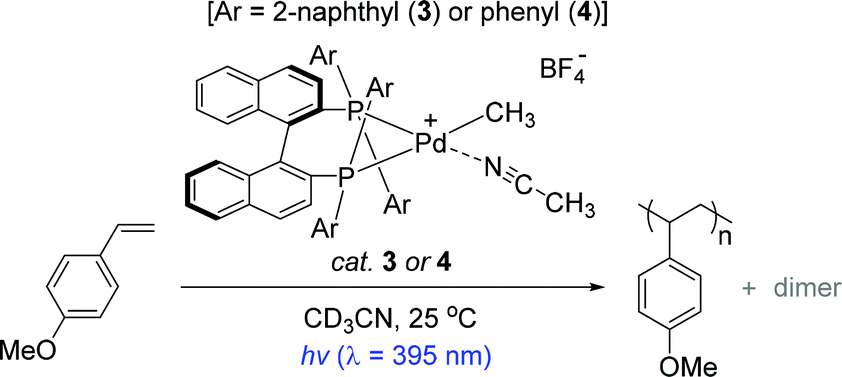
The Inagaki and Akita group previously reported a series of bimetallic photocatalysts composed of bipyridyl-ligated Ru-based photosensitizer units and a Pd reactive center linked by a bipyrimidyl ligand, where the naphthyl substituents installed on the ligands played a key role in extending the lifetimes of the metal to ligand charge transfer (MLCT) excited states under light irradiation.80,81 Based on the photophysical insights into the Ru–Pd photocatalysts and the known photosensitizing ability of BINAP,82–85 the same group developed the naphthyl-substituted BINAP-Pd(II) complex 3 as a catalyst for the preassumed photo-induced styrene polymerization (Scheme 3).86
 | ||
| Scheme 3 Polymerization of 4-methoxystyrene catalyzed by a cationic Pd(II)-BINAP complex. | ||
To understand the effects of a naphthyl substituent in terms of photophysical properties and catalytic activity, the Inagaki group prepared a bidentate (R)-BINAP Pd complex that has two phenyl groups on each phosphine atom (4) for comparison. The polymerization reactions of a handful of styrenes under UV light were examined in the presence of catalyst 3 (1 mol%) to reveal that an electron-rich substrate 4-methoxystyrene underwent chain reaction to give poly(4-methoxystyrene) with a Mn of 14000 (Mw/Mn = 1.50) and small amounts of dimers. This Pd catalysis was perfectly photoswitchable: the reaction stopped under dark conditions, and resumed upon photo-irradiation. It is noteworthy that the overall polymerization outcomes under the light ON–OFF switching sequence were substantially similar to those resulting from continuous light-ON for the same period of time. These results strongly suggest a single site propagating Pd species during polymer chain growth in the light. In stark contrast, however, catalyst 4 exhibited lower polymerization performance when compared to the catalysis by 3 under identical conditions [polymer yields in 10 h ∼60% (cat.3) vs. ∼20% (cat.4)]. This result implied that the naphthyl substituent in catalyst 3 serves as a key photochromic moiety to lead to light-mediated styrene polymerization.
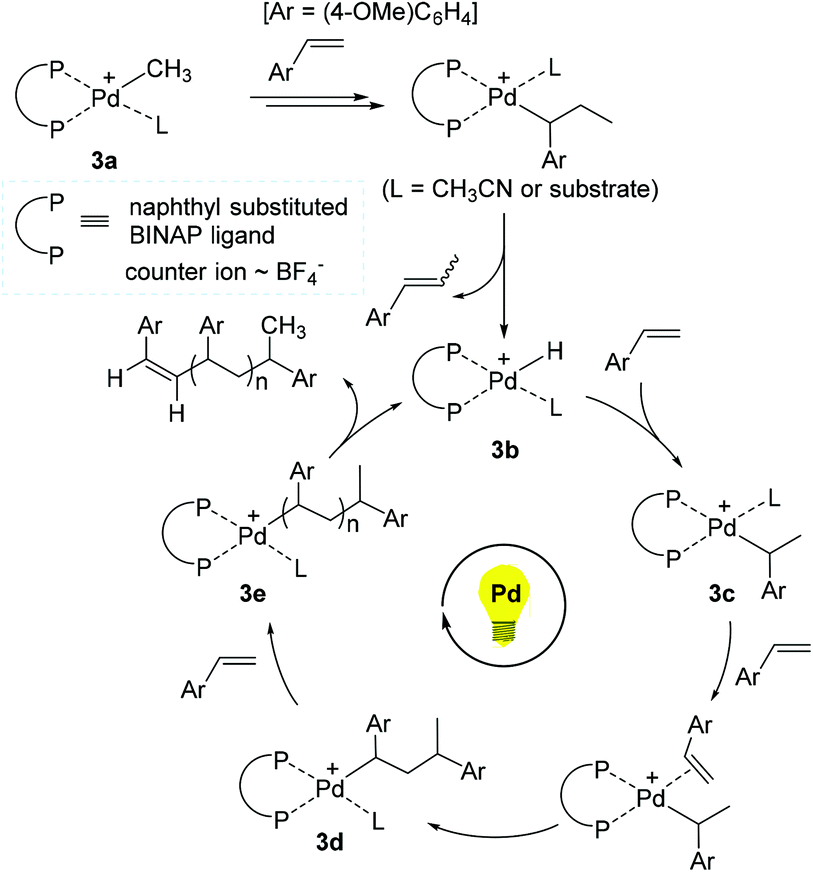
On the basis of experimental and theoretical studies, a reaction pathway for the 3-promoted polymerization of 4-methoxystyrene was proposed (Scheme 4). Initially, the Pd methyl species 3a is assumed to undergo 2,1-insertion of 4-methoxystyrene, followed by β-hydrogen elimination to generate the Pd hydride 3b as an active species. 3b is presumed to consecutively react with 4-methoxystyrene for chain propagation via a series of Pd alkyl intermediates such as 3c, 3d, and 3e. Indeed, molecular ions corresponding to intermediates 3c (m/z = 1063) and 3d (m/z = 1197) were detected by electrospray ionization mass spectrometry (ESI-MS), supporting a non-radical, coordination–insertion mechanism. A Pd propagating species with a polymer chain such as 3e likely liberates the corresponding polymer upon β-H elimination with the regeneration of 3b. Photoirradiation is proposed to accelerate the insertion of 4-methoxystyrene, and thus to make the overall polymerization reaction facile, while the released polymer bearing alkene chain-ends can be reinserted into the Pd–H bond of 3b for further chain growth under (repeated) photo-irradiation. Given the fact that styrene and 2-norbornene did not react, while ethyl vinyl ether and methyl acrylate reacted to give the corresponding polymers under the photoirradiation conditions, the polymerization mechanism might vary from coordination to cationic or radical pathways depending on the type of the olefin monomers.
 | ||
| Scheme 4 Proposed pathway for the 3-catalyzed polymerization of 4-methoxystyrene under UV light. | ||
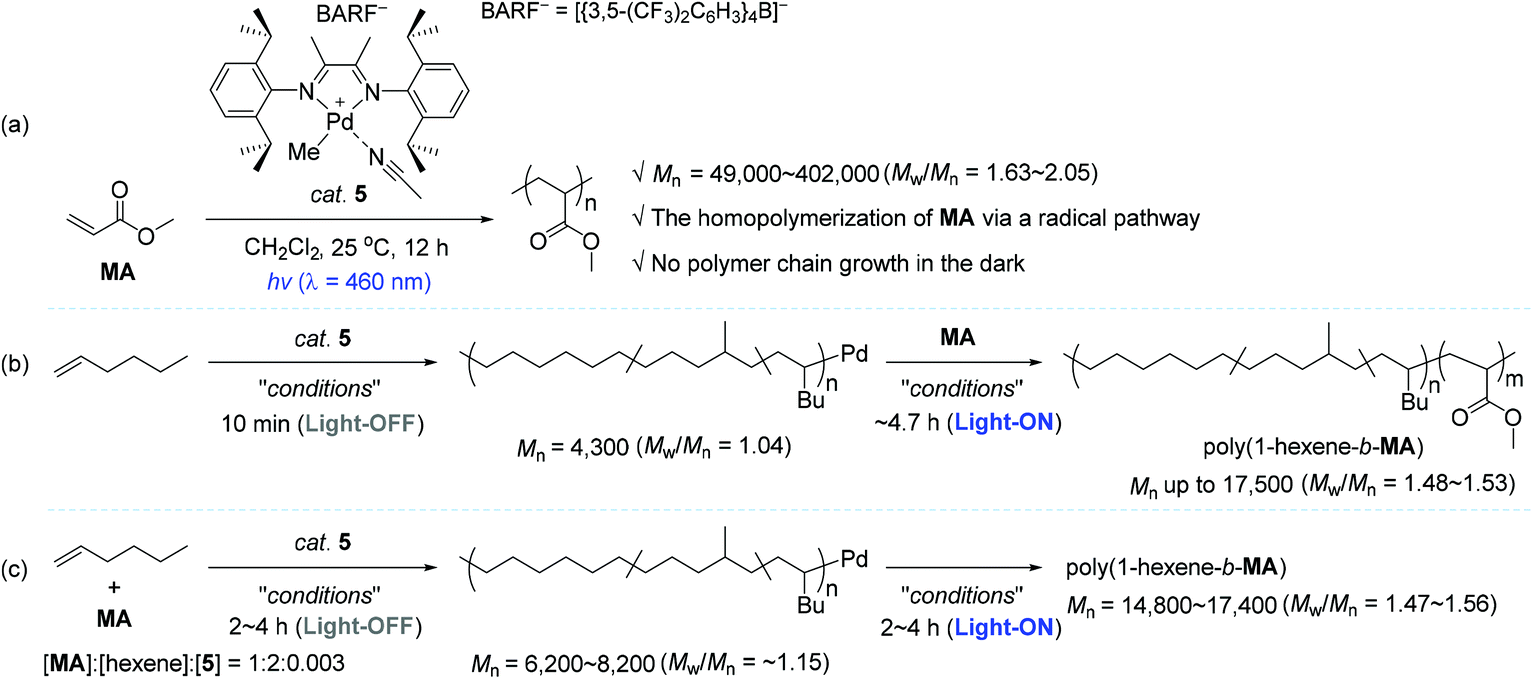
In the context of coordination–insertion polymerization, despite the significant efforts to increase the incorporation of polar comonomer unit into the polymer backbone under an insertion mechanism, the copolymerization of non-polar olefins with polar olefins generally brings about low comonomer incorporation in the resultant polar functionalized polyolefins. Such low contents of polar comonomer units in the polymer backbone are mainly owing to the stable chelation structure of metallacycles that are in situ generated upon the insertion of polar olefin comonomers, followed by isomerization. The chelation structure eventually inhibits the consecutive insertions of the polar comonomers.25,28–30,87–91 Being aware of the intrinsic limitation of insertion polymerization in terms of the synthesis of polar polyolefins, a range of research groups applied a radical polymerization strategy for the synthesis of homo- and copolymers of vinyl polar monomers using well-defined organometallic catalysts based on Pd, Ni, and Co.92–98 Based on this background, Harth and coworkers conceived the development of a light-sensitive catalyst system for olefin polymerization, in which the catalytic working mode is photoswitchable between coordination–insertion and radical mechanisms. It turned out that an α-diimine Pd complex 5 worked well in such a photoswitchable catalytic mode for the target homo- and copolymerization reactions (Scheme 5).99 The 5-catalyzed homopolymerization reactions of methyl acrylate (MA) in various concentrations were conducted under blue light (λ = 460 nm) to produce poly(methyl acrylate) (poly-MA) with a Mn of 49000–402000 (Mw/Mn = 1.63–2.05). It should be noted that the linear conversion of MA was achieved even through alternating the light irradiation, while no polymerization of MA occurred in the dark cycles (Scheme 5a).
 | ||
| Scheme 5 Photocontrolled homopolymerization and block copolymerization catalyzed by 5. | ||
Harth and coworkers were next able to accomplish the synthesis of block copolymers of 1-hexene and MA using photoswitchable catalyst 5 under suitable light manipulation (ON/OFF process): the MA block was propagated upon photoirradiation via a radical process, during which the coordination–insertion mechanism remains non-operative, while the hexene block was only enchained in the dark via a coordination–insertion polymerization. This work represents the first example of diblock polymer synthesis via a photoswitchable dual catalytic polymerization pathway. Two polymerization approaches were viable for the block copolymer synthesis. As one approach, the living polymerization of 1-hexene was first conducted in the dark to furnish poly(1-hexene) with a narrow polymer dispersity (Mn = 4300, Mw/Mn = 1.04), which was subsequently applied as a macroinitiator to create a MA block in the light, yielding the desired block copolymer poly(1-hexene-b-MA) with a Mn of up to 17500 (Mw/Mn = 1.48–1.53) (Scheme 5b). Notably, hexene polymerization was not substantially observed during the growth of a MA block under the UV light conditions. As another strategy leading to poly(1-hexene-b-MA), the polymerization was performed in the presence of both olefin monomers, and catalyst 5 in a ratio of 1:2:0.003 ([MA]:[hexene]:[5]) under alternating dark and light conditions. As a result, three different compositions of block copolymers (hexene/MA units = 25/75, 50/50, and 75/25) with Mn values of 14800–17400 (Mw/Mn = 1.47–1.56) were obtained simply by tuning the period of time of the dark and light cycles (2 h in the dark/4 h in light; 4 h in the dark/4 h in light; 4 h in the dark/2 h in light) (Scheme 5c). The resultant polymeric materials were analyzed by small-angle X-ray scattering (SAXS), which was able to confirm the existence of copolymer blocks.
