
Insertion of sulfur dioxide via a radical process: an efficient route to sulfonyl compounds
Guanyinsheng
Qiu
*a,
Kaida
Zhou
b,
Liang
Gao
c and
Jie
Wu
 *b
*b
aCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China. E-mail: qiuguanyinsheng@mail.zjxu.edu.cn
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn
cDepartment of Natural Science and Computer Science, Ganzhou Teacher's College, Ganzhou, 341000, China
First published on 12th December 2017
Abstract
This review is focused on the recent advances in the chemistry of sulfur dioxide fixation through a radical process. Diverse sulfonyl compounds can be obtained efficiently under mild conditions. In general, aryl radicals, vinyl radicals, alkyl radicals, and nitrogen-centered radicals can react with sulfur dioxide, giving rise to sulfonyl radical intermediates. The resulting sulfonyl radicals can be captured by nucleophiles, olefins, and alkynes. The sources of aryl radicals from aryldiazonium salts, aryl halides, and diaryliodonium salts are demonstrated. It is believed that more amazing SO2-based achievements will be developed in the near future.
 Guanyinsheng Qiu | Guanyinsheng Qiu was born in Jiangxi (China) in 1984. He received his BS (2008) and MS (2011) from the Department of Chemistry at Jiangxi Normal University. In 2011, he moved to Fudan University to pursue his Ph.D. under the guidance of Professor Jie Wu. After obtaining his Ph.D. degree in 2014, he joined Prof. Jieping Zhu's group at École Polytechnique Fédérale de Lausanne as a postdoctoral fellow. One year later, he joined Jiaxing University as a lecturer. In 2016, he was promoted as an associate professor. His current research interest mainly focuses on the methods development for the synthesis of natural product-like compounds under palladium catalysis. |
 Kaida Zhou | Kaida Zhou was born in Changzhou (China) in 1993. He received his BS (2015) from the Department of Chemistry at Soochow University. He continued his studies as a Ph.D. candidate at Fudan University, under the guidance of Professor Jie Wu. His current research interest mainly focuses on the methods development for the synthesis of sulfonyl compounds through the insertion of sulfur dioxide. |
 Liang Gao | Liang Gao was born in Ganzhou (China) in 1989. She received her BS (2010) and MS (2013) from the Department of Chemistry at Jiangxi Normal University. During 2011–2013, she studied at Fudan University under the guidance of Professor Jie Wu. In 2013, she joined Ganzhou Teachers’ College as a lecturer. |
 Jie Wu | Jie Wu received his B.Sc. in chemistry from Jiangxi Normal University in 1995 and he pursued his Ph.D. studies at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the supervision of Prof. Xue-Long Hou (1995–2000). After working as a postdoctoral fellow at Harvard University (2000–2001), he worked as a visiting scientist at the Aaron Diamond AIDS Research Center (2001–2002) and a staff scientist at VivoQuest, Inc. (2002–2004). In 2004, he moved to the Department of Chemistry at Fudan University and was promoted as a professor two years later. He received the Thieme Chemistry Journals Award in 2010 and the Dow Innovation Challenge Award in 2013. He serves as a member of the Editorial Advisory Board of ACS Combinatorial Science. |
1. Introduction
Sulfones and related sulfonyl compounds are ubiquitous in many bioactive natural products and marketed pharmaceuticals.1 As classic tools, sulfonyl groups are always installed by electrophilic aromatic substitution with chlorosulfonic acid and nucleophilic sulfonylation of organometallics.2 It is well known that for sulfonylation with organometallic reagents, low reaction temperature is often required and the substrate scope is limited, thus restricting its synthetic applications. On the other hand, carbon dioxide-based carboxylation has made significant progress.3 Among these established achievements, numerous elegant examples have illustrated that low-cost transitional metal catalysis (especially copper salt) would enable the carbon dioxide-based carboxylation at a mild temperature and pressure, even at atmospheric pressure. The exploitation of synthetic application suggested that carbon dioxide would be a valuable C1 source towards