
pH-Dependent morphology and optical properties of lysine-derived molecular biodynamers†
Sangeun
Lee
 ab,
Cansu
Kaya
ac,
Hongje
Jang
d,
Marcus
Koch
e,
Brigitta
Loretz
b,
Eric
Buhler
f,
Claus-Michael
Lehr
*bc and
Anna Katharina Herta
Hirsch
*ac
ab,
Cansu
Kaya
ac,
Hongje
Jang
d,
Marcus
Koch
e,
Brigitta
Loretz
b,
Eric
Buhler
f,
Claus-Michael
Lehr
*bc and
Anna Katharina Herta
Hirsch
*ac
aDepartment for Drug Design and Optimization, Helmholtz Institute of Pharmaceutical Research Saarland (HIPS) – Helmholtz Centre for Infection Research (HZI), Campus E 8.1, 66123 Saarbrücken, Germany. E-mail: Anna.Hirsch@helmholtz-hips.de
bDepartment for Drug Delivery, HIPS – HZI, Campus E8.1, 66123 Saarbrücken, Germany. E-mail: Claus-Michael.Lehr@helmholtz-hips.de
cDepartment of Pharmacy, Saarland University, 66123, Saarbrücken, Germany
dThird Institute of Physics, Georg-August-Universität, Friedrich-Hund-Platz 1, 37077 Göttingen, Germany
eINM-Leibniz-Institute for New Materials, Campus D2.2, Saarland University, 66123 Saarbrücken, Germany
fLaboratoire Matière et Systèmes Complexes (MSC), UMR 7057, University of Paris (Paris Diderot), Bâtiment Condorcet, 75205 Paris Cedex 13, France
First published on 27th December 2019
Abstract
Polymerization of carbazole dicarboxaldehydes and lysine derivatives by imine and acylhydrazone formation afforded peptide-derived molecular biodynamers. Characterization of their physicochemical properties revealed an interesting morphology change upon polymerization from monomers forming submicrometer spherical micelles to nanometer-sized rigid-rod-shaped polymeric particles. A combination of light-scattering methods, small-angle neutron scattering, and transmission electron microscopy enabled a detailed investigation of this morphological change. Moreover, we investigated by dynamic and static light scattering how the pH affects the fluorescence and size of the biodynamers. These morphological and pH-dependent changes are expected to open the door to a myriad of applications of molecular biodynamers.
Molecular biodynamers combine the advantages of biopolymers and dynamic covalent chemistry.1 Monomers are connected through covalent bonds that are reversible under specific conditions such as different pH and temperature. The resulting materials are stimuli-responsive systems and self-healing.2,3 Therefore, the polymerization and degradation of the molecular biodynamers are reversible depending on conditions. Moreover, when the biodynamers mimic biological biopolymers such as nucleotides, peptides, and polysaccharides, these molecular biodynamers are expected to feature advantages inherent to biopolymers (biocompatibility, biodegradability, and biofunctionality).4,5
In a previous study, we reported a biodynamer composed of carbazole dicarboxaldehyde 1 and lysine hydrazide 2 (Fig. 1A).6 Acylhydrazone and imine bond formation between hexaethylene glycol conjugated carbazole dicarboxaldehyde and lysine hydrazide, both of which are water-soluble, afforded proteoid biodynamers at slightly acidic conditions. Interestingly, the resulting biodynamers form rigid nanorods in aqueous solution due to numerous π–π-stacking interactions. A number of features of each monomer make this biodynamer particularly attractive for various applications. The carbazole is a fluorophore, thus, its derivatives are widely used for bioimaging and sensors.7,8 The other monomer L-lysine hydrazide, is a derivative of an amino acid, which is used as a building block for pharmaceutical ingredients, as food additive, and cosmetics. Especially, polymerized L-lysines (poly-L-lysine) are versatile biomedical materials and are widely used for tissue culture and drug delivery.9 In this study, we have evaluated the physicochemical properties of the biodynamers, and examined pH-dependent changes in view of further application of the polymer.
