
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall-molecule fluorescent probes and their design
Yanhua Fu and
Nathaniel S. Finney *
*
School of Pharmaceutical Sciences and Technology, Health Sciences Platform, Tianjin University, 92 Weijin Road, Nankai District, Tianjin, 300072, China. E-mail: nfinney@tju.edu.cn
First published on 14th August 2018
Abstract
Small-molecule fluorescent probes have become powerful tools for using light to advance the study of cell biology, discover new drugs, detect environmental contaminants, and further the detection of cancer. These applications correlate with the expansion of the fluorescent probe research community – small in the late 20th century, now a collection of more than a hundred research groups world-wide. This expansion required the entry of adventurous scientists from many other fields. This tutorial review introduces some important concepts related to fluorescent probe development. It is hoped that it will facilitate further expansion of the field by demystifying it.
Overview
Small-molecule fluorescent probes are molecules that change their fluorescence emission in response to a binding event, chemical reaction, or change in their immediate environment.1 They are now widely used in drug discovery, cellular imaging, environmental analysis, and various medical applications.1 Among the reasons for this are: fluorescence emission can be measured with great sensitivity; fluorescence microscopy allows remarkable spatial and temporal resolution in cell imaging; and, many fluorophores are now readily accessible. There is precedent for many applications, so fluorescent probes can be used by those who are not actively engaged in the research field. Probe structure can be varied to control excitation and emission wavelengths, target-binding affinity, chemical reactivity, and subcellular localization.2 Fluorescence is also amenable to high throughput analysis for screening applications,3 construction of fiber optics for medical and environmental measurements,4 and visual detection of analytes.5While much of this is appreciated in a general sense, there is still a significant activation barrier to entering the field. Scholarly articles are written to make results and conclusions accessible, but the underlying details of probe design and mechanism are seldom discussed. Thus, while the importance of fluorescent probes may be readily apparent, making the transition to designing and synthesizing new probes for use in one's own research remains, at the least, challenging.
The purpose of this Tutorial Review is to introduce the “intelligent non-expert” to the fundamentals of small-molecule fluorescent probe function. We present several central concepts, with representative examples. Organization of the review is based on three common mechanisms by which binding to, or reaction with, an analyte of interest can be converted to a change in fluorescence. We hope that understanding how these mechanisms are coupled to probe design will empower non-expert scientists to invent new probes.
This is not intended as a comprehensive review. Illustrative examples have been selected from recent literature, with a single exception. Apologies are given in advance to the pioneers of the field, and the many researchers whose work is not directly addressed.
We begin with a brief summary of some essential aspects of fluorescence. Following, three widely-used mechanisms for “fluorescent signal transduction” are described. Simplified explanations of the relevant electronic processes are provided, to accompany the examples. The final section provides a short overview of some commonly-used fluorophores, the ways in which they can be easily modified, and their advantages and limitations.
Summary of fluorescence fundamentals
The fluorescence of organic molecules is closely associated with delocalized electronic structure. Conjugated π systems absorb UV or visible light. Deletion of specific absorbed wavelengths from reflected visible light leads to our perception of color. However, a very small fraction of conjugated systems convert the absorbed energy into re-emission of light-fluorescence.6In slightly more detail, absorbance of light by a conjugated π system is the result of the energy of incoming UV and/or visible light matching the π/π* energy gap. This allows excitation of a HOMO π electron to the π* orbital. (Prior to excitation, the π* orbital would be denoted as the LUMO.) This generates a high-energy (excited) state of the molecule, where one electron populates the antibonding π* orbital, and one electron remains in what was the fully-bonding π orbital (Fig. 1). Throughout the rest of the paper, we simply describe the π HOMO and the π* LUMO, and the corresponding excited state orbitals, as the “π” and “π*” orbitals.
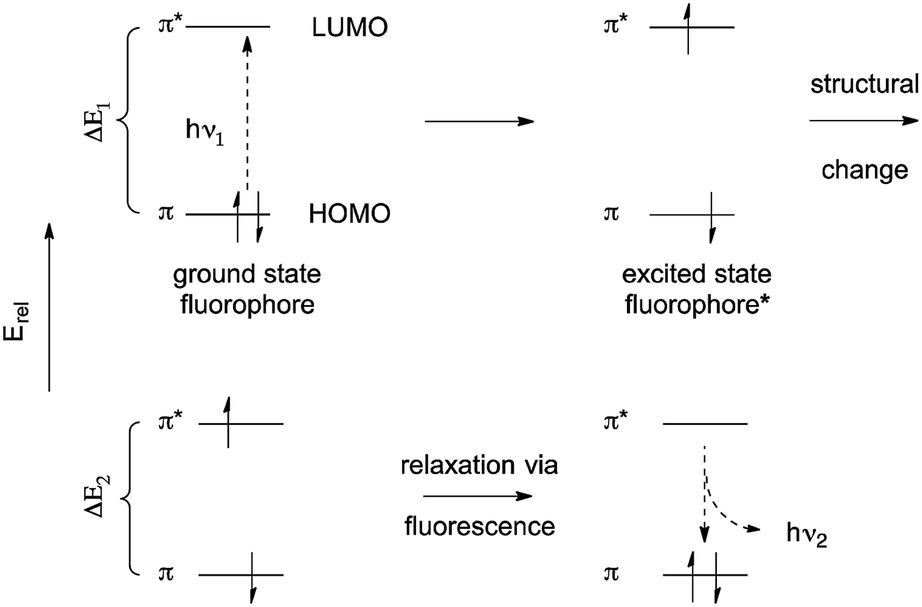 | ||
| Fig. 1 Simple scheme for excitation and emission in conjugated π systems.a,b,c aAs a matter of convention, excited states are usually denoted with an asterisk(*). Absorbed or emitted light is routinely abbreviated as hν, where ν denotes frequency, and hν gives the energy of the radiation. bThe initial excited state is formed with the electrons still spin-paired. cTerminology for ground states and excited states is deliberately minimized in this review. For more comprehensive discussion, see ref. 2. | ||
The excited state is higher in energy than the ground state, so there is a thermodynamic driving force for returning to the fully bonded ground state. There are three primary mechanisms for dissipating excited state energy: internal conversion (IC), the mechanical dissipation of energy; intersystem crossing (ISC), the lowering of energy by inverting the spin of one of the electrons; and fluorescence. Fluorescence is the emission of a photon of the exact energy separating the excited state at a given configuration, and allows direct relaxation.
The utility of fluorescence originates with the difference between the excitation and emission wavelengths. Because the excitation and emission wavelengths are different, emission intensity can be measured with minimized interference from the incoming excitation light. Without this, it would impossible to distinguish input and output. (There are other factors involved, but they all originate with this one.)
The difference between excitation and emission wavelengths depends on two factors. The first has to do with the difference between the lowest energy structure of the excited state and the lowest-energy structure of the ground state. Excitation occurs on a timescale (∼1013 s−1) much shorter than structural relaxation, so one can think of the fluorophore as being “frozen” during transition from the ground state to the excited state. This means that the excited state is initially formed at the optimal structure for the ground state – not the excited state. Excited state molecules persist long enough (∼5–10 ns) to undergo structural relaxation.6,7 Subsequent emission then necessarily leads to formation of the ground state that has the optimal structure for the excited state, not the ground state. The combination of these effects narrow the energetic separation of the ground and excited state. This energetic loss leads to emission at longer wavelength than absorption, given E = hc/λ (see Fig. 1; ΔE1 > ΔE2). The difference the excitation and emission λ values is called the “Stokes shift”.6,8 We neglect here a discussion of vibrational energy levels, although this is very important.6,7
For most organic π systems, the non-radiative IC and/or ISC processes are much faster than fluorescence emission, which is why most molecules do not fluoresce. IC involves mechanical dissipation of energy. For example, this is why black clothing gets hot in sunlight – the absorbed energy is converted to heat. ISC can only be described quantum mechanically – it does not have a clear counterpart in classical mechanics. This makes it conceptually inaccessible to most organic and biomolecular chemists. ISC involves an electron spin inversion, leading to a “triplet” state with two spin-unpaired electrons. The triplet state is always lower in energy than the excited singlet state.6,9 At room temperature, this triplet state relaxes non-radiatively to the ground state.10 Fluorescence, too, can only be well described in quantum mechanical terms. However, it is easier to conceptualize than ISC: emission is the reverse of the absorption process. Once the electronic basis for absorption leading the appearance of color has been accepted, accepting the reverse is less mystifying.
