
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Krypton-derivatization highlights O2-channeling in a four-electron reducing oxidase†
Sylvain
Engilberge‡
 a,
Tristan
Wagner‡
b,
Philippe
Carpentier
cd,
Eric
Girard
*e and
Seigo
Shima
*f
a,
Tristan
Wagner‡
b,
Philippe
Carpentier
cd,
Eric
Girard
*e and
Seigo
Shima
*f
aPaul Scherrer Institut, Forschungsstrasse 111, 5232 Villigen PSI, Switzerland
bMicrobial Metabolism Group, Max-Planck-Institut für Marine Mikrobiologie, Celsiusstraße 1, 28359, Bremen, Germany
cInstitut de Recherche Interdisciplinaire de Grenoble (IRIG), Laboratoire Chimie et Biologie des Métaux (LCBM), Université Grenoble Alpes, CNRS, CEA, Grenoble, France
dEuropean Synchrotron Radiation Facility (ESRF), Grenoble, France
eUniv. Grenoble Alpes, CEA, CNRS, IBS, F-38000 Grenoble, France. E-mail: eric.girard@ibs.fr
fMicrobial Protein Structure Group, Max Planck Institute for Terrestrial Microbiology, Karl-von-Frisch Straße 10, 35043 Marburg, Germany. E-mail: shima@mpi-marburg.mpg.de
First published on 3rd August 2020
Abstract
F420H2-oxidase (FprA) catalyses the four-electron reduction of O2 to 2H2O using the reduced form of F420 as electron donor. The hydrophobic O2-channel detected by Kr-derivatization and the concerted movement of a gating loop could contribute to prevent unwanted side-reaction between the catalytic intermediates and solvents, therefore preventing reactive oxygen species formation.
Electrochemical reactions require electro catalysts functioning as a cathode and anode.1 Metal-containing redox-enzymes are considered as blueprints to design mimic complexes for the development of efficient electro catalysts. For example, some oxidases (e.g. laccase) harbour cathode catalyst property by using O2 as an electron acceptor and the chemical compounds mimicking their active-site are used as electrocatalysts.2 For future application of bio-inspired electrochemical processes, we need to learn more from the chemical reaction, structure and function of different groups of O2-reducing enzymes. Flavodiiron proteins (FDPs) belong to an enzyme family harbouring a di-nuclear iron centre and flavin mononucleotide (FMN). FDPs catalyse four-electron reduction of O2 to form water.3,4 The active site of FDPs represents an excellent template to design O2 reducing catalysts because of its affinity towards O2, high-turnover and specific reaction path avoiding reactive oxygen species formation. The latter would damage the catalysts in the electrochemical systems.
FprA has been found in many anaerobic organisms (Fig. S1, ESI†), initially in Desulfovibrio gigas5 and later also in methanogens6,7 and clostridia.8,9 It has even been identified in the parasite Giardia intestinalis.10 Structural studies have been performed on these FprAs to understand the physiological electron donor for the reaction and highlight high fold similarity between homologous enzymes (Fig. S2, ESI†).7,9–12 In methanogenic archaea, the electron donor of the reaction (Fig. 1a) is reduced F420 (F420H2).6 F420 is a deazaflavin derivative, which functions as electron carrier. Affinity of FprA against O2 is 2 μM,6 which is lower than those of laccases (Km > 20 μM).13
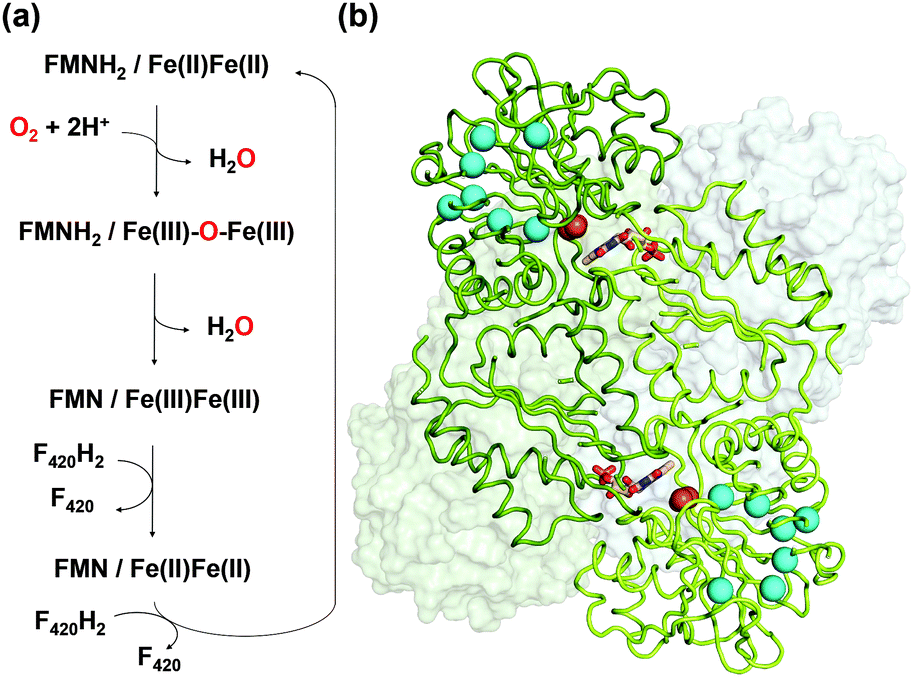 | ||
| Fig. 1 (a) Reaction scheme of FprA. The tFprAanox and tFprAoxo forms correspond to the redox state at Fe(II)Fe(II) and Fe(III)–O–Fe(III), respectively. (b) Quaternary structure of tFprA organized as a dimer of homodimer. A first homodimer is displayed in the back as white surface. The second one in front (green ribbon) highlights the di-iron centre as orange spheres and FMN in sticks. Kr atoms in the tFprAoxo-Kr structure after derivatization with 125 bars of krypton are depicted in cyan spheres. | ||