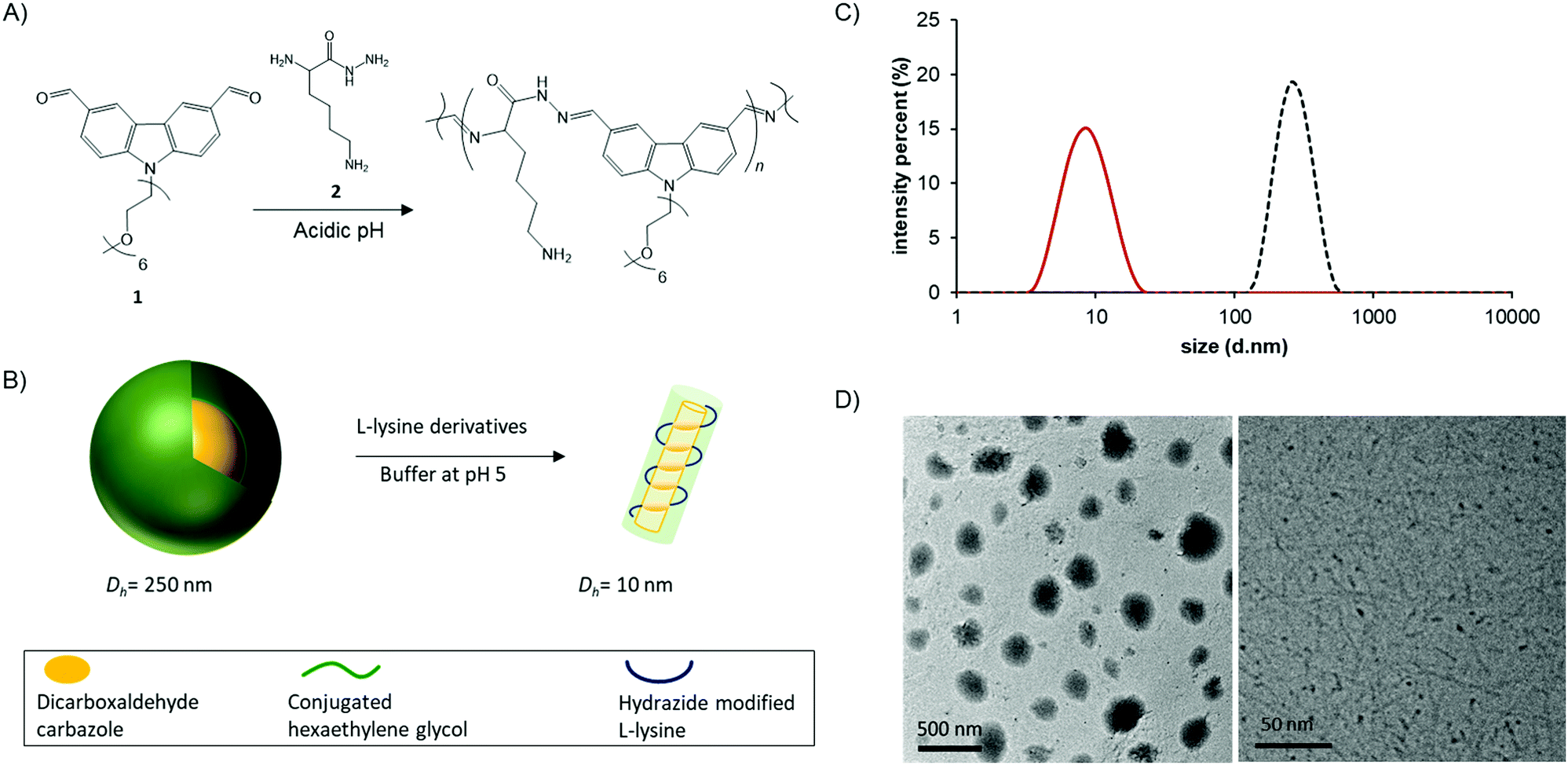 | ||
| Fig. 1 Chemical structure of the monomers 1 and 2 and the corresponding molecular biodynamer (A) and illustration of carbazole-based dialdehyde 1 micelles (Dh = 250 nm) transferring to biodynamer (Dh = 10 nm) (B). Size of the carbazole micelle (dotted line, Zave = 255 nm, polydispersity index (PDI) = 0.077) and biodynamers (solid line, Zave = 8 nm, PDI = 0.099) obtained by DLS (C). TEM image of carbazole-based dialdehyde 1 micelles (D, left) and cryo-TEM image of biodynamers (D, right). | ||
We set out to investigate the morphological and optical changes upon polymerization. To do so, we synthesized each monomer following the reported methods (Fig. S1 and S2, ESI†).10,11 Each of the monomers was dissolved in 100 mM acetate buffer with a final concentration of 10 mM for biodynamer synthesis. After incubation at r.t. for 19 hours, we observed successful polymerization confirmed by following the disappearance of the aldehyde protons by 1H-NMR spectroscopy (Fig. S2, ESI†). After filtration, we monitored the chemical, optical, and morphological changes.