Hereafter, we will focus primarily on fluorescence. IC and ISC are fundamentally important, but controlling them is the basis for a minority of fluorescent probes.11 Most fluorescent probes are based on other mechanisms by which fluorescence emission can be controlled. The three mechanisms that will be discussed are: suppression of photoinduced electron transfer (PET); modulation of intramolecular charge transfer (ICT); and, Förster resonance energy transfer (FRET). Another important concept, blended into the examples shown below, is the chemical transformation of a probe to change its emission properties.12 This will not be discussed as a separate topic.
Photoinduced electron transfer (PET)
Suppression of photoinduced electron transfer (PET) is one of the most commonly used methods for converting a non-fluorescent molecule into a fluorescent molecule.1,6 PET involves electron transfer from a donor (D) to the excited state of a fluorophore. A requirement is that, energetically, the donor electron's energy must lie between the energies of the π and π* orbitals. PET lowers the net energy of the excited state, and blocks the π* → π relaxation that leads to fluorescence (Fig. 2).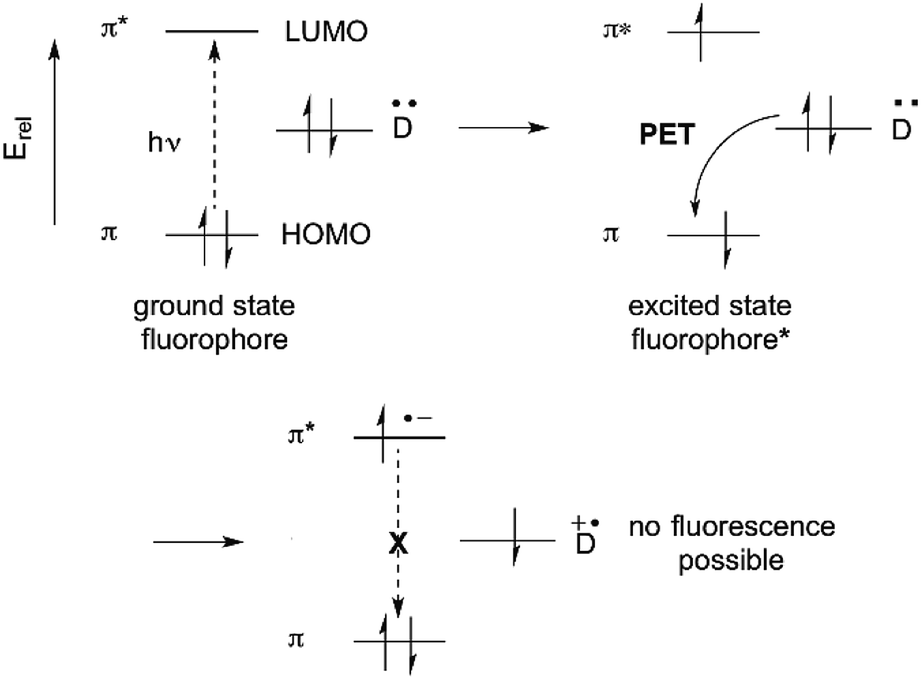 | ||
| Fig. 2 Photoinduced electron transfer (PET) quenching by a donor (D). | ||
Historically, benzylic nitrogen atoms are among the most common donors. PET from the N atom lone pair quenches fluorescence.1,13,14 Metal ion coordination or protonation turns the lone pair into a σ-bonding electron pair, which is far lower than the π-orbital in energy. PET is suppressed, and fluorescence is restored. This remains a powerful approach to metal ion detection and imaging, and for pH-responsive fluorescent probes.15 An important feature of this approach is reversible binding, which allows measurement of both increases and decreases of analyte concentration.
An alternative is to use an electron-rich heterocycle as a donor. If the donor π orbital is higher in energy that the fluorophore π orbital, PET can also be very efficient. Covalent modification induced by the analyte can reduce the electron density of the donor π system, eliminating PET. This approach has proven very important in detecting reactive molecules such as reactive oxygen species (ROS).16 Covalent modification is almost always irreversible. Thus, this approach measures the cumulative presence of a reactive analyte; decreases in concentration cannot be detected.
Two illustrative examples are shown below (Fig. 3 and 4).17,18
The first probe (Fig. 3) is based on a fluorescein-like fluorophore.17 In its excited state, the fluorescence emission is quenched by PET from the tertiary amine. Two substituents of the amine are thioethers, which are known high-affinity Cu2+ chelating groups. Upon binding to Cu2+ in aqueous media, the lone pair of the amine is converted to a bonding electron pair via coordination. This eliminates PET quenching, and allows the probe to become emissive. Selective detection of Cu2+ in biological and environmental systems is important, and this particular probe has been used extensively in biological imaging.
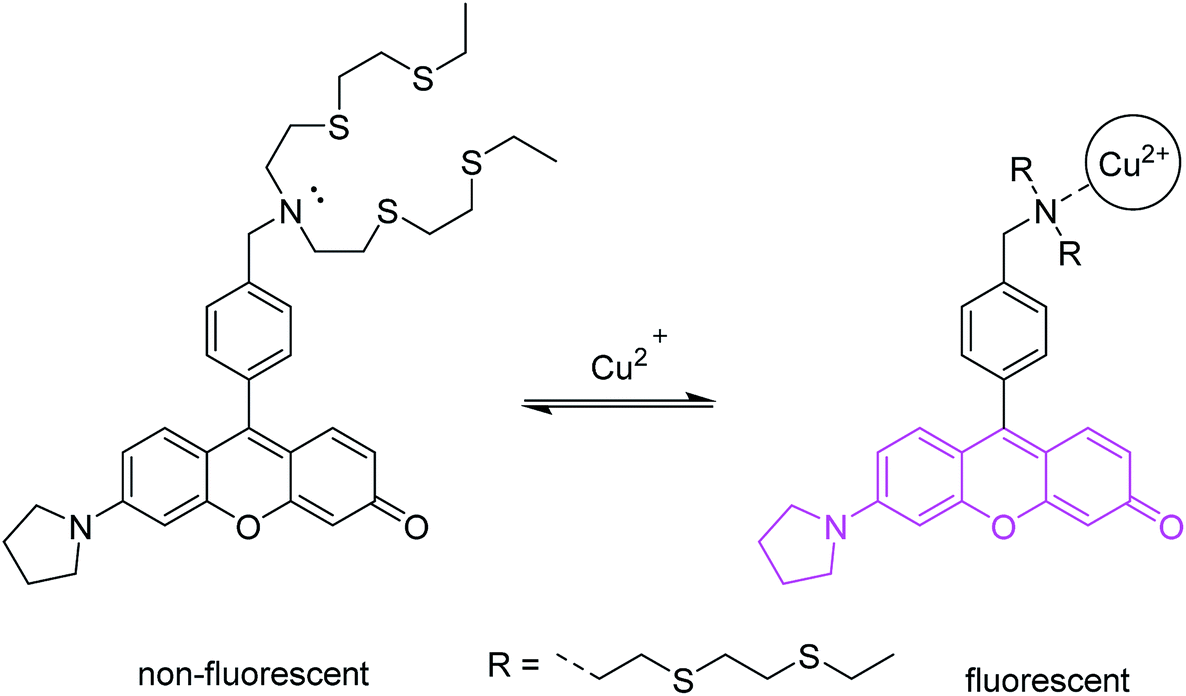 | ||
| Fig. 3 A PET probe for trace cellular Cu2+. a Structure of Cu2+ coordination complex not shown in detail. | ||
The second probe (Fig. 4) is highly selective for the detection of hypochlorite.18 Hypochlorite is among the reactive oxygen species (ROS) generated in cells as the result of the leakage of even more energetic ROS from the mitochondria.18 Hypochlorite can damage proteins, DNA, and RNA. These oxidative events are associated with numerous diseases, and the process of aging. The probe is based on a “BODIPY” fluorophore.19 (See final section on common fluorophores.) In the initial state, emission from the BODIPY unit is quenched by PET from the electron rich pyrrole. The pyrrole can be oxidized by hypochlorite to a ketone. The “pyrrol-3-one” is no longer electron-donating enough to quench emission via PET, which leads to fluorescence enhancement.
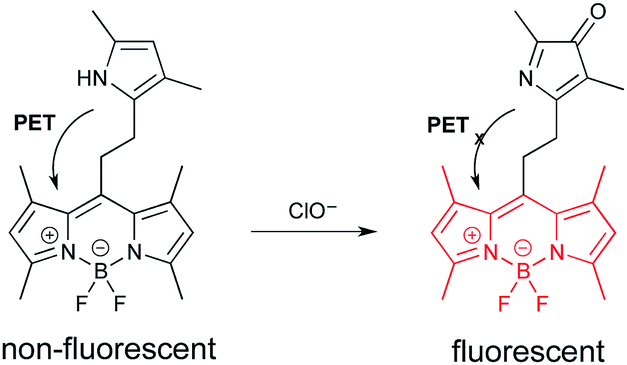 | ||
| Fig. 4 A PET probe for cellular hypochlorite based on oxidation of a pyrrole. | ||
Intramolecular charge transfer (ICT)
Along with suppression of PET, modulation of intramolecular charge transfer (ICT) is one of the most common signaling mechanisms used in fluorescent probe design. Fluorophores that alter their emission via changes in ICT usually have an electron donor (D) on one end of the fluorophore, and an electron acceptor (A) on the other end.1 In some cases, the fluorophore itself serves as the donor or the acceptor. That is, the fluorophore bears only a donor or an acceptor group.The excited state of ICT systems most often has a stronger dipole moment than the ground state.20 Simplified explanations of the relevant electronic processes are provided, to accompany the examples. (Fig. 5 and 6).
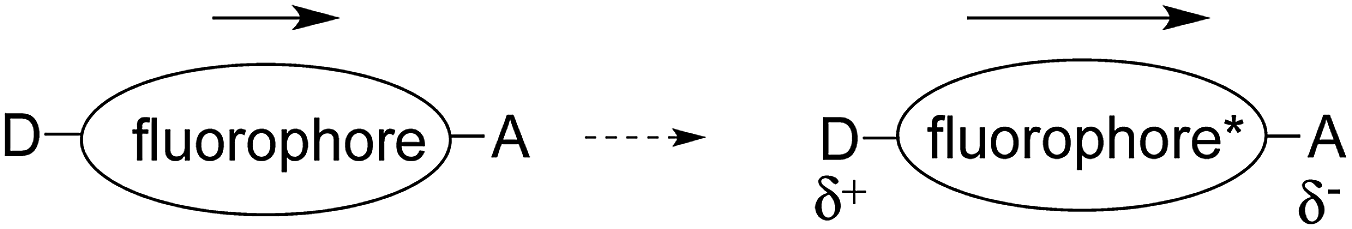 | ||
| Fig. 5 Increase in dipole moment upon excitation to the excited state. | ||
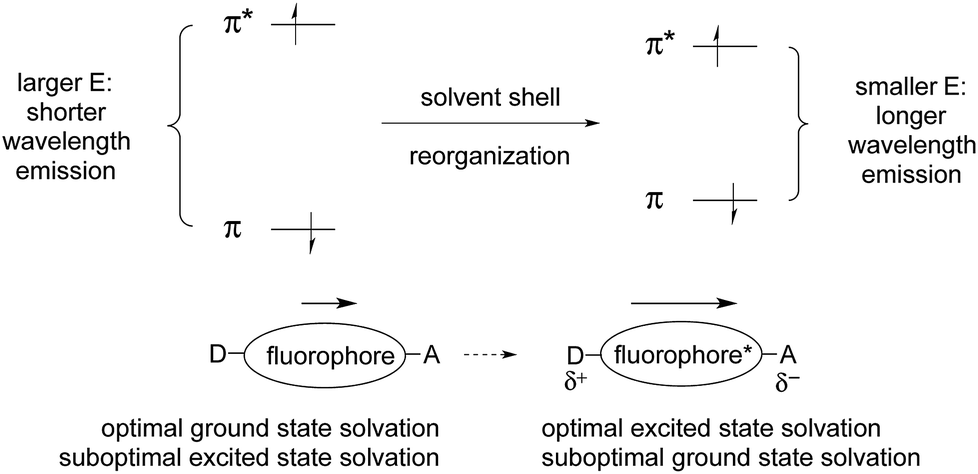 | ||
| Fig. 6 Energy diagram correlating ICT with solvation, in polar solvents. | ||
As a result of this polarization change, the solvation that was optimal for the ground state is not optimal for the excited state. The excited state lasts long enough to allow rearrangement of the solvation shell to optimize solvation. This lowers the energy of the excited state. But fluorescence emission is so fast that there is not enough time for the solvent shell to rearrange when returning to the ground state. This results in the formation of the ground state with sub-optimal solvation. This, in turn, raises the relative energy of the initially formed ground state, which further reduces the π/π*energy gap. These effects lead to longer wavelength emission (E = hc/λ).
Coordination of a metal ion, or covalent reaction, can perturb the polarization of the ground and excited states, which in turn changes the energetic separation of the excited and ground states. This leads to a shift in emission wavelength, as discussed above. If ICT is reduced, the emission will shift to shorter wavelength. If ICT is increased, the emission will shift to longer wavelength.
Two relevant examples are shown below (Fig. 7 and 8).21,22 The first is an exception to this review's focus on very recent literature. In a pioneering example, “fura-2” was developed as a highly-selective fluorescent probe for intracellular Ca2+.21 Upon reversible binding of Ca2+, coordination to the anilinic nitrogen blocks ICT, because conjugation of the donor nitrogen to the fluorophore is broken. Emission then shifts to shorter wavelength, as expected upon reduction of the excited state dipole. This allows the emission intensity of fura-2 and its Ca2+ complex to be measured separately, so that changes in [Ca2+] can readily be measured. While this was not the first Ca2+-responsive probe, it was functionally superior to previous probes, and helped revolutionize the study of Ca2+-dependent cellular processes using fluorescence microscopy.
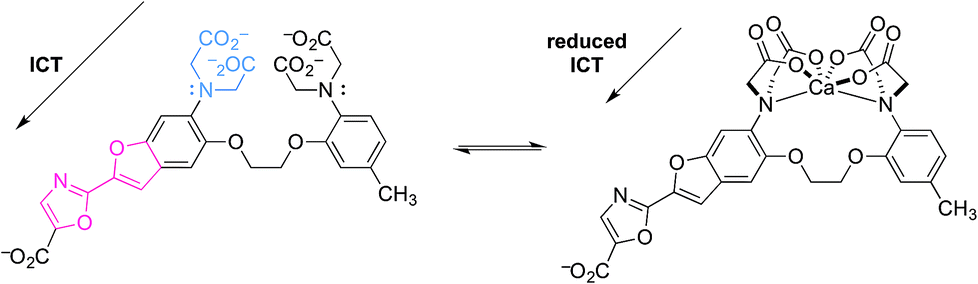 | ||
| Fig. 7 A pioneering ICT-based probe for cellular Ca2+ imaging, fura-2. | ||
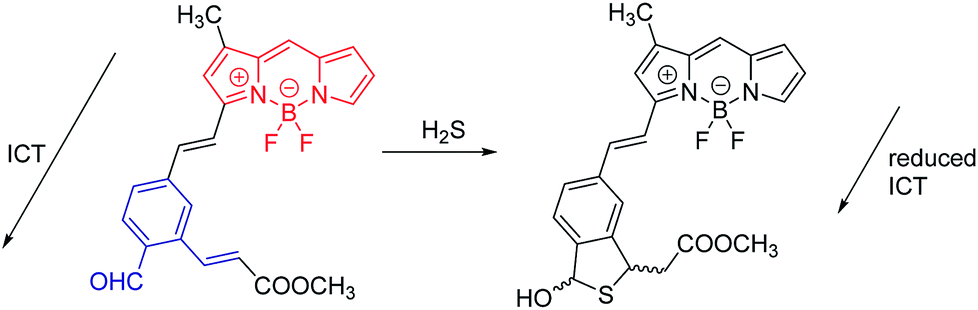 | ||
| Fig. 8 An ICT-based probe for H2S in cells. | ||
The second example (Fig. 8) is a probe for H2S. Remarkably, H2S proves to be a trace contributor to intracellular signaling.22 This area of research is still in its early stages, but has been dramatically advanced by the development of fluorescent probes such the one shown. Here, the initial ICT state is non-radiative, because of conjugation to the aldehyde. In the initial state, the BODIPY donor and the aldehyde acceptor represent a very strong D–A system. In a strong D–A system, the S0/S1 energy gap can become so small that internal conversion (IC) becomes very efficient, and fluorescence is quenched. When the probe reacts with H2S, ICT is eliminated, and the fluorescence of the BODIPY is recovered. While these two examples happen to embody reduction of ICT; enhancement of ICT is also commonly exploited.