Crystal structure of FprA from Methanothermobacter marburgensis (mFprA) was solved in three states; active reduced state, active oxidized state and inactive oxidized state at the resolutions of 2.30 Å, 1.7 Å and 2.26 Å, respectively.7 Based on the different conformations observed, the importance of a switch-loop (residues 148–153 in mFprA) for the Fe coordination and possibly for F420 interaction has been highlighted. In the reduced state, the active site is closed and O2 could bind to the di-iron centre to be reduced by two electrons. During this event, the first water is formed and the oxidized di-iron site contains an oxygen bridging ligand; Fe(III)–O–Fe(III). Probably, the first water product is extruded by a transient opening of the catalytic cavity, although there is no experimental evidence. The next two electrons from FMNH2 reduce the bridging oxygen to form the second water molecule in the closed state. In this mechanism, no reactive oxygen species are produced7 as observed in the enzyme assay.6 To perform such catalytic reaction, the hydrophobic environment towards the di-iron is crucial for the electron transfer and proton donation in a concerted manner, which avoids disturbance caused by the solvent molecules and prevents any side-reaction leading to reactive oxygen species. The O2 supply into the closed active site pocket is however puzzling, since the catalytic chamber is not directly exposed to the solvent but somehow concealed inside the enzyme. Sequence alignments and computation predicted that there is an organized channel in FDPs (Fig. S1 and S3, ESI†).14 This hydrophobic channel would allow O2 transport from the bulk solvent to the active site. The experimental proof of the O2 channel must be established because properties of the channel is ascertained only by its exact geometry and composition.
Krypton (Kr) or Xenon (Xe) soaking are powerful methods to study hydrophobic channels in proteins.15,16 Successful examples have been reported on heme–copper oxidases17 and laccase.18 To investigate hydrophobic channels in FprA, we have chosen Kr which offers a smaller probe radius and an accessible X-ray absorption edge compared to Xe. We report here two crystal structures of FprA from Methanothermococcus thermolithotrophicus (tFprA) (Table S1, ESI†), a marine thermophilic methanogen belonging to Methanococcales, a distinct group from the terrestrial methanogen M. marburgensis, whose FprA was structurally analysed before.7 In our previous work, native tFprA structure was solved under aerobic condition with a crystallophore derivative.19 In contrast, the first crystal structure of this report presents tFprA (tFprAanox) was obtained in strict anaerobic conditions after native purification. The second structure was obtained aerobically in the presence of 125 bars of Kr and is refered to as tFprAoxo-Kr. As Kr derivatization requested several trials and an important number of crystals, tFprA was heterologously produced in Escherichia coli and crystallized in the presence of oxygen. Derivatization with Kr was performed using a “soak and freeze” methodology developed at the ESRF, Grenoble – France.16
FprA is composed of a dimer of homodimer. However, the functional unit is a homodimer; the catalytic pocket is formed at the dimeric interface, where the di-iron site located in the N-terminal β-lactamase-like domain is in front of the FMN, itself bound to the C-terminal flavodoxin-like domain side. tFprA structures present the same overall structure and composition as that described for mFprA. The crystal structure of tFprAanox obtained at 1.83 Å resolution is in an active reduced state comparable to that observed in mFprA structure (PDB code: 2OHI). tFprA and mFprA have a sequence identity of 70%. The tFprAoxo-Kr structure at 1.80 Å resolution is similar to the active oxidized state of mFprA (PDB code: 2OHH) (RMSD: 0.88 Å) and tFprAanox superposes well with mFprA in the active reduced state (PDB code: 2OHI) (RMSD: of 0.77 Å). The unusual non-prolyl cis-peptide bond between Leu150 and His151 is observed in both structure tFprAanox and tFprAoxo-Kr.