Using dynamic light scattering (DLS) and transmission electron microscopy (TEM), we examined the size and morphology changes upon polymerization (Fig. 1B). The dialdehyde monomer 1 itself formed spherical micelles (Dh = 255 nm; polydispersity index (PDI): 0.077) (Fig. 1C) in aqueous solution (Fig. 1D, right). This micelle formation via self-assembly is driven by hydrophobic and π–π-stacking interactions between the monomers and stabilized in the aqueous solution by the hydrophilic shells of the hexaethylene glycol substituents. Upon addition of hydrazide 2, the nanoparticle size decreased to 8 nm (Dh, PDI: 0.099) (Fig. 1C) as a result of the polymerization, affording biodynamers, in line with the nanorods we had previously observed. In the cryo-TEM image of the biodynamers (Fig. 1D, right), the polymer has a rod-like shape, similar to reported supramolecular polymers.12 The shown morphology also corresponds well with our previously reported biodynamers.6,13 Note that, the darker parts of the nanorods in the images are the results of the increased density due to the vertically oriented nanorods.14,15 However, the cryo-TEM image gives limited information on its morphology. Therefore, the morphology, such as the shape of the section, the stacking, and the local structures were further investigated.
Solution-state polymer morphologies were investigated in depth using complementary small-angle neutron (SANS, beamline D11 at ILL)16 and static light scattering (SLS) experiments. A significant advantage of this combination is its ability to provide information on characteristic sizes, local structure and overall shape of objects in solution across a broad range of length scales varying between ∼1 and 300 nm. Fig. 2 shows the variation of the product MWP(q) as a function of the scattering vector q for a 10 mg mL−1 solution at pD 7.4, where MW and P(q) are respectively the weight-averaged molecular weight and the form factor of the objects. The SLS and SANS scattered intensities are here normalized by their respective contrast terms and concentration, allowing a representation of MWP(q) without an arbitrary shift to scale SLS data on SANS data (see Methods in ESI†). The formation of cylindrical structures identified using cryo-TEM is in agreement with the overall behavior observed here. The following sequence describes the behavior: (i) a low-q plateau in the SLS q-range associated with the finite mass of the polymers and allowing a direct determination of MW by extrapolation to zero-q, followed by (ii) a Guinier regime associated to the finite size (radius of gyration Rg) of the objects, (iii) a small intermediate rod-like regime, in which the variation of the signal can be described by a power-law with an exponent close to −1, (iv) another Guinier regime at the upper q-range, corresponding to the cross-section of the cylinders, and finally (v) an oscillation associated with the shape-dependent form factor of the section of the cylinders.
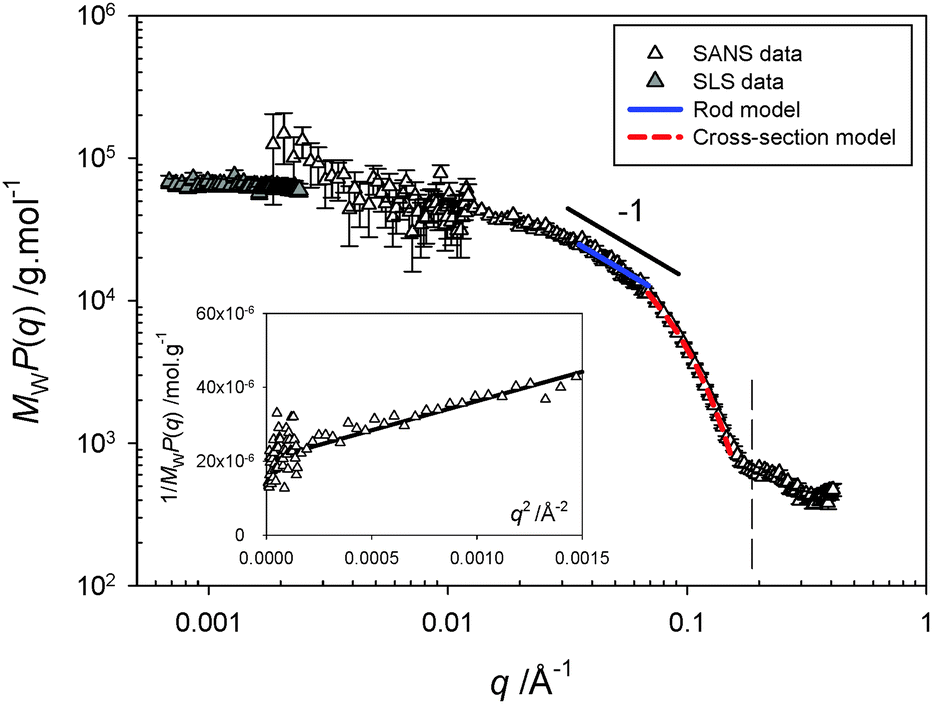 | ||
| Fig. 2 Scattering profile obtained from combined static light (SLS) and small-angle neutron scattering (SANS) experiments for a 10 mg mL−1 solution at pD 7.4. The product of the weight-averaged molecular weight and form factor, MWP(q), is obtained by normalization of the scattered intensity by concentration and contrast term of the technique (see ESI†), allowing a rescaling of the SLS and SANS data without an arbitrary shift. The continuous and dashed lines represent the best fit of the data in the intermediate q−1 regime by a rod model and in the high-q range by a Guinier expression for the form factor of the cross-section, respectively. The inset represents the variation of 1/MWP(q) with q2 in the low-q SANS domain. | ||