In a continuous effort to understand in depth of the relationship between the α-diimine ligand structure and polymerization behavior for the light-mediated polymerization of vinyl monomers, Harth and coworkers systematically examined a series of Pd diimine complexes as the polymerization catalyst under both dark and light conditions (Scheme 6).100 The polymerization of MA with catalyst 6D (∼0.1 mol%) proceeded upon blue light irradiation to attain 63% MA conversion, providing poly-MA with a Mn of 92000 (Mw/Mn = 1.76) in 16 h. However, methyl methacrylate (MMA) was less reactive (conversion = 27%) relative to MA in the presence of 5 (= 5A) as the best working catalyst to give poly-MMA [Mn 87000 (Mw/Mn = 1.74)]. It is interesting to see that the polymerization reactivity of acrylamides, including n-isopropyl acrylamide (NIPAm) and dimethyl acrylamide (DMAm), was highly dependent on the α-diimine ligand structure in the light: catalysts 5C–5D and 6C–6D did not react at all with acrylamides, while other ligand variations of the Pd catalysts allowed for the polymerization of acrylamides, albeit in low yields (e.g. 19% conversion of NIPAm in 24 h with catalyst 7C). The polymerization of styrene (St) proceeded in low conversions irrespective of the presence of light (e.g. 26% conversion of St in 24 h with 8D). Isobutyl vinyl ether (IBVE) was highly reactive to all Pd catalysts examined in both the light and dark to afford poly-(IBVE) in high yields (e.g. quantitative conversion of IBVE in 16 h with 6B). These results indicate that St and IBVE as olefin monomers undergo chain propagation via radical and/or cationic mechanisms within the Pd diimine catalyst system.
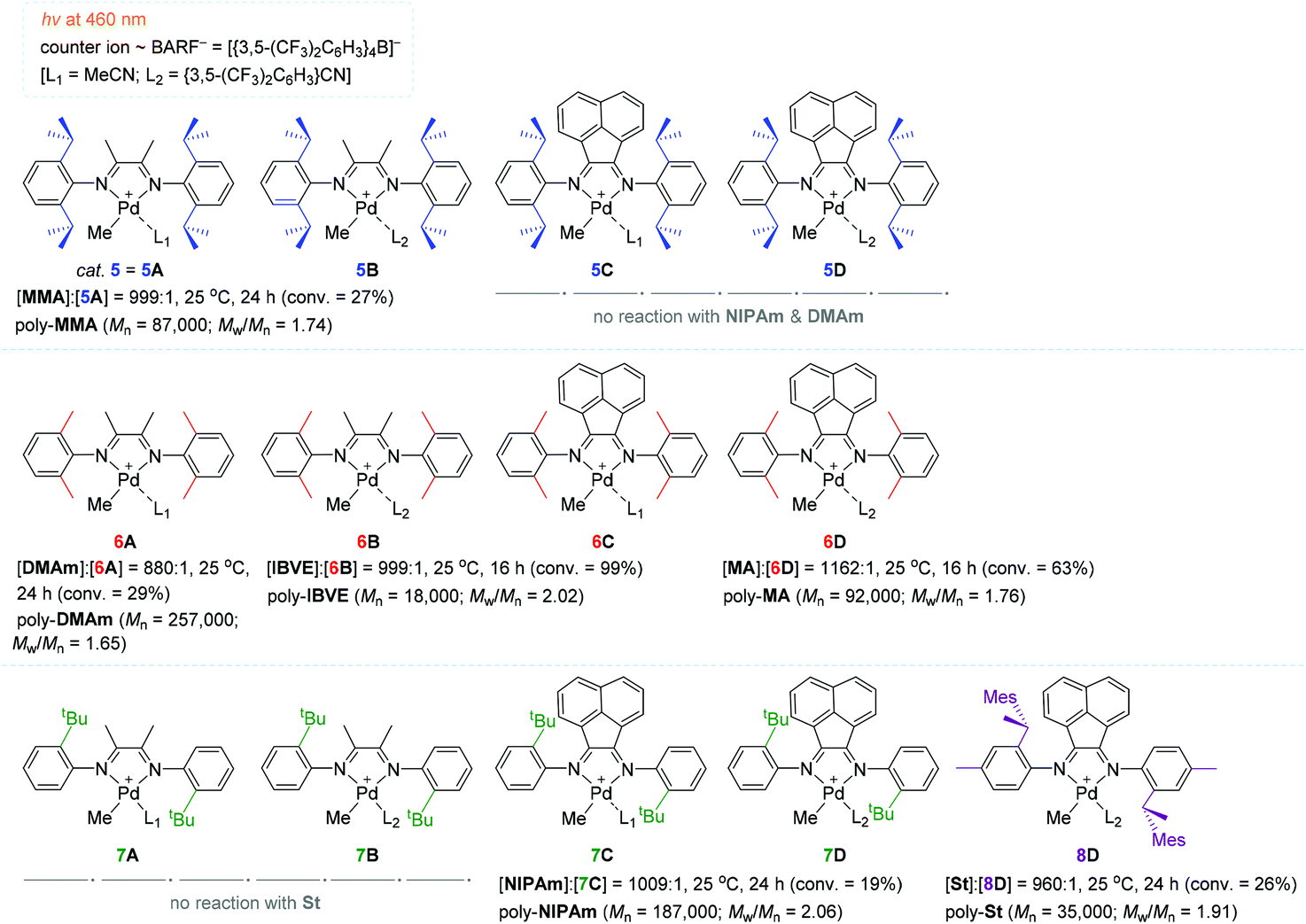 | ||
| Scheme 6 The α-diimine-Pd catalysts tested for the (photo)-polymerization of vinyl monomers. | ||
In order to gain insights into the photoexcitation energies of an array of α-diimine Pd complexes, computational studies [density functional theory (DFT) and time-dependent DFT (TD-DFT)] were carried out to show that MLCT transitions from the Pd–Me σ-bonding into the diimine ligand π-bonding orbitals readily occur upon visible light irradiation, causing loss of electron density in the σ-bonding (weakening the Pd–Me bond) and then homolytic cleavage of the Pd–carbon bond to generate a carbon-centered radical capable of initiating radical polymerization.101–104 In fact, catalyst 5 was found to decompose upon irradiation with blue light to result in the loss of the methyl group in 5, while the polymerization of MA did not take place at all in the presence of a radical scavenger (galvinoxyl) under light irradiation. These observations again support the MLCT-led homolysis of a metal–carbon bond essential for radical polymerization. This result corroborated the radical mechanism as a predominant working mode under visible light conditions. Notably, the simulated HOMO–LUMO gaps were all comparable for the Pd complexes (5A, 5C–5D, 6A–6B, 7C) subjected to these calculations, implying that the ligand structure had no significant influence on the photoinitiation process.
3. Bimetallic catalytic system
In a photo-responsible bimetallic polymerization catalyst system, the two metal centers employed play their own catalytic roles: one metal center serves as a photocatalyst to absorb light photons for charge and/or energy transfer, while another metal unit acts as a chain propagating species to bring about polymerization turnovers under the electronic effects derived from an excited state of the photocatalytic species.72 Importantly, the light-driven electrochemical alteration at the active metal center by the (catalytic)photoredox species significantly influences the overall catalytic behavior to give rise to unique microstructures of polyolefins that are not achievable via conventional coordination polymerization strategies.Inagaki and coworkers synthesized a new family of bimetallic Ru–Pd complexes supported by a range of bipyridyl (bpy) and bipyrimidinyl (bpm) ligands as photoactive olefin polymerization catalysts [9A–9D, and 9E (control)], where 2-naphthyl groups installed on the ligands were envisioned to lengthen the excited-state lifetimes of the Ru–Pd bimetallic species (Scheme 7).80,81 The photophysical properties of these complexes were investigated via a series of spectroscopic analysis, including UV-vis absorption and luminescence spectroscopies (Fig. 1). Based on the UV-vis spectra of the dinuclear complexes, the naphthyl group as an additional chromophore unit on the bpy or bpm ligands were assumed to enhance light-harvesting capability to cover a wide spectral range (from ultraviolet to visible light) for efficient transfer of the absorbed light energy to the Ru(II) center (Fig. 1a). The luminescence spectra of the complexes 9A–9D provided key insights into the emission lifetimes of the excited state Ru–Pd species: (i) the metal-to-bpynaph charge transfer (CT) states in 9A and 9C have relatively long lifetimes; (ii) the emissions for 9B and 9D are dominant in the metal-to-bpm CT states; (iii) the direct connection of a naphthyl group on the bpm ligand in 9D results in longer lifetimes for the metal-to-bpm CT state relative to that in the parent complex 9E, which lacks the naphthyl moiety (Fig. 1b).
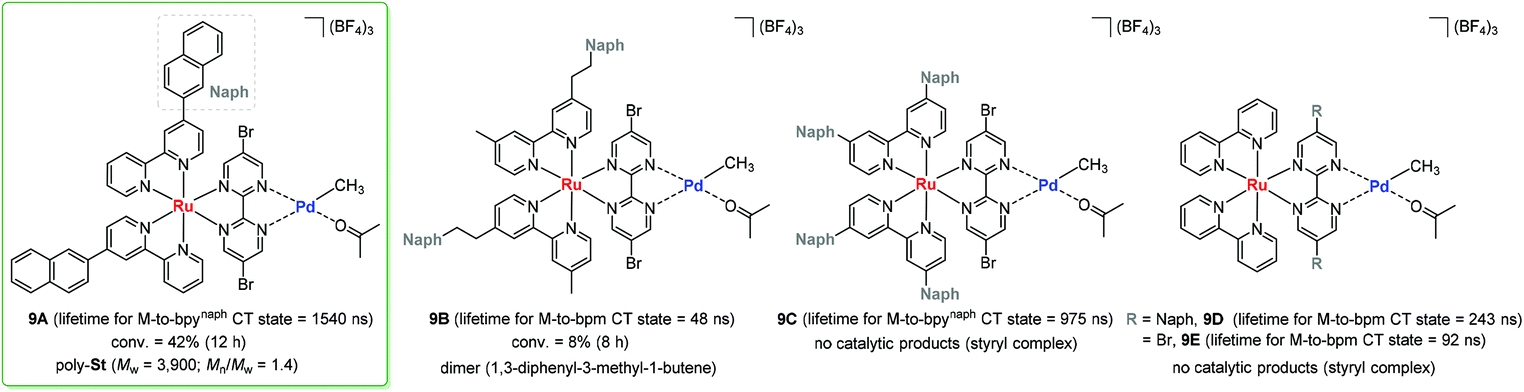 | ||
| Scheme 7 Bimetallic Ru–Pd complexes and their catalytic reactivities towards styrene polymerization under visible light. | ||
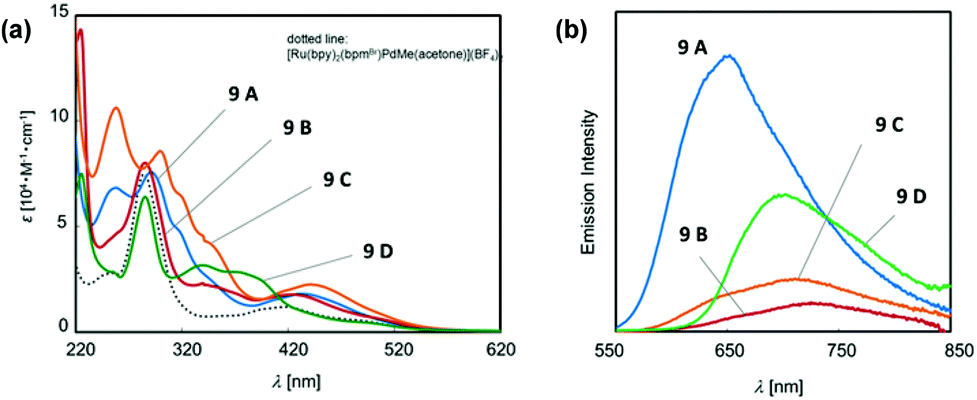 | ||
| Fig. 1 (a) UV-vis absorption and (b) luminescence spectra of Ru–Pd complexes 9A–9D.81 | ||
Based on the photophysical properties of 9A–9D, Inagaki examined their photocatalytic activities with respect to styrene polymerization under visible light conditions.80,81 As a result, 9A turned out to be active in producing poly-St (Mw = 3900, Mw/Mn = 1.4), albeit with incomplete conversion (40%), probably due to catalyst decomposition. It is noteworthy that 9B brought about only styrene dimers under otherwise the same photocatalytic conditions. These contrastive reaction outcomes indicated that the naphthyl unit needs to be directly bonded to the bipyridine ligand via a σ bond for effective styrene polymerization. Control experiments involving a set of [9E + naphthalene] or {(bpynaph)2Ru + [(bpm)PdMe(Me2CO)]BF4} again implied that the π-conjugated direct linkage of the naphthyl unit and bridged binding of the Pd-methyl moiety are crucial requirements for intramolecularly photosensitized polymerization. Unexpectedly, 9C and 9D did not display catalytic activities at all, and rather formed stable styryl complexes. In the case of 9C, the installed two naphthyl substituents on each bpy ligand were presumed to act as an electron-withdrawing factor, which hinders the desired direction of MLCT and thus results in a lack of polymerization activity. 9D has an excited state lifetime of ∼200 ns for metal-to-bpm CT as a major contribution, which is much shorter than that of the bpynaph-localized MLCT state in 9A.
