
Charge transfer in DHICA eumelanin-like oligomers: role of hydrogen bonds†
Arpan
Choudhury
and
Debashree
Ghosh
 *
*
School of Chemical Science, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, India. E-mail: pcdg@iacs.res.in
First published on 30th July 2020
Abstract
Eumelanin is a versatile bio-polymer with a heterogeneous structure and atypical functionalities. The constituent parts of eumelanin can be used to engineer systems with spectacular properties. We will describe some design principles to accentuate the charge transfer properties in eumelanin, especially those containing dihydroxyindole carboxylic acid (DHICA). We will show that the oligomers of DHICA can form isomers (atropisomers) of almost equivalent stability and the difference in charge transfer properties between these isomers is strongly dependent on the presence of inter-monomer hydrogen bonds.
Eumelanin is the brownish-black pigment present in human skin, as well as other plant and animal species.1 It is best known for its photo-protective properties and is characterized by a broadband monotonic spectrum spanning most of the UV and visible part of the spectrum. Much of the research to date has been involved in understanding the origin of this atypical spectrum and its subsequent photo-protection properties.2–4
However, of late, research has been focused on its spectacular charge and energy transfer properties.5–7 Several studies based on EPR techniques have identified the importance of protons in the electrical conductivity of eumelanin and the role of free radicals.8,9 There are efforts towards leveraging the atypical properties of eumelanin molecules to engineer desired functionalities. Semiconductors and supercapacitors based on eumelanin building blocks have been attempted10 and the charge transfer properties of certain types of eumelanin have been studied both experimentally and theoretically.11 Here, it is pertinent to mention that eumelanin is a heterogeneous structure, made up of poly-disperse oligomers of dihydroxyindole (DHI) and dihydroxyindole carboxylic acid (DHICA).12 There are 3 sites of polymerization in DHI (as shown in Fig. 1a). However, in the case of DHICA, one of the sites is blocked by the carboxylic acid substitution. Furthermore, due to the electron withdrawing nature of the carboxylic acid substitution, the two sites in DHICA are more effective as polymerization sites (as shown in Fig. 1b).
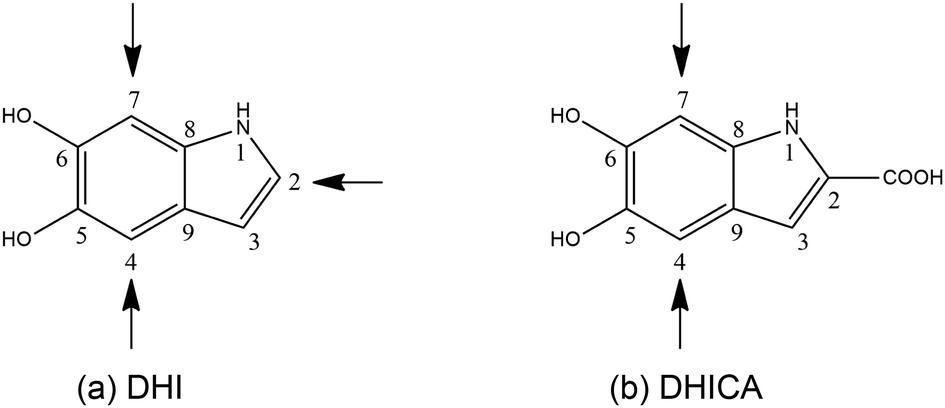 | ||
| Fig. 1 The constituent monomers of eumelanin with the positions of polymerization marked by arrows. | ||
The heterogeneity in the eumelanin structure is central to the structure–function relationship of eumelanin.13,14 Despite the extreme heterogeneity in natural eumelanin, the polymers of DHI and DHICA are increasingly being used to form synthetic eumelanin-like oligomers with desired properties. Since the DHICA moiety has two sites of polymerization, oligomers of DHICA are expected to form linear chains, which have extreme dipole moments. Within the generalized Mulliken–Hush theory, the charge transfer coupling between the monomers can be defined as,
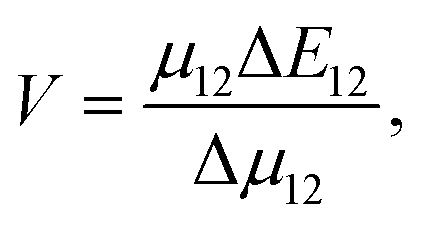 | (1) |
It is pertinent to mention that DNA bases and their different sequences have been extensively studied as efficient conducting species of biological origin. There is considerable controversy about the nature of these species, ranging from insulators to conductors to superconductors. The mechanism of charge transfer in DNA has also been intensely researched.15–18
In the case of DHICA-oligomers, the distances between the monomers are far lower than in DNA, and therefore, orbital overlap can potentially play a role. However, we have found that due to the inter-monomer angles and the resultant non-planarity in the structure, it is mainly the hydrogen bond network that seems to be of importance. This is in line with the experimental observations of proton mediated charge transfer in eumelanin.