carboxyl-containing architectures. Encouraged by the findings of carbon dioxide chemistry and considering the structural similarity of carbon dioxide and sulfur dioxide, transitional metal-catalyzed aminosulfonylation attracted the interest of synthetic chemists with an expectation to achieve a series of sulfonamides, a “privileged” structural core in many top-selling drugs such as Tipranavir and Celecoxib. However, the handling problem and safety of gaseous sulfur dioxide limited its applications. An initiative report from Willis and co-workers represented a breakthrough for the palladium-catalyzed aminosulfonylation of aryl iodides, hydrazines, and 1,4-diazabicyclo[2.2.2]octane-sulfur dioxide (DABCO·(SO2)2).4 The generation of sulfonamides from sulfur dioxide was promising, although the amino of sulfonamides was restrictively derived from hydrazines.4 This pioneer work witnessed a claim of DABCO·(SO2)2 as a bench-stable and harmless source of sulfur dioxide.To understand the reaction mechanism and expand the scope of the transition metal-catalyzed aminosulfonylation, subsequent efforts were focused intensively on expanding other aromatic electrophiles,5 developing various transition-metal catalysis,6 and exploiting other sulfur dioxide sources.7 Related publications were well documented by Wu's group, Blanchard's group and Willis’ group, respectively.8 The advances of sulfur dioxide chemistry were summarized before 2015, mainly focusing on the transition metal-catalyzed transformations. Additionally, to explore the synthetic applications of aminosulfonylation, many research groups investigated the N-aminosulfonamide-based transformations for the synthesis of sulfones and their analogs.9 For example, since 2010, Wu and co-workers have explored the sulfur dioxide chemistry extensively.5–7 To date, the palladium-catalyzed aminosulfonylation of aryl halides,7a aryl boronic acids,5a,6b and arylnonaflates5c with hydrazines for the synthesis of N-aminosulfonamides has been developed by using DABCO·(SO2)2 or inorganic sulfites as the source of sulfur dioxide. By altering to aryl triethoxylsilanes, both copper catalysis6d and cobalt catalysis6e could enable the insertion of sulfur dioxide, followed by nucleophilic substitution with electrophiles to construct sulfone derivatives. Behaving as a well-recognised source of sulfur dioxide, DABCO·(SO2)2 exhibited excellent reaction efficiency for transition metal-catalyzed sulfonylation. Interestingly, potassium metabisulfite (K2S2O5) also proved to be an efficient candidate for the sulfur dioxide source in the palladium-catalyzed aminosulfonylation of aryl halides and hydrazines.7a
Besides the transition metal-catalyzed sulfonylation, the radical process of sulfur dioxide provides an alternative pathway to sulfonyl compounds. The radical copolymerizations involving sulfur dioxide have been known with simple alkyl radicals since the 1940s.10 However, the reversible process was usually recognized as a method for the formation of carbon radicals. As an early example, the Reed reaction was used for the generation of alkylsulfonyl chlorides from alkanes and sulfur dioxide via a radical process.11 However, the regioselectivity limited its further applications. The synthesis of arylsulfonyl chlorides through the Sandmeyer reaction was described by Meerwein and co-workers.12a The in situ generated aryl radical would be trapped by sulfur dioxide, leading to an arylsulfonyl radical. Recently, the use of thionyl chloride/water as a replacement for the sulfur dioxide source was developed.12b The generation of thiosulfonates was reported through the photolysis of a thiohydroxamic ester and decarboxylative sulfonylation with sulfur dioxide. In the reaction process, the sulfonyl radical generated in situ would react with the thiocarbonyl moiety, providing various thiosulfonates.13 Spirosulfones could be obtained through an irradiation of a p-benzochinon/alkene mixture with sulfur dioxide.14 The chlorosulfonylation of diazonium salts with sulfur dioxide in MeCN via a radical process by using a continuous flow reactor was achieved.15 Large excess of gaseous sulfur dioxide had to be utilized in the above transformations, which brought the safety problem.
As an expansion of the palladium-catalyzed aminosulfonylation, Wu and co-workers conducted the aminosulfonylation of aryldiazonium tetrafluoroborates and hydrazines with DABCO·(SO2)2.16 Surprisingly, a blank experiment in the absence of palladium catalysis showed that the aminosulfonylation of aryldiazonium tetrafluoroborates could proceed smoothly (Scheme 1). Radical trapping experiments and theoretical DFT calculations implied that the metal-free aminosulfonylation was ascribed to a radical process. Hydrazine probably played a triple role in the reaction: (1) as a reductant to facilitate aryldiazonium tetrafluoroborate to produce an aryl radical through a single electron transfer pathway; (2) as a stabilizer to exchange sulfur dioxide from 1,4-diazabicyclo[2.2.2]octane (DABCO) to form an pivotal intermediate; and (3) as a reactant to produce N-aminosulfonamide. In the reaction, the resulting aryldiazo radical would convert into an aryl radical by releasing one molecular nitrogen. Similar to radical carbonylation, the radical fixation of sulfur dioxide occurred to an aryl radical. Sulfur dioxide would be introduced in a fashion of 1,1-radical insertion to offer the arylsulfonyl radical. This metal-free aminosulfonylation of aryldiazonium tetrafluoroborates with sulfur dioxide opened a new window in the radical insertion of sulfur dioxide. To gain more experimental evidence and insights into the mechanism, many groups developed numerous radical sulfonylative reactions by using DABCO·(SO2)2 or inorganic sulfites as the source of sulfur dioxide. So far, many amazing findings have been established. However, there is no related review summarizing the recent advances of the radical insertion of sulfur dioxide. Therefore, it is highly desirable to present a review on the chemistry of sulfur dioxide insertion via a radical process. For convenience, this paper is organized according to the type of radical precursor.
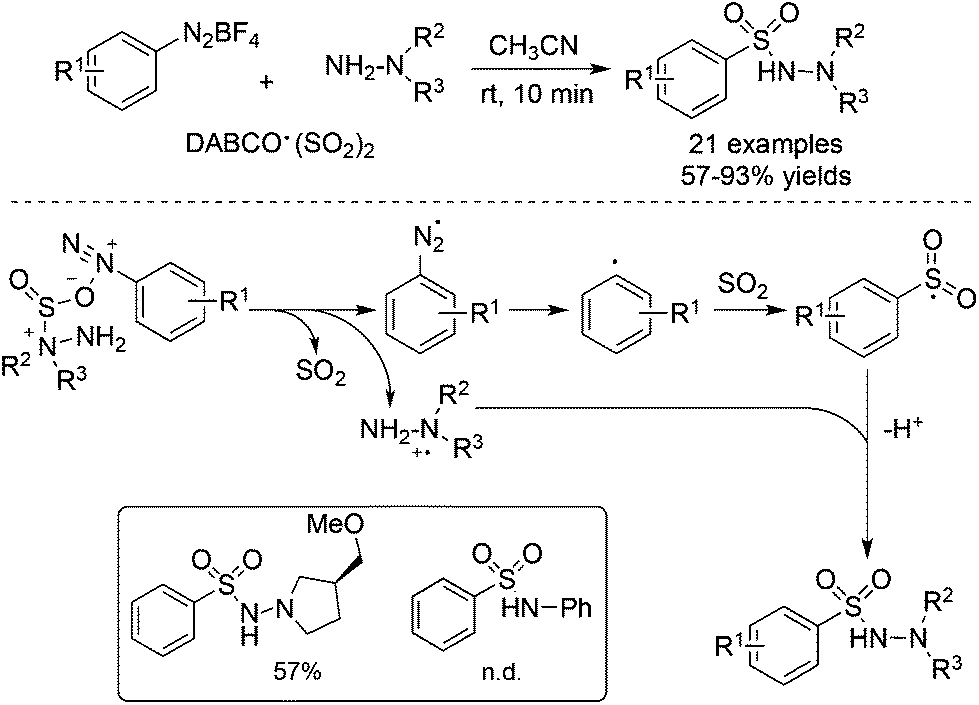 | ||
| Scheme 1 Metal-free aminosulfonylation of aryldiazonium salts, sulfur dioxide, and hydrazines. | ||
2. Insertion of sulfur dioxide with aryldiazonium tetrafluoroborates
Since an aryldiazonium cation could be generated in situ from aromatic amines, a coupling of aromatic amines (including heteroaromatic amines) with DABCO·(SO2)2 and hydrazines under mild conditions was then described (Scheme 2).17 The expected aryl N-aminosulfonamides could be formed in good to excellent yields. Sensitive functional groups including ester, hydroxyl, chloro, and trifluoromethyl groups could be tolerated under the conditions. Moreover, heteroaromatic amines were also workable in this transformation. This chemistry with the removal of the amino group of anilines provided an alternative for the aryl source for the insertion of sulfur dioxide. Subsequently, 2-(allyloxy)anilines were employed in the reaction of sulfur dioxide and hydrazines under mild conditions (Scheme 2).18a Various 1-(2,3-dihydrobenzofuran-3-yl)-methanesulfonohydrazides were prepared in good yields. The key step was the formation of an aryl radical and intramolecular 5-exo-cyclization, which provided an alkyl radical intermediate. This chemistry also demonstrated that an alkyl radical could be involved in the insertion of sulfur dioxide as well, producing an alkylsulfonyl radical. This approach was extended to the synthesis of benzo[b]thiophene 1,1-dioxides through a reaction of 2-alkynylaryldiazonium tetrafluoroborates with sulfur dioxide in the presence of morpholin-4-amine catalyzed by copper(I) bromide.18b Qiu and co-workers applied the above chemistry for the synthesis of trifluoromethyl thiolsulfonates (Scheme 2) starting from anilines, sulfur dioxide, and trifluoromethanesulfanylamide in the presence of bismuth(III) chloride.18c Aminosulfonylation occurred firstly by using N-aminomorphine as an additive, providing a sulfonate intermediate. In the meantime, the presence of bismuth(III) chloride promoted the formation of “CF3S+”, which underwent an electrophilic reaction with sulfonate to produce the corresponding trifluoromethyl thiolsulfonate. Further application of this strategy for the synthesis of 2-arylsulfonyl hydrazones through a four-component reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, 1,2-dibromoethane, and hydrazines under metal-free conditions was developed.18d Additionally, various sulfones could be generated through a reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and aryliodonium tetrafluoroborates under metal-free conditions. A radical process was involved in the presence of morpholin-4-amine, and N-aminosulfonamide acted as the intermediate, proceeding through sulfinate in the presence of a base.18e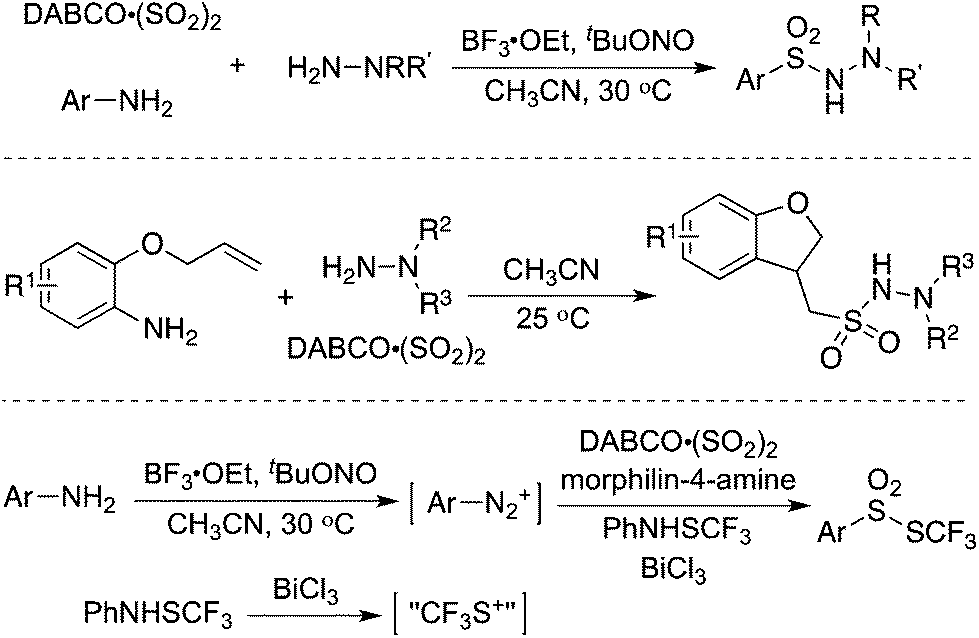 | ||
| Scheme 2 Aminosulfonylation with the insertion of sulfur dioxide by using anilines as the aryl source. | ||
As mentioned above, the aryl radical derived from aryldiazonium tetrafluoroborate would be an efficient means for the capture of sulfur dioxide. Not only anilines but also 1-aryl triazenes19 and hydrazines20 were all good precursors for the generation of aryl radicals. For instance, Wu's group in Germany reported an aminosulfonylation starting from 1-aryl triazenes and sulfur dioxide (Scheme 3).19 Pan and Han described the aminosulfonylation of aryl hydrazines, potassium metabisulfite (K2S2O5), and hydrazines under an air atmosphere (Scheme 3).20 In the reaction process, aryl hydrazines would be oxidized by air, giving rise to aryl radicals, and potassium metabisulfite was used as the source of sulfur dioxide.
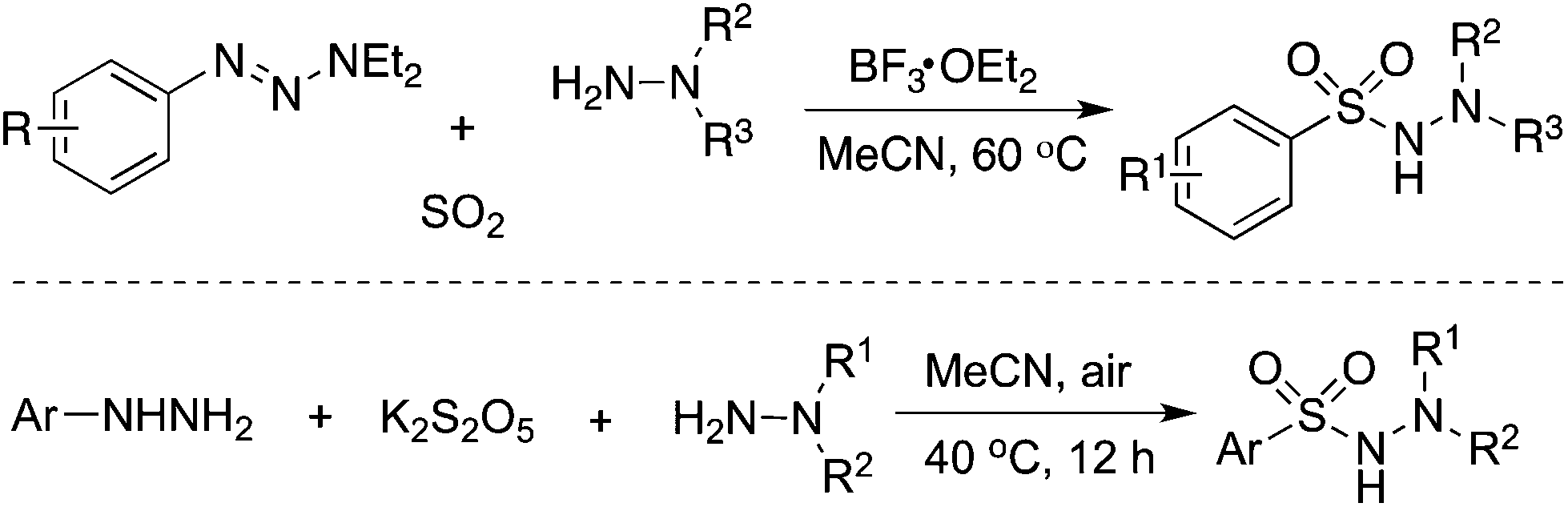 | ||
| Scheme 3 Aminosulfonylation with the insertion of sulfur dioxide by using aryl hydrazines as the aryl source. | ||
From mechanistic investigations, it seemed that the use of hydrazines contributed to the radical generation to provide N-aminosulfonamides. Moreover, reaction scope screening revealed that replacing hydrazines with anilines under standard conditions completely hampered the reaction, thus failing to produce the corresponding sulfonamide (Scheme 1). This discrepancy pushed chemists to further explore the exact roles of hydrazines in the reaction. As a consequence, Wu's group developed a radical hydroxylsulfonylation of aryldiazonium tetrafluoroborates, sulfur dioxide, and hydroxylamines (Scheme 4).21 It was surprising to find that the radical insertion of sulfur dioxide worked well without the assistance of hydrazines, and a series of hydroxylamine enabled the hydroxylsulfonylation to provide O-aminosulfonates in 50–93% yields. A distinctive pathway including the radical insertion of sulfur dioxide and hydrogen atom transfer could account for the above hydroxylsulfonylation. As presented in Scheme 4, it was assumed that DABCO in DABCO·(SO2)2 served as an electron donor to reduce aryldiazonium tetrafluoroborate to an aryl radical. In the meantime, DABCO was oxidized into a DABCO radical cation (DABCO˙+). Based on the DFT theoretical calculation, hydrogen atom transfer took place between hydroxylamine and the DABCO radical cation, thus providing an O-centered radical. The coupling with the resulting arylsulfonyl radical would produce O-aminosulfonate.
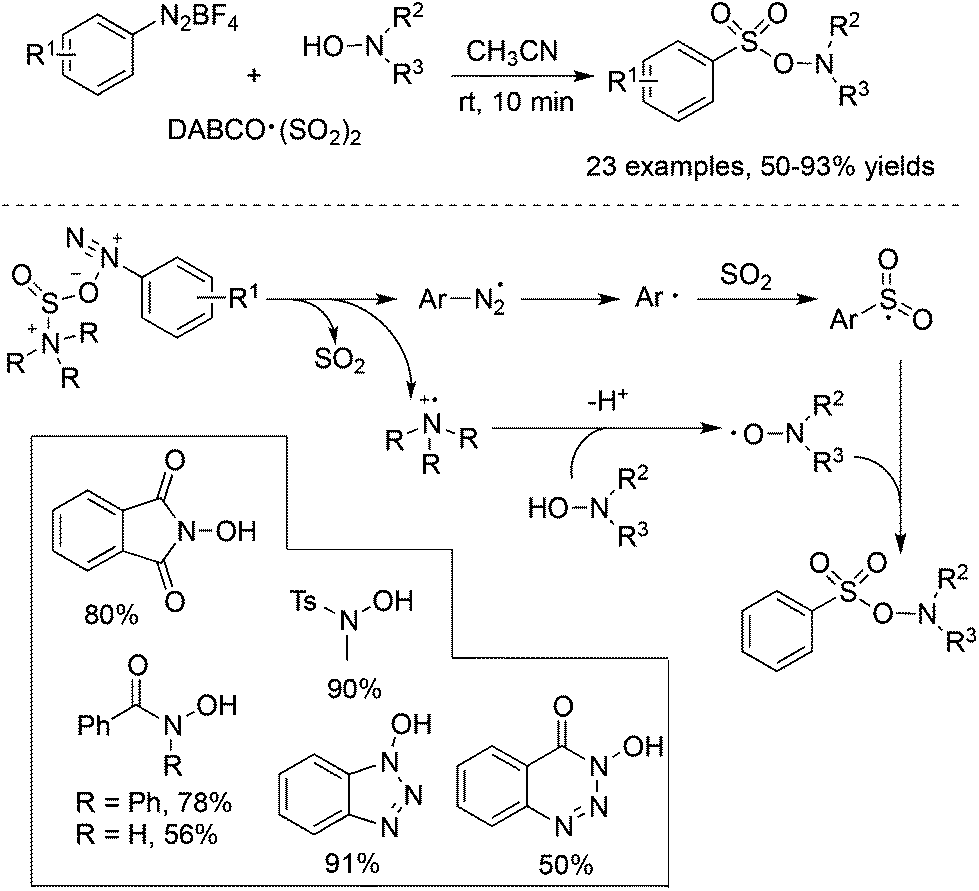 | ||
| Scheme 4 Metal-free aminosulfonylation of aryldiazonium salts, sulfur dioxide, and N-hydroxylamines. | ||
Based on the above results, a conclusion was drawn that arylsulfonyl radicals could be directly generated from aryldiazonium salts by treatment with DABCO·(SO2)2. To obtain experimental evidence, efforts were made to capture the in situ generated arylsulfonyl radicals. Both alkynes and olefins were employed to trap the arylsulfonyl radicals. In this section, the progress on the addition of arylsulfonyl radicals to alkynes and olefins was mainly summarized.
2.1 Reactions of aryldiazonium tetrafluoroborates, sulfur dioxide, and alkynes
As described in Scheme 5, the arylsulfonyl radical could be captured by various alkynes. An initiated example developed by Wu's group was focused on the reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and alkynoates (Scheme 5).22 As expected, the above targeted reaction proceeded smoothly without any catalysts or additives. Interestingly, mechanistic investigation suggested that ester migration occurred in the process, thus causing a formal migration of the methyl in alkynoates from the para-site to the meta-site to provide 3-arylsulfonyl-7-methylcoumarin in a 93% yield (Scheme 5). Radical trapping reaction demonstrated that a radical process was involved in the reaction. The whole process was constituted by the radical insertion of sulfur dioxide, radical cyclization, and ester migration, as presented in Scheme 6. This method was further applied for the synthesis of 3-((2,3-dihydrobenzofuran-3-yl)methyl)sulfonyl coumarins through the reaction of 2-(allyloxy)anilines, DABCO-bis(sulfur dioxide), and aryl propiolates.23 In the reaction process, a 2-(allyloxy)aryl radical would be formed, which would attack the intramolecular double bond to provide an alkyl radical intermediate.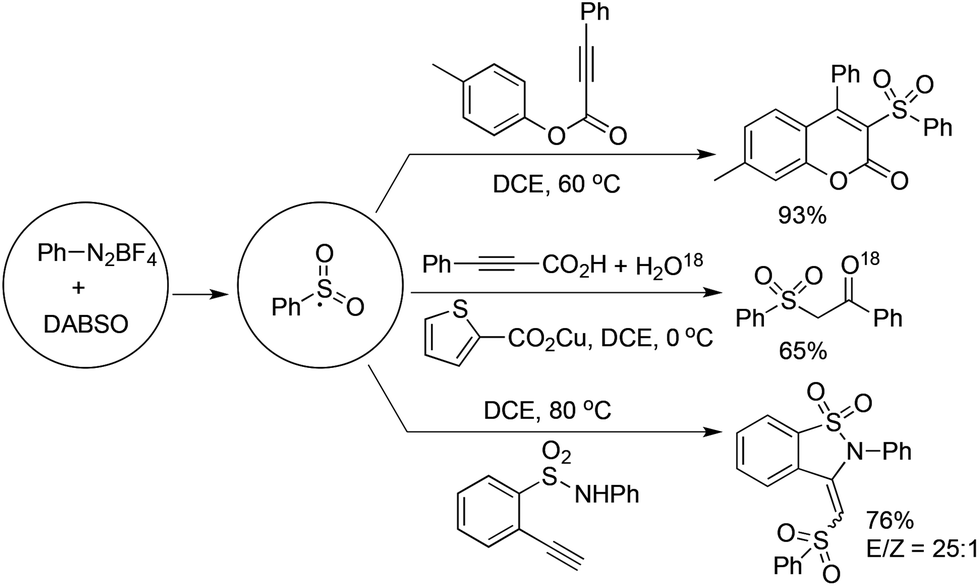 | ||
| Scheme 5 Reactions of aryldiazonium tetrafluoroborates, DABCO·(SO2)2, and alkynes. | ||