One main difference between the reduced and oxidized states is the loop 24 to 28 pointing from a β-hairpin in the β-lactamase-like domain which is unique in FDPs and covers the active site.20 This loop is observed in the closed position in both tFprA structures as observed in the active reduced state of mFprA (Fig. S4, ESI†).7 In the oxidised state (tFprAoxo-Kr), the loop 24–28 shows an average B-factor higher than in the reduced states (tFprAanox) suggesting a higher loop flexibility in the oxidized form although this feature is not observed in mFprA structures (Table S2, ESI†). These inconsistencies could be explained by differences of iron occupancy in mFprA structures. A close analysis of the variation of the loop 24–28 unravelled a flip of Y28 close to L203 in tFprAanox structure (Fig. S4, ESI†) which probably acts as a gate to control solvent access or sealing.
In mFprA and tFprA structures, the di-iron centre is coordinated by four histidines, one aspartate, one glutamate and one bridging aspartate residues, which are also perfectly conserved in other FDPs (Fig. S1, ESI†). In mFprA (PDB codes: 2OHH and 2OHI) and tFprA structures (tFprAanox and tFprAoxo-Kr) the distance between the two irons is between 3.3 to 3.5 Å, which corresponds to van-der-Waals contacts. Iron atom occupancies are close to 100% in tFprAanox and slightly lower in tFprAoxo-Kr (between 0.7 to 0.9). In the case of mFprA structures (PDB codes: 2OHH and 2OHI), one iron of the catalytic centre is present at low occupancy (0.4) that provokes a high increase of flexibility for the switch loop 149–154 coordinating the Fe (Fig. S5, ESI†). tFprAoxo-Kr structure, exposed to oxygen during the purification and crystallization, exhibits the classic oxo-bridging ligand, which is a chemical proof of the oxidized state because such bridging ligand was also observed in one active site of the mFprA tetramer in the active oxidized state (PDB 2OHH, chain E). This reaction intermediate Fe(III)–O–Fe(III) is produced after the first reduction step. In the reduced active state, tFprAanox does not have such bridging oxygen, or at low occupancy (Fig. S6, ESI†). FMN in tFprAanox and tFprAoxo-Kr structures are in a partial butterfly conformation (Fig. S7, ESI†). The distance between N5 of FMN and an iron of the di-iron centre is ∼9.3 Å in both tFprA structures, which is identical to that observed in mFprA structures. Such distance would allow a direct electron transfer for the reduction reaction.
O2 supply from the enzyme surface to the active site seems unrealistic by the FMN entrance since this region is highly hydrated and would therefore repulse O2 molecules, which have an hydrophobic nature. Thus, a hydrophobic channel would make more sense to capture O2 molecules with high specificity and efficiency, as it was predicted and calculated by Romão and co-workers.20 To characterize experimentally this possible gas channel, we produced Kr-derivatives of oxidized tFprA by flash cooling crystals under gas pressure and collected datasets below and above the Kr K-edge (SI Method and Table S1, ESI†). Anomalous signal coming unambiguously from Kr atoms have been detected in an anomalous Fourier map at similar positions in each of the eight monomers constituting the asymmetric unit (Fig. S8, ESI†). Up to six Kr were localized in a hydrophobic channel (Fig. 1b and 2b, c, Fig. S8, ESI†). The narrow channel is ∼30 Å long and has an average radius of 1.2 Å, which is in accordance with the size of O2 molecule. The channel is built at the conserved interface (Fig. S2, ESI†) between the anti-parallel β-sheets β13β14β15 and the α-helices 6 and 7. It starts from the surface and ends at the di-iron site. The hydrophobic channel simulated by the CAVER software in tFprAanox structure (Fig. 2c and Fig. S3, ESI†) perfectly coincides with the Kr positions. Comparison between tFprAanox and tFprAoxo-Kr does not indicate any channel distortion or size modification upon pressurisation and Kr binding. Kr4,5 and Kr6 highlight two possible entrances for the O2 (Fig. 1b, 2c and Fig. S8, ESI†). Interestingly, a modulation in the Kr atoms occupancies along the channel is observed. Two main Kr atoms Kr2 and Kr3 are trapped in a high hydrophobic/rigid part of the channel formed by three phenylalanine's side chains (F158, F173, F215) (Fig. 2c). These two Kr atoms present high occupancies (0.75) compared to the four other ones (Kr1, Kr4, Kr5 and Kr6). This might suggest a lower affinity for Kr and therefore also for O2 in the outer part of the tunnel (Kr4,5 and Kr6) and at that close to the iron centre (Kr1) in the oxidized state. The high Kr affinity part of the tunnel also probably acts as a “retention cavity” and could play a role in the kinetics of O2 transport. Similar variation in Kr-binding were also observed in other O2 hydrophobic tunnels.21 The Kr1 atom (occupancy 0.1) observed near the di-iron site points out the final destination of O2 from the gas channel, which could be the O2-binding site, where O2 is in interaction with the di-iron site centre. Kr7 atom is located outside of the channel in a hydrophobic pocket. The hydrophobic residues lining the channel are very well conserved in methanogens and could be extended to other dinuclear oxidases such as those from Giardia intestinalis10 and Moorella thermoactica9 (Fig. S1 and S2, ESI†). CAVER simulation argues for a common O2 route in all structural homologues available (Fig. S3, ESI†). This finding highlights the universal needs of such hydrophobic channel to specifically react with O2 or NO. CAVER analysis also identified a hydrophilic channel in the oxidized state, likely the channel involved in water release. This second channel (Fig. 2) starts from the di-iron site at the end of the O2 channel, includes the loop 24–28 and finishes in the large hydrophilic cavity formed by the FprA tetrameric interface.
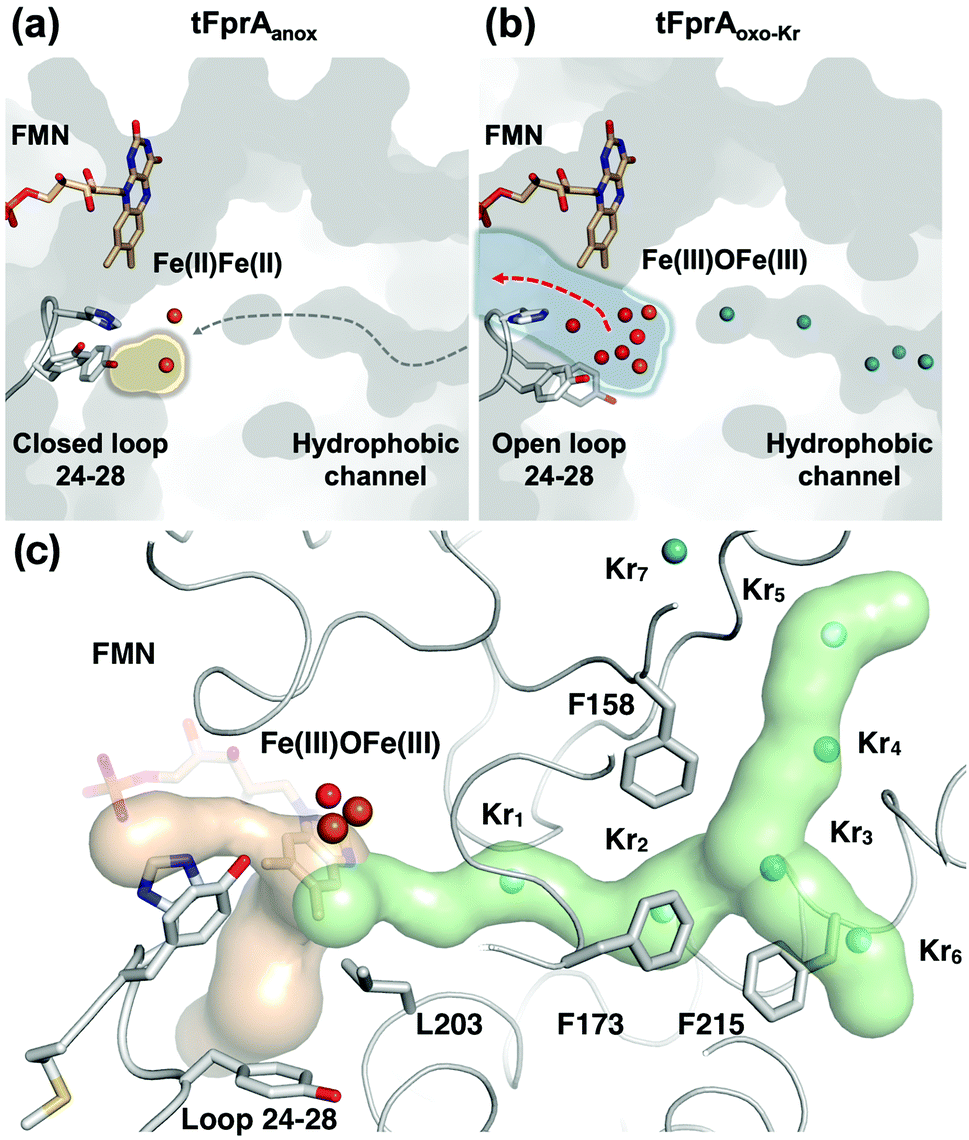 | ||
| Fig. 2 Structure of the active site and a hydrophobic O2-channel. (a) tFprAanox structure, in which the loop 24–28 is fixed as a closed conformation and isolates the catalytic cavity highlighted by a yellow halo. The predicted O2 pathway is indicated with a gray dashed arrow. (b) tFprAoxo-Kr structure after derivatization with 125 bars of krypton, in which the loop 24–28 is flexible compared to tFprAanox. The water channel is depicted by light blue halo and a red arrow indicates solvent exchange. Kr are depicted as cyan balls. (c) tFprA channels identified by CAVER in tFprAanox structure. Hydrophobic part of the channel is depicted in light green. Kr atoms from tFprAoxo-Kr structures are superimposed as cyan spheres. Hydrophilic part of the channel involved in water release after O2 reduction is depicted in light orange. Di-iron centre is highlighted as orange spheres, FMN and the loop 24–28 in sticks. Y28 in tFprAanox exists as a double conformation and only the one at highest occupancy (70%) is displayed. | ||
The FprA reaction is performed by a ping-pong mechanism using two molecules of F420H2. When the di-iron centre and FMN are completely reduced to Fe(II)Fe(II) and FMNH2, the loop 24–28 would lock the entrance of the water channel. In tFprAanox, Y28 might contribute to bridge the hydrophilic gap and prevent unwanted side reactions by moving close to L203, which is one of the last amino acid of the hydrophobic channel (Fig. 2c). The channel ends-up in front of the empty di-iron site (tFprAanox), where O2 could bind, as previously predicted by the analysis of mFprA. The first two-electron reduction is performed by the Fe(II)Fe(II). O2 is first bound to the dinuclear Fe site, which forms transient bridged Fe(III) peroxo intermediate7,22,23 and then form a water molecule with the first oxgen atom, while the second one is trapped at the di-iron site to form an oxo-bridge Fe(III)–O–Fe(III). Protons required for the water-forming reactions are supplied from the histidine side-chains (H84–H152 and H89–H234), which coordinate the irons. The formed water molecule is repelled from the hydrophobic region when H26 and Y28 become more flexible, and open the hydrophilic channel (Fig. 3). After removal of the first water molecule from the active site pocket, the loop 24–28 is restabilized, probably upon the reduction event by FMNH2. In the closed structure, reduction of oxygen bound to the dinuclear iron site is performed in the absence of water molecule. After that, the water channel opens again and the water molecule is released. The driving-force able to guide the closure and opening of the 24–28 loop to prevent or allow solvent accessibility could be the FMN state or even the F420H2 binding that could lead to a local rearrangement. For instance, the isoalloxazine group is in van der Waals contact with the loop 24–28 (3.4–4.2 Å) and could contribute to stabilise or destabilise the loop depending on its state.
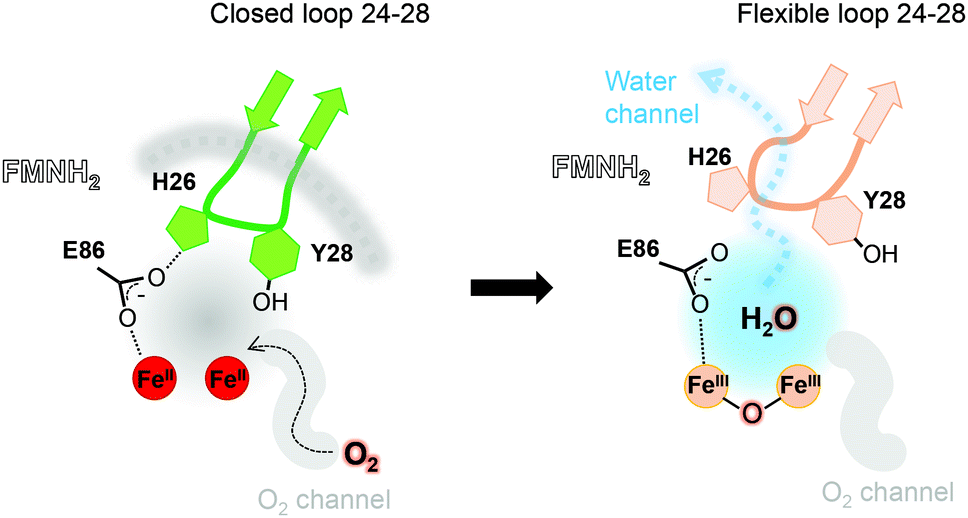 | ||
| Fig. 3 Contribution of the hydrophobic O2-channel, loop 24–28 and water channel in the catalytic mechanism. The hydrophobic O2-channel (gray) and hydrophilic water-channels (light blue) are indicated. In the reduced state, the loop is closed and fixed (green and gray dashed line). After the first water is produced, the switch loop is still closed but becomes flexible (orange), where the water is released to bulk solvent. | ||
In conclusion, previous biochemical studies indicated that FprA catalyses reduction of O2 with four electrons supplied by two molecules of F420H2. The reaction avoids the generation of reactive oxygen species, hydrogen peroxide and superoxide anion. Seedorf et al. proposed a catalytic mechanism involving a Fe(III)–O–Fe(III), which does not produce reactive oxygen species as an intermediate. However, the mechanism did not explain how to supply O2 into the active site while stopping water intrusion. The shape and position of the hydrophobic channel allowed us to draw a reaction scheme taking into account a water repulsion system to avoid unwanted reaction. Such reaction scheme would require a gating system by surrounding loops coordinating the reactivity of the catalytic chamber, which is a conserved system retained by life in numerous organisms dealing with oxidative stress.
The authors acknowledge funding by Agence Nationale de la Recherche (ANR) grant (program Ln23 ANR-13-BS07-0007-01), Swiss National Science Foundation grant (200021-182369), Deutsche Forschungsgemeinschaft, (SH 87/1-1) and the Max Planck Society. This work used the platforms of the Grenoble Instruct center (ISBG; UMS 3518 CNRS-CEA-UJF-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). We thank ESRF synchrotron (id29 beamline) and SOLEIL synchrotron (PXII beamline) for beam time allocation and the respective beamline staffs for assistance with data collection. We also acknowledge access to the ESRF MX high-pressure laboratory (HPMX). IBS acknowledges integration into the Interdisciplinary Research Institute of Grenoble (IRIG, CEA).
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- C. F. Blanford, C. E. Foster, R. S. Heath and F. A. Armstrong, Faraday Discuss., 2009, 140, 319–335 RSC.
- F. Folgosa, M. C. Martins and M. Teixeira, FEMS Microbiol. Lett., 2018, 365, fnx267 CrossRef PubMed.
- K. A. Brown, Z. J. Guo, M. Tokmina-Lukaszewska, L. W. Scott, C. E. Lubner, S. Smolinski, D. W. Mulder, B. Bothner and P. W. King, Sustainable Energy Fuels, 2019, 3, 3191–3200 RSC.
- L. Chen, M. Y. Liu, J. Legall, P. Fareleira, H. Santos and A. V. Xavier, Biochem. Biophys. Res. Commun., 1993, 193, 100–105 CrossRef CAS PubMed.
- H. Seedorf, A. Dreisbach, R. Hedderich, S. Shima and R. K. Thauer, Arch. Microbiol., 2004, 182, 126–137 CrossRef CAS PubMed.
- H. Seedorf, C. H. Hagemeier, S. Shima, R. K. Thauer, E. Warkentin and U. Ermler, FEBS J., 2007, 274, 1588–1599 CrossRef CAS PubMed.
- A. Das, R. Silaghi-Dumitrescu, L. G. Ljungdahl and D. M. Kurtz Jr, J. Bacteriol., 2005, 187, 2020–2029 CrossRef CAS PubMed.
- R. Silaghi-Dumitrescu, D. M. Kurtz Jr, L. G. Ljungdahl and W. N. Lanzilotta, Biochemistry, 2005, 44, 6492–6501 CrossRef CAS PubMed.
- A. Di Matteo, F. M. Scandurra, F. Testa, E. Forte, P. Sarti, M. Brunori and A. Giuffre, J. Biol. Chem., 2008, 283, 4061–4068 CrossRef CAS PubMed.
- H. Fang, J. D. Caranto, R. Mendoza, A. B. Taylor, P. J. Hart and D. M. Kurtz Jr, J. Biol. Inorg. Chem., 2012, 17, 1231–1239 CrossRef CAS PubMed.
- C. Frazão, G. Silva, C. M. Gomes, P. Matias, R. Coelho, L. Sieker, S. Macedo, M. Y. Liu, S. Oliveira, M. Teixeira, A. V. Xavier, C. Rodrigues-Pousada, M. A. Carrondo and J. Le Gall, Nat. Struct. Biol., 2000, 7, 1041–1045 CrossRef PubMed.
- F. Xu, Appl. Biochem. Biotechnol., 2001, 95, 125–133 CrossRef CAS PubMed.
- B. L. Victor, A. M. Baptista and C. M. Soares, J. Biol. Inorg. Chem., 2009, 14, 853–862 CrossRef CAS PubMed.
- N. Colloc'h, P. Carpentier, L. C. Montemiglio, B. Vallone and T. Prange, Biophys. J., 2017, 113, 2199–2206 CrossRef PubMed.
- B. Lafumat, C. Mueller-Dieckmann, G. Leonard, N. Colloc'h, T. Prange, T. Giraud, F. Dobias, A. Royant, P. van der Linden and P. Carpentier, J. Appl. Crystallogr., 2016, 49, 1478–1487 CrossRef CAS.
- V. M. Luna, Y. Chen, J. A. Fee and C. D. Stout, Biochemistry, 2008, 47, 4657–4665 CrossRef CAS PubMed.
- J. P. Kallio, J. Rouvinen, K. Kruus and N. Hakulinen, Biochemistry, 2011, 50, 4396–4398 CrossRef CAS PubMed.
- S. Engilberge, T. Wagner, G. Santoni, C. Breyton, S. Shima, B. Franzetti, F. Riobé, O. Maury and E. Girard, J. Appl. Crystallogr., 2019, 52, 722–731 CrossRef CAS PubMed.
- C. V. Romão, J. B. Vicente, P. T. Borges, B. L. Victor, P. Lamosa, E. Silva, L. Pereira, T. M. Bandeiras, C. M. Soares, M. A. Carrondo, D. Turner, M. Teixeira and C. Frazão, J. Mol. Biol., 2016, 428, 4686–4707 CrossRef PubMed.
- J. Kalms, A. Schmidt, S. Frielingsdorf, P. van der Linden, D. von Stetten, O. Lenz, P. Carpentier and P. Scheerer, Angew. Chem., Int. Ed., 2016, 55, 5586–5590 CrossRef CAS PubMed.
- M. Costas, C. W. Cady, S. V. Kryatov, M. Ray, M. J. Ryan, E. V. Rybak-Akimova and L. Que Jr, Inorg. Chem., 2003, 42, 7519–7530 CrossRef CAS PubMed.
- S. V. Kryatov, E. V. Rybak-Akimova, V. L. MacMurdo and L. Que Jr, Inorg. Chem., 2001, 40, 2220–2228 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, Fig. S1–S8, Tables S1 and S2 and references. See DOI: 10.1039/d0cc04557h |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |