
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
19F and 1H quantitative-NMR spectroscopic analysis of fluorinated third-generation synthetic cannabinoids†
Husain A.
Naqi
,
Timothy J.
Woodman
 ,
Stephen M.
Husbands
and
Ian S.
Blagbrough
*
,
Stephen M.
Husbands
and
Ian S.
Blagbrough
*
Department of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: prsisb@bath.ac.uk; Tel: +44 (0)1225 386795
First published on 1st May 2019
Abstract
Quantitative nuclear magnetic resonance (q-NMR) spectroscopy is a robust and reliable analytical method that possesses many advantages over conventional chromatographic techniques used in drug analysis. In this paper, the application of 19F and 1H NMR spectroscopy to quantify the amounts of synthetic cannabinoids (SCs), AM-694 and 5F-ADB, in herbal incense packages is discussed. These SC samples, seized in the South West of England in the summers of 2016 and 2017, are part of a growing illicit drug problem in the UK. For accurate quantitative analysis using 19F observe, the data acquisition and the NMR processing parameters, such as spectral width, the centre point of the spectrum, nuclear Overhauser effect (NOE) enhancement and relaxation delay, are discussed together with cross-method validation. The reproducibility, simplicity, high speed, and non-destructive nature provide reliable quantitative analysis and, by using 19F NMR, there is essentially no background interference. This quantitation is without resorting to the use of (often unavailable) standards as reference materials or to lengthy sample preparation, which are the norm in many analytical chromatographic techniques. The NMR methods allowed a direct comparison between 1H and 19F NMR, revealing the robustness and the effectiveness of 19F NMR for application as a rapid (∼8 min), quantitative analytical method for fluorinated SCs which are now being seized with an increasing frequency and are highly toxic.
Introduction
Synthetic cannabinoids (SCs), also known by their street name “spice”, are potent agonists binding to the cannabinoid receptors CB1 and CB2 distributed throughout the central nervous system (CNS) and immune system, respectively, producing psychoactive effects similar to, and in most cases more potent than, the mainstream drugs they are mimicking, e.g. Δ9-tetrahydrocannabinol (THC).1 Unlike Δ9-THC, a partial agonist with low affinity for the CB1 receptor, SCs are full receptor agonists with high affinity binding to CB1 and moreover they also possess CB2 receptor affinity.2 These pharmacological characteristics result in drug users/abusers having severe physical and psychiatric episodes, not present with traditional cannabis smoking. These effects are described as the “cannabinoid tetrad”, which are hypothermia, analgesia, catalepsy, and locomotor activities, leading to symptoms ranging from excited delirium to kidney damage.2In 2008, the first generation of synthetic cannabinoids hit the streets,3 such as the Pfizer compound CP 47,497 and the John W. Huffman designed JWH-018 (Fig. 1). Typically these ligands were designed and developed as medicinal chemistry compounds, intended to exploit the pathological implications of the CB receptors in many diseases, but they were side-tracked to the illicit clandestine designer-drug market.4,5 The following generations of SC were based initially on JWH-018, but they have evolved with variations of fluoroalkyls (AM-694), indazoles (5F-ADB), quinoline (5F-PB22) and amides (PX-1) integrated into their structures, replacing the naphthoylindole of JWH-018.6,7 The continuous and rapid change in substituents on the available SCs makes them a moving target posing many analytical challenges. The Korean National Forensic Services reported that from 2008 to 2010 most of the SCs seized were first generation non-fluorinated compounds,8e.g. JWH-018, CP 47,497, and UR-144 (Fig. 1). In 2012, fluorinated analogues started emerging such as XLR-11, a fluoropentyl analogue of UR-144, and by 2013 approximately 90% of the SCs seized were fluorinated.8 It is believed that the growing trend in the bioisosteric fluorine introduction into SCs was inspired by a Makriyannis patent,9 where he demonstrated a much higher potency of AM-2201 than that of non-fluorinated analogues, e.g. JWH-018 (Fig. 1). Initially, AM-2201 was identified in herbal blends, and this has escalated into many SCs with no precedent in the scientific literature, e.g. 5F-ADB-PINACA, 5F-AB-PICA, and 5F-PB-22.5 The third-generation SCs include fluorinated AM-694 and 5F-ADB (Fig. 1). Also, besides the enhanced potency of fluorinated analogues, the addition of a fluorine substituent was possibly intended to circumvent legal restrictions imposed on specified SCs.5,10
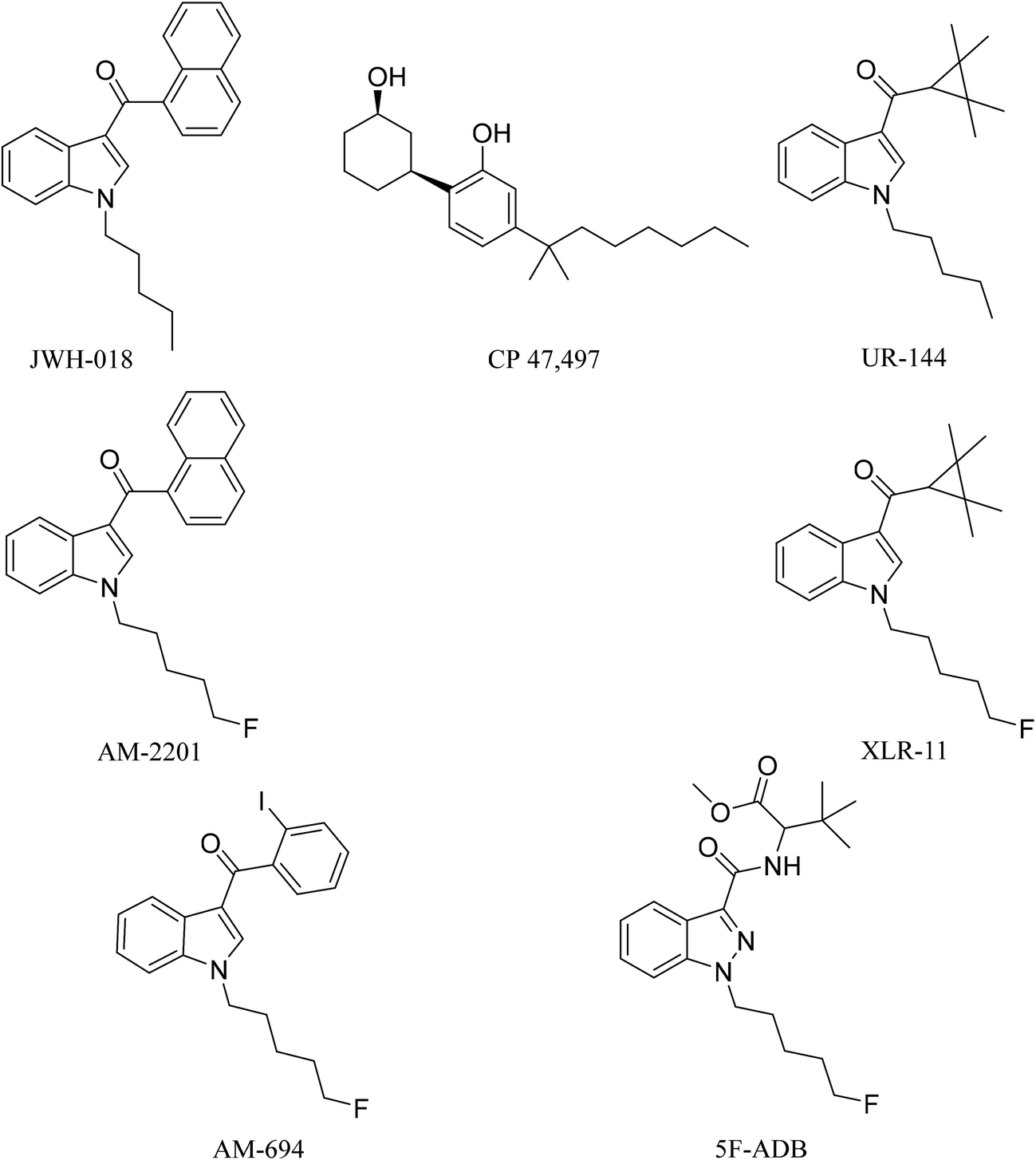 | ||
| Fig. 1 1st, 2nd, and 3rd generation SCs. | ||
1H-NMR is inherently quantitative, as the integrated functional group signals are directly proportional to the number of spins generated by the signals in question. Nevertheless, NMR is only quantitative if the appropriate acquisition and processing parameters are determined by experiment and then implemented. Early applications of quantitative NMR (q-NMR), using low-field instruments, required considerably large amounts of sample and Internal Standard (IS).11 The development of high-field NMR spectrometers facilitated improved sensitivity meaning that impurities could be quantified at less than 0.1% of the total sample, demonstrating that NMR is comparable with chromatographic methods for quantitative analysis.12–15 NMR has more advantages than other analytical approaches such as those that are chromatography based. NMR does not require the use of a high purity reference standard for the construction of the required calibration curve. Such a standard is expensive and often unavailable, especially for newer, more recently identified SCs.14 NMR also has the advantage of less sample preparation being required. No serial dilution is required to run the sample and no mobile phase has to be prepared. Also, as there is no interaction with a column, no blank samples are required to be chromatographed in order to avoid carry-over that could affect the analysis. NMR is not subject to problems from small compounds and impurities with no chromophore or a different UV response which pose challenges to chromatographic and UV methods.14,15