Studies have already shown that the DHICA-oligomers in eumelanin form linear chains with the inter-monomer angles of about 40–50°.19,20 The presence of atropisomerism due to hindered rotation between these monomers is well known. The relative importance of these atropisomers is evaluated at the ωB97x-D/6-31G* level of theory and is comparable to the MP2/6-31G* level of theory (included in the ESI†).
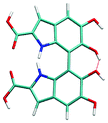
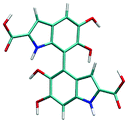
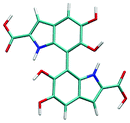
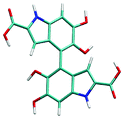
Atropisomerism forms the cornerstone of relative abundance between the possible dimers of DHICA. There are 3 possible isomers, solely from the interconnection patterns. We will call these 44′, 77′ and 47′ isomers (shown in Table 1). However, due to hindered rotation there are cis and trans like conformations for each of these isomers. The relative energies of these isomers (Table 1) show that the most stable 3 isomers are the cis forms. This is somewhat expected given the strong inter- and intra-molecular H-bonds between the phenolic OH groups. However, what is surprising is the higher than expected stability of the trans isomers, where these H-bonds are not possible. The reasons for this stability are the favorable interactions between the NH group and phenolic OH groups in the trans conformation. H-Bonds between these moieties are noticed in the trans configurations. Therefore, at room temperature, the possibility of these conformers should not be underestimated. Dipole moments of these isomers show that the cis isomers have a higher dipole moment than the trans isomers. The dipole moments of all the cis isomers are somewhat comparable. Another important point to note is that the dipole moment on each monomer depends on the directionality of the phenolic OH bonds and not along the long axis of the molecule. Therefore, one would expect correlation between the dipole moment of the oligomers and their charge transfer properties.
| Species | Rel. energy (kcal mol−1) | Dipole (Debye) | |
|---|---|---|---|
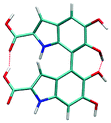
|
4–7′ cis | 0 | 7.57 |
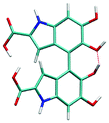
|
4–4′ cis | 0.63 | 8.54 |
|
7–7′ cis | 0.97 | 7.78 |
|
4–7′ trans | 4.01 | 3.98 |
|
7–7′ trans | 4.85 | 2.12 |
|
4–4′ trans | 8.40 | 4.08 |
Apart from the atropisomers with hindered inter-monomer rotations shown in the table, there can be conformers with rotated phenolic OH bonds, where the inter-molecular H-bonding is absent. These conformers are 2–3 kcal mol−1 higher in energy than the more stable forms (with inter-molecular H-bonds). This OH rotation is facile and is expected to occur at room temperature, as also noticed from the molecular dynamics simulations of the oligomers.
Since the atropisomers are connected to each other via hindered rotations, we calculate the energetics of these rotations and evaluate the properties of the conformations along the axis of rotation. Fig. 2 shows the energies and dipole moments of the three DHICA isomers with respect to the inter-monomer rotation angles. The hindered rotation shows the activation energy barriers of 4–5 kcal mol−1 for the 47′, 44′ and 77′ isomers. The minima of the energies (i.e., the most probable structure) have an angle of 40–50° (cis conformer) or 125° (trans conformer). Thus, the structures are significantly away from planarity. The fully planar structures (cis and trans) are extremely high in energy and do not exist at room temperature. This lack of planarity in the most stable forms of DHICA oligomers has been noted both experimentally and theoretically. The dipole moments follow the opposite pattern, i.e., the lowest energy cis structures have the highest dipole moments and these dipole moments decrease with rotation. The trans structures have low dipole moments.
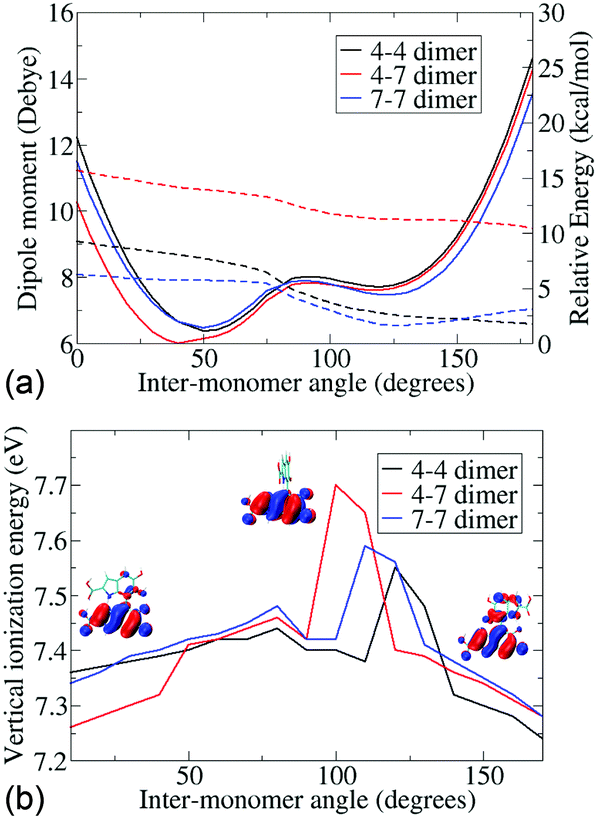 | ||
| Fig. 2 (a) The relative energies, in kcal mol−1 (marked with solid lines), and dipole moments, in Debye (marked with dashes lines) of the 3 dimers with respect to the inter-monomer angles. (b) The vertical ionization energies (in eV) of the 3 isomers with respect to inter-monomer angles. The orbital from which ionization occurs is shown at 0°, 120° and 170°. | ||
The lowest vertical ionization energies (VIEs) of the isomers fall between 7.3 and 7.5 eV and are close to the lowest VIE of the monomer. This is due to the non-planarity between the monomers, which localizes the positive charge on one or the other monomer, as corroborated from the highest occupied molecular orbital (HOMO). Thus, an orbital overlap mediated charge transfer process is unlikely in these dimers. Despite this, a very high charge transfer coupling is noticed in these dimers (see Table 2). Charge transfer coupling matrix elements in the diabatic basis have been calculated within a constrained DFT configuration interaction (CDFT-CI) framework.21 This is the coupling between a charge separated state and a charge neutral state, and therefore, shows the propensity of charge transfer between the monomers. We notice that apart from the 4–4′ trans dimer, all the others have a very similar coupling element. From the structural information, we further notice that apart from the 4–4′ trans dimer all other dimers have at least one inter-monomer H-bond.
To corroborate this hypothesis, the coupling elements are calculated for some modified dimers without H-bonds (shown in the ESI,† Tables S8 and S9). It is important to note that theoretical work has previously alluded to the importance of H-bonding in the charge transfer of DHICA dimers.20 However, their arguments were based predominantly on the basis of dipole moments and H-bonds between the phenolic OH groups. By this hypothesis, the trans isomers would be the least favorable in the charge transfer processes. However, what we notice is that the trans isomers can be as efficient at charge transfer (or charge separation) as the cis isomers and even more efficient at times. We believe that this trans isomer charge transfer proceeds via the NH–OH H-bonding channels.
Moreover, it is noteworthy that the phenolic OH can undergo facile rotations which can make or break these H-bond networks. Therefore, we constructed rotated phenolic OH configurations and we again found that the absence of H-bond network leads to a remarkable decrease in the charge transfer coupling elements.
The knowledge amassed from the dimer studies shows that the predominant mode of charge transfer is connected with the H-bonded network of the phenolic OH groups and proceeds via a hopping mechanism. Furthermore, the frontier orbitals of the dimers are strongly localized on different monomers, mainly due to the non-planarity in the structures and ionization processes are, therefore, expected to be predominantly on a single monomer. This is further corroborated by the small change in the ionization energies noticed with change in the inter-monomer angles.
However, eumelanin chemistry involves other aspects of heterogeneity, such as the oxidation states. The dihydroxy indole can also exist in its oxidized forms, the mono-keto indole (MKI) and di-keto indole (DKI). Oxidized DHICA counterparts are, therefore, called MKICA and DKICA, respectively. The frontier orbitals of the mono-keto and di-keto forms are significantly different from that of the dihydroxy indole form. Therefore, we expect different electronic properties for these moieties. The VIEs of MKI and DKI are higher than DHI.22 Therefore, the VIEs of the heterodimers are somewhat higher than those of DHICA dimers (given in the ESI†). However, the charge localization is quite different between the hetero and homo-dimers. The hole is predominantly localized on the DHICA moiety, as expected from the relative HOMO energies. From a similar argument about the LUMO energies, we expect excess electrons to be localized on the oxidized MKICA and DKICA monomers. Furthermore, calculation of the coupling matrix elements shows a remarkable lowering of charge transfer coupling and, therefore, charge mobility around the oxidized forms. Thus, one can expect electron and hole trapping to occur near the sites with these oxidized monomers.
Trimer and tetramer structures also show similar trends of stability to the dimers. The cis structures are typically more stable (the most stable trimer and tetramer structures are included in the ESI†), due to the presence of multiple H-bonds and favorable interactions. They have an angle of 40–50° between adjacent monomers. However, there are a few oligomers with adjacent trans monomers that are within 4–6 kcal mol−1 of the most stable oligomer. This is stabilized due to favorable OH–NH interactions. The ionization energies of the trimers are remarkably similar to those of the dimers due to localized ionization. Therefore, one can expect similar charge transfer mechanisms and efficiency in these higher oligomers.
Due to persistently similar trends in stability between the dimers, trimers and tetramers, higher oligomers – pentamers and octamers, are constructed in their most stable conformers, i.e., cis forms. It is important to note that for higher oligomers there are two cis forms that can be constructed, the zigzag (with the inter-monomer angles alternating at +50° and −50°) and spiral structures (with an inter-monomer angle of −50°). The energies and electronic properties as deduced from the frontier orbitals are very similar. For the pentamers and octamers both these forms are constructed for further dynamical studies. The similarity in the distribution of the inter-monomer angles between the two forms also shows the similarity of the dynamical features of these oligomers. Fig. 3a shows the distribution of the inter-monomer angles in a spiral octamer (the same distribution for the zigzag octamer is included in the ESI† for comparison). We notice that along the trajectory, the oligomers show a symmetrical motion around its minimum energy configuration. Fig. 3b shows the number of H-bonds present along the trajectory of both zigzag and spiral octamers. We notice that there are on average 9.25 and 8.97 H-bonds present throughout the trajectory for the two isomers. It, therefore, shows that on average there are inter-monomer H-bonds that are present between all adjacent monomers. Due to the importance of H-bonding in the charge transfer process along these DHICA strands and the large number of H-bonds present along the trajectory, we expect these oligomers to have high electrical conductivity.
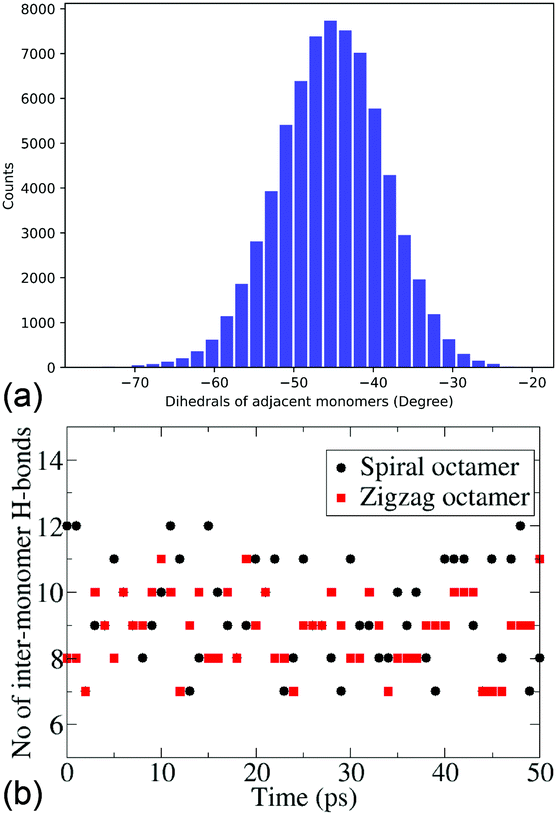 | ||
| Fig. 3 (a) The distribution of inter-monomer angles in a spiral octamer structure. (b) The number of inter-monomer H-bonds in spiral and zigzag octamers along the trajectory. The average number of H-bonds is 9.25 and 8.97, respectively. | ||
To summarize, due to the non-planarity between monomers and the lack of overlap between the orbitals residing on the monomers, the charge transfer processes mediated by orbital overlap are not expected in these molecules. On the other hand, the correlation between inter-monomer H-bonds and higher charge transfer coupling matrices shows the importance of H-bonds as conduits for charge transfer. Therefore, a hopping mechanism via the H-bonding networks is expected to play a major role. Such importance of H-bonding in electron and hole transfer has been noticed in other bio-systems, such as in blue copper proteins.23,24 We notice no clear correlation between charge transfer and molecular dipole moments, especially since the lower dipole trans structures can have favorable H-bond pathways for charge transport. Since the H-bonds between monomers are found to be the driving force behind the charge transfer process, a simple screening protocol for such molecules can be derived. The inter-monomer H-bond interaction energies can be calculated from a simple point charge model and can be used as a screening tool to identify highly conducting molecules. The importance of dynamics should be appreciated from the change in the number of H-bonds along trajectories. This is also expected to be dependent on the temperature of the system.
Due to the importance of H-bonding networks, we analyze the structures and ionization energies of larger oligomers. We notice that in most room temperature structures, the presence of H-bonds is almost ubiquitous, and therefore, we expect the most stable forms of DHICA oligomers to be efficient at charge transfer across their backbones. Furthermore, the presence of oxidized forms gives rise to significant changes in local ionization energies and might act as either barriers to charge transfer pathways or as sites for electron and hole recombination.
We would like to thank DST-SERB for funding (EMR/2017/001054) and IACS for computational facilities. A. C. thanks DST-INSPIRE for his junior research fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Prota, Melanins and Melanogenesis, Academic Press, San Diego, 2012 Search PubMed.
- J.-P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef PubMed.
- F. Solano, Polym. Int., 2016, 65, 1276–1287 CrossRef.
- A. Huijser, A. Pezzella and V. Sundström, Phys. Chem. Chem. Phys., 2011, 13, 9119–9127 RSC.
- L. Migliaccio, P. Manini, D. Altamura, C. Giannini, P. Tassini, M. G. Maglione, C. Minarini and A. Pezzella, Front. Chem., 2019, 7, 162 CrossRef PubMed.
- M. d'Ischia, K. Wakamatsu, F. Cicoira, E. Di Mauro, J. Garcia-Borron, S. Commo, I. Galvan, G. Ghanem, K. Kenzo and P. Meredith, Pigm. Cell Melanoma Res., 2015, 28, 520–544 CrossRef PubMed.
- A. B. Mostert, B. J. Powell, F. L. Pratt, G. R. Hanson, T. Sarna, I. R. Gentle and P. Meredith, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8943–8947 CrossRef CAS PubMed.
- J. Wünsche, Y. Deng, P. Kumar, E. Di Mauro, E. Josberger, J. Sayago, A. Pezzella, F. Soavi, F. Cicoira, M. Rolandi and C. Santato, Chem. Mater., 2015, 27, 436–442 CrossRef.
- S. B. Rienecker, A. B. Mostert, G. Schenk, G. R. Hanson and P. Meredith, J. Phys. Chem. B, 2015, 119, 14994–15000 CrossRef CAS PubMed.
- P. Kumar, E. Di Mauro, S. Zhang, A. Pezzella, F. Soavi, C. Santato and F. Cicoira, J. Mater. Chem. C, 2016, 4, 9516–9525 RSC.
- J. Wunsche, F. Cicoira, C. Graeffe and C. Sanato, J. Mater. Chem. B, 2013, 1, 3836 RSC.
- K. Bochenek and E. Gudowska-Nowak, Chem. Phys. Lett., 2003, 373, 532–538 CrossRef CAS.
- C.-T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5(1), 1–10 Search PubMed.
- J. D. Simon, D. Peles, K. Wakamatsu and S. Ito, Pigm. Cell Melanoma Res., 2009, 22, 563–579 CrossRef CAS PubMed.
- F. C. Grozema, S. Tonzani, Y. A. Berlin, G. C. Schatz, L. D. A. Siebbeles and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 5157–5166 CrossRef CAS PubMed.
- M. Bixon, B. Giese, S. Wessely, T. Langenbacher, M. E. Michel-Beyerle and J. Jortner, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11713–11716 CrossRef CAS PubMed.
- E. M. Boon, A. L. Livingston, N. H. Chmiel, S. S. David and J. K. Barton, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12543–12547 CrossRef CAS PubMed.
- T. Kubař, P. B. Woiczikowski, G. Cuniberti and M. Elstner, J. Phys. Chem. B, 2008, 112, 7937–7947 CrossRef PubMed.
- R. Micillo, L. Panzella, K. Koike, G. Monfrecola, A. Napolitano and M. D'Ischia, Int. J. Mol. Sci., 2016, 17, 746 CrossRef PubMed.
- M. Matta, A. Pezzella and A. Troisi, J. Phys. Chem. Lett., 2020, 11, 1045 CrossRef CAS PubMed.
- Q. Wu and T. Van Voorhis, J. Chem. Phys., 2006, 125, 164105 CrossRef PubMed.
- M. Mandal, T. Das, B. K. Grewal and D. Ghosh, J. Phys. Chem. B, 2015, 119, 13288–13293 CrossRef CAS PubMed.
- J. S. Kretchmer, N. Boekelheide, J. J. Warren, J. R. Winkler, H. B. Gray and T. F. Miller, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(24), 6129–6134 CrossRef CAS PubMed.
- T. Cheng, D. Shen, S. Mallick, L. Cao, N. Patmore, H. L. Zhang, S. F. Zou, H. W. Chen, Y. Qin, Y. Y. Wu and C. Liu, Nat. Commun., 2019, 10, 1531 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc04702c |
| This journal is © The Royal Society of Chemistry 2020 |