A reaction pathway for 9A-initiated styrene polymerization was proposed (Scheme 8). The initial step is suggested to involve the 2,1-insertion of styrene into the cationic Pd–Me bond in 9A to form a Pd-secondary alkyl species (9f). The alkyl species 9f can undergo consecutive insertions of styrene monomers to furnish poly-Stvia a photo-excited species [9f]* in light. The existence of the presupposed intermediate 9f, was in fact corroborated by the observation of β-methylstyrene and the presence of polymer chains with 14 mass difference, ascribed to the two active species bearing Pd–CH3 and Pd–H moieties. Upon β-hydrogen elimination at 9f, another active species 9g is likely formed to undergo a subsequent styrene insertion. The key species leading to chain propagation upon light irradiation/absorption are Pd-alkyl species such as 9f and 9i, where the bpynaph-localized MLCT states (e.g. [9f*], [9i]*) with long lifetimes are supposed to render the Pd center more electrophilic to delay the β-H elimination. From a sterics aspect, the excited state's structure in Pd-alkyl species such as [9i]* can be altered to have the β-H distant from the Pd center, which can be another factor to prevent β-H elimination/chain transfer.105,106
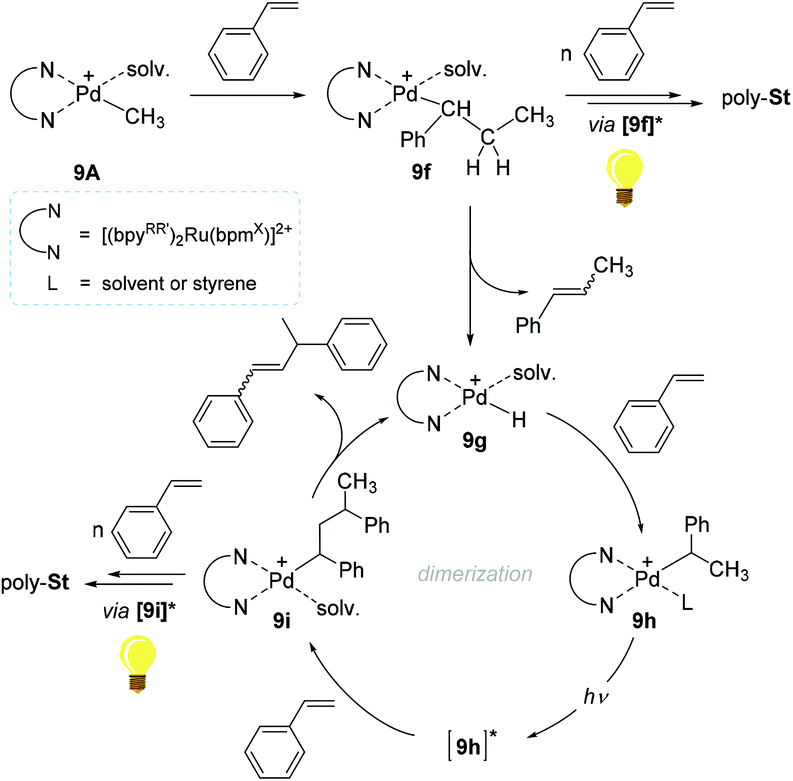 | ||
| Scheme 8 Proposed pathway for the polymerization of styrene catalyzed by 9A under visible light. | ||
In consideration of the bichromophore-led long MLCT lifetimes (up to 1540 ns) and insufficient polymerization activity observed in the catalytic system of 9A, Inagaki and coworkers synthesized the naphthyl group-connected bichromophoric Ir–Pd complex 10 as an advanced polymerization catalyst, where the Ir(III) center is supported by both σ-bonds and N-coordination from 2-phenylpyridyl (2-PhPy) and bipyrimidinyl ligands (Scheme 9). Preliminary spectroscopic studies revealed that 10 and its bichromophoric Ir unit 10′ have longer MLCT lifetimes when compared to those of the Ru(–Pd) complexes 9A–9E.
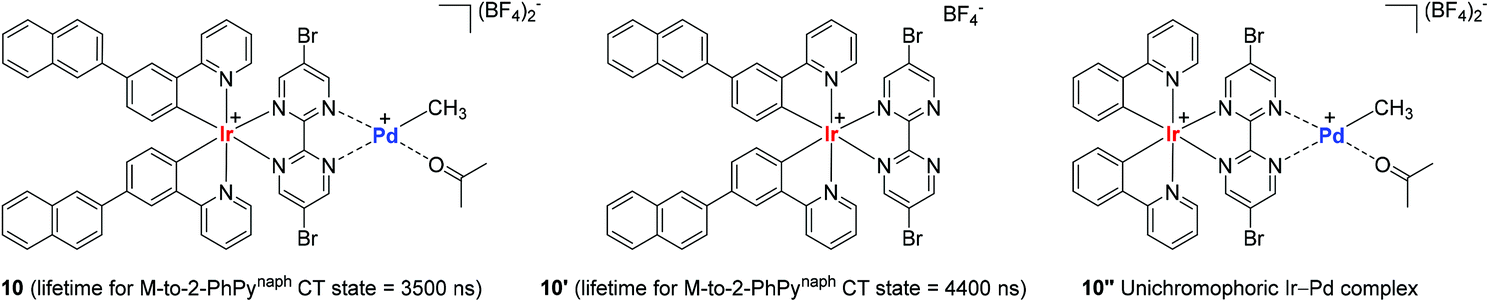 | ||
| Scheme 9 Bi- or unichromophoric Ir(III) cyclometallated complexes and their MLCT lifetimes. | ||
Encouraged by the observed longer MLCT lifetime for 10, homopolymerization reactions of a range of functional α-olefins, including styrene, and copolymerization of styrene with 2,2,2-trifluoroethyl vinyl ether (TFEVE) or pentafluorophenylbenzyl vinyl ether (PFBnVE) under various photo-irradiation conditions were conducted in the presence of catalytic 10 (1–2 mol%) at 25 °C (Scheme 10).107,108 The homopolymerization of styrene proceeded under light irradiation to attain the quantitative conversion in 8 h with the formation of poly-St (Mw = 6200, Mw/Mn = 2.3), whereas only dimerization affording trans-1,3-diphenyl-1-butene took place in the dark. This indicated that light as an external stimulus is essential for chain propagation (Scheme 10a). Interestingly, the Ir–Pd complex bearing unichromophoric ligand set 10′′ catalyzed styrene dimerization under photocatalytic conditions, suggesting the importance of the bichromophore in the Ir(III) cyclometallated ligand system for polymerization. The observed living fashioned photo-polymerization of 4-fluorostyrene (not shown) and constant growth of polymer chains even in the presence of a 2,6-di-tert-butyl-p-cresol (BHT) radical scavenger strongly supported a non-radical, single site coordination–insertion mechanism.
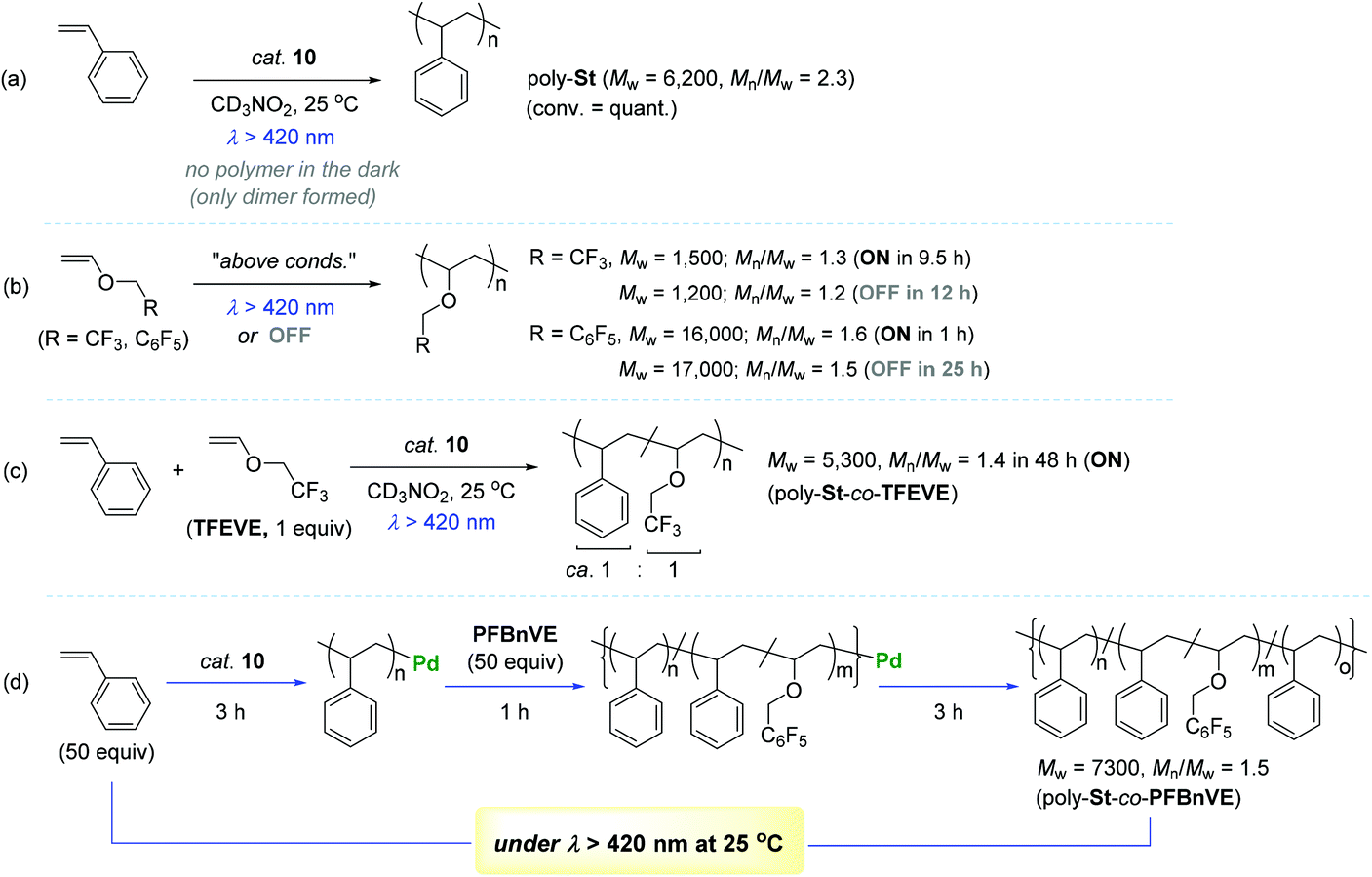 | ||
| Scheme 10 Catalytic behavior of 10 towards the (co)polymerization of styrenes and vinyl ethers under variable photo-irradiation conditions. | ||
Unlike the case of styrene, TFEVE and PFBnVE underwent homopolymerization reactions under both light and dark conditions to give poly-TFEVE [Mw ∼ 1500 (Mw/Mn ∼ 1.3)] and poly-PFBnVE [Mw ∼ 17000 (Mw/Mn ∼ 1.5)], respectively (Scheme 10b). To further extend the catalytic utility of 10, the photocatalytic copolymerization of styrene and TFEVE was examined [10 (1 mol%), 50 equiv. of St and TFEVE at 25 °C in CD3NO2 under hv > 420 nm]. As a result, the desired copolymerization proceeded with almost the same consumption rates of the two monomers to yield a random copolymer of styrene and TFEVE (poly-St-co-TFEVE) with a ca. 1:1 ratio of comonomer incorporation [Mw = 5300 (Mw/Mn = 1.4)] (Scheme 10c). In an attempt to synthesize multi-block copolymers consisting of poly-St and poly(St-co-PFBnVE) blocks, homopolymerization of styrene (50 equiv.) in light was conducted for 3 h at 25 °C to attain ∼30% conversion of styrene, into which PFBnVE (50 equiv.) was subsequently added to propagate a copolymeric chain as another block on the polystyrene backbone. After 1 h of polymerization, the initially loaded PFBnVE was found to be completely consumed, while ∼35% of the styrene monomer was still left in the crude polymerization solution. This copolymerization was quenched following another 3 h of polymerization reaction in the light to deliver an unprecedented triblock copolymer [Mw = 7300 (Mw/Mn = 1.5)] with two distinctive Tg temperatures at 22 and 64 °C (Scheme 10d). Notably, the observed two Tg values were assigned to the polymer blocks poly-St and poly(St-co-PFBnVE) in a single polymer chain.
In order to gain more insights into the kinetic behavior of each of the monomers during (co)polymerization, the consumption rates for each monomer in both the homo- and copolymerization reactions were measured to disclose the following insights: (i) the (co)polymerization reactions are generally accelerated upon visible-light irradiation; (ii) the relative reactivity of vinyl monomers under the photocatalytic homopolymerization conditions increases in the order of PFBnVE ≫ TFEVE ≥ St; and (iii) the consumption rate of styrene upon irradiation is most dramatically enhanced in the homopolymerization when compared with vinyl ether monomers. Two representative stoichiometric reactions of 10 with St or PFBnVE were carried out in parallel to show that the reaction with styrene generates trans-β-methyl styrene as a β-H elimination product with the formation of a stable π-benzyl complex, while the reaction with PFBnVE formed the Pd-alkyl complex through double consecutive insertions of PFBnVE, as evidenced by an m/z value of 1680.2 observed in its ESI mass spectrum (Scheme 11). These results again corroborated a coordination–insertion mechanism rather than a radical pathway. Given the fact that vinyl ethers undergo polymerization in the presence of catalyst 10 regardless of photoirradiation, the polymerization reactions of a series of vinyl ethers were examined in order to gain more insights into the catalytic working mode (coordination vs. cationic or radical mechanisms). As a result, ethyl vinyl ether and N-vinylcarbazole were converted to the corresponding homopolymers, whereas the polymerization of phenyl vinyl ether did not take place under either irradiation or dark conditions. This observed reactivity trend was found to be inconsistent with the vinyl substituent's electronics (e or Q values), with the conclusion that the polymerization of vinyl ethers by 10 proceeds not via a cationic or radical mechanism, but via a coordination–insertion mechanism.
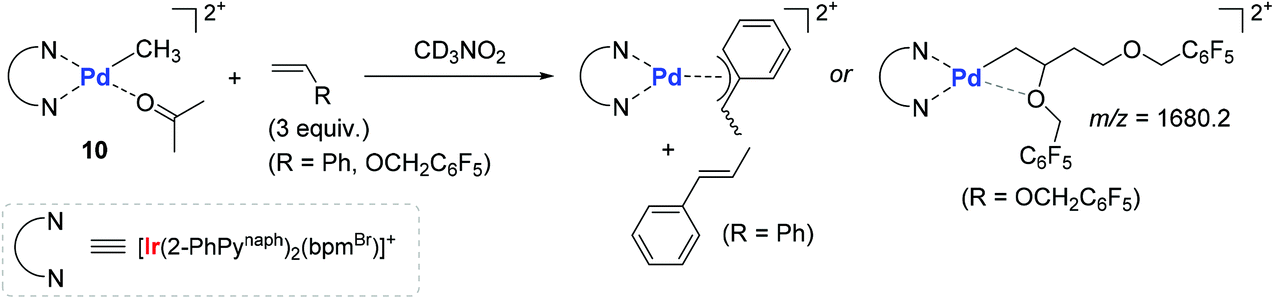 | ||
| Scheme 11 Stoichiometric reactions of 10 with styrene or PFBnVE in a 1:3 ratio. | ||
Based on stoichiometric observations, a mechanism for the 10-catalyzed copolymerization of styrene with PFBnVE was proposed (Scheme 12). The initial step likely involves the 2,1-insertion of vinyl monomer, followed by rapid β-hydride elimination to form an active species Pd–H (10a) with β-methylstyrene. This initial step has an induction period where styrene is initially inserted into the Pd-methyl bond of 10. The in situ generated 10a is assumed to undergo consecutive insertions of styrene and/or vinyl ether, during which photo-irradiation greatly accelerates the turnover-limiting monomer insertion step, thus promoting the overall polymerization process.107,108 The measured long MLCT lifetimes of the excited-state species109 and the increased stability from the Ir–C σ-bonded cyclometalate ligand in 10 are considered as the main factors responsible for the observed facile and living polymerization behavior.
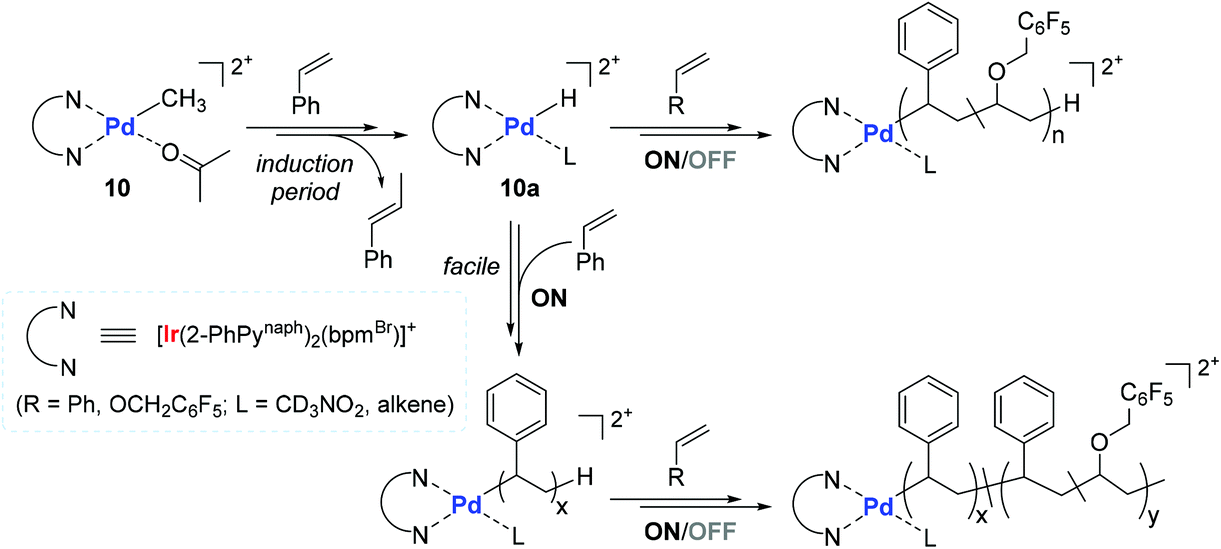 | ||
| Scheme 12 Proposed mechanism of the 10-catalyzed copolymerization of styrene with PFBnVE under light ON/OFF irradiation conditions. | ||
The reaction behaviors of well-defined olefin polymerization catalysts based on group 10 transition metals have been known to be influenced by not only reaction parameters (e.g. temperature, ethylene feed pressure),46,53,110,111 but also ligand-based electronics,40 being able to provide polyolefins bearing highly varied microstructures. Inspired by the observed electronic effects of ligands on the polymerization profiles, a handful of research groups recently developed new olefin polymerization catalysts possessing redox-active ligands in an effort to control the branching contents and/or microstructures of the resultant polyolefins via in situ alterations of the electronic nature of the catalytically active species.112–116 As one example, in 2016, Long and coworkers demonstrated that the microstructure of polyethylene (PE) can be modulated in terms of branching density and branching identity by employing a reductant as an additive in the presence of the Ni-based Brookhart catalyst 11 [E1/2 = −0.80 V (vs. FeII/FeIII)] (Scheme 13).117,118 For instance, the PE branching density observed via the Ni catalysis was decreased in a linear relationship to the equivalents of the cobaltocene additive [E1/2 = −1.33 V], and sec-butyl branching was not substantially observed in the resultant polyethylene. These drastic effects of the cobaltocene reductant indicate that the in situ Ni species generated upon reduction by cobaltocene is redox-active and capable of providing a less-branched microstructure of PE (Scheme 13a).117 Based on this precedence, Long next envisioned that the chemical reductant cobaltocene could be replaced with a suitable photoreductant for modulation of the PE microstructure. Considering its similar reduction potential to cobaltocene and its chemical compatibility with 11, tris(2-phenylpyridinato-C2,N) iridium(III) [fac-Ir(2-PhPy)3] possessing a photo-excited reduction potential of 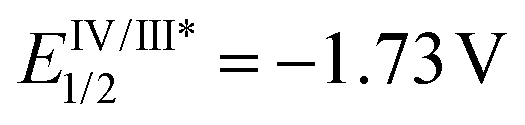 was chosen as a photo-reducing agent for 11-catalyzed ethylene polymerization under visible light irradiation.118
was chosen as a photo-reducing agent for 11-catalyzed ethylene polymerization under visible light irradiation.118
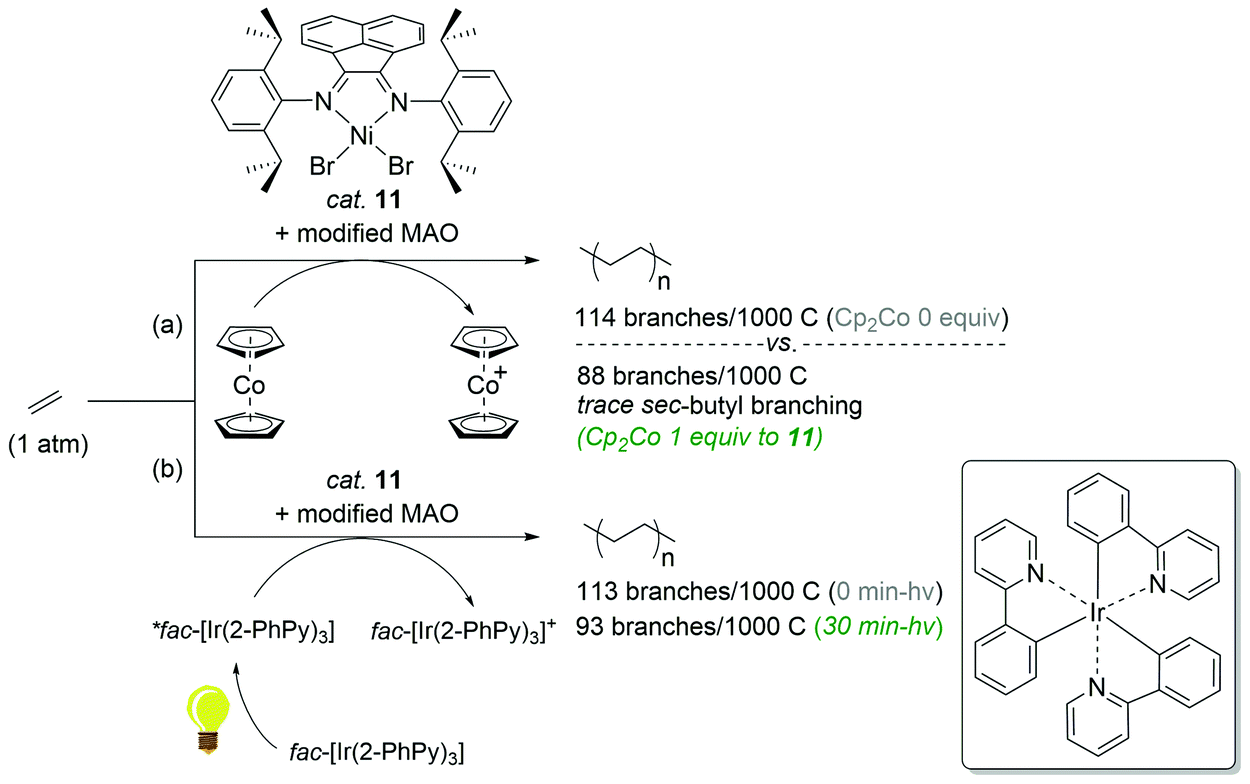 | ||
| Scheme 13 Polymerization of ethylene catalyzed by 11 in combination with the (photo)chemical reductants [(a) Cp2Co or (b) fac-Ir(2-PhPy)3]. | ||
The Ni-catalyzed polymerization of ethylene was generally performed under 1 atm of ethylene at 20 °C for 30 min in the light (3 W blue LED) with equimolar photoreductant fac-Ir(2-PhPy)3. The catalytic activity of 11 was found to be constant regardless of both light irradiation (<30 min) and the presence of fac-Ir(2-PhPy)3, leading to polyethylene products in similar yields (2.20–2.50 g), whereas the polymer dispersity increased from 1.54 to 1.97 with irradiation time. This trend was also observed when Cp2Co was used as a reductant,117 suggesting similar roles of fac-Ir(2-PhPy)3 and Cp2Co in the Ni-catalyzed ethylene polymerization. It is noteworthy that the branching content in the resultant polyethylene remarkably decreased to 93/1000C from 113/1000C upon polymerization under blue LED irradiation (30 min) (Scheme 13b). This outcome showcases the first example of the photo-mediated modulation of the polyethylene microstructure. Similar to the precedence using the reductant Cp2Co, long exposure times during the Ni catalysis led to a dramatic reduction in sec-butyl branching from 6.1 to 1.4%. Such prominent changes in the branching density of PE as a function of light exposure in the presence of fac-Ir(2-PhPy)3 were attributable to the delayed chain-walking process during the polymerization via the action of a reduced, more electron-rich propagating Ni species.32 Such a hypothetical relationship between the electron density of the active metal species and the rate of chain-walking was supported by Guan's work, in which catalysts with electron-donating α-diimine ligands exhibited higher rates of ethylene insertion relative to the rates of chain-walking.40
In 2016, Rovis reported the photoswitchable [2 + 2 + 2] cycloaddition of alkynes using a catalyst set of a Co(II) complex and a photoredox catalyst under visible light, in which the reaction clearly responded to the light ON/OFF process during catalytic turnover.119 In 2011, Okamoto succeeded in the Co-catalyzed chain-growth polymerization of triynes based on a conventional [2 + 2 + 2] cyclotrimerization reaction.120 Motivated by these findings, Rovis and coworkers recently developed the photocontrolled, Co-catalyzed cycloaddition polymerization of triynes. This work represents the first example of the photocontrolled cycloaddition polymerization of alkynes. The catalytic system was composed of a phosphine-ligated Co(II) complex as a precatalyst [CoBr2(PCy3)2, 12], a fluorinated polypyridyl Ir(III) photocatalyst (PC), and diisopropylethylamine (DIPEA) as a sacrificial reductant (Scheme 14).121 A standard triyne monomer possessing a malonate moiety (12a) was subjected to the preassumed photocontrolled cycloaddition polymerization to find the optimal reaction conditions within a certain catalytic reaction system [12 (3 mol%), PC (0.5 mol%), DIPEA (33 mol%), MeCN/DCE cosolvent, and additive (3–10 mol%) under visible light at 24 °C]. Without an alkyne additive, the catalytic system afforded a mixture of oligomers and polymers in 90% yield in 16 h (Mn = 4700; Mw/Mn = 2.17). Interestingly, the introduction of an alkyne additive (12b or 12c) along with minor deviations from the above catalytic reaction system gave rise to the corresponding cycloaddition polymers with relatively reduced Mw/Mn values ranging from 1.28 to 1.39 (poly-12a, Mn = 2700–3300) (Scheme 14a). The most efficient polymerization (Mn = 3300, Mw/Mn = 1.28, and 82% yield in 3 h) was observed, when 12c (3 mol%) was used as an additive, in particularly with 15 min of preirradiation prior to the addition of the monomer. The light ON–OFF process under the standard polymerization conditions was highly responsible for increasing (ON) and stalling (OFF) monomer conversion and the polymer molecular weight. Such observed precise photo-responsiveness of the present catalytic system implied that the initiation efficiency, involving a Co(I) species in situ generated under the light from a combination of 12 and an Ir-based PC, is high.122–125
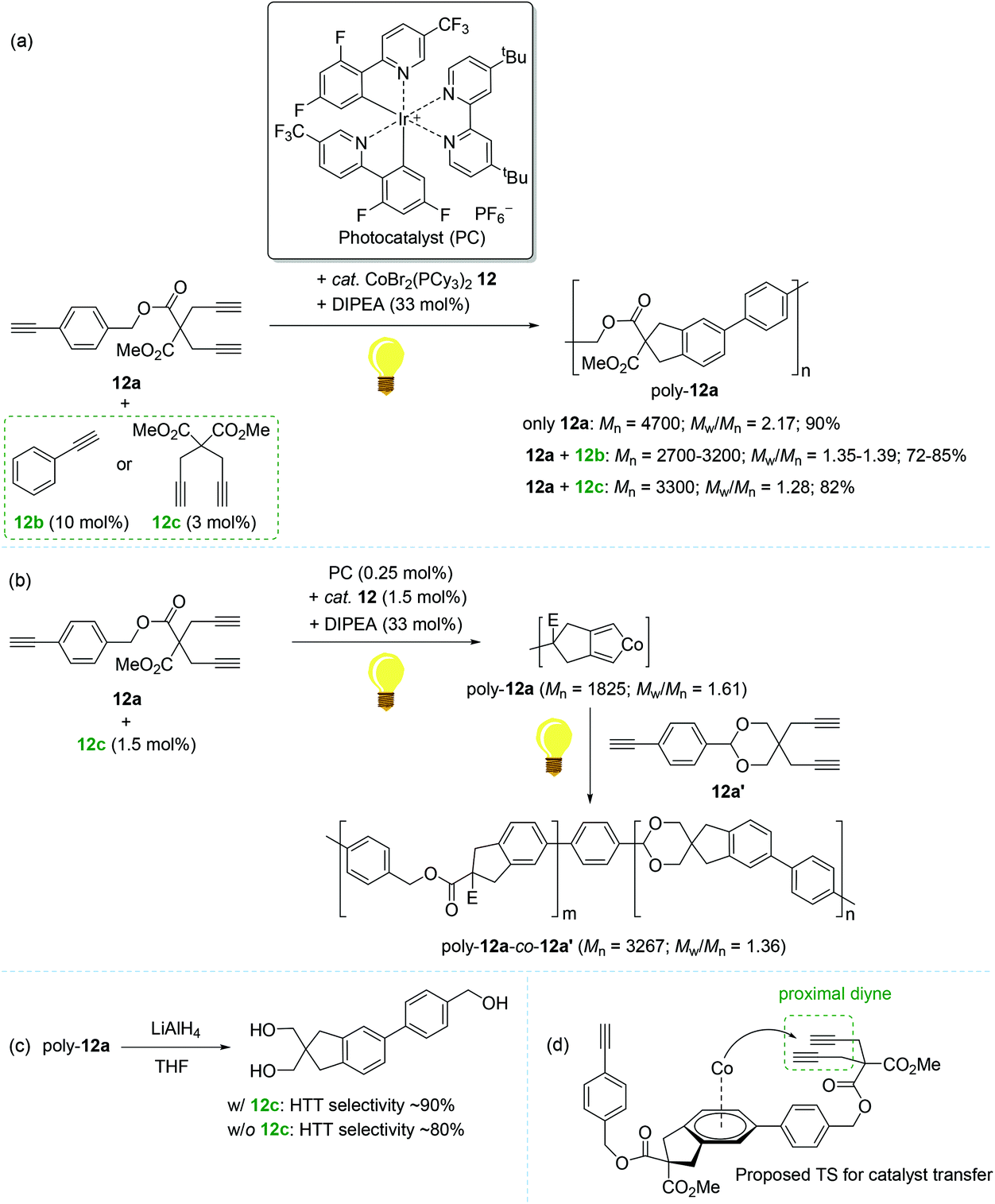 | ||
| Scheme 14 Photo-controlled cycloaddition polymerization of triynes catalyzed by 12 in combination with the Ir-based photocatalyst. | ||
Being aware of the differing molecular dispersities (Mw/Mn ∼ 2.2 vs. 1.3) between the polymers obtained with and without an alkyne additive, Rovis assumed that two mechanisms (step-growth vs. chain growth) of chain propagation could be operating depending on the addition of an alkyne additive. To gain more insights into the plausible dual polymerization pathways, the light-mediated polymerizations with and without 12c as an additive were monitored, revealing that the Mn of poly-12a increased exponentially as a function of conversion in the absence of 12c, while a linear relationship was observed between Mn and monomer conversion with the maintaining of narrow molecular dispersities (Mw/Mn) ∼ 1.25 in the presence of 12c. These results strongly indicated that the alkyne additive allows for living chain-growth polymerization.
By taking such a living-fashioned polymerization behavior approach, a block copolymer was able to be synthesized through the sequential addition of two monomers 12a and 12a′: the Mn ∼ 1800 of the first homopolymer (poly-12a) obviously increased to ∼3300 as the block copolymer (poly-12a-co-12a′) formed after the addition of the second monomer 12a′ (Scheme 14b). This sequential polymer growth supported a chain-growth mechanism, where the monomer is added primarily to the ends of existing chains. It is noteworthy that the LiAlH4-reduction of poly-12a synthesized with the 12c additive afforded a slightly larger amount of head-to-tail (HTT) product when compared to the polymer prepared without 12c (Scheme 14c). The enchainment of the HTT monomer unit was regarded as further evidence of a chain-growth mechanism. Based on the observation of the HTT product, a catalyst transfer transition state during chain propagation was proposed: the Co-benzene η6-complex formed upon reductive elimination from the catalytic cycle interacts with a proximal diyne moiety on a HTT polymer chain to cause the transfer of the Co center onto the growing chain end (Scheme 14d).126–128
Experimental observations strongly implied the existence of two distinct redox-active catalytic cycles: (i) a step-growth pathway involving Co(I)/Co(III) species and (ii) a chain-growth mechanism involving Co(0)/Co(II) species (Scheme 15). With the pre-reduction process using an alkyne additive such as 12c in light, precatalyst 12 is assumed to be initially converted to a Co(II) species 12evia a Co(III) species 12d to undergo HTT-oriented chain-growth polymerization; meanwhile, 12 is presumed to be initially reduced to Co(I)Br, followed by oxidative addition of a triyne to generate Co(III) 12d. In the absence of an additive, excess alkyne monomers such as 12a are suggested to displace a phosphine ligand on 12d to generate an alkyne adduct 12f upon energy transfer by the photoexcited Ir species (IrIII*), which undergoes less HTT-selective, step-growth polymerization accompanied by the release of Co(I) species upon reductive elimination events.
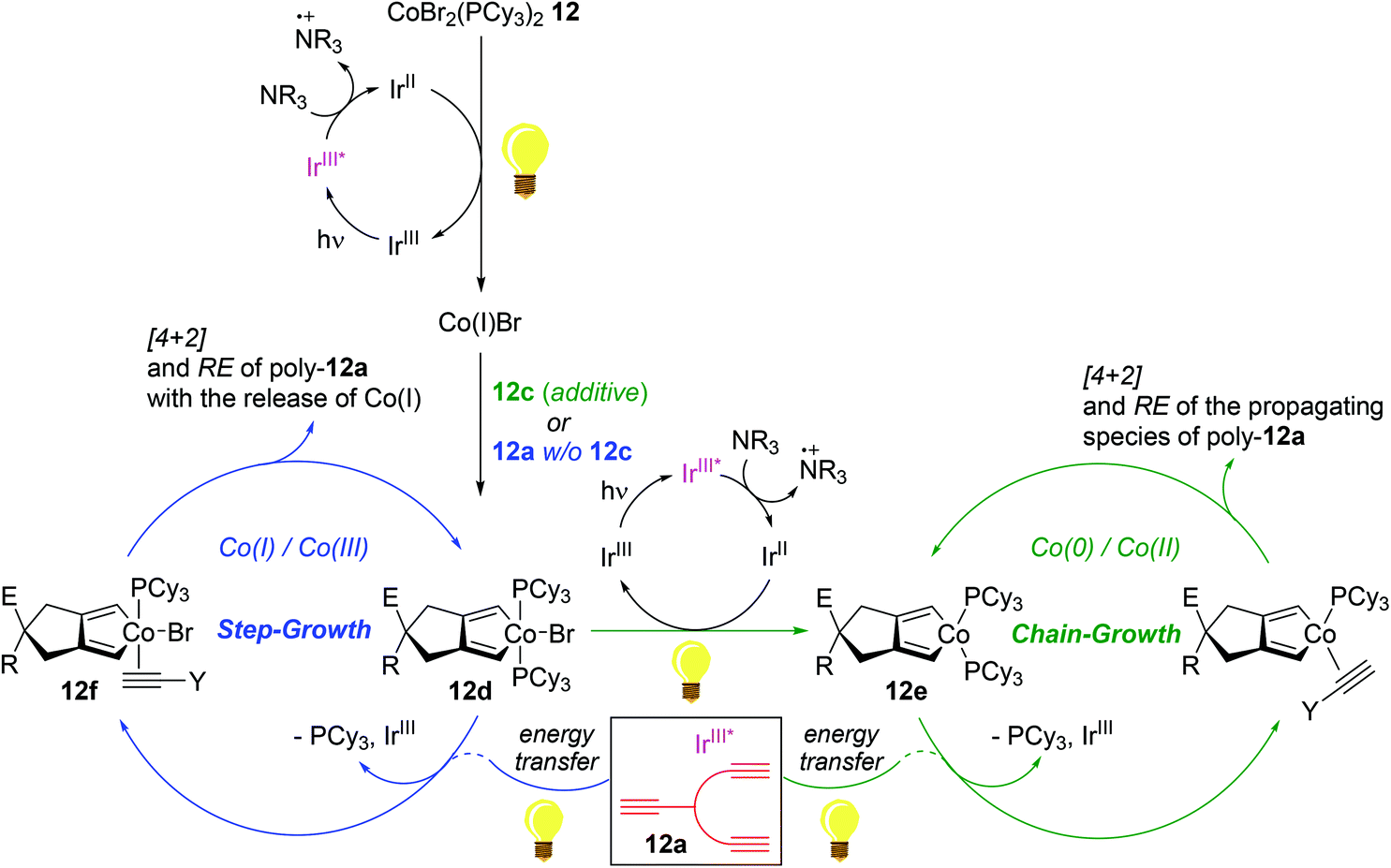 | ||
| Scheme 15 Proposed mechanism of the 12-catalyzed photo-cycloaddition polymerization of triynes. | ||
4. Heterogeneous catalytic systems
TiO2-based nanostructured materials have application potential in pollution remediation, photovoltaics, and photocatalytic hydrogen generation due to their low toxicity, low cost, and chemical and thermal stability.129,130 However, applications utilizing the surface chemical properties of TiO2 have been limited in the context of olefin polymerization as it is known that TiO2 is generally inert toward ethylene polymerization. Groppo and coworkers previously reported the thermal reduction of TiO2 under H2 atmosphere as a reducing agent to enable ethylene polymerization without the need of an activator,131 but this method requires harsh conditions and thus causes undesirable structural modifications.132,133 In this regard, Mino developed, for the first time, the light-mediated generation of surface-reduced Ti sites on TiO2 (13) under H2 or ethylene atmosphere at 25 °C.134 The surface-reduced Ti species were found to be capable of polymerizing ethylene at 25 °C without the need for any pre-reduction step (Scheme 16). A range of analytical tools such as electron paramagnetic resonance (EPR), ultraviolet/visible (UV/Vis), near infrared (NIR), and Fourier-transform infrared (FT-IR) spectroscopies were used to observe the effects of UV-light irradiation on TiO2 in the presence of H2 or ethylene. In the case of H2-photoreduced TiO2 materials, H2 splitting and the formation of (sub)surface Ti3+ centers were observed on the TiO2, and this reduced surface eventually polymerized ethylene upon long contact times to produce high-density polyethylene (HDPE). Interestingly, during ethylene polymerization, ethylene oxidization products such as formaldehyde and formates were detected and suggested to remain absorbed on the surface of TiO2.135,136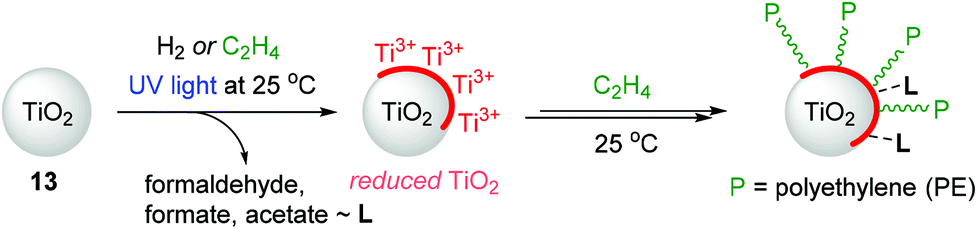 | ||
| Scheme 16 Ethylene polymerization catalyzed by photoreduced 13. | ||
In the system of ethylene-photoreduced TiO2, ethylene was shown to act as a reducing agent to generate Ti3+ species in TiO2, which simultaneously initiated ethylene polymerization via a one-pot process at 25 °C under UV light. Notably, the Ti3+ species reduced from TiO2 by ethylene were mainly generated on the surface rather than the subsurface or bulk lattice Ti3+ centers, which were mainly obtained via a H2-photoreduced process under otherwise identical conditions. The FT-IR analysis showed that the formation of polyethylene at the surface of the TiO2 sample accompanies the products of ethylene oxidation, as similarly observed in the reaction of ethylene on H2-photoreduced TiO2.
With a successful example of photoreduced TiO2-mediated ethylene polymerization, Mino and coworkers applied a photoinduced polymerization strategy to the Phillips catalyst system, CrVI/SiO2 (14) (Scheme 17),137 which is one of the most utilized catalysts in the petrochemical industry.138,139 Photoreduction of CrVI/SiO2 occurred under 0.1 atm of either CO or ethylene at 25 °C. The FT-IR spectra of CrVI/SiO2 upon UV irradiation under a CO atmosphere showed that a fraction of the CrVI sites were gradually reduced via a CrIV intermediate to CrII as an active species for ethylene polymerization. It is noteworthy that the relative amount of highly uncoordinated and thus accessible CrII sites formed via the photoreduction of CrVI was much higher when compared with the amount obtained through the thermal reduction in CO at high temperature (350 °C). The UV-vis spectroscopic analysis suggested that the reduction of the CrVI sites (d0 species) to low-valent Cr sites (dn species) was carried out by ethylene upon a short period of UV-vis irradiation (less than 50 s). The ethylene polymerization began to take place after only after 50 s of irradiation, and efficiently progressed with prolonged photo-irradiation time. The FT-IR analysis suggested that surface byproducts containing methylformate and ethylene oxide as a result of ethylene oxidation are formed during the initial steps of photoinduced ethylene polymerization, and remain in the coordination sphere of the reduced CrII sites, contributing towards defining the ligand sphere around the active Cr sites. The amount of polyethylene produced using this Cr catalysis was too small to conduct a more thorough product characterization.
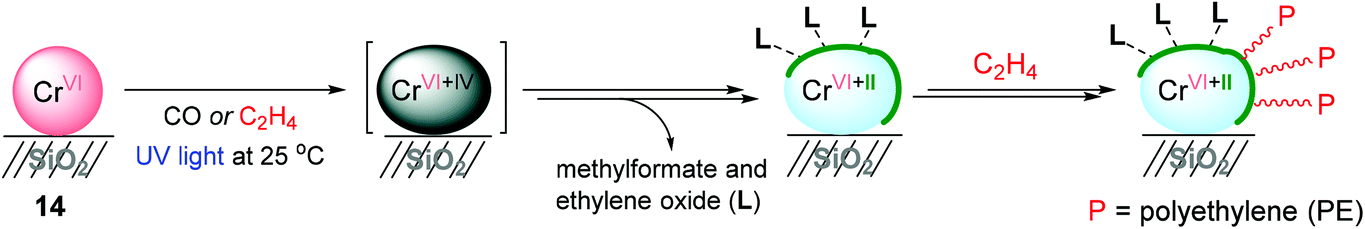 | ||
| Scheme 17 Photoinduced ethylene polymerization catalyzed by photoreduced 14. | ||
5. Conclusions
This Review describes the catalytic systems for light-mediated olefin coordination polymerization, enabling photo-controlled and/or -initiated polymer chain propagation. The catalytic reaction systems are all based on transition metals, and encompass monometallic and bimetallic complexes as well as homogeneous and heterogeneous catalytic systems. Most of the catalytic metal species undergo significant alterations in their electronic states via charge and/or energy transfer under suitable wavelengths of light irradiation, whereas a single example involves the photoswitchable steric changes of the ligand to influence the coordination sphere of the active metal site for the olefin coordination–insertion process.In the monometallic catalyst system, Aida disclosed the cyclopolymerization of a diallylmalonate catalyzed by an α-diimine Pd complex possessing a photoisomerizable azobenzene moiety, in which the installed azobenzene group as a photosensitized steric factor was for the first time shown to have an effect on the stereochemistry-determining step of the polymerization under light conditions. On the other hand, Inagaki synthesized a naphthyl-substituted BINAP-Pd complex for photoresponsive styrene polymerization, where the naphthyl moieties were found to improve the excited-state lifetime and stability of the Pd complex. Harth and Beezer showed that photoirradiation as an external stimulus in a series of Pd-based Brookhart catalysts switches the catalytic working mode from a conventional coordination mechanism to a radical mechanism for olefin polymerization. This photo-responsiveness allowed the synthesis of not only homopolymers of acrylates, but also block copolymers of α-olefins and acrylates using Brookhart catalysts under photoswitchable conditions.
In terms of bimetallic systems, the Inagaki and Akita group, as major players in this area, contributed towards developing a range of new heterodinuclear organometallic complexes as photosensitizing olefin polymerization catalysts, where one center based on Ru or Ir supported by naphthyl-substituted (2-phenyl)-pyridinyl and bipyrimidinyl ligands works as a photocatalyst in an intramolecular charge transfer process, while another bridged center is a Pd active species for polymer chain propagation. The Long group first revealed that an Ni-based Brookhart catalyst polymerizes ethylene to produce relatively less branched polyethylene, which is attributed to the action of a propagating Ni species photochemically reduced by an Ir-based photosensitizing complex. Rovis and coworkers applied the Co-catalyzed [2 + 2 + 2] photo-cycloaddition reaction of alkynes to the photoswitchable polymerization of triyne monomers. The polymerization proceeds in a living fashion, being able to synthesize cycloaddition block copolymers via regulating their Mn values under visible light.
In a heterogeneous catalytic system, Mino and coworkers for the first time showed that the photoreduction of inert TiO2 under UV light with ethylene as a reducing agent generates Ti3+ species on the surface of TiO2 nanoparticles, which promote ethylene polymerization in a one-pot reaction without the need for alkylating reagents under milder conditions. In a similar manner, the ethylene-photoreduction method was applied to the Phillips catalyst (CrVI/SiO2) system, in which a fraction of the CrVI sites are reduced to the active species CrII by ethylene upon UV-vis irradiation to trigger the polymerization of ethylene.
Overall, the majority of coordination polymerization catalytic systems combined with photosensitizing metal species and/or photochromic moieties are highly responsive to external light irradiation to influence the polymerization activity and/or catalytic working mode as an outcome of electronic alteration on the active propagating metal species. Since the propagating metal center undergoes light-mediated charge and/or energy transfer within a short period of time under mild conditions, good photo-controllability and switchability of the active metal species over the chain growth process are often observed. Despite diverse catalytic systems being known for photocontrolled olefin coordination polymerization, there have been no (systematic) studies on olefin polymerization catalyzed by transition metal complexes with a sterically photoswitchable ligand system. In this regard, developing a new family of olefin polymerization catalysts bearing modular and photoswitchable steric moieties of ligands would be a promising task distinctive from the precedence involving ambiguous photoresponsive electronic effects on the catalytic polymerization behavior. Photoswitchable steric groups installed on the ligand system towards transition metal-catalyzed photocontrolled olefin polymerization will allow the isolation of possible isomeric metal complexes as thermodynamic catalytic intermediates, and each isomeric metal species will provide straightforward and fundamental insights into the relationship between the ligand's geometric structure (e.g. cis vs. trans) and polymerization behavior such as regioselectivity for α-olefin insertion, and chain-walking and chain transfer processes that determine the polymer molecular weight and dispersity. This review will hopefully inspire organometallic chemists to develop new polymerization catalyst systems that enable light-mediated steric and electronic control over the active propagating metal species for olefin coordination polymerization.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank Yuhang Zhang for his valuable comments.Notes and references
- M. Stürzel, S. Mihan and R. Mülhaupt, From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- J. K. Fink, Handbook of Engineering and Specialty Thermoplastics, Polyolefins and Styrenics, Wiley, Somerset, 2010 Search PubMed.
- T. Kyu and D. Nwabunma, Polyolefin composites, Wiley-Interscience, Hoboken, 2008 Search PubMed.
- H. R. Kricheldorf, Polycondensation: History and New Results, Springer, Heidelberg, 2013 Search PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Stille Polycondensation for Synthesis of Functional Materials, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- S. K. Gupta and A. Kumar, Reaction engineering of step growth polymerization, Plenum Press, New York, 1987 Search PubMed.
- Y. N. Zhou, J. J. Li, Y. Y. Wu and Z. H. Luo, Role of External Field in Polymerization: Mechanism and Kinetics, Chem. Rev., 2020, 120, 2950–3048 CrossRef PubMed.
- E. Saldivar-Guerra and E. Vivaldo-Lima, Handbook of Polymer Synthesis, Characterization, and Processing, Wiley, Somerset, 1st edn, 2013 Search PubMed.
- K. C. Seavey and Y. A. Liu, Step-growth polymerization process modeling and product design, Wiley, Hoboken, 2008 Search PubMed.
- D. W. Sauter, M. Taoufik and C. Boisson, Polyolefins, a Success Story, Polymers, 2017, 9, 185 CrossRef PubMed.
- M. A. AlMaadeed and I. Krupa, Polyolefin Compounds and Materials: Fundamentals and Industrial Applications, Springer, Cham, 2016 Search PubMed.
- S. C. O. Ugbolue, Polyolefin fibres: Structure, properties and industrial applications, Woodhead, 2nd edn, 2017 Search PubMed.
- J. M. Lagaron, Multifunctional and nanoreinforced polymers for food packaging, Woodhead, Cambridge, 2011.
- A. C. Albertsson, Long-term properties of polyolefins, Springer, New York, 2004 Search PubMed.
- W. Kaminsky, Metalorganic Catalysts for Synthesis and Polymerization: Recent Results by Ziegler-Natta and Metallocene Investigations, Springer, Heidelberg, 1999 Search PubMed.
- P. Pino and R. Mülhaupt, Stereospecific Polymerization of Propylene: An Outlook 25 Years after Its Discovery, Angew. Chem., Int. Ed. Engl., 1980, 19, 857–875 CrossRef.
- J. Boor, Ziegler-Natta catalysts and polymerizations, Academic Press, New York, 1979 Search PubMed.
- P. Cossee, Ziegler-Natta catalysis I. Mechanism of polymerization of α-olefins with Ziegler-Natta catalysts, J. Catal., 1964, 3, 80–88 CrossRef CAS.
- H. Sinn, W. Kaminsky, H. J. Vollmer and R. Woldt, “Living polymers” on polymerization with extremely productive Ziegler catalysts, Angew. Chem., Int. Ed. Engl., 1980, 19, 390–392 CrossRef.
- M. Atiqullah, M. Tinkl, R. Pfaendner, M. N. Akhtar and I. Hussain, Synthesis of Functional Polyolefins using Metallocenes: A Comprehensive Review, Polym. Rev., 2010, 50, 178–230 CrossRef CAS.
- W. Kaminsky, The discovery of metallocene catalysts and their present state of the art, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3911–3921 CrossRef CAS.
- W. Kaminsky, Highly active metallocene catalysts for olefin polymerization, J. Chem. Soc., Dalton Trans., 1998, 1413–1418 RSC.
- L. K. Johnson, C. M. Killian and M. Brookhart, New Pd(II)-and Ni(II)-based catalysts for polymerization of ethylene and. alpha.-olefins, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(II) catalysts, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, Mechanistic Studies of the Palladium-Catalyzed Copolymerization of Ethylene and α-Olefins with Methyl Acrylate, J. Am. Chem. Soc., 1998, 120, 888–899 CrossRef CAS.
- F. Wang and C. Chen, A continuing legend: the Brookhart-type α-diimine nickel and palladium catalysts, Polym. Chem., 2019, 10, 2354–2369 RSC.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Late-Metal Catalysts for Ethylene Homo- and Copolymerization, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed.
- C. Chen, Designing catalysts for olefin polymerization and copolymerization: beyond electronic and steric tuning, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- L. Guo, W. Liu and C. Chen, Late transition metal catalyzed α-olefin polymerization and copolymerization with polar monomers, Mater. Chem. Front., 2017, 1, 2487–2494 RSC.
- A. Nakamura, S. Ito and K. Nozaki, Coordination−Insertion Copolymerization of Fundamental Polar Monomers, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed.
- W. Kaminsky, Polyolefins: 50 Years after Ziegler and Natta II : Polyolefins by Metallocenes and Other Single-Site Catalysts, Springer, Heidelberg, 2013 Search PubMed.
- L. Guo, S. Dai, X. Sui and C. Chen, Palladium and nickel catalyzed chain walking olefin polymerization and copolymerization, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- G. W. Coates, Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS PubMed.
- E. Y.-X. Chen and T. J. Marks, Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure−Activity Relationships, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- C. Tan and C. Chen, Emerging Palladium and Nickel Catalysts for Copolymerization of Olefins with Polar Monomers, Angew. Chem., Int. Ed., 2019, 58, 7192–7200 CrossRef CAS PubMed.
- C. J. Stephenson, J. P. McInnis, C. Chen, M. P. Weberski Jr., A. Motta, M. Delferro and T. J. Marks, Ni(II) phenoxyiminato olefin polymerization catalysis: Striking coordinative modulation of hyperbranched polymer microstructure and stability by a proximate sulfonyl group, ACS Catal., 2014, 4, 999–1003 CrossRef CAS.
- C. S. Popeney, C. M. Levins and Z. Guan, Systematic investigation of ligand substitution effects in cyclophane-based nickel(II) and palladium(II) olefin polymerization catalysts, Organometallics, 2011, 30, 2432–2452 CrossRef CAS.
- C. S. Popeney and Z. Guan, Effect of Ligand Electronics on the Stability and Chain Transfer Rates of Substituted Pd(II) α-Diimine Catalysts, Macromolecules, 2010, 43, 4091–4097 CrossRef CAS.
- J. Liu, Y. Li, Y. Li and N. Hu, Ethylene polymerization by (α-diimine)nickel(II) complexes bearing different substituents on para-position of imines activated with MMAO, J. Appl. Polym. Sci., 2008, 109, 700–707 CrossRef CAS.
- C. Popeney and Z. Guan, Ligand electronic effects on late transition metal polymerization catalysts, Organometallics, 2005, 24, 1145–1155 CrossRef CAS.
- N. E. Mitchell and B. K. Long, Recent advances in thermally robust, late transition metal-catalyzed olefin polymerization, Polym. Int., 2019, 68, 14–26 CrossRef CAS.
- S. Dai and C. Chen, Direct Synthesis of Functionalized High-Molecular-Weight Polyethylene by Copolymerization of Ethylene with Polar Monomers, Angew. Chem., Int. Ed., 2016, 55, 13281–13285 CrossRef CAS PubMed.
- S. Dai, X. Sui and C. Chen, Highly Robust Palladium(II) α-Diimine Catalysts for Slow-Chain-Walking Polymerization of Ethylene and Copolymerization with Methyl Acrylate, Angew. Chem., Int. Ed., 2015, 54, 9948–9953 CrossRef CAS PubMed.
- F.-S. Liu, H.-B. Hu, Y. Xu, L.-H. Guo, S.-B. Zai, K.-M. Song, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Thermostable α-diimine nickel(II) catalyst for ethylene polymerization: effects of the substituted backbone structure on catalytic properties and branching structure of polyethylene, Macromolecules, 2009, 42, 7789–7796 CrossRef CAS.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Cyclophane-Based Highly Active Late-Transition-Metal Catalysts for Ethylene Polymerization, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Synthesis of branched polyethylene using (α-diimine) nickel(II) catalysts: influence of temperature, ethylene pressure, and ligand structure on polymer properties, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, Living Polymerization of Ethylene and Copolymerization of Ethylene/Methyl Acrylate Using “Sandwich” Diimine Palladium Catalysts, ACS Catal., 2015, 5, 456–464 CrossRef CAS.
- A. C. Gottfried and M. Brookhart, Living and Block Copolymerization of Ethylene and α-Olefins Using Palladium(II)−α-Diimine Catalysts, Macromolecules, 2003, 36, 3085–3100 CrossRef.
- A. C. Gottfried and M. Brookhart, Living Polymerization of Ethylene Using Pd(II) α-Diimine Catalysts, Macromolecules, 2001, 34, 1140–1142 CrossRef CAS.
- Z. Guan and C. S. Popeney, Recent Progress in Late Transition Metal α-Diimine Catalysts for Olefin Polymerization, Top. Organomet. Chem., 2009, 2, 179–220 CrossRef.
- J. M. Rose, A. E. Cherian and G. W. Coates, Living Polymerization of α-Olefins with an α-Diimine Ni(II) Catalyst: Formation of Well-Defined Ethylene−Propylene Copolymers through Controlled Chain-Walking, J. Am. Chem. Soc., 2006, 128, 4186–4187 CrossRef CAS PubMed.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, A C2-Symmetric, Living α-Diimine Ni(II) Catalyst: Regioblock Copolymers from Propylene, J. Am. Chem. Soc., 2005, 127, 13770–13771 CrossRef CAS PubMed.
- Z. Guan, P. Cotts, E. McCord and S. McLain, Chain walking: a new strategy to control polymer topology, Science, 1999, 283, 2059–2062 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Photoinduced electron transfer reactions for macromolecular syntheses, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Photocatalysis in organic and polymer synthesis, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- J. Phommalysack-Lovan, Y. Chu, C. Boyer and J. Xu, PET-RAFT polymerisation: towards green and precision polymer manufacturing, Chem. Commun., 2018, 54, 6591–6606 RSC.
- Q. Michaudel, V. Kottisch and B. P. Fors, Cationic polymerization: from photoinitiation to photocontrol, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, External regulation of controlled polymerizations, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS.
- K. A. Ogawa, A. E. Goetz and A. J. Boydston, Metal-free ring-opening metathesis polymerization, J. Am. Chem. Soc., 2015, 137, 1400–1403 CrossRef CAS PubMed.
- A. J. Teator, H. Shao, G. Lu, P. Liu and C. W. Bielawski, A photoswitchable olefin metathesis catalyst, Organometallics, 2017, 36, 490–497 CrossRef CAS.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, A photoswitchable catalyst system for remote-controlled (co) polymerization in situ, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- M. Li, P. Zhang and C. Chen, Light-Controlled Switchable Ring Opening Polymerization, Macromolecules, 2019, 52, 5646–5651 CrossRef CAS.
- C. Fu, J. Xu and C. Boyer, Photoacid-mediated ring opening polymerization driven by visible light, Chem. Commun., 2016, 52, 7126–7129 RSC.
- B. M. Neilson and C. W. Bielawski, Photoswitchable NHC-promoted ring-opening polymerizations, Chem. Commun., 2013, 49, 5453–5455 RSC.
- M. Chen and C. Chen, Controlling the Ring-Opening Polymerization Process Using External Stimuli, Chin. J. Chem., 2020, 38, 282–286 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Control of a Living Radical Polymerization of Methacrylates by Light, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Organocatalyzed atom transfer radical polymerization driven by visible light, Science, 2016, 352, 1082–1086 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, Metal-free atom transfer radical polymerization, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- E. Murtezi and Y. Yagci, Simultaneous Photoinduced ATRP and CuAAC Reactions for the Synthesis of Block Copolymers, Macromol. Rapid Commun., 2014, 35, 1782–1787 CrossRef CAS PubMed.
- S. Doran and Y. Yagci, Graft polymer growth using tandem photoinduced photoinitiator-free CuAAC/ATRP, Polym. Chem., 2015, 6, 946–952 RSC.
- Y. Kwak and K. Matyjaszewski, Photoirradiated Atom Transfer Radical Polymerization with an Alkyl Dithiocarbamate at Ambient Temperature, Macromolecules, 2010, 43, 5180–5183 CrossRef CAS.
- A. Inagaki and M. Akita, Visible-light promoted bimetallic catalysis, Coord. Chem. Rev., 2010, 254, 1220–1239 CrossRef CAS.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Switchable polymerization catalysts, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- S. P. Ihrig, F. Eisenreich and S. Hecht, Photoswitchable polymerization catalysis: state of the art, challenges, and perspectives, Chem. Commun., 2019, 55, 4290–4298 RSC.
- It should be noted that a range of catalytic systems in this review may have possibilities to operate via a cationic or radical mechanism against the originally proposed coordination insertion pathway depending on certain reaction systems. In order to avoid such arguable discussions, but to reflect the experimental mechanistic insights, we present the proposed catalytic working modes together with the experimental observations throughout this review article as the original papers describe.
- S. Park, D. Takeuchi and K. Osakada, Pd Complex-Promoted Cyclopolymerization of Functionalized α,ω-Dienes and Copolymerization with Ethylene to Afford Polymers with Cyclic Repeating Units, J. Am. Chem. Soc., 2006, 128, 3510–3511 CrossRef CAS PubMed.
- S. Park, T. Okada, D. Takeuchi and K. Osakada, Cyclopolymerization and Copolymerization of Functionalized 1,6-Heptadienes Catalyzed by Pd Complexes: Mechanism and Application to Physical-Gel Formation, Chem. – Eur. J., 2010, 16, 8662–8678 CrossRef CAS PubMed.
- D. Takeuchi, S. Park, T. Okada, R. Matsuura and K. Osakada, Novel Precision Cyclopolymerization of Dienes by Late Transition Metal Catalysts, Kobunshi Ronbunshu, 2007, 64, 597–606 CrossRef CAS.
- Y. Miyamura, K. Kinbara, Y. Yamamoto, V. K. Praveen, K. Kato, M. Takata, A. Takano, Y. Matsushita, E. Lee, M. Lee and T. Aida, Shape-Directed Assembly of a “Macromolecular Barb” into Nanofibers: Stereospecific Cyclopolymerization of Isopropylidene Diallylmalonate, J. Am. Chem. Soc., 2010, 132, 3292–3294 CrossRef CAS PubMed.
- K. Murata, M. Ito, A. Inagaki and M. Akita, Photocatalytic Styrene Polymerization by Novel Bichromophoric Pd Catalyst Having Long Excited-state Lifetime, Chem. Lett., 2010, 39, 915–917 CrossRef CAS.
- K. Murata, M. Araki, A. Inagaki and M. Akita, Syntheses, photophysical properties, and reactivities of novel bichromophoric Pd complexes composed of Ru(II)–polypyridyl and naphthyl moieties, Dalton Trans., 2013, 42, 6989–7001 RSC.
- T. Tsubomura, Y. Ito, S. Inoue, Y. Tanaka, K. Matsumoto and T. Tsukuda, Strongly luminescent palladium (0) and platinum (0) diphosphine complexes, Inorg. Chem., 2008, 47, 481–486 CrossRef CAS PubMed.
- Z. Abedin-Siddique, T. Ohno, K. Nozaki and T. Tsubomura, Intense Fluorescence of Metal-to-Ligand Charge Transfer in [Pt (0)(binap)2][binap = 2,2′-Bis (diphenylphosphino)-1,1′-binaphthyl], Inorg. Chem., 2004, 43, 663–673 CrossRef CAS PubMed.
- T. Ohkubo, K. Takao and T. Tsubomura, Blue to orange emitters; palladium (0) monodentate phosphine complexes, Inorg. Chem. Commun., 2012, 20, 27–29 CrossRef CAS.
- T. Tsubomura, H. Murota and K. Takao, Luminescent palladium (0) complexes bearing N-heterocyclic carbene and phosphine ligands, Inorg. Chem. Commun., 2013, 35, 110–112 CrossRef CAS.
- C. Son and A. Inagaki, Synthesis and photocatalytic activity of a naphthyl-substituted photosensitizing BINAP–palladium complex, Dalton Trans., 2016, 45, 1331–1334 RSC.
- G. Zhang, C. Nam, L. Petersson, J. Jämbeck, H. Hillborg and T. M. Chung, Increasing polypropylene high temperature stability by blending polypropylene-bonded hindered phenol antioxidant, Macromolecules, 2018, 51, 1927–1936 CrossRef CAS.
- C. Hong, X. Wang and C. Chen, Palladium-catalyzed dimerization of vinyl ethers: mechanism, catalyst optimization, and polymerization applications, Macromolecules, 2019, 52, 7123–7129 CrossRef CAS.
- T. Liang, S. B. Goudari and C. Chen, A simple and versatile nickel platform for the generation of branched high molecular weight polyolefins, Nat. Commun., 2020, 11, 372 CrossRef CAS PubMed.
- C. Zou and C. Chen, Polar-Functionalized, Crosslinkable, Self-Healing, and Photoresponsive Polyolefins, Angew. Chem., 2020, 59, 395–402 CrossRef CAS.
- M. Chen and C. Chen, Direct and Tandem Routes for the Copolymerization of Ethylene with Polar Functionalized Internal Olefins, Angew. Chem., 2020, 59, 1206–1210 CrossRef CAS PubMed.
- G. Tian, H. W. Boone and B. M. Novak, Neutral Palladium Complexes as Catalysts for Olefin− Methyl Acrylate Copolymerization: A Cautionary Tale, Macromolecules, 2001, 34, 7656–7663 CrossRef CAS.
- A. Leblanc, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Synthesis of copolymers of ethylene and (meth) acrylates or styrene by an original dual radical/catalytic mechanism, Pure Appl. Chem., 2012, 84, 2113–2120 CAS.
- A. Leblanc, E. Grau, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Homo-and copolymerizations of (meth) acrylates with olefins (styrene, ethylene) using neutral nickel complexes: a dual radical/catalytic pathway, Macromolecules, 2011, 44, 3293–3301 CrossRef CAS.
- A. Kermagoret, A. Debuigne, C. Jérôme and C. Detrembleur, Precision design of ethylene-and polar-monomer-based copolymers by organometallic-mediated radical polymerization, Nat. Chem., 2014, 6, 179–187 CrossRef CAS PubMed.
- R. López-Fernández, N. Carrera, A. C. Albéniz and P. Espinet, Dual behavior of cationic palladium pentafluorophenyl complexes as catalysts for the homopolymerization of acrylates and of nonpolar olefins, Organometallics, 2009, 28, 4996–5001 CrossRef.
- P. Xiang and Z. Ye, Homo-and Co-polymerization of norbornene and methyl acrylate with Pd–diimine catalysts, J. Organomet. Chem., 2015, 798, 429–436 CrossRef CAS.
- M. Nagel and A. Sen, Intermediacy of Radicals in Rearrangement and Decomposition of Metal− Alkyl Species: Relevance to Metal-Mediated Polymerization of Polar Vinyl Monomers, Organometallics, 2006, 25, 4722–4724 CrossRef CAS.
- A. Keyes, H. E. Basbug Alhan, U. Ha, Y.-S. Liu, S. K. Smith, T. S. Teets, D. B. Beezer and E. Harth, Light as a Catalytic Switch for Block Copolymer Architectures: Metal–Organic Insertion/Light Initiated Radical (MILRad) Polymerization, Macromolecules, 2018, 51, 7224–7232 CrossRef CAS.
- A. Keyes, H. Dau, H. E. Basbug Alhan, U. Ha, E. Ordonez, G. R. Jones, Y.-S. Liu, E. Tsogtgerel, B. Loftin, Z. Wen, J. I. Wu, D. B. Beezer and E. Harth, Metal–organic insertion light initiated radical (MILRad) polymerization: photo-initiated radical polymerization of vinyl polar monomers with various palladium diimine catalysts, Polym. Chem., 2019, 10, 3040–3047 RSC.
- K. A. Smoll, W. Kaminsky and K. I. Goldberg, Photolysis of Pincer-Ligated PdII−Me Complexes in the Presence of Molecular Oxygen, Organometallics, 2017, 36, 1213–1216 CrossRef CAS.
- P. Huo, T. Chen, J.-L. Hou, L. Yu, Q.-Y. Zhu and J. Dai, Ligand-to-Ligand Charge Transfer within Metal–Organic Frameworks Based on Manganese Coordination Polymers with Tetrathiafulvalene-Bicarboxylate and Bipyridine Ligands, Inorg. Chem., 2016, 55, 6496–6503 CrossRef CAS PubMed.
- J. van Slageren, A. Klein and S. Záliš, Ligand-to-ligand charge transfer states and photochemical bond homolysis in metal−carbon bonded platinum complexes, Coord. Chem. Rev., 2002, 230, 193–211 CrossRef CAS.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, Long-lived charge-transfer states of nickel(II) aryl halide complexes facilitate bimolecular photoinduced electron transfer, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Iron and Cobalt Complexes of 2,3,7,8-Tetrahydroacridine-4,5(1H,6H)-diimine Sterically Modulated by Substituted Aryl Rings for the Selective Oligomerization to Polymerization of Ethylene, Organometallics, 2011, 30, 2285–2294 CrossRef CAS.
- A. Igarashi, S. Zhang and K. Nomura, Ethylene Dimerization/Polymerization Catalyzed by (Adamantylimido)vanadium(V) Complexes Containing (2-Anilidomethyl)pyridine Ligands: Factors Affecting the Ethylene Reactivity, Organometallics, 2012, 31, 3575–3581 CrossRef CAS.
- K. Murata, K. Saito, S. Kikuchi, M. Akita and A. Inagaki, Visible-light-controlled homo-and copolymerization of styrenes by a bichromophoric Ir–Pd catalyst, Chem. Commun., 2015, 51, 5717–5720 RSC.
- S. Kikuchi, K. Saito, M. Akita and A. Inagaki, Nonradical Light-Controlled Polymerization of Styrene and Vinyl Ethers Catalyzed by an Iridium–Palladium Photocatalyst, Organometallics, 2018, 37, 359–366 CrossRef CAS.
- K. Murata, A. Inagaki, M. Akita, J.-F. Halet and K. Costuas, Revelation of the Photoactive Species in the Photocatalytic Dimerization of α-Methylstyrene by a Dinuclear Ruthenium–Palladium Complex, Inorg. Chem., 2013, 52, 8030–8039 CrossRef CAS.
- Z. Guan, Recent Progress of Catalytic Polymerization for Controlling Polymer Topology, Chem. – Asian J., 2010, 5, 1058–1070 CrossRef CAS PubMed.
- Y. Xu, P. Xiang, Z. Ye and W.-J. Wang, Hyperbranched−linear polyethylene block polymers constructed with chain blocks of hybrid chain topologies via one-pot stagewise chain walking ethylene “living” polymerization, Macromolecules, 2010, 43, 8026–8038 CrossRef CAS.
- M. Chen, B. Yang and C. Chen, Redox-Controlled Olefin (Co)Polymerization Catalyzed by Ferrocene-Bridged Phosphine-Sulfonate Palladium Complexes, Angew. Chem., Int. Ed., 2015, 54, 15520–15524 CrossRef CAS PubMed.
- W. C. Anderson, S. H. Park, L. A. Brown, J. M. Kaiser and B. K. Long, Accessing multiple polyethylene grades via a single redox-active olefin polymerization catalyst, Inorg. Chem. Front., 2017, 4, 1108–1112 RSC.
- W. C. Anderson Jr. and B. K. Long, Modulating polyolefin copolymer composition via redox-active olefin polymerization catalysts, ACS Macro Lett., 2016, 5, 1029–1033 CrossRef.
- M. Zhao and C. Chen, Accessing multiple catalytically active states in redox-controlled olefin polymerization, ACS Catal., 2017, 7, 7490–7494 CrossRef CAS.
- C. Chen, Redox-Controlled Polymerization and Copolymerization, ACS Catal., 2018, 8, 5506–5514 CrossRef CAS.
- W. C. Anderson Jr., J. L. Rhinehart, A. G. Tennyson and B. K. Long, Redox-active ligands: an advanced tool to modulate polyethylene microstructure, J. Am. Chem. Soc., 2016, 138, 774–777 CrossRef PubMed.
- J. M. Kaiser, W. C. Anderson and B. K. Long, Photochemical regulation of a redox-active olefin polymerization catalyst: controlling polyethylene microstructure with visible light, Polym. Chem., 2018, 9, 1567–1570 RSC.
- K. E. Ruhl and T. Rovis, Visible light-gated cobalt catalysis for a spatially and temporally resolved [2 + 2 + 2] cycloaddition, J. Am. Chem. Soc., 2016, 138, 15527–15530 CrossRef CAS PubMed.
- Y.-k. Sugiyama, R. Kato, T. Sakurada and S. Okamoto, Chain-growth cycloaddition polymerization via a catalytic alkyne [2 + 2 + 2] cyclotrimerization reaction and its application to one-shot spontaneous block copolymerization, J. Am. Chem. Soc., 2011, 133, 9712–9715 CrossRef CAS PubMed.
- B. D. Ravetz, K. E. Ruhl and T. Rovis, External Regulation of Cobalt-Catalyzed Cycloaddition Polymerization with Visible Light, ACS Catal., 2018, 8, 5323–5327 CrossRef CAS.
- W. G. L. Aalbersberg, A. J. Barkovich, R. L. Funk, R. L. Hillard and K. P. C. Vollhardt, Transition metal catalyzed acetylene cyclizations. 4,5-Bis(trimethylsilyl)benzocyclobutene, a highly strained, versatile synthetic intermediate, J. Am. Chem. Soc., 1975, 97, 5600–5602 CrossRef CAS.
- K. P. C. Vollhardt, Cobalt-Mediated [2 + 2 + 2]-Cycloadditions: A Maturing Synthetic Strategy [New Synthetic Methods (43)], Angew. Chem., Int. Ed. Engl., 1984, 23, 539–556 CrossRef.
- H. Bönnemann, Organocobalt Compounds in the Synthesis of Pyridines–An Example of Structure-Effectivity Relationships in Homogeneous Catalýsis, Angew. Chem., Int. Ed. Engl., 1985, 24, 248–262 CrossRef.
- N. Saino, F. Amemiya, E. Tanabe, K. Kase and S. Okamoto, A Highly Practical Instant Catalyst for Cyclotrimerization of Alkynes to Substituted Benzenes, Org. Lett., 2006, 8, 1439–1442 CrossRef CAS PubMed.
- Z. J. Bryan and A. J. McNeil, Conjugated Polymer Synthesis via Catalyst-Transfer Polycondensation (CTP): Mechanism, Scope, and Applications, Macromolecules, 2013, 46, 8395–8405 CrossRef CAS.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Chain Growth Mechanism for Regioregular Nickel-Initiated Cross-Coupling Polymerizations, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- R. Miyakoshi, A. Yokoyama and T. Yokozawa, Catalyst-Transfer Polycondensation. Mechanism of Ni-Catalyzed Chain-Growth Polymerization Leading to Well-Defined Poly(3-hexylthiophene), J. Am. Chem. Soc., 2005, 127, 17542–17547 CrossRef CAS PubMed.
- M. Grätzel, Dye-sensitized solar cells, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- X. Chen and S. S. Mao, Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- C. Barzan, E. Groppo, S. Bordiga and A. Zecchina, Defect sites in H2-reduced TiO2 convert ethylene to high density polyethylene without activator, ACS Catal., 2014, 4, 986–989 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals, Science, 2011, 331, 746–750 CrossRef CAS.
- X. Chen, L. Liu and F. Huang, Black titanium dioxide (TiO2) nanomaterials, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- C. Barzan, L. Mino, E. Morra, E. Groppo, M. Chiesa and G. Spoto, Photoinduced Ethylene Polymerization on Titania Nanoparticles, ChemCatChem, 2017, 9, 4324–4327 CrossRef CAS.
- B. Schumacher, V. Plzak, M. Kinne and R. J. Behm, Highly Active Au/TiO2 Catalysts for Low-Temperature CO Oxidation: Preparation, Conditioning and Stability, Catal. Lett., 2003, 89, 109–114 CrossRef CAS.
- J. Raskó and J. Kiss, Adsorption and surface reactions of acetaldehyde on TiO2, CeO2 and Al2O3, Appl. Catal., A, 2005, 287, 252–260 CrossRef.
- L. Mino, C. Barzan, G. A. Martino, A. Piovano, G. Spoto, A. Zecchina and E. Groppo, Photoinduced Ethylene Polymerization on the CrVI/SiO2 Phillips Catalyst, J. Phys. Chem. C, 2018, 123, 8145–8152 CrossRef.
- T. E. Nowlin, Business and Technology of the Global Polyethylene Industry, Scrivener Publishing LLC, New York, 2014 Search PubMed.
- M. P. McDaniel, in Advances in Catalysis, Academic Press, 2010, vol. 53, pp. 123–606 Search PubMed.
| This journal is © the Partner Organisations 2020 |