Förster resonance energy transfer (FRET)
Förster resonance energy transfer (FRET) is based on the ability of a donor fluorophore to transfer its excited state energy to an acceptor fluorophore with a lower energy excited state, provided that there is a matching acceptor excited state vibrational level available.6 This energy transfer is not a photon emission/absorption phenomenon, and is thus a non-radiative process.23 Rather, it can be thought of as transfer of donor excited state energy to the acceptor. This energy transfer “calls up” a ground state acceptor electron to the excited state level into which the donor energy is transferred. Following rapid vibrational relaxation of the acceptor excited state, this represents a reduction of the total energy of the system24 (see Fig. 9).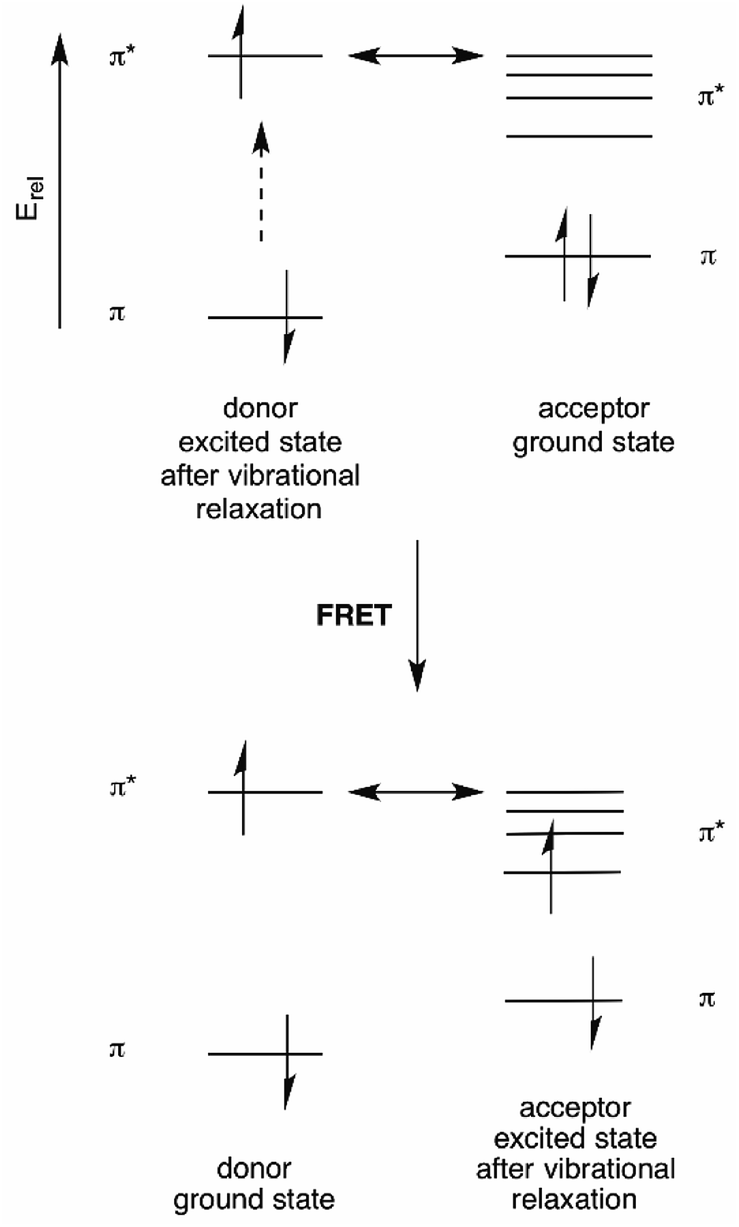 | ||
| Fig. 9 Energy diagram for FRET. aThe double-headed arrow denotes the energy-matching of the lowest energy vibrational state of the donor excited state with a high energy vibrational level of the acceptor excited state. This is equivalent to the absorption/emission overlap shown in Fig. 10. | ||
For a utilitarian understanding of FRET, one can focus on two essential factors that determine FRET efficiency: the distance between the donor and acceptor molecules strongly influences the efficiency of FRET, which scales with 1/r6, where r is the average spatial separation; and, the “spectral overlap” of the “spectral overlap” of donor emission and acceptor absorption, which controls the degree to which the excited states can “talk to each other”. The final brightness of the FRET pair is also controlled by the intrinsic brightness of the donor and acceptor fluorophores (see Fig. 10).
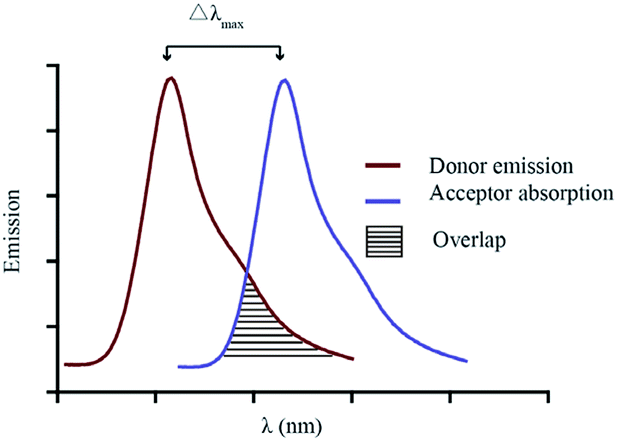 | ||
| Fig. 10 Emission overlap vs. λmax separation in a hypothetical FRET pair. | ||
Because the donor and acceptor have different absorption and emission maxima, selective excitation and emission measurements allow separate measurement of the “FRET state” (donor → acceptor emission) and the “non-FRET state,” in the form of donor-only or acceptor-only emission. This in turn increases the precision with which fluorescence changes can be measured.25
Choosing a donor/acceptor pair for FRET probes is a balancing act. On the one hand, maximizing the overlap of donor emission and acceptor absorption increases the efficiency of FRET (Fig. 10). On the other hand, keeping the donor and acceptor emission maxima far apart facilitates separate measurement of the two emission bands. In small molecules, it is very difficult to change to donor/acceptor distance enough to alter FRET efficiency without cleavage of the covalent linkage between the two. Such cleavage-based signaling has been very important in developing probes for, e.g., protease activity. However, there are many other ways to turn FRET “on and off”. These usually involve generating or deactivating one half of the donor acceptor pair.26 Both reversible binding and irreversible chemical reactions can be used to induce these changes. These concepts are illustrated by the two systems shown below (Fig. 11 and 12).27,29
 | ||
| Fig. 11 FRET-based probe for measurement of pH in lysosomes. | ||
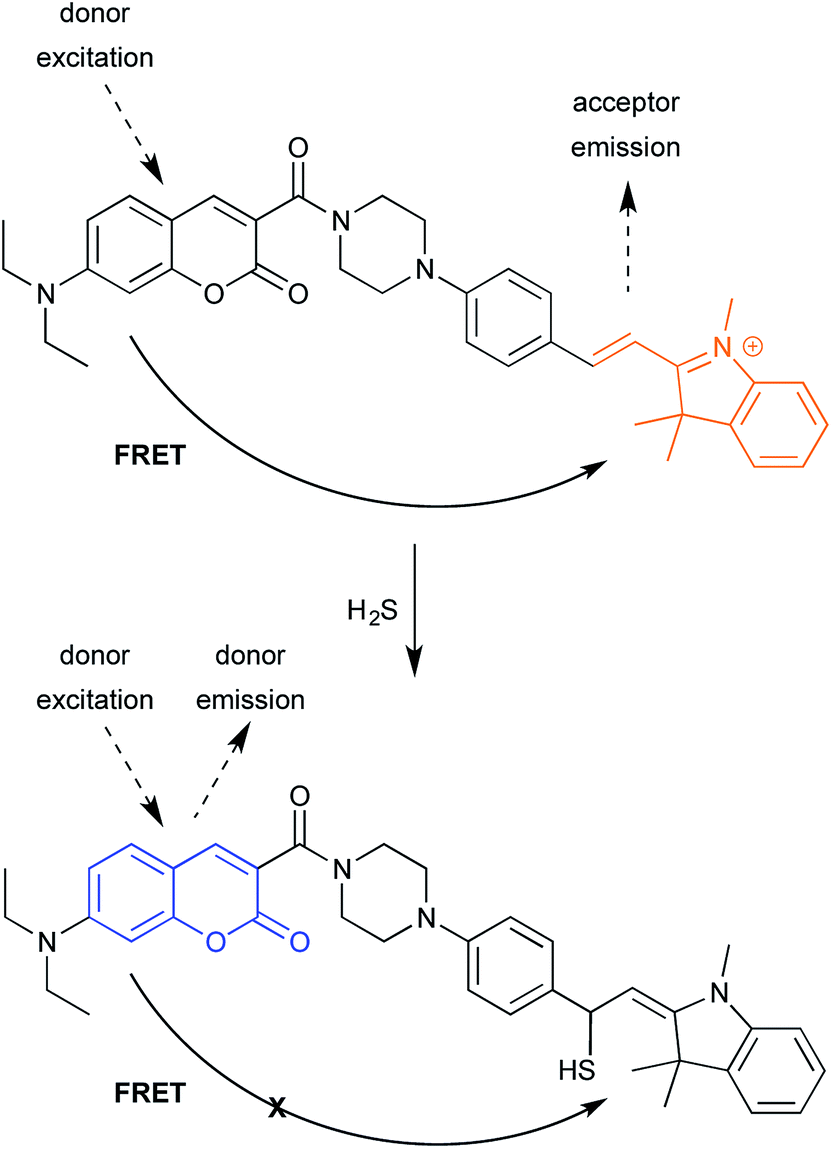 | ||
| Fig. 12 FRET-based probe for H2S detection in cells. | ||
The first example uses a coumarin donor and a rhodamine acceptor (Fig. 11).27 Coumarins have higher energy excited states, and shorter excitation/emission wavelengths, than rhodamines.28 (See final section on common fluorophores.) In lactam form, the rhodamine fragment is unconjugated, and has no relevant absorbance or emission. Thus, donor excitation leads to donor emission. Protonation converts the lactam form of the rhodamine to the ring-opened, fully conjugated emissive isomer. This turns on coumarin/rhodamine FRET.