The data obtained at low q corresponding to large spatial scales have been fitted by a classical Guinier law (see eqn (S5) in ESI†). From the best linear fit to the curve representing 1/MWP(q) versus q2 (see inset of Fig. 2), one obtains a radius of gyration equal to Rg = 54.4 Å giving a relatively short contour length of L = √12 × Rg = 188 Å for the rods and explaining the narrowness of the q−1 regime. In the intermediate q regime, the scattering profile can be fitted satisfactorily using a rigid-rod model using the form factor derived for rod-shaped particles, P(q) = π/qL. The high q data can be fitted by a Guinier expression for the form factor of the section, VpP(q) = πac/q × exp(−q2rc2/2). By fitting the two equations above to the experimental data, one can determine the linear mass density (μ), the cross-sectional area (ac), and the Rg of the section of the cylinders (rc). From the fits of Fig. 2, we obtain μ = 279.4 g mol−1 Å−1, ac = 444 Å2, and rc = 14 Å. The contour length of the cylinders can also be obtained from the value of MW and μ: L = MW/μ = 253 Å, in a relatively good agreement with that determined directly from the Rg.
The cylindrical morphology is also confirmed by the so-called ρ ratio derived from the combination of SANS and DLS. The correlation function characterized by a single diffuse relaxation (i.e., single population, see ESI,† Fig. S3) allows determination of the hydrodynamic radius Rh of the objects. Therefore, the ratio showing a value, ρ = Rg/Rh = 1.52, larger than one is a characteristic of elongated objects such as cylinders. While Rg, Rh, and L give information on the overall structure of the cylinders, the experimental values of μ, ac, and rc provide important indications on the local particle topology and molecular stacking. Assuming a full disk-shaped cross-section with ac = πR2 leads to conflicting results as one obtains a geometrical radius R = 11.9 Å for the disk smaller than rc = 14 Å. A possible explanation is the formation of a core–shell structure with a cross-sectional area given by ac = π(Rext2 − Rint2) and rc = √2/2 × √(Rext2 + Rint2), where Rext and Rint are the external and internal radii of the section of the cylinders, respectively. Using the experimental values for ac and rc, one finds Rext = 16.3 Å and Rint = 11.2 Å; the thickness of such a shell section being equal to Rext − Rint = 5.1 Å. Such a structure is compatible with the stacking of the carbazole moieties in the core and hexaethylene glycol as a shell in agreement with the linear mass density value of 279.4 g mol−1 Å−1.
Lastly, we observed optical changes of the biodynamers by polymerization. The carbazole monomer 1 has shown absorption and emission at λabs/λem = 350/470 nm (Fig. S5, ESI†). By adding the lysine monomers, the absorption around 425 nm increased. Due to the changes in absorption, the observed color of the polymer solution changed from yellow to orange over 24 hours (Fig. 3A). Besides, the intensity of the emission spectrum of the polymeric solution decreased in the first 4 hours of polymerization. The decrease is presumably ascribed to a substantially reduced distance between the carbazoles upon polymerization, resulting in quenching. However, after 10 hours of polymerization, the emission intensity slightly increased. A possible explanation is that the initially randomly stacked and quenched carbazole moieties reorganized during nanorod formation. This rearrangement and nanorod formation possibly increase the distance between the carbazole moieties by cation-π interaction between the carbazole and lysine and by steric repulsion of hexaethylene glycols, both leading to a slight recovery of the quenched fluorescence.
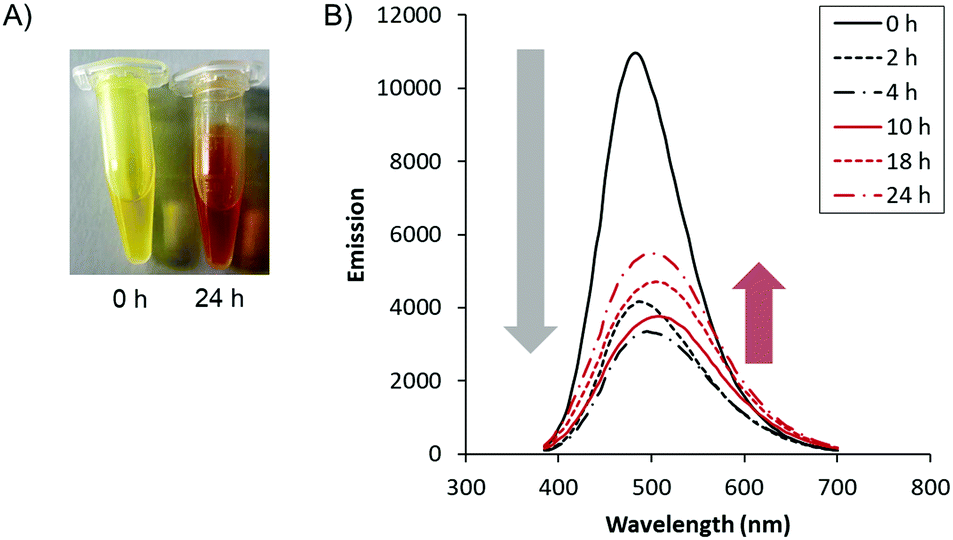 | ||
| Fig. 3 Optical changes by molecular biodynamer formation. (A) The colorimetric changes of monomer mixture by reaction time (left: 0 hours, right: 24 hours at r.t.) (B) emission changes as a function of reaction time. The peak top shifted from 485 nm to 505 nm. | ||
Having confirmed the polymerization, we evaluated the pH effect on the optical properties of the polymer. We prepared the polymer solution (1 mg mL−1) at pH 3 to 13 (pH 3, 5, 6, 7.4, 9, 11, 13) and measured the absorption and emission of the polymeric solutions at different pH values (Fig. 4A). As shown in the spectra, λmax of the emissions shifts from 470 nm at pH 3 to 535 nm at pH 13 (Fig. 4B), corresponding to a gradual redshift as the pH increase. Interestingly, each of the emission peaks shifts by a ratio change between two different emission peaks of λmax = 470 nm and λmax = 535 nm. It is more obvious at pH 9 and 11 that the peaks have two peak tops composed of two emission peaks. It suggests that a ratio change of the two components in the polymer affects the fluorescence. Considering the peak shifts are pH-dependent, the proton concentration affects the fluorophores, i.e., the carbazole groups, in the polymer. We speculate that protonation of the tertiary amine of the carbazole induced the emission peak shifts. The protonation of the tertiary amine shortens the conjugated system of the fluorophore. On the other hand, the unprotonated form has a longer conjugated system, resulting in an increased redshift. Another explanation is that pH-dependent protonation on the lysine side chains affects the electron conjugation on the fluorophore by photo-induced electron transfer (PET). However, even if the fluorophore can behave either as an electron acceptor (a-PET) or a donor (d-PET) towards the side chains, the PET affects the emission intensity by pH but not the emission shifts. Moreover, considering the pKa of the L-lysine side chain, the gradual peak shift between pH 3 to 13 is not explainable by side chain protonation. (Note that, the pKa value of the L-lysine side chain is 10.53). Therefore, we believe that the blueshift under acidic conditions is a result of protonation on the backbone.
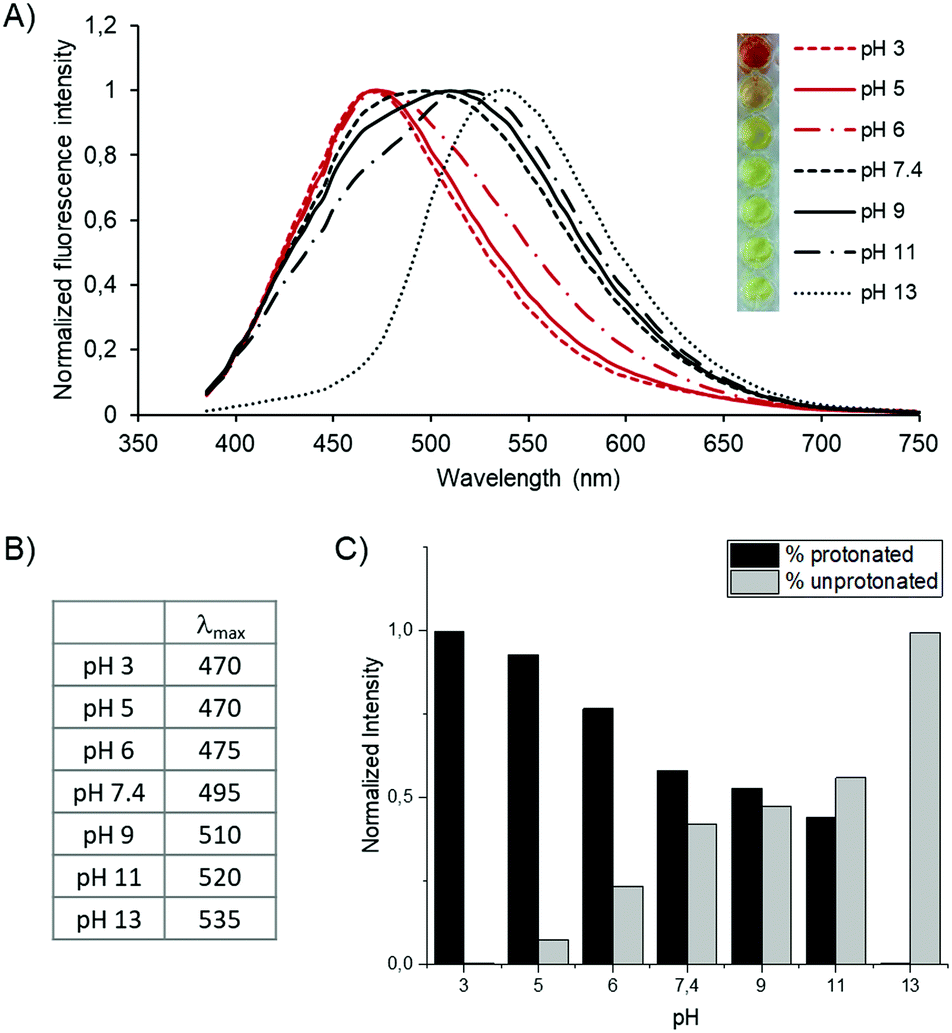 | ||
| Fig. 4 pH-dependent optical changes (A) emission shift of the biodynamers as a function of pH. The concentration of the polymer is set to 100 μg mL−1. 100 mM Acetate solution was used for pH 3, and pH 5. 100 mM Phosphate solution was used for pH 7.4 to pH 13. The colors of each solution are shown on the left. (B) Table of the λmax obtained from the emission spectrum. (C) The intensity ratio of two fluorescence components at various pH. The ratio is calculated based on the fitting process using two model curves. | ||
To evaluate the ratio of these two components (protonated/unprotonated) at different pH, we calculated it with a fluorescence peak deconvolution method (Fig. 4C). The two spectra at pH 3 and 13 are utilized as model spectra of the protonated and deprotonated states of the molecule. Each spectrum was composed of two Gaussian curves to fit the curves precisely. The height and width ratio of the Gaussian functions of the model spectrum was fixed to keep the shape of each basic spectrum. By a linear combination of the two model spectra of pH 3 and 13, the protonation ratio at other pH conditions was calculated from each spectrum. Consequently, we revealed clearly that the emission spectrum between pH 5 and 11 is composed of the summation of the two primary signals. The ratio of unprotonated carbazole gradually increases when increasing the pH and eventually is reversed between pH 9 and 11. Note that we obtained the ratio based on the assumption that the emission peak at pH 3 is purely protonated, and it is an entirely unprotonated spectrum at pH 13. Therefore, further investigation is required to quantify absolute protonation values.
We expected that this pH-dependent protonation also affects the size of the polymeric nanorods. To verify it, we measured the size of the polymer at pH 3, 5, 7.4, and 13 by DLS using the cumulant method, based on the backscattering at 173° (Fig. 5A and B). As a result, the size of the nanorods was larger in a more acidic environment. The size of the nanorods at pH 3 was 14.5 nm, while it was 8.8 nm at pH 13. This means that the hydrodynamic radius of the biodynamer increased by up to 65% upon increasing the proton concentration 1010 times. However, measurement using a single scattering angle may not be sufficient to support these changes and also does not allow a mass determination.
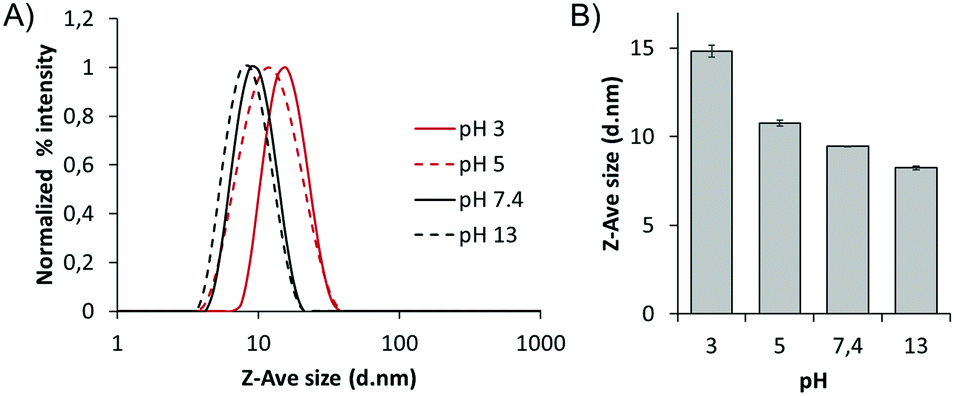 | ||
| Fig. 5 pH-dependent size changes (A) normalized percent intensity of the size of the biodynamers (1 mg mL−1) as a function of pH. (B) Zeta average (Z-Ave) size difference of the biodynamer (1 mg mL−1) at pH 3, 5, 7.4, and 13. Each size value is the mean value of DLS measurements performed in triplicate. | ||
Therefore, we have also confirmed the pH-dependent size and mass differences using combined DLS and SLS with scattering angles from 30° to 140°. From the SLS data, the MW of the polymer has shown a negligible difference between pH 5 and pH 7.4 (Table 1). At pH 5, the measured molecular weight of the polymer is 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 939 g mol−1, and the polymerization degree derived from the MW is about 109. At pH 7.4, the MW is 66552 g mol−1, and the degree of polymerization is 106, which is not significantly different compared to the values obtained at pH 5. In both of the calculation methods, however, the size of the polymer at pH 5 is 35% (by the Contin method, and 31% by the cumulant method) larger, indicating a change within the structure and the compactness of the objects. In other words, the nanorod size changed as a function of pH despite the molecular weight remaining consistent. A possible explanation is that the increment in electrostatic repulsion upon protonation on the polymer backbone affects the packing of the nanostructure.
939 g mol−1, and the polymerization degree derived from the MW is about 109. At pH 7.4, the MW is 66552 g mol−1, and the degree of polymerization is 106, which is not significantly different compared to the values obtained at pH 5. In both of the calculation methods, however, the size of the polymer at pH 5 is 35% (by the Contin method, and 31% by the cumulant method) larger, indicating a change within the structure and the compactness of the objects. In other words, the nanorod size changed as a function of pH despite the molecular weight remaining consistent. A possible explanation is that the increment in electrostatic repulsion upon protonation on the polymer backbone affects the packing of the nanostructure.
| Buffer acidity | c | ϕ | M W | DP | R g | R h a | R h b | k 2/k12 | ρ = Rg/Rh | r c | a c | μ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c = polymer concentration (mg mL−1), ϕ = c/d volume fraction (monomer density d = 1.46 g cm−3), MW = weight-averaged molecular weight (g mol−1), DP = degree of polymerization, Rg = radius of gyration (Å), Rh = hydrodynamic radius obtained using the Contina and the cumulantb analysis (Å), k2/k12 = polydispersity index, rc = cross-sectional radius of gyration (Å), ac = cross-sectional area (Å2), μ = linear mass density of cylinders (g mol−1 Å−1). (SLS, DLS and SANS measurements determined the values with ∼10% error, see ESI.) | ||||||||||||
| pD 7.4 | 10 | 6.83 × 10−3 | 70570 |
113 | 54.4 | 39.4 | 35.7 | 0.095 | 1.52 | 14 | 444 | 279.4 |
| pH 7.4 | 1 | 6.83 × 10−4 | 66552 |
106 | — | 39.8 | 32.1 | 0.1153 | — | — | — | — |
| pH 5 | 1 | 6.83 × 10−4 | 67939 |
109 | — | 61.5 | 47 | 0.1434 | — | — | — | — |
In this study, we have investigated molecular biodynamers composed of carbazole and lysine derivatives. We evaluated morphological, optical-property, and size changes of the biodynamers upon polymerization under acidic conditions. The spherical micelles of the carbazole monomer transformed into rigid nanorods in the course of the polymerization, as shown through a thorough analysis by SANS and SLS. pH-dependent changes in fluorescence emission and size of the biodynamers are expected to broaden the further application of the molecular biodynamers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors deeply acknowledge the Institut Laue Langevin (ILL Grenoble, France) for beamtime allocation as well as G. Mariani and S. Prevost for their precious help during the SANS experiments, and Jörg Schmauch at Saarland University, Experimentalphysik INM-Leibniz-Institute for New Materials for the TEM experiments. Funding from the Helmholtz Association's Initiative and Networking Fund (A. K. H. H.) and the SFB937 Collective behavior of soft and biological matter (A11, H. J.) is gratefully acknowledged. Dedicated to J.-M. Lehn on the occasion of his 80th birthday.References
- E. Kolomiets and J. M. Lehn, Double dynamers: molecular and supramolecular double dynamic polymers, Chem. Commun., 2005, 1519–1521 RSC.
- H. Ying, Y. Zhang and J. Cheng, Dynamic urea bond for the design of reversible and self-healing polymers, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- F. Garcia and M. M. Smulders, Dynamic covalent polymers, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(22), 3551–3577 CrossRef CAS PubMed.
- Y. Liu, M. C. A. Stuart, E. Buhler and A. K. H. Hirsch, Dynamic Proteoids Generated From Dipeptide-Based Monomers, Macromol. Rapid Commun., 2018, 39(13), 1800099 CrossRef PubMed.
- Y. Liu, M. C. A. Stuart, M. D. Witte, E. Buhler and A. K. H. Hirsch, Saccharide-Containing Dynamic Proteoids, Chem. – Eur. J., 2017, 23(64), 16162–16166 CrossRef CAS PubMed.
- Y. Liu, M. C. A. Stuart, E. Buhler, J.-M. Lehn and A. K. H. Hirsch, Proteoid Dynamers with Tunable Properties, Adv. Funct. Mater., 2016, 26(34), 6297–6305 CrossRef CAS.
- Y. Liu, J. M. Lehn and A. K. Hirsch, Molecular Biodynamers: Dynamic Covalent Analogues of Biopolymers, Acc. Chem. Res., 2017, 50(2), 376–386 CrossRef CAS PubMed.
- X. J. Feng, P. L. Wu, F. Bolze, H. W. C. Leung, K. F. Li, N. K. Mak, D. W. J. Kwong, J.-F. Nicoud, K. W. Cheah and M. S. Wong, Cyanines as New Fluorescent Probes for DNA Detection and Two-Photon Excited Bioimaging, Org. Lett., 2010, 12(10), 2194–2197 CrossRef CAS PubMed.
- I. L. Shih, Y. T. Van and M. H. Shen, Biomedical applications of chemically and microbiologically synthesized poly(glutamic acid) and poly(lysine), Mini-Rev. Med. Chem., 2004, 4(2), 179–188 CrossRef CAS PubMed.
- J. F. Folmer-Andersen, E. Buhler, S.-J. Candau, S. Joulie, M. Schmutz and J.-M. Lehn, Cooperative, bottom-up generation of rigid-rod nanostructures through dynamic polymer chemistry, Polym. Int., 2010, 59(11), 1477–1491 CrossRef CAS.
- D. A. Patrick, D. W. Boykin, W. D. Wilson, F. A. Tanious, J. Spychala, B. C. Bender, J. E. Hall, C. C. Dykstra, K. A. Ohemeng and R. R. Tidwell, Anti-Pneumocystis carinii pneumonia activity of dicationic carbazoles, Eur. J. Med. Chem., 1997, 32(10), 781–793 CrossRef CAS.
- P. Besenius, G. Portale, P. H. Bomans, H. M. Janssen, A. R. Palmans and E. W. Meijer, Controlling the growth and shape of chiral supramolecular polymers in water, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(42), 17888–17893 CrossRef CAS PubMed.
- A. K. Hirsch, E. Buhler and J. M. Lehn, Biodynamers: self-organization-driven formation of doubly dynamic proteoids, J. Am. Chem. Soc., 2012, 134(9), 4177–4183 CrossRef CAS PubMed.
- E. Buhler, S. J. Candau, J. Schmidt, Y. Talmon, E. Kolomiets and J.-M. Lehn, Fibrillar structure of self-assemblies formed from heterocomplementary monomers linked through sextuple hydrogen-bonding arrays, J. Polym. Sci., Part B: Polym. Phys., 2007, 45(1), 103–115 CrossRef CAS.
- Y. Ruff, E. Buhler, S. J. Candau, E. Kesselman, Y. Talmon and J. M. Lehn, Glycodynamers: dynamic polymers bearing oligosaccharides residues--generation, structure, physicochemical, component exchange, and lectin binding properties, J. Am. Chem. Soc., 2010, 132(8), 2573–2584 CrossRef CAS.
- G. Marinani, E. Buhler, J.-R. Colard-Itté, D. Dattler, N. Giuseppone, E. Moulin and S. Prevost, Effects of polymer molecular weight on the structure and on the efficiency of light-driven contractile gels, Institut Laue-Langevin (ILL), 2019, DOI:10.5291/ILL-DATA.9-11-1845.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00651f |
| This journal is © the Partner Organisations 2020 |