Quantitative analysis of SCs in herbal blends has been reported using some analytical techniques, mostly chromatography and MS-based ones.16 GC/MS showed qualitative and quantitative variations among SCs in herbal-blend brands in 2014.171H q-NMR reports on SC quantitation are scarce, but there is a report on purchased herbal blends containing SCs using maleic acid (MA) as an internal standard.18 A study on the extraction efficiency of common solvents, e.g. acetone, acetonitrile, chloroform, and methanol, using 1H q-NMR with 1,3,5-trimethoxybenzene as an internal standard and GC/MS on seized herbal blends and in-house preparations found no significant difference between the solvents used for the SC extraction.19
The 19F nucleus is attractive for q-NMR spectroscopy, mainly due to the sensitivity of the nucleus (its relative sensitivity is 83.4% of 1H) and its natural abundancy of 100%.20 Additionally, the wide range of chemical shift (500 ppm) reduces the chance of overlapping signals, and the absence of fluorinated impurities inherently means that there is less background noise.2119F q-NMR has been applied to analyse fluorinated Active Pharmaceutical Ingredients (API).22 Nevertheless, for quantitative results more NMR parameters have to be addressed than for 1H q-NMR,21e.g. equal excitation for the signals across the entire spectral width must be achieved, otherwise the integration values will suffer which in turn affects the analytical results. This is achieved by setting the centre point of the spectral window midway between the signal of the internal standard and the compound, using a 90° pulse angle followed by a sufficient relaxation delay of 5 × T1 to recover the magnetization to 99.3% of its size. The use of a suitable relaxation delay is common with 1H q-NMR. If the 19F spectrum is acquired with broadband 1H decoupling, then NOE enhancement of the signals may arise. In order to avoid this, an inverse-gated decoupling sequence is used.21
A validated 19F q-NMR spectroscopic method is reported for the first time to quantify fluorinated SCs, e.g. AM-694 and 5-F-ADB (Fig. 1), in herbal blends recently seized in the South West of England. The technique was compared to both 1H q-NMR and UHPLC for accurate quantification and was shown to be in good agreement. Moreover, quantitative differences between seized sample batches are discussed. This investigation of the acquisition parameters associated with 19F q-NMR will help drug analysts to run a fast and robust quantitative analysis for fluorinated (illicit) drugs with minimal background interference and signal overlap. It is important because such highly toxic SCs are currently being found with increasing frequency and outbreaks of zombification caused by AMB-FUBINACA have been reported in NEJM,23 and in various UK cities in the popular press.
Experimental section
Chemicals and sample preparation
Extraction solvents (all 99.9% anhydrous) chloroform, methanol, and acetonitrile were purchased from Fisher Scientific (UK) and ACROS Organics (UK). Deuterated solvents (CDCl3, CD3OD, and CD3CN) were purchased from Cambridge Isotope Laboratories (Goss Scientific, UK). NMR internal standards (IS) 2-chloro-4-fluorotoluene, dimethyl sulfone (DMS), and maleic acid (MA) are TraceCERT certified reference materials purchased from Sigma-Aldrich (UK). [1-(5-Fluoropentyl)-1H-indol-3-yl](2-iodophenyl)-methanone (AM-694) 10.0 mg, N-(1-adamantyl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide (5F-AKB-48) 1.0 mg mL−1 in a 1.0 mL vial, and (1-pentyl-1H-indol-3-yl)-1-naphthalenyl-methanone (JWH-018) 100 μg mL−1 in a 1.0 mL vial were purchased from LGC (Teddington, UK). N-Methyltrifluoroacetamide (N-methyl-TFA) >98.0% was purchased from Tokyo Chemical Industry (TCI, Tokyo, Japan). SC samples were provided by the Drug Expert Action Team (DEAT), Avon and Somerset Constabulary, from recent (2016–2017) seizures. The samples were in the form of herbal blends (1.0–3.0 g) as commercially packaged brands (Exodus, Loco Elite). Turnera diffusa (damiana) dried herb (illicit-drug free) was purchased from Spiceworks (Hereford, UK).All standards and samples were weighed using a SE2F Sartorius analytical balance, between 1.0 and 2.0 mg mL−1 IS was used. Preliminary analysis of non-homogenized herbal-blend samples yielded large variations in the amounts of the SCs sprayed on the carrier plant materials between samples tested by NMR. Therefore, two approaches were employed for the homogenization of the herbal-blend samples. Either they were ground to a fine powder with 100 grit sandpaper24 or they were frozen in liquid nitrogen, followed by grinding to a fine powder using a mortar and pestle. For sample preparation for UHPLC and NMR analyses, homogenized plant materials (100 mg) were extracted with methanol (2 × 4.0 mL) with sonication (30 min) at 20 °C, centrifuged, and then the supernatant extract was decanted and the pellet (plant material) discarded. The extract was then evaporated to dryness under reduced pressure and reconstituted in deuterated solvent (1.0 mL) containing the IS (DMS, MA or 2-chloro-4-fluorotoluene) for NMR spectroscopic analysis. For UHPLC analysis, samples were diluted 100-fold in UHPLC solvent to bring them within the calibration range. AM-694 was quantified using a 7-point calibration curve between 1.25 and 80 μg mL−1 with JWH-018 as the IS. 5F-ADB was quantified using a 6-point calibration curve between 1.25 and 40 μg mL−1 with 5F-AKB48 as the IS (10.0 μg mL−1). The response was calculated as the ratio of the area under the curve of the compounds to that of the respective IS. Data analysis was conducted using the Microsoft Excel data analysis tool pack.
Instrumentation
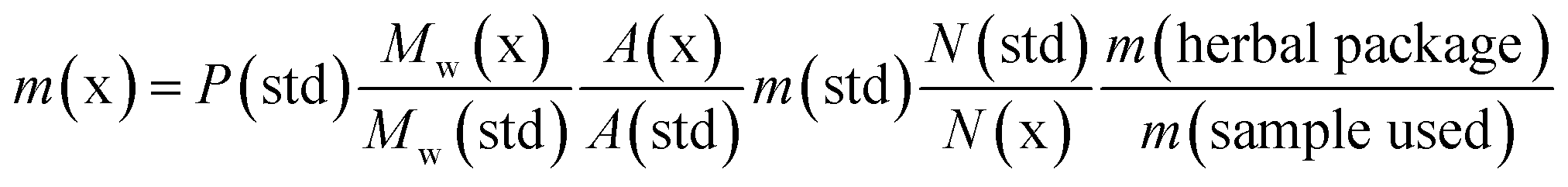 | (1) |
Mobile phase A consisted of MS grade water with 0.1% trifluoroacetic acid (v/v), and mobile phase B consisted of acetonitrile with 0.1% trifluoroacetic acid (v/v). For AM-694 and 5F-ADB calibration curves and quantitation the following solvent gradient 1 was used: the gradient started from 1% B for 2.0 min followed by a linear increase to 100% B at 5.0 min, held for 3 min, followed by a return to 1% B at 8.1 min, where it was held for equilibration for 3.9 min, with a total run time of 12.0 min. For 5F-ADB purity determination, the flow rate was 0.4 mL min−1, and the column temperature was 25.0 °C. Gradient 2 started with 1% B until 2.0 min followed by a linear increase to 100% B at 20.0 min, held for 4.0 min, followed by a return to 10% B at 24.1 min where it was held for 10.9 min with a total run time of 35.0 min. Data analysis used the Bruker data and quant analysis 4.3 package.
Results and discussion
The 5F-ADB reference material was extracted from a seized sample (1.3 g) with CHCl3 (2 × 25.0 mL) with sonication for 30 min each time. The combined extracts were passed through a 0.25 μm syringe filter. The filtrate was evaporated to dryness under reduced pressure yielding ∼90 mg of residue which was purified by flash-column normal phase silica chromatography, followed by semi-preparative RP HPLC, resulting in pure 5F-ADB (38.0 mg). Purity and confirmation of the structure were obtained by NMR, UHPLC, and HRMS (Fig. 2).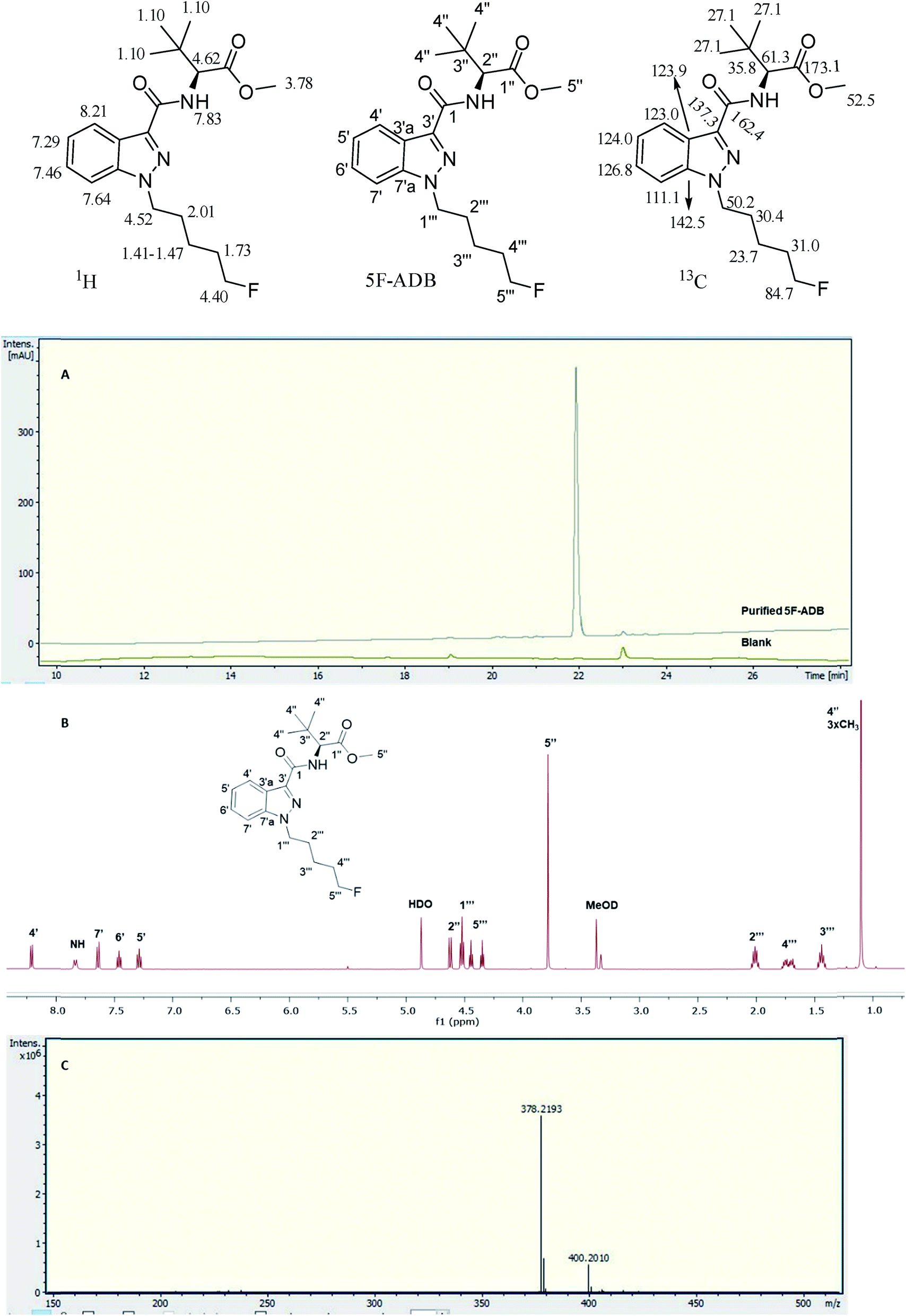 | ||
| Fig. 2 Analytical purity of 5F-ADB as tested by (A) UHPLC with RT = 22.0 min, (B) 1H-NMR in CD3OD with assignments, and (C) HRMS showing [M + H]+ and [M + Na]+. | ||
1H NMR (500 MHz in CD3OD): δ 8.21 (4′) (1H, dd J = 8.0, 2.0 Hz), 7.83 (NH) (1H, br d J = 9.5 Hz), 7.64 (7′) (1H, dd J = 8.5, 1.5 Hz), 7.46 (6′) (1H, td J = 8.5, 1.5 Hz), 7.29 (5′) (1H, td J = 8.5, 2.0 Hz), 4.62 (2′′) (1H, d J = 9.5 Hz), 4.52 (1′′′) (2H, t J = 7.5 Hz), 4.40 (5′′′) (2H, dt J = 47.5, 6.0 Hz), 3.78 (5′′) (3H, s), 2.01 (2′′′) (2H, quintet J = 7.5 Hz), 1.73 (4′′′) (2H, d quintet J = 26.0, 6.0 Hz), 1.41–1.47 (3′′′) (2H, m), 1.10 (4′′) (9H, s).
13C NMR (125.8 MHz in CD3OD): δ 173.1 (1′′), 164.2 (1), 142.5 (7′a), 137.3 (3′), 126.8 (6′), 124.0 (5′), 123.9 (3′a), 123.0 (4′), 111.1 (7′), 84.7 (5′′′, d 1JCF = 164.0 Hz), 61.3 (2′′), 52.5 (5′′), 50.2 (1′′′), 35.8 (3′′), 31.0 (4′′′, d 2JCF = 19.5 Hz), 30.4 (2′′′), 27.1 (4′′), 23.7 (3′′′ d, 3JCF = 5.5 Hz); 19F observe δ −220.3 (5′′′F, tt 2JHF = 47.5, 3JHF = 26.0 Hz). HRMS found [M + H]+ 378.2193 m/z for C20H29FN3O3 requires 378.2187, and found [M + Na]+ 400.2010 m/z for C20H28FN3O3Na requires 400.2006 (Fig. 2).
5F-ADB quantified in seized herbal blends
N-[[1-(5-Fluoropentyl)-1H-indazol-3-yl]carbonyl]-3-methyl-L-valine methyl ester (5F-ADB) was identified in seized herbal blend samples branded as “Exodus”. Identification was achieved by interpretation of 2D NMR data and the LC-MS/MS fragmentation pattern. Results are confirmed by comparison with the literature with only minor differences in the NMR, due to solvent effects.25,26 The 19F signal for q-NMR analysis is on the N-pentyl tail with its chemical shift of δ = −220 ppm assigned as a triplet of triplets, 2JHF 47.5 Hz coupling to methylene protons on position 5′′′ and 3JHF 26.0 Hz coupling to methylene at position 4′′′.The extraction was evaluated in chloroform, methanol, and acetonitrile. The signals used for quantification in methanol were the indazole protons 4′ at 8.22 ppm, 7′ at 7.64 ppm, 6′ at 7.46 ppm, and 5′ at 7.31 ppm. In acetonitrile, the same protons were used except 6′ due to an overlapping impurity. In chloroform, H-5′ was excluded due to the overlap with the residual chloroform H-solvent signal; nevertheless chloroform gave a cleaner spectrum, with fewer impurities and no sugars from the matrix component (as found when methanol was the solvent of extraction) with additional signals available for integration such as the fluoropentyl methylenes 1′′′ and 5′′′. The DMS singlet at δ = 3.00 ppm integrating for six protons was used as an IS in CDCl3.
In 19F q-NMR with N-methyltrifluoroacetamide, apparently significantly lower amounts of SC, using Anova two factor analysis, were obtained than in a contemporaneous analysis by 1H q-NMR using maleic acid (IS) in methanol and acetonitrile, DMS (IS) in chloroform, and then N-methyltrifluoroacetamide in chloroform (Table 1). The reason behind this apparently lower assay result is the resonance (chemical shift) of the N-methyltrifluoroacetamide 19F signal at δ = −75.9 compared to the 19F signal of 5F-ADB at δ = −220.2 ppm, resulting in unequal excitation. Uniform excitation across the spectrum has to be achieved in order for all the signals to get the same magnetization in the pulse sequence, thus making the centre point of the spectral window a crucial parameter when accurate and reproducible quantitative results are to be achieved for 19F q-NMR.
| Nucleus, solvent (IS) | 1H in CD3OD (MA) | 1H in CD3CN (MA) | 1H in CDCl3 (DMS) | 1H in CDCl3 (N-Me-TFA) | 19F in CDCl3 (N-Me-TFA) |
|---|---|---|---|---|---|
| Amount (mg g−1) | 11.84 ± 0.28 | 11.06 ± 0.16 | 11.03 ± 0.14 | 11.48 ± 0.12 | 8.86 ± 0.19 |
| RSD (%) | 2.34 | 1.45 | 1.30 | 1.01 | 2.18 |
When N-methyltrifluoroacetamide was evaluated as an IS for 1H q-NMR it was shown to be as useful an IS as MA or DMS. Although apparently attractive for 19F NMR with its 3 equivalent fluorine atoms, its wide chemical shift separation from the analyte signal made it a poor choice. Rather, 2-chloro-4-fluorotoluene was used as a 19F q-NMR IS with (protio) methanol as the extraction solvent and CD3OD as the NMR solvent, resulting in a good agreement with the data from 1H q-NMR using maleic acid (MA) as the IS. This 19F NMR IS signal has a chemical shift of δ = −117.8 ppm. As 5F-ADB 19F resonates at δ = −220.2 ppm, the central point (Bruker's O1P) was therefore set at δ = −165 ppm approximately equally between both resonances resulting in equal excitation of both fluorine signals. The 19F q-NMR (proton coupled) results of 5F-ADB are in agreement with the 1H q-NMR results using maleic acid (MA) (10.4 ± 0.2 mg g−1, RSD 1.6%, n = 5) as the IS and 9.8 ± 0.8 mg g−1 (RSD 7.9%, n = 5) was observed with 2-chloro-4-fluorotoluene as the IS, and 9.4 ± 0.7 mg g−1 (RSD 7.3%, n = 5) with 19F NMR. The effect of changing the O1P was tested using the plant material (100.0 mg) containing 5F-ADB with 2-chloro-4-fluorotoluene as the IS, and setting the O1P approximately in the middle of the two signals (−165 ppm). This resulted in quantitative results in agreement with 1H q-NMR results. Not unexpectedly, shifting the O1P to −220 and −117 ppm resulted in significantly lower and higher integration values, respectively, and, consequently, significantly altered quantitative results as tested by t-tests (p < 0.05) (Fig. S1†). The need to set the spectral midpoint as the excitation frequency is an important parameter.
A seized sample (HN Exodus5) containing 5F-ADB was analysed using inverse-gated decoupling 19F NMR in order to eliminate the nuclear Overhauser effect (NOE), and for providing the added benefit of an enhanced signal to noise (S/N) ratio by collapsing the 19F signals to singlets. The results were compared with proton-coupled 19F NMR; the results from the 19F proton coupled and 19F proton decoupled methods are in good agreement (Fig. S2†). 1H q-NMR showed 7.1 ± 0.11 mg g−1 (RSD of 1.57%), 19F proton-coupled q-NMR showed 6.9 ± 0.02 mg g−1 (RSD of 0.24%), and 19F inverse-gated decoupled q-NMR showed 6.8 ± 0.08 mg g−1 (RSD of 0.78%).
Two batches of seized “Exodus” brand, 8 seized in 2016 and 7 seized in 2017, were subjected to quantitative analysis using 19F proton coupled/decoupled and 1H NMR using the IS 2-chloro-4-fluorotoluene, and also by UHPLC. Confirmation of the quantitation by 19F q-NMR was achieved with UHPLC using the purified 5F-ADB as the reference standard to construct a calibration curve (gradient 1), using λ = 298 nm wavelength where the indazole absorbs strongly, resulting in RT = 6.8 min. Initially, 1-methylindazole-3-carboxylic acid was used as a UHPLC IS, but this was abandoned due to the overlap of the 1-methylindazole-3-carboxylic acid peak with a plant matrix component at RT = 5.6 min (Fig. 3). 5F-AKB48 (RT = 7.2 min) was chosen instead as an IS in UHPLC analysis due to its similar chromophore to 5F-ADB (indazole), and the presence of an N-adamantanyl substituent provided sufficient hydrophobicity to be separated from the peak of 5F-ADB (Fig. 3). The 5F-ADB UHPLC calibration curve using 5F-AKB48 as an IS was in the range of 1.25–40.00 μg mL−1 giving excellent linearity, R2 = 0.9999, and an IS RSD of 4.8% (Fig. 4).
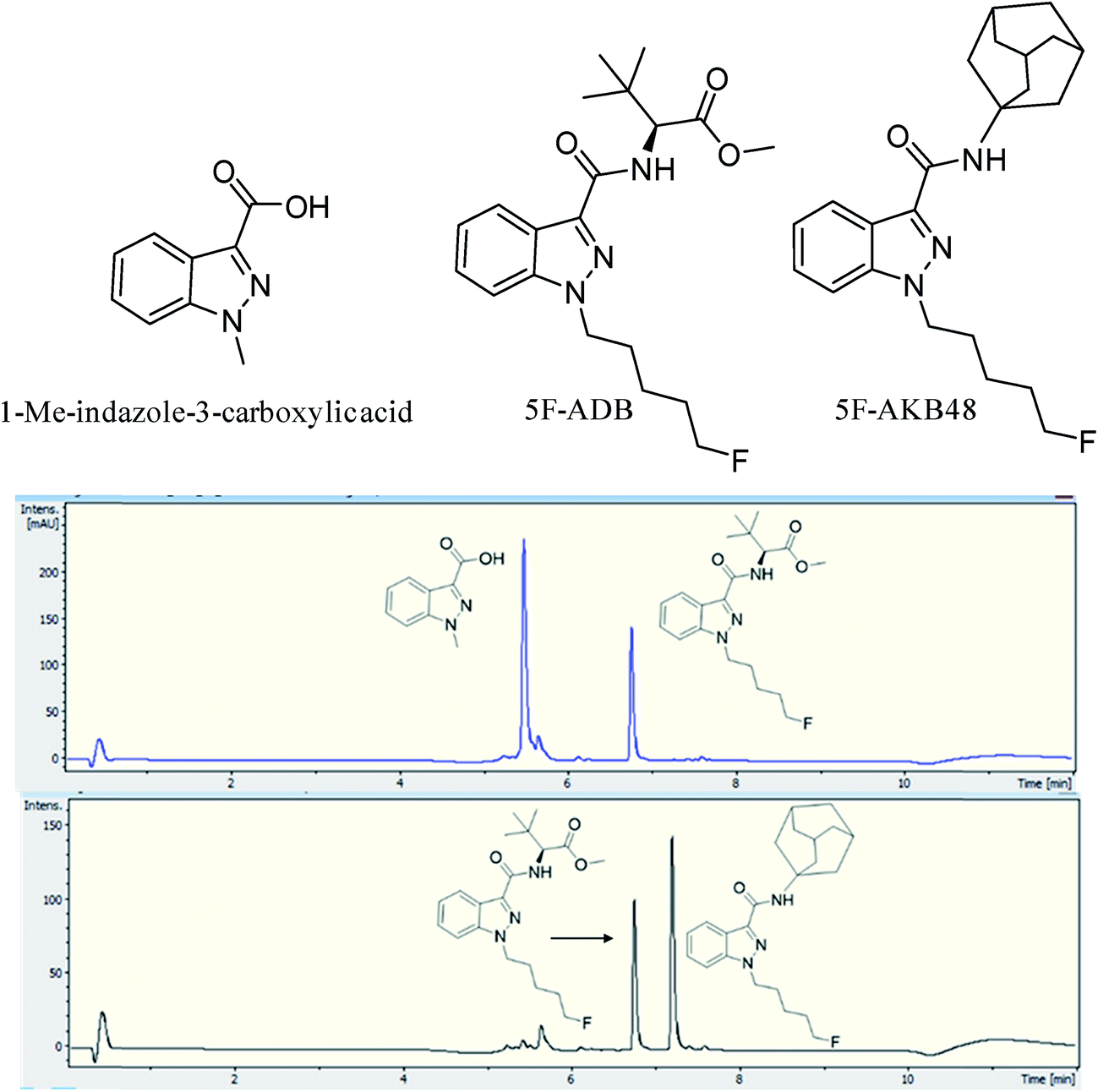 | ||
| Fig. 3 UHPLC chromatograms (λ = 298 nm) for an “Exodus” sample containing 5F-ADB, RT = 6.8 min, (upper) using 1-methylindazole-3-carboxylic acid as the IS, RT = 5.6 min, showing overlap with a matrix component, (lower) using 5F-AKB48 as the IS, RT = 7.2 min. | ||
 | ||
| Fig. 4 (Upper) stacked UHPLC chromatogram concentrations from 1.25 to 40.0 μg mL−1 (λ = 298 nm) of 5F-ADB (RT = 6.8 min) and 5F-AKB48 IS (RT = 7.2 min); (lower) calibration curve of 5F-ADB against 5F-AKB48 (IS), R2 = 0.9999. | ||
2016 seized “Exodus” sample analyses revealed a consistent dose of 5F-ADB across all 8 samples with an acceptable precision (RSD %) of less than 10% for the analysis of samples in the herbal form (Table 2).27 Furthermore, analysis using ANOVA two-factor with replication analysis of the four groups (19F coupled, 19F decoupled, 1H NMR, and UHPLC) revealed no statistically significant differences (p > 0.05). However, seven 2017 “Exodus” samples containing 5F-ADB revealed different quantitative results (Table 3). 5 packs of the 7 contained a similar dose of 5F-ADB to the 2016 samples, but samples 5 and 7 contained from 1.5 to more than double the dose of 5F-ADB, with good precision in most of the samples. The presence of such a large quantitative variation in the 2017 samples is alarming, especially as this recently identified SC (5F-ADB) is toxic, being implicated in 10 deaths in Japan,28,29 and it is comparable to similar analogues which have approximately 220-fold potency of that of THC, e.g. 5F-ADBICA EC50 = 0.77 nM compared to THC EC50 = 172 nM.1 The wide deviation and lack of homogeneity of the levels of 5F-ADB both within and between sample packages varied 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold from 0.8 μg g−1 to 49 mg g−1.28 An easy and robust quantitative analysis of fluorinated SCs is clearly important. This technique has the potential to be applied in the rapid analysis of herbal blends sprayed with fluorinated SCs, gaining in importance with the annual increase in the occurrence of such fluorinated third-generation SCs seen in 2016–2019.29–32 This analysis is of importance to users/abusers, health professionals and law enforcement to determine how much SC is in the sample. It also clearly demonstrates how there is no quality control of the “Exodus” preparations.
000-fold from 0.8 μg g−1 to 49 mg g−1.28 An easy and robust quantitative analysis of fluorinated SCs is clearly important. This technique has the potential to be applied in the rapid analysis of herbal blends sprayed with fluorinated SCs, gaining in importance with the annual increase in the occurrence of such fluorinated third-generation SCs seen in 2016–2019.29–32 This analysis is of importance to users/abusers, health professionals and law enforcement to determine how much SC is in the sample. It also clearly demonstrates how there is no quality control of the “Exodus” preparations.
| Sample | NMR n = 3 | UHPLC n = 4 | ||||||
|---|---|---|---|---|---|---|---|---|
| 1H | RSD % | 19F coupled | RSD % | 19F decoupled | RSD % | UHPLC | RSD % | |
| Exodus9 | 7.39 ± 0.20 | 2.74 | 7.34 ± 0.27 | 3.73 | 7.12 ± 0.24 | 3.36 | 6.96 ± 0.12 | 1.67 |
| Exodus10 | 8.04 ± 0.43 | 5.35 | 7.87 ± 0.37 | 4.69 | 7.87 ± 0.47 | 5.94 | 7.92 ± 0.38 | 4.79 |
| Exodus11 | 8.24 ± 0.17 | 2.00 | 8.01 ± 0.12 | 1.48 | 8.05 ± 0.06 | 0.79 | 7.87 ± 0.11 | 1.46 |
| Exodus12 | 8.04 ± 0.12 | 1.49 | 7.89 ± 0.07 | 0.88 | 7.93 ± 0.14 | 1.81 | 7.97 ± 0.10 | 1.20 |
| Exodus13 | 7.78 ± 0.02 | 0.21 | 7.71 ± 0.04 | 0.54 | 7.72 ± 0.05 | 0.65 | 8.19 ± 0.07 | 0.79 |
| Exodus14 | 7.65 ± 0.08 | 1.00 | 7.61 ± 0.07 | 0.90 | 7.66 ± 0.11 | 1.45 | 7.91 ± 0.12 | 1.52 |
| Exodus15 | 7.57 ± 0.22 | 2.93 | 7.52 ± 0.24 | 3.14 | 7.50 ± 0.17 | 2.26 | 7.54 ± 0.09 | 1.17 |
| Exodus16 | 7.43 ± 0.18 | 2.46 | 7.45 ± 0.21 | 2.75 | 7.48 ± 0.24 | 3.18 | 7.94 ± 0.14 | 1.82 |
| Sample | NMR n = 3 | UHPLC n = 4 | ||||||
|---|---|---|---|---|---|---|---|---|
| 1H | RSD % | 19F coupled | RSD % | 19F decoupled | RSD % | UHPLC | RSD % | |
| Exodus1 | 10.48 ± 0.29 | 2.78 | 10.65 ± 0.11 | 1.08 | 10.68 ± 0.12 | 1.11 | 12.44 ± 0.63 | 5.03 |
| Exodus2 | 7.81 ± 0.12 | 1.51 | 7.75 ± 0.07 | 0.92 | 7.77 ± 0.16 | 2.09 | 8.83 ± 0.03 | 0.34 |
| Exodus3 | 9.17 ± 0.13 | 1.41 | 9.07 ± 0.19 | 2.13 | 9.12 ± 0.23 | 2.55 | 11.10 ± 1.29 | 11.60 |
| Exodus4 | 8.69 ± 0.10 | 1.17 | 8.66 ± 0.13 | 1.49 | 8.60 ± 0.10 | 1.12 | 8.35 ± 0.15 | 1.80 |
| Exodus5 | 17.50 ± 0.09 | 0.53 | 17.48 ± 0.23 | 1.31 | 17.71 ± 0.05 | 0.26 | 20.22 ± 0.25 | 1.23 |
| Exodus6 | 8.70 ± 0.08 | 0.93 | 8.57 ± 0.09 | 1.01 | 8.59 ± 0.11 | 1.33 | 8.78 ± 0.07 | 0.83 |
| Exodus7 | 14.35 ± 0.10 | 0.69 | 13.88 ± 0.09 | 0.66 | 13.45 ± 0.53 | 3.92 | 16.08 ± 0.17 | 1.06 |
AM-694 quantified in seized herbal blends
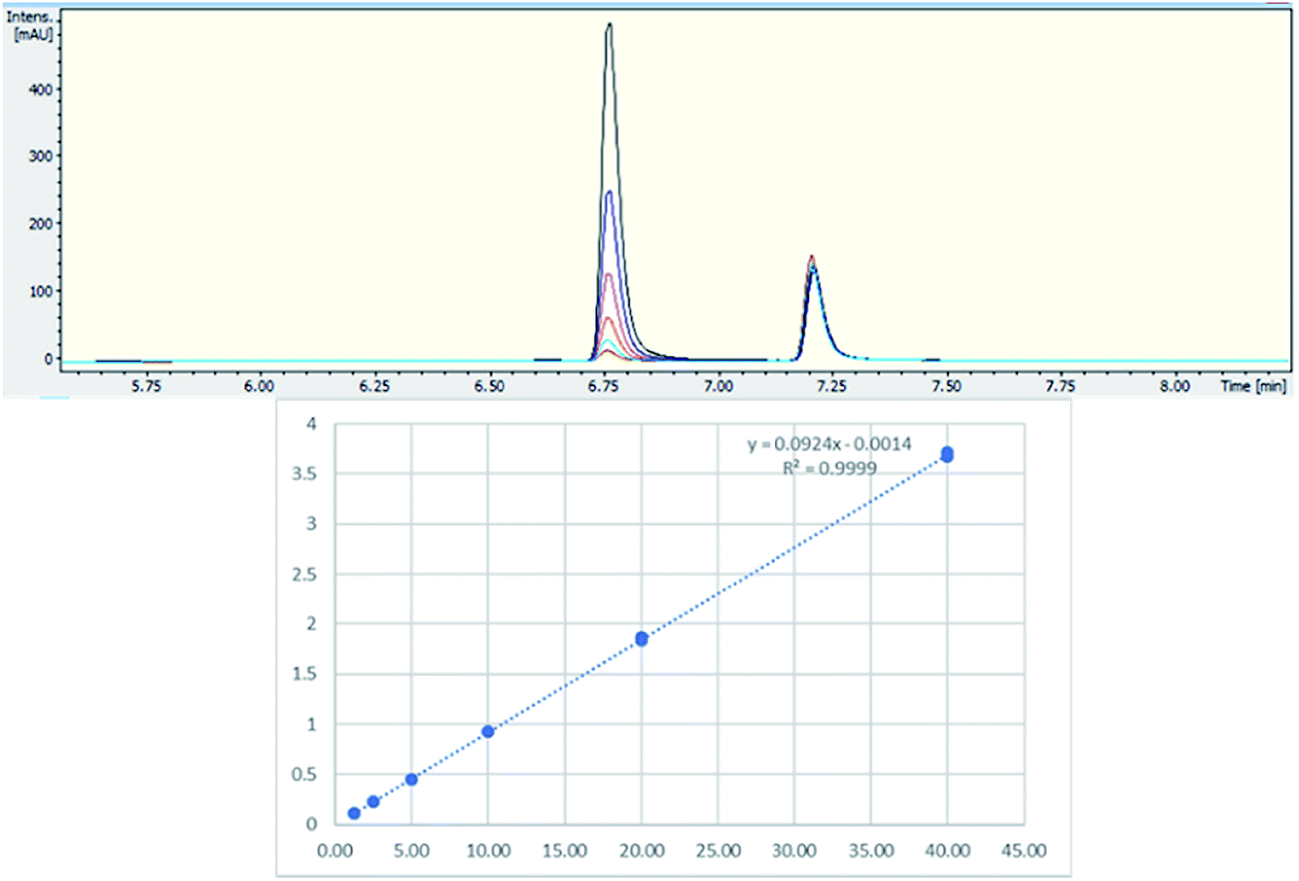
[1-(5-Fluoropentyl)-1H-indol-3-yl](2-iodophenyl)-methanone (AM-694) was isolated from seized herbal blends (3.0 g) branded as “Loco elite”. Identification was achieved through 2D NMR spectroscopy and its LC-MS/MS fragmentation pattern. Results were confirmed by comparison with the published literature of the first analytical characterization of illicit AM-694 from seizures.6 Candidate signals for integration are 3′′, 4′′, 5′′, and 6′′ of the 2-iodophenyl substituent, 2′ and 7′ of the indole core, and 1′′′ and 5′′′ of the fluoropentyl chain. The impact of relaxation delay in 19F NMR was investigated, and it was found that using only a short relaxation delay (<15 s) significantly affected the quantitative results. 15 s and 30 s relaxation delays were sufficient to achieve reproducible quantitative results. Moreover, such relaxation delays still allowed fast overall sample run-times of 8 and 10 min, respectively. 1H q-NMR and 19F q-NMR showed consistent results when using 2-chloro-4-fluorotoluene as the IS (Fig. 5). Furthermore, cross-method validation was demonstrated using UHPLC with reference standard AM-694, RT = 6.9 min, and JWH-018 (IS), RT = 7.3 min, constructing a seven-point calibration curve between 1.25 and 80.0 μg with R2 = 0.997 and IS RSD = 4.4% (Fig. 6).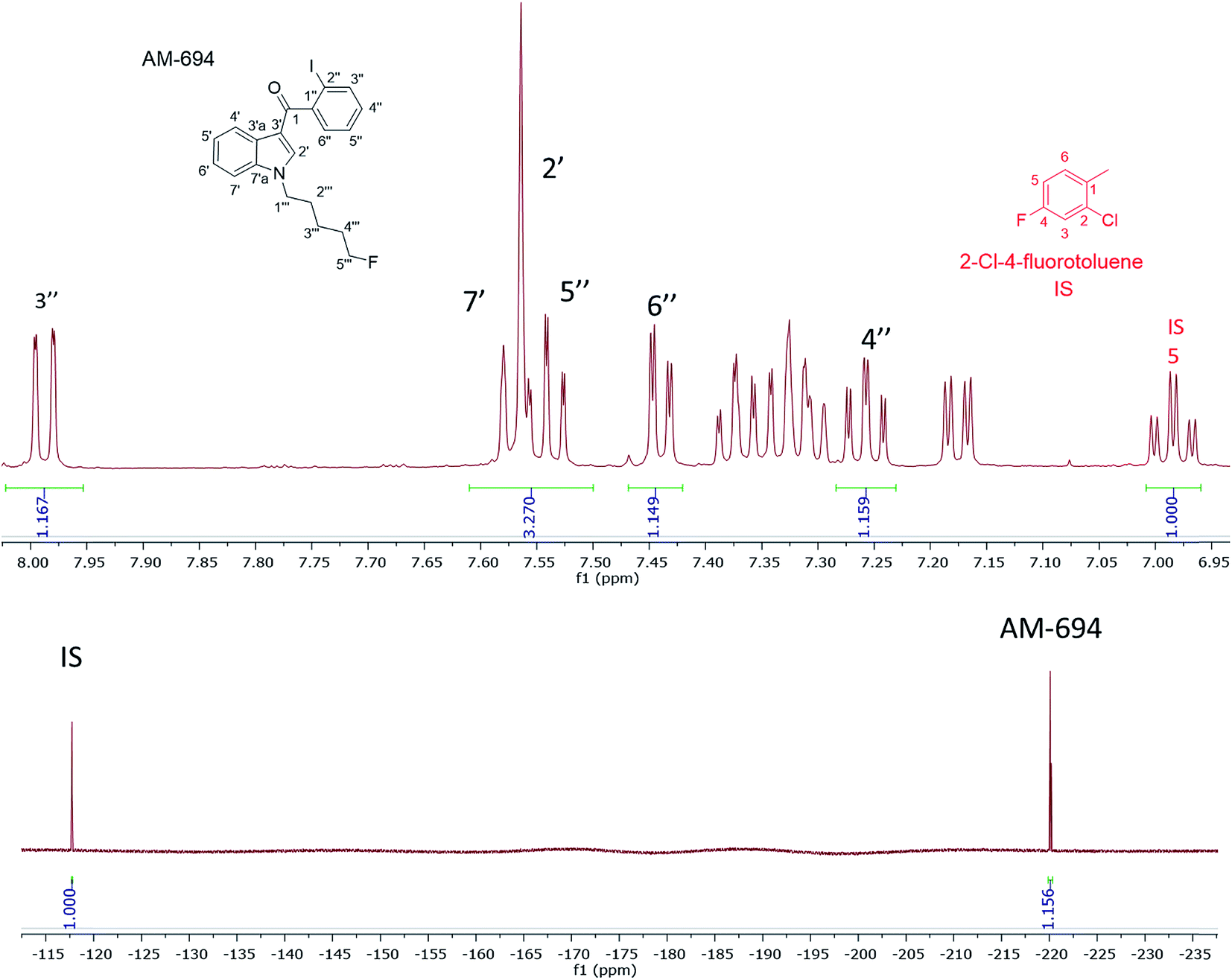 | ||
| Fig. 5 (Upper) expansion of the 1H NMR aromatic region of AM-694 in CD3OD showing signals used for quantification where the integration of the 2-chloro-4-fluorotoluene IS H5 signal (td) is normalized (1.00 H); (lower) 19F NMR signals of AM-694 at −220 ppm and the same IS 19F signal at −117 ppm also normalized (1.00 F). | ||
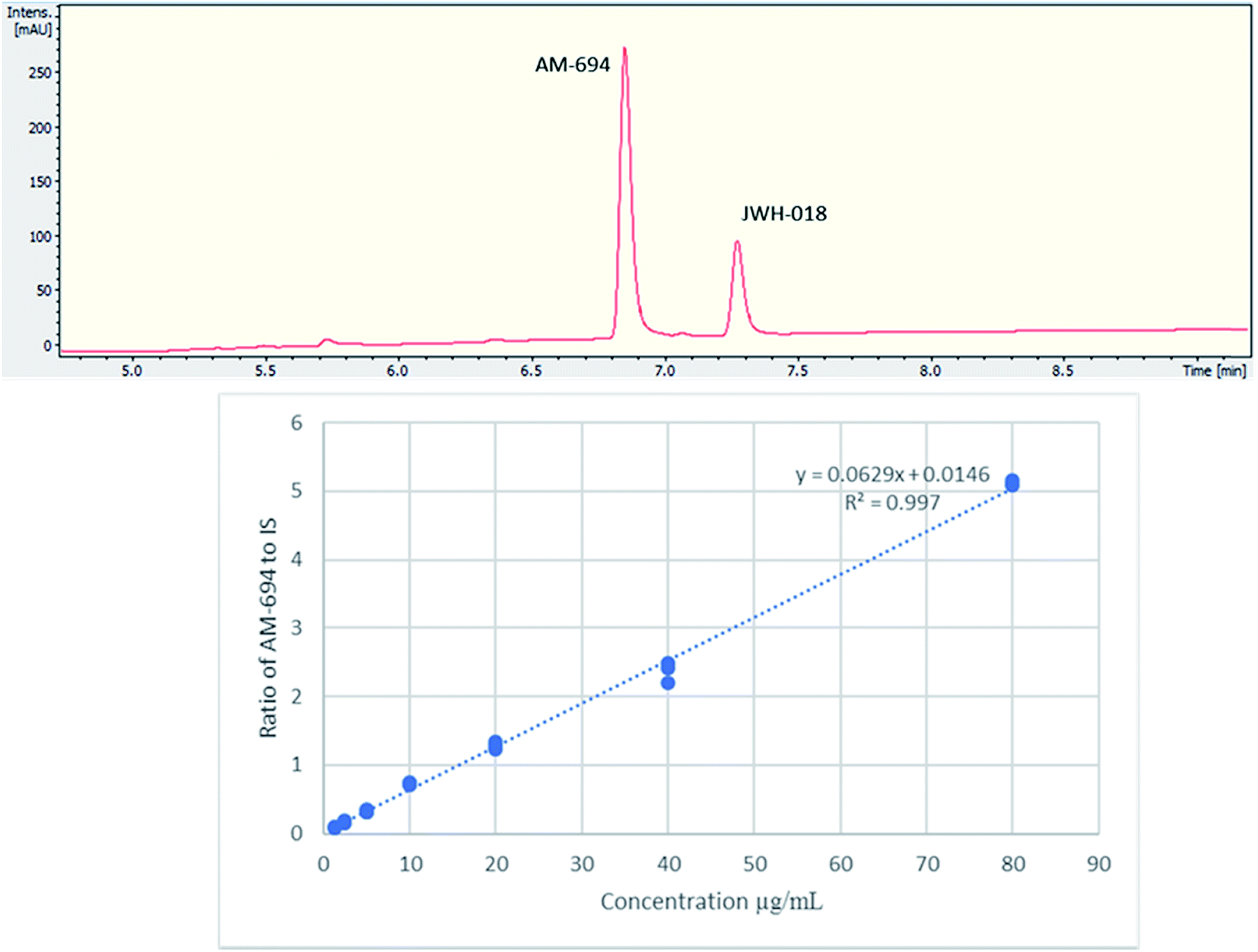 | ||
| Fig. 6 (Upper) UHPLC chromatogram (λ = 254 nm) of AM-694 (RT = 6.9 min) and JWH-018 (RT = 7.3 min); (lower) calibration curve of AM-694 against JWH-018 (IS), R2 = 0.997. | ||
Two samples were quantified, with significant differences (p < 0.05) in their AM-694 content. Sample 1 analysis using (only) 1H NMR spectroscopy, with maleic acid as the IS, showed 57.0 ± 2.9 mg g−1 of plant material, compared to the value for Sample 2 of 37.5 ± 1.1 mg. The latter 1H NMR quantification of Sample 2 was shown to be consistent when analysed by 1H NMR spectroscopy against both maleic acid and 2-chloro-4-fluorotoluene as the IS, and by 19F q-NMR, and also in agreement with UHPLC results, showing no significant differences between these methods using Anova two-factor analysis (p > 0.05) (Table 4).
In the 1H NMR when using 2-chloro-4-fluorotoluene as the IS, its H6 signal overlapped with 5′ of the indole and indazole SC. Nevertheless, other AM-694 signals such as 4′′, 6′′, and 7′ were resolved and used as candidate quantitative signals in CD3OD. 5-Fluoropentyl signals 1′′′ and 5′′′ were resolved when CDCl3 or CD3CN was used as the NMR solvent, providing further options for q-NMR analysis.
Conclusions
In this study, 19F-NMR spectroscopy has been applied for the first time to seized herbal blends containing fluorinated 3rd generation SCs to provide a fast (∼8 min), accurate and robust quantitative analytical method with no background interference from the plant-material matrix. This analytical technique requires almost no method development (beyond the NMR acquisition parameters) compared to chromatographic methods. There is no need to resort to any lengthy chromatographic analysis. 2-Chloro-4-fluorotoluene was used as an IS in 19F q-NMR, resulting in a method with close agreement with 1H q-NMR results using two different ISs, and cross-method validation was performed using UHPLC.Acquisition parameters such as the centre point of the spectral window and the relaxation delay have to be chosen carefully for accurate and precise outcomes. An inverse-gated decoupling NMR experiment was employed to improve the S/N ratio and to remove any NOE enhancement. That such analytical data are important is underlined by the analysis of packets of the “Exodus” brand containing 5F-ADB which revealed quantitative differences between 2016 and 2017 seizures in the dose of 5F-ADB, with some packets having double the dose compared to others.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Drug Expert Action Team (DEAT), Avon and Somerset Constabulary, for the provision of seized samples, and the Government of Kuwait (a fully funded studentship to HAN).References
- S. D. Banister, M. Moir, J. Stuart, R. C. Kevin, K. E. Wood, M. Longworth, S. M. Wilkinson, C. Beinat, A. S. Buchanan, M. Glass, M. Connor, I. S. McGregor and M. Kassiou, Pharmacology of indole and indazole synthetic cannabinoid designer drugs AB-FUBINACA, ADB-FUBINACA, AB-PINACA, ADBPINACA, 5F-AB-PINACA, 5F-ADB-PINACA, ADBICA, and 5F-ADBICA, ACS Chem. Neurosci., 2015, 6, 1546–1559 CrossRef CAS PubMed.
- S. Tai and W. E. Fantegrossi, Synthetic cannabinoids: Pharmacology, behavioral effects, and abuse potential, Current Addiction Reports, 2014, 1, 129–136 CrossRef PubMed.
- V. Auwärter, S. Dresen, W. Weinmann, M. Muller, M. Putz and N. Ferreiros, ‘Spice’ and other herbal blends: harmless incense or cannabinoid designer drugs?, J. Mass Spectrom., 2009, 44, 832–837, DOI:10.1002/jms.1558.
- J. W. Huffman, D. Dai, B. R. Martin and D. R. Compton, Design, synthesis, and pharmacology of cannabimimetic indoles, Bioorg. Med. Chem. Lett., 1994, 4, 563–566 CrossRef.
- S. D. Banister, J. Stuart, R. C. Kevin, A. Edington, M. Longworth, S. M. Wilkinson, C. Beinat, A. S. Buchanan, D. E. Hibbs, M. Glass, M. Connor, I. S. McGregor and M. Kassiou, Effects of bioisosteric fluorine in synthetic cannabinoid designer drugs JWH-018, AM-2201, UR-144, XLR-11, PB-22, 5F-PB-22, APICA, and STS-135, ACS Chem. Neurosci., 2015, 6, 1445–1458 CrossRef CAS PubMed.
- J. I. Nakajima, M. Takahashi, T. Seto, C. Kanai, J. Suzuki, M. Yoshida and T. Hamano, Identification and quantitation of two benzoylindoles AM-694 and (4-methoxyphenyl)(1-pentyl-1H-indol-3-yl)methanone, and three cannabimimetic naphthoylindoles JWH-210, JWH-122, and JWH-019 as adulterants in illegal products obtained via the internet, Forensic Toxicol., 2011, 29, 95–110 CrossRef CAS.
- N. Uchiyama, Y. Shimokawa, R. Kikura-Hanajiri, Y. Demizu, Y. Goda and T. Hakamatsuka, A synthetic cannabinoid FDU-NNEI, two 2H-indazole isomers of synthetic cannabinoids AB-CHMINACA and NNEI indazole analog (MN-18), a phenethylamine derivative N-OH-EDMA, and a cathinone derivative dimethoxy-alpha-PHP, newly identified in illegal products, Forensic Toxicol., 2015, 33, 244–259, DOI:10.1007/s11419-015-0268-7.
- H. Chung, H. Choi, S. Heo, K. Eunmi and J. Lee, Synthetic cannabinoids abused in South Korea: drug identifications by the National Forensic Service from 2009 to June 2013, Forensic Toxicol., 2014, 32, 82–88 CrossRef CAS.
- A. Makriyannis and H. Deng, University of Connecticut. Cannabimimetic indole derivatives, Patent US6900236B1, 2005, https://patentimages.storage.googleapis.com/9d/ec/b4/ea8af239e287da/US6900236.pdf, accessed 17/04/19.
- S. M. Wilkinson, S. D. Banister and M. Kassiou, Bioisosteric fluorine in the clandestine design of synthetic cannabinoids, Aust. J. Chem., 2015, 68, 4–8 CrossRef CAS.
- A. Zivkovic, J. J. Bandolik, A. J. Skerhut, C. Coesfeld, M. Prascevic, L. Zivkovic and H. Stark, Quantitative analysis of multicomponent mixtures of over-the counter pain killer drugs by low-field NMR spectroscopy, J. Chem. Educ., 2017, 94, 121–125, DOI:10.1021/acs.jchemed.6b00105.
- G. Maniara, K. Rajamoorthi, S. Rajan and G. W. Stockton, Method performance and validation for quantitative analysis by 1H and 31P NMR spectroscopy. Applications to analytical standards and agricultural chemicals, Anal. Chem., 1998, 70, 4921–4928 CrossRef CAS PubMed.
- U. Holzgrabe, Quantitative NMR spectroscopy in pharmaceutical applications, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 229–240 CrossRef CAS PubMed.
- P. A. Hays, Proton nuclear magnetic resonance spectroscopy (NMR) methods for determining the purity of reference drug standards and illicit forensic drug seizures, J. Forensic Sci., 2005, 50, 1342–1360 CrossRef CAS.
- F. Malz and H. Jancke, Validation of quantitative NMR, J. Pharm. Biomed. Anal., 2005, 38, 813–823 CrossRef CAS PubMed.
- J. P. Smith, O. B. Sutcliffe and C. E. Banks, An overview of recent developments in the analytical detection of New Psychoactive Substances (NPSs), Analyst, 2015, 140, 4932–4948 RSC.
- A. Frinculescu, C. L. Lyall, J. Ramsey and B. Miserez, Variation in commercial smoking mixtures containing third-generation synthetic cannabinoids, Drug Test. Anal., 2017, 9, 327–333 CrossRef CAS PubMed.
- F. Fowler, B. Voyer, M. Marino, J. Finzel, M. Veltri, N. M. Wachter and L. Huang, Rapid screening and quantification of synthetic cannabinoids in herbal products with NMR spectroscopic methods, Anal. Methods, 2015, 7, 7907–7916 RSC.
- S. J. Dunne and J. P. Rosengren-Holmberg, Quantification of synthetic cannabinoids in herbal smoking blends using NMR, Drug Test. Anal., 2017, 9, 734–743 CrossRef CAS PubMed.
- G. F. Pauli, B. U. Jaki and D. C. Lankin, Quantitative 1H NMR: development and potential of a method for natural products analysis, J. Nat. Prod., 2005, 68, 133–149 CrossRef CAS PubMed.
- R. Martino, V. Gilard, F. Desmoulin and M. Malet-Martino, Fluorine-19 or phosphorus-31 NMR spectroscopy: A suitable analytical technique for quantitative in vitro metabolic studies of fluorinated or phosphorylated drugs, J. Pharm. Biomed. Anal., 2005, 38, 871–891 CrossRef CAS PubMed.
- S. J. Barry, T. N. Pham, P. J. Borman, A. J. Edwards and S. A. Watson, A risk-based statistical investigation of the quantification of polymorphic purity of a pharmaceutical candidate by solid-state 19F NMR, Anal. Chim. Acta, 2012, 712, 30–36, DOI:10.1016/j.aca.2011.10.064.
- A. J. Adams, S. D. Banister, L. Irizarry, J. Trecki, M. Schwartz and R. Gerona, “Zombie” outbreak caused by the synthetic cannabinoid AMB-FUBINACA in New York, N. Engl. J. Med., 2017, 376, 235–242, DOI:10.1056/nejmoa1610300.
- B. K. Logan, L. E. Reinhold, A. Xu and F. X. Diamond, Identification of synthetic cannabinoids in herbal incense blends in the United States, J. Forensic Sci., 2012, 57, 1168–1180 CrossRef CAS PubMed.
- V. Shevyrin, V. Melkozerov, A. Nevero, O. Eltsov, Y. Shafran, Y. Morzherin and A. T. Lebedev, Identification and analytical characteristics of synthetic cannabinoids with an indazole-3-carboxamide structure bearing a N-1-methoxycarbonylalkyl group, Anal. Bioanal. Chem., 2015, 407, 6301–6315, DOI:10.1007/s00216-015-8612-7.
- S. Akamatsu and M. Yoshida, Fragmentation of synthetic cannabinoids with an isopropyl group or a tert-butyl group ionized by electron impact and electrospray, J. Mass Spectrom., 2016, 51, 28–32 CrossRef CAS PubMed.
- European Network of Forensic Science Institutes - Drugs Working Group, Guidelines on sampling of illicit drugs for quantitative analysis, 2014, http://enfsi.eu/wp-content/uploads/2016/09/guidelines_quant_sampling_dwg_printing_vf4.pdf accessed 17/04/19 Search PubMed.
- K. Hasegawa, A. Wurita, K. Minakata, K. Gonmori, I. Yamagishi, H. Nozawa, K. Watanabe and O. Suzuki, Identification and quantitation of 5-fluoro-ADB, one of the most dangerous synthetic cannabinoids, in the stomach contents and solid tissues of a human cadaver and in some herbal products, Forensic Toxicol., 2015, 33, 112–121, DOI:10.1007/s11419-014-0259-0.
- S. D. Banister, M. Longworth, R. Kevin, S. Sachdev, M. Santiago, J. Stuart, J. B. C. Mack, M. Glass, I. S. McGregor, M. Connor and M. Kassiou, Pharmacology of valinate and tert-leucinate synthetic cannabinoids 5F-AMBICA, 5F-AMB, 5F-ADB, AMB-FUBINACA, MDMB-FUBINACA, MDMB-CHMICA, and their analogues, ACS Chem. Neurosci., 2016, 7, 1241–1254, DOI:10.1021/acschemneuro.6b00137.
- M. D. P. Risseeuw, P. Blanckaert, V. Coopman, S. Van Quekelberghe, S. Van Calenbergh and J. Cordonnier, Identification of a new tert-leucinate class synthetic cannabinoid in powder and “spice-like” herbal incenses: methyl 2-[[1-(5-fluoropentyl)indole-3-carbonyl]amino]-3,3-dimethyl-butanoate (5F-MDMB-PICA), Forensic Sci. Int., 2017, 273, 45–52, DOI:10.1016/j.forsciint.2017.01.023.
- L. Mogler, F. Franz, D. Rentsch, V. Angerer, G. Weinfurtner, M. Longworth, S. D. Banister, M. Kassiou, B. Moosmann and V. Auwärter, Detection of the recently emerged synthetic cannabinoid 5F-MDMB-PICA in ‘legal high’ products and human urine samples, Drug Test. Anal., 2018, 10, 196–205, DOI:10.1002/dta.2201.
- S. D. Banister, A. Adams, R. C. Kevin, C. Macdonald, M. Glass, R. Boyd, M. Connor, I. S. McGregor, C. M. Havel, S. J. Bright, M. V. Vilamala, C. G. Lladanosa, M. J. Barratt and R. R. Gerona, Synthesis and pharmacology of new psychoactive substance 5F-CUMYL-P7AICA, a scaffold-hopping analog of synthetic cannabinoid receptor agonists 5F-CUMYL-PICA and 5F-CUMYL-PINACA, Drug Test. Anal., 2019, 11, 279–291, DOI:10.1002/dta.2491.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows the 19F-NMR (1H coupled) stacked spectra of 5F-ADB with its 19F signal at −220.2 ppm and the IS 2-chloro-4-fluorotoluene (−117.8 ppm) with different O1P set at (A) −165 ppm, (B) −220 ppm, and (C) −117 ppm, with (inset) expansion of the of 5F-ADB 19F signal. Fig. S2 shows 5F-ADB quantified in a sample using 1H, 19F proton coupled, and 19F inverse-gated decoupled spectroscopy. See DOI: 10.1039/c9ay00814d |
| This journal is © The Royal Society of Chemistry 2019 |