The first example uses a coumarin donor and a rhodamine acceptor (Fig. 11).27 Coumarins have higher energy excited states, and shorter excitation/emission wavelengths, than rhodamines.28 (See final section on common fluorophores.) In lactam form, the rhodamine fragment is unconjugated, and has no relevant absorbance or emission. Thus, donor excitation leads to donor emission. Protonation converts the lactam form of the rhodamine to the ring-opened, fully conjugated emissive isomer. This turns on coumarin/rhodamine FRET.
This is an equilibrium process, controlled by pH, which allows the probe to be used for fluorescence imaging of pH fluctuations. The probe localizes in the lysosome, which is normally weakly acidic. But the intra-lysosomal pH of cancerous cells is lower than that of normal cells. Thus, optical measurement of lysosomal pH can be used to differentiate them.
The second example relies, in contrast, on turning FRET off (Fig. 12).29 As noted in the ICT example shown in Fig. 8, fluorescent probes for detecting H2S have helped advance the study of H2S-mediated intracellular processes. Here, a coumarin donor transfers its excited state energy to a merocyanine receptor. (Merocyanines are related the common cyanine dyes; see final section on common fluorophores.) This allows coumarin excitation to produce merocyanine emission. Conjugate of H2S to the merocyanine iminium ion disrupts conjugation, eliminating emission. Following, shorter-wavelength donor emission is observed, which is easily distinguished from merocyanine emission. While in current form, the reaction is irreversible, one can imagine that changes in probe structure could make the addition reversible.
Overview of some common fluorophores used in fluorescent probes
Numerous fluorophore scaffolds have been used in the preparation of fluorescent probes. Structures with new or improved properties are routinely developed, and this remains a very active area of research. We begin with a summary of some practically important fluorophore. We then provide specific examples of: coumarins; naphthalimides; fluorescein and its analogs; rhodamine and its analogs; BODIPYs; and cyanines. We finish with a pictorial summary of how and where these fluorophores can – and have been – modified to tune optical properties (Fig. 13–15).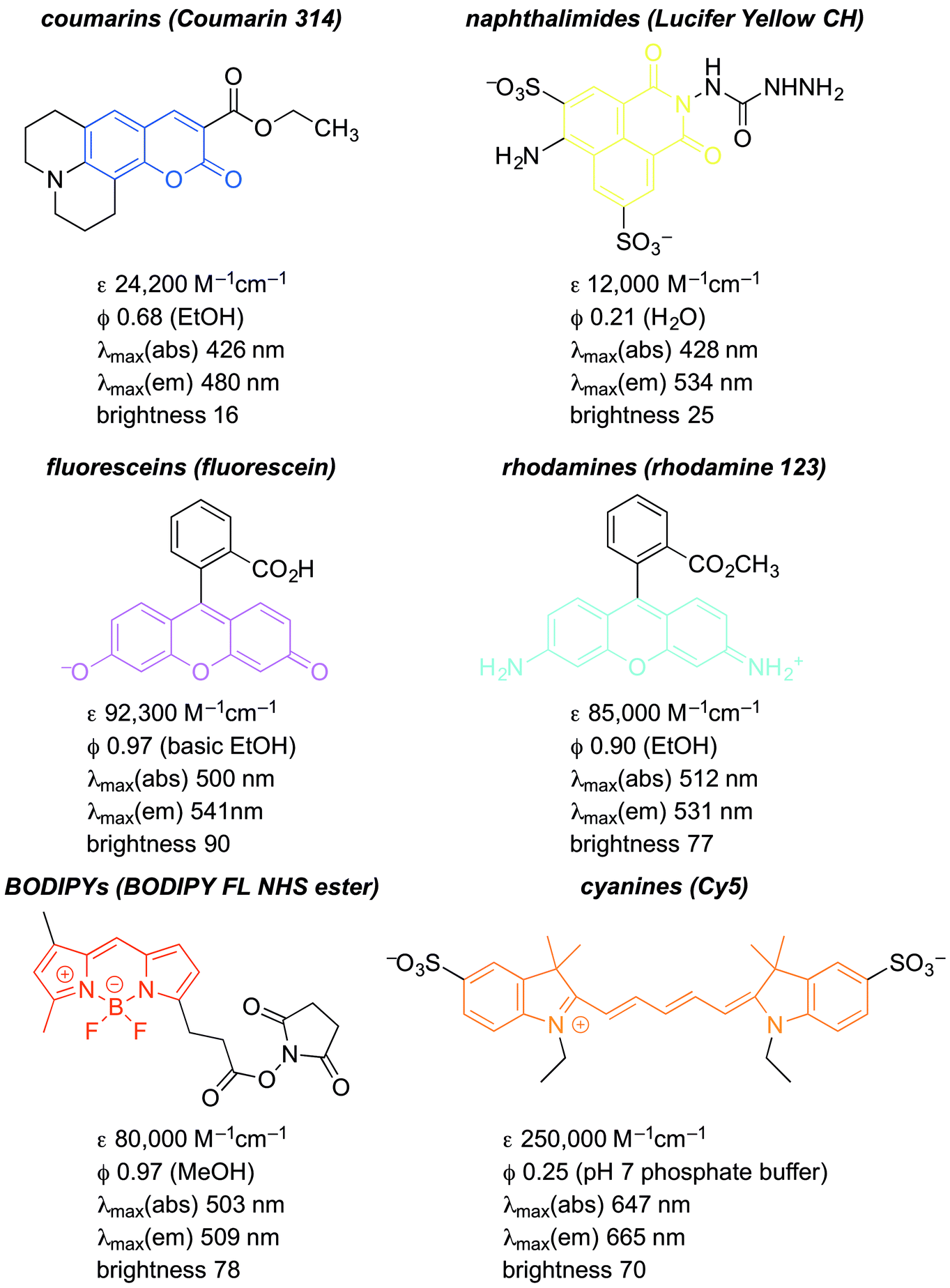 | ||
| Fig. 13 Representative molecules from some important fluorophore classes. | ||
 | ||
| Fig. 14 Interplay between emissive open-form and non-fluorescent spirocyclic closed-form of fluorescein and rhodamine derivatives.6 | ||
 | ||
| Fig. 15 Visual summary of how substituents in some important fluorophore classes can be modified to alter properties and function. | ||
Regarding optical properties, the important considerations to come down to controlling light going in and light coming out. The efficiency with which incoming light is absorbed is characterized by the extinction coefficient (molar absorptivity), ε. This value derives from the Beer-Lambert law A = εcl, ε has units of M−1 cm−1. The maximum absorbance wavelength is given as λmax(abs). In absorption spectroscopy, this is simply the most intense absorption wavelength.
The other important absorbance parameter is the excitation wavelength. In principle, it would seem simplest to excite at λmax(exc), the excitation wavelength that produces the maximum fluorescence emission. For fluorophores with sufficiently large Stokes shifts – that is, minimal overlap of absorption and emission, this is usually the case. However, it must be noted that for fluorophores with multiple absorption bands, λmax(abs) is rarely the same as λmax(exc). In fact, λmax(exc) typically corresponds to the longest wavelength, well-resolved absorption band. This absorption band corresponds to the π to π* (S0 to S1) transition that has been discussed throughout this review. Shorter wavelength absorptions correspond to the formation of excited states with higher energy. These higher energy excited states routinely have greater access to ISC deactivation, leading to reduced emission intensity.6
An important and contrary situation is that of fluorophores with small Stokes shifts, such as BODIPY and cyanine dyes. One of the keys to sensitive fluorescence measurement is that excess excitation light passes directly through the sample, while fluorescence emission is typically randomly oriented. Because the detector of bench-top instruments is placed at a 90° angle relative to the excitation beam, this minimizes the inevitable excitation scattering that overlaps with the emission (the optics for fluorescence microscopy are more complicated). With a large Stokes shift, such scattering lies outside the detection window, and is rarely problematic. But with a small Stokes shift this is not so: the scattering begins to overlap with the emission. To minimize scattering background, the fluorophore must then be excited at wavelengths < λmax(exc). This necessarily reduces the intensity of fluorophore emission.
Outgoing light (fluorescence) is also (mostly) described by two metrics. The maximum emission wavelength is given as λmax(em). The efficiency with which excitation energy is converted to emission is given by the quantum yield, ϕ, which is the dimensionless ratio of (photons absorbed)/(photons emitted), and has values of 0–1.0, with 1.0 representing a “perfect fluorophore.” As a cautionary note, the fluorescent probe literature is often deficient in reporting optical parameters, especially ε and ϕ.
An additional important parameter for probe design is brightness. Brightness is the multiple εϕ, usually divided by 1000.30 It allows rapid comparison of fluorophores, weighted for both excitation and emission efficiency. It has the units of ε, although these are often left out for convenience. To our knowledge, there is not a standard symbol for brightness.
There are important physicochemical issues for fluorescent probes that the reader should be aware of. These include: short-wavelength excitation leading to background autofluorescence from cells, and/or causing cell damage; λ-cutoffs of common commercial imaging filter sets, many of which are designed for “old school” fluorophores, restricting viable excitation and emission wavelengths for microscopy; many probes are solvatofluorochromic – that is, their emission is influenced by the polarity of their immediate environment; fluorophore photostability (and, for cyanine dyes, ozone stability) often being problematic; and, membrane permeability and intracellular localization are strongly influenced by probe structure. Finally, there has been increasing demand for fluorophores that emit in the far-red to near IR (NIR) region of the emission spectrum, where biological samples (including tissue in live animals) are effectively transparent.31
The structures and optical properties of representative fluorophores are shown below (Fig. 13).32–38
Coumarins
Coumarins are among the oldest and most easily synthesized fluorophores.39 They usually have short wavelength (UV) excitation, making them non-ideal for cellular assays or imaging (see coumarin 314, Fig. 13). However, they are useful as FRET probes, especially in enzyme assays. Consider a FRET pair cleaved by an enzymatic reaction: D–A + enzyme → D∼ + A∼ + enzyme. Cleavage of the D–A connection eliminates FRET, and provides a readout of enzymatic activity. Such assays are valuable for the discovery of new enzyme inhibitors. Coumarins have limited brightness, because they do not absorb strongly, due to the relatively minimal fluorescence scaffold. (The authors' generalization is that ε ≥ 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 constitutes a highly absorbent fluorophore.) However, they are very photostable. That is, continued excitation leads to minimal fluorophore degradation. In addition, as ICT fluorophores, they have very large Stokes shifts.
000 M−1 cm−1 constitutes a highly absorbent fluorophore.) However, they are very photostable. That is, continued excitation leads to minimal fluorophore degradation. In addition, as ICT fluorophores, they have very large Stokes shifts.
Naphthalimides
Naphthalimides usually considered short wavelength fluorophores, although there are now derivatives with long wavelength excitation and emission as far out as the red/NIR.40 (This remains underappreciated.) Their brightness is superior to that of coumarins. Their synthesis is generally versatile and predictable, and they are very robust fluorophores, highly resistant to photobleaching (see Lucifer yellow CH, Fig. 13). Like coumarins, as ICT fluorophores, they have very large Stokes shifts.Fluoresceins and rhodamines derivatives
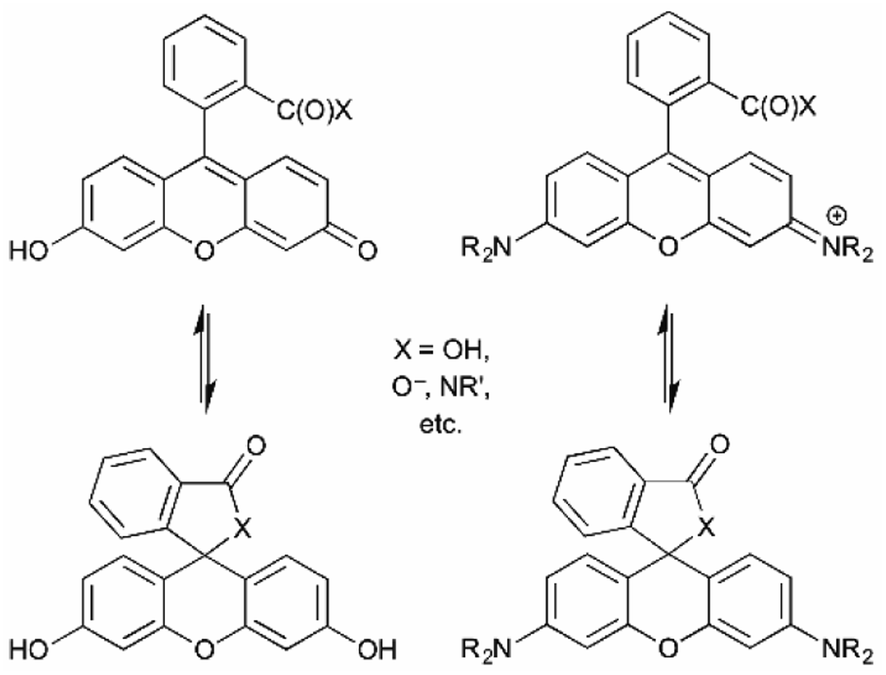
Fluoresceins and rhodamines remain among the most widely-recognized fluorophores.41 (Fluorescein and rhodamine 123 are representative; Fig. 13.) The synthesis of the core fluorophores dates back to the late 1800s. They remain among the brightest fluorophores in common use, and new derivatives have shifted available emission wavelengths into the red/NIR part of the spectrum. They remain “workhorse” fluorophores for chemical, biochemical, biological, and medical applications. However, they also remain problematic in that the synthesis of derivatives is often low-yielding, and purification is usually laborious. However, there are so many established synthetic methods that the preparation of fluorescein and rhodamine derivatives is relatively predictable, despite sometimes being tedious.41As a cautionary note, a limitation of fluoresceins and rhodamines is their limited photostability.42 An hour of irradiation can lead to significant photobleaching, via oxidative degradation of the fluorophore in its excited state. Important, but perhaps less surprising, is that permeability, cellular localization, and intracellular aggregation are very dependent on the specific structure of the fluorophore. The structure influences not only these properties, but also the interconversion between the fluorescent “open form” and the non-fluorescent spirocyclic “closed form” (Fig. 14).6 The position of this equilibrium is pH dependent, and the pH dependence can vary with fluorophore structure. It then also varies with the pH of the local environment, which can be important in biological imaging. This can be, but is not always, advantageous. Still, the versatility and remarkable brightness of these fluorophores guarantees their continued importance.
BODIPYs
BODIPY fluorophores are comparatively new, initial reports dating back to 1968.15–20 They are notable in several respects. They have high quantum yields, and sharp absorption and emission bands. This is a result of their structural rigidity. Such rigidity limits the density of vibrational states that can lead to absorption and emission line-broadening, and diminishes IC deactivation of the excited state. While they are very bright fluorophores, this rigidity also provides limitation, in that the Stokes shifts are very small. This can make resolving excitation and emission problematic. The narrow emission lines can make BODIPY/BODIPY FRET pairs difficult to develop as well. A distinctive feature of BODIPYs is that they are “self-contained”, and exhibit very limited solvatofluorochromism. They are well suited for PET and FRET probes. As with all of the scaffolds described here, there are many commercially-available derivatives for direct conjugation to biomolecules. For example, the BODIPY NHS ester shown in Fig. 13 is an excellent tool for selective labelling of lysine residues on protein surfaces.Cyanines
Finally, cyanine fluorophores such as Cy5 (Fig. 13) have proven to be very important.43 While their quantum yields are typically low (≤0.25), their very high extinction coefficients make them among the brightest known fluorophores. They were some of the earliest dyes developed that gave red/NIR emission, and remain highly popular for this reason. A significant limitation is the ease with which they are oxidized. This is not limited to the problematic photo-oxidation common in many fluorophores. Derivatives of Cy5 and Cy7 (Fig. 13), undergo background oxidation with O2, and even the minute amounts of O3 present in the atmosphere.42 This makes it difficult to use these fluorophores in studies that require long measurement periods. However, new cyanine derivatives have been developed with increased quantum yields and improve oxidation resistance. Most of these improvements are based on an understanding of the origins of excited state deactivation and oxidative degradation. This underscores the value of mechanistic analysis in moving the fluorescent probe field forward.Visual summary of substituent modification of common fluorophores
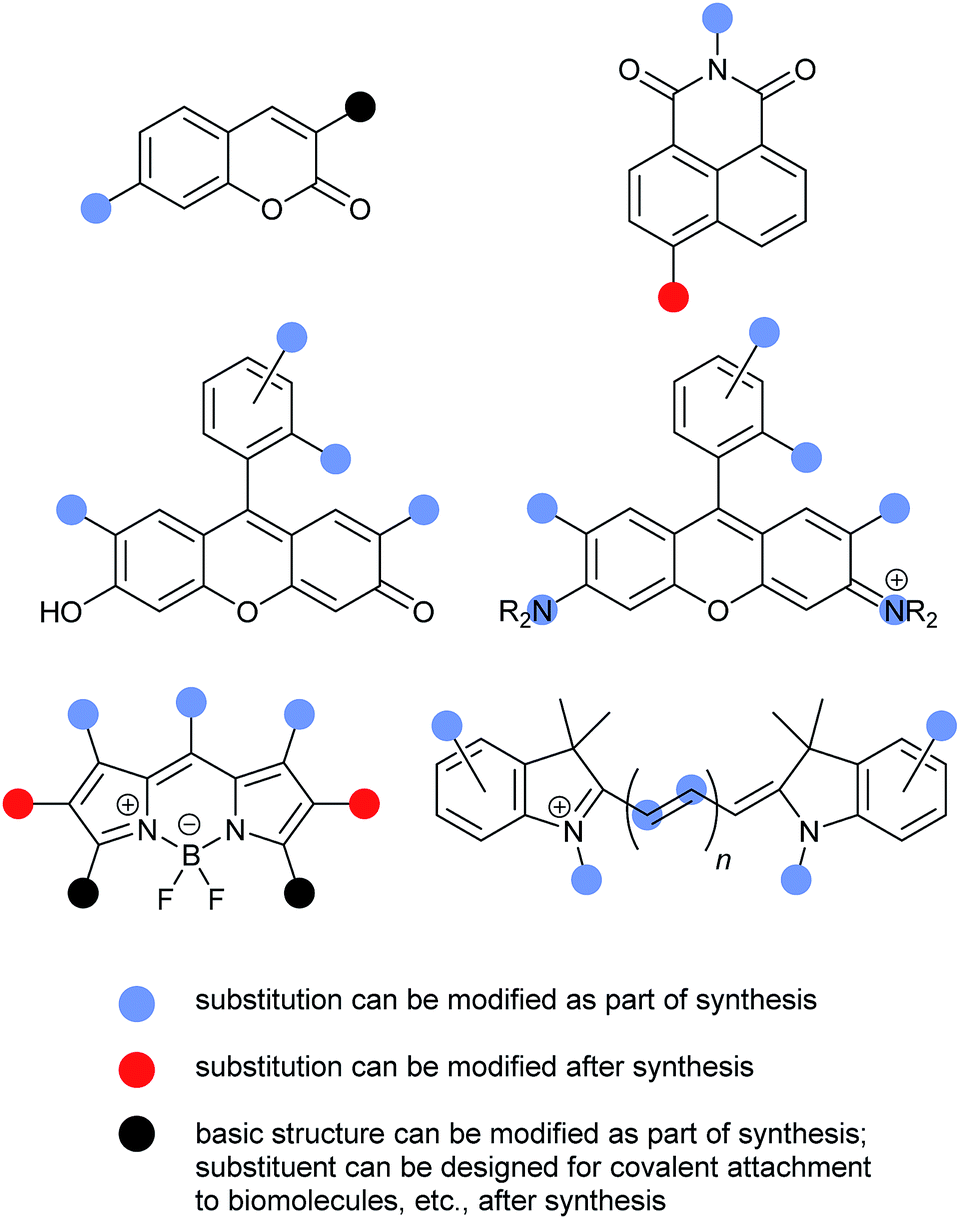
A detailed discussion of possible substitution patterns for the common fluorophores described above is beyond the scope of this review. (See individual fluorophore sections for synthetic references.) However, the following graphical summary provides an overview of some of the most common sites for substituent variation, and when the substituents are typically introduced during fluorescent probe synthesis (Fig. 15).Conclusions
There has been exceptional progress in the development and application of fluorescent probes over the last two decades. This has been an incredibly productive and exciting period. Of course, there remain many challenges to address. Examples include: developing probes for the reversible detection of reactive oxygen/nitrogen/sulfur species; broader application of probes for environmental detection; improved probes for guiding real-time surgical procedures; practical methods for imaging lithium cation; and finding more general strategies for fluorescence-based detection of target molecules in a biological milieu.These ambitious objectives will take significant effort to attain. Note, however, that many prior advances have derived from the growth of the field. Twenty years ago, there were comparatively few researchers in the area. Now, there are well over a hundred groups engaged in probe research. This expansion necessarily involved drawing people in from a wide array of other backgrounds, such as medicine, biology, biochemistry, and all areas of chemistry. In turn, this provided new perspectives, new challenges, and new inspired solutions. Future progress will surely be accelerated by engaging additional motivated, curious and creative “intelligent non-expert” scientists. We hope this review facilitates this process, to the benefit of all aspects of the probe development field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (grant 21672160) and Tianjin University.Notes and references
- For representative contemporary reviews, see: D. Wu, A. C. Sedgwick, T. Gunnlaugsson, E. U. Akkaya, J. Yoon and T. D. James, Chem. Soc. Rev., 2017, 46, 7105–7123 RSC; L. B. Daly and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC; J. Wu, B. Kwon, W. Liu, E. V. Anslyn, P. Wang and J. S. Kim, Chem. Rev., 2015, 11, 7893–7943 CrossRef PubMed; L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef PubMed; M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC; X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590–695 CrossRef PubMed For representative earlier reviews of fluorescent chemosensors and probes, see: M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2012, 256, 170–192 CrossRef; D. G. Cho and J. L. Sessler, Chem. Soc. Rev., 2009, 38, 1647–1662 RSC; A. V. Tsukanov, A. D. Dubanosov, V. A. Bren and V. I. Minkin, Chem. Heterocycl. Compd., 2008, 44, 899–923 CrossRef; J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef; T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 Search PubMed; K. Rurack and U. G. Resch, Chem. Soc. Rev., 2002, 31, 116–127 RSC; V. A. Bren, Russ. Chem. Rev., 2001, 70, 1017–1036 CrossRef; A. P. de Silva, H. Q. N. Gunarathe, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 515–527 CrossRef; Fluorescent Chemosensors for Ion and Molecule Recognition, ed. A. W. Czarnik, American Chemical Society, Washington, D. C., 1994 Search PubMed.
- B. Valeu and M. N. Berberan-Santos, J. Chem. Educ., 2011, 88, 731–738 CrossRef.
- L. Liu, X. Zhou, R. Ma, M. He, H. Shi and Q. Yi, Sens. Actuators, B, 2018, 259, 888–893 CrossRef.
- D. DiBerardino, R. J. Rafac, S. Boone, V. P. Gerginov and C. E. Tanner, Opt. Commun., 2002, 210, 233–243 CrossRef.
- W. Zhang, J. Kang, P. Li, H. Wang and B. Tang, Anal. Chem., 2015, 87, 8964–8969 CrossRef PubMed.
- For several valuable text resources, see: N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, 2008 Search PubMed; Handbook of Photochemistry, M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, CRC Press, New York, 3rd edn, 2006 Search PubMed; J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer, New York, 2nd edn, 1999 Search PubMed; M. Klessinger and J. Michl, Excited States and Photo-Chemistry of Organic Molecules, John Wiley & Sons, 1995 Search PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- The terms singlet and triplet derive from quantum mechanical descriptions of the net electron spin of a molecule. The spin quantum number, S, is given as S = (2s + 1) s = ± 1/2. In the initially-formed excited state, the spins are still paired and 2s = 0. Thus, S = 1, hence the term singlet. In the triplet state S = 3, explaining the notation.
- Without known exception, the triplet excited states of conjugated π systems are lower in energy than the singlet excited states. At a simplified level, the two dominant factors involved are: Increased entropy in the triplet state (which has two possible net-spin configurations, as opposed to one for the singlet); and, a manifestation of Hund’s Rule and the Pauli Exclusion Principle. One accessible way of restating this is that, in the absence of the bonding energy associated with a spin paired covalent bond, electrostatic repulsion is reduced in the triplet state. The triplet state forces the electrons to occupy different orbitals, minimizing their interaction.
- A consequence of the excited state triplet being lower in energy than the excited state singlet is that the vibrationally relaxed excited state triplet is much closer in energy to the ground state singlet. This routinely allows efficient energy-matching of the vibrationally relaxed triplet with a high-energy vibrational level of the ground state singlet. This is one of the requirements for rapid ISC at room temperature. There are also dynamic effects (conformational distortions) that are essential for ISC being possible. At 77 K, in a frozen matrix, the dynamic contribution to ISC is eliminated. Then triplet can/must then radiatively return to the singlet ground state. This process is referred to as phosphorescence.
- The efficiency of IC and ISC is closely tied to the overlap of vibrational levels between the excited state and the ground state. The energy and spacing of these levels depends on how much a molecule can distort. Twisting and pyramidalization are especially important, because they alter orbital overlap and vibration the most. While some physical and physical organic chemists have recognized this, the idea that excited state conformational restriction could be used to control fluorescence is relatively new. For a recent overview of one group’s important work, see: J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC (The Corresponding Author declines to self-cite). For representative references from our own work, see: S. A. McFarland and N. S. Finney, J. Am. Chem. Soc., 2001, 123, 1260–1261 CrossRef PubMed; J. V. Mello and N. S. Finney, Angew. Chem., Int. Ed. Engl., 2001, 40, 1536–1538 CrossRef PubMed; S. A. McFarland and N. S. Finney, J. Am. Chem. Soc., 2002, 124, 1178–1179 CrossRef PubMed.
- This approach has proven particularly valuable in developing probes for reactive oxygen species (ROS). For a recent overview, see: Z. Lou, P. Li and K. Han, Acc. Chem. Res., 2015, 48, 1358–1368 CrossRef PubMed.
- Other donating heteroatoms, such as S and P, can be used for PET systems as well.
- It should be noted that electron transfer efficiency has an inverse exponential dependence on the distance between the donor and the fluorophore. Taking amine donors as an example, benzylic amines can quench fluorescence very efficiently. The quenching efficiency falls off very rapidly with the length of the linker between the nitrogen and the fluorophore, so that even systems with a C2 linker are rarely useful. For early demonstrations of this, see: J. C. Beeson, M. E. Huston, D. A. Pollard, T. K. Venkatachalam and A. W. Czarnik, J. Fluoresc., 1993, 3, 65–68 CrossRef PubMed; H. F. Ji, R. Dabestani, G. M. Brown and R. L. Hettich, J. Photochem. Photobiol., A, 1999, 69, 513–516 CrossRef.
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef PubMed.
- Reactive oxygen species (ROS) play manifold important roles in cell biology. For representative reviews: T. Finkel, J. Cell Biol., 2011, 194, 7–15 CrossRef PubMed; P. D. Ray, B. H. Huang and Y. T. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef PubMed; L. A. Sena and N. S. Chandel, Mol. Cell, 2012, 48, 158–167 CrossRef PubMed; M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–R462 CrossRef PubMed.
- H. Zhu, J. Fan, J. Wang, H. Mu and X. Peng, J. Am. Chem. Soc., 2014, 136, 12820–12823 CrossRef PubMed.
- P. Li, X. Duan, Z. Chen, Y. Liu, T. Xie, L. Fang, X. Li, M. Yin and B. Tang, Chem. Commun., 2011, 47, 7755–7757 RSC.
- For a comprehensive review of the synthesis and application of BODIPY dyes, see: A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef PubMed For their application in photodynamic therapy, see: A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- C. Zhang, Z. Han, M. Wang, Z. Yang, X. Ran and W. He, Dalton Trans., 2018, 47, 2285–2291 RSC.
- G. Grynkiewicz, M. Poenie and R. Tsien, J. Biol. Chem., 1985, 260, 3440–3450 Search PubMed . Cited with great respect for the late Roger Yongchien Tsien (1952-2016) – a brilliant and inspirational scientist, pioneer of the field, and Nobel Laureate (2008).
- X. Li, S. Zhang, J. Cao, N. Xie, T. Liu, B. Yang, Q. He and Y. Hu, Chem. Commun., 2013, 49, 8656–8658 RSC.
- P. Taya, B. Maiti, V. Kumar, P. De and S. Satapathi, Sens. Actuators, B, 2018, 255, 2628–2634 CrossRef.
- A. D. Hughes, I. C. Glenn, A. D. Patrick, A. Ellington and E. V. Anslyn, Chem.–Eur. J., 2008, 14, 1822–1827 CrossRef PubMed.
- For a recent review discussing the benefits of ratiometric probes, see: J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- M. E. Dempsey, H. D. Marble, L. Tun, N. L. Fawzi and E. M. Darling, Bioconjugate Chem., 2018, 29, 335–342 CrossRef PubMed.
- X. F. Zhang, T. Zhang, S. L. Shen, J. Y. Miao and B. X. Zhao, RSC Adv., 2015, 5, 49115–49121 RSC.
- V. Kumar and E. V. Anslyn, J. Am. Chem. Soc., 2013, 135, 6338–6344 CrossRef PubMed.
- X. Feng, T. Zhang, J. T. Liu, J. Y. Miao and B. X. Zhao, Chem. Commun., 2016, 52, 3131–3134 RSC . (In the manuscript, the authors propose and illustrate 1,2-addition of H2S to the iminium ion. Based on literature precedent, we have chosen to show 1,4-addition).
- For an informative visual summary, see Fig. 2 in: L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef PubMed.
- See, for example: S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef PubMed; Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC; A. Haque, S. Haque, F. Jahangir, A. Rather and M. S. Khan, Bioorg. Med. Chem., 2017, 25, 2017–2034 CrossRef PubMed.
- Coumarin 314 Data: https://omlc.org/spectra/PhotochemCAD/html/002.html. (Accessed 24 February, 2018).
- Lucifer Yellow CH Data: https://omlc.org/spectra/PhotochemCAD/html/065.html. (Accessed 24 February, 2018).
- Fluorescein Data: https://omlc.org/spectra/PhotochemCAD/html/037.html. (Accessed 24 February, 2018).
- Rhodamine 123 Data: https://omlc.org/spectra/PhotochemCAD/html/008.html. (Accessed 24 February, 2018).
- The PhotoChemCAD database of J. Lindsey, et al., is readily available online. It provides carefully measured optical parameters for many common fluorophores. These data can be found at: https://omlc.org/spectra/PhotochemCAD/. (Accessed 24 February, 2018).
- BODIPY FL NHS ester Data: https://www.lumiprobe.com/p/bodipy-fl-nhs-ester. (Accessed 24 February, 2018).
- Cy5 Data:http://www.fluorophores.tugraz.at/substance/423 R. B. Ratnakar, L. A. Ernst, S. R. Mujumdar, C. J. Lewis and A. S. Waggoner, Bioconjugate Chem., 1993, 4, 105–111 CrossRef.
- For one overview of coumarin synthesis, see: K. A. Kumar, N. Renuka, G. Pavithra and G. V. Kumar, J. Chem. Pharm. Res., 2015, 7, 67–81 Search PubMed.
- Leading references on naphthalimide synthesis: see ref. 1 and R. M. Duke, E. B. Weale, F. M. Pfeffer, P. E. Kruger and T. Gunnlauggson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- For an overview of fluorescein and rhodamine syntheses: M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC; Y. Duan, M. Liu, W. Sun, M. Wang, S. Liu and Q. X. Li, Mini-Rev. Org. Chem., 2009, 6, 35–43 CrossRef.
- Discussions of photobleaching, and ways it can be addressed: Q. Zheng and L. D. Lavis, Curr. Opin. Chem. Biol., 2017, 39, 32–38 CrossRef PubMed.
- For a recent overview of cyanine chemistry, see: H. A. Shindy, Dyes Pigm., 2017, 145, 505–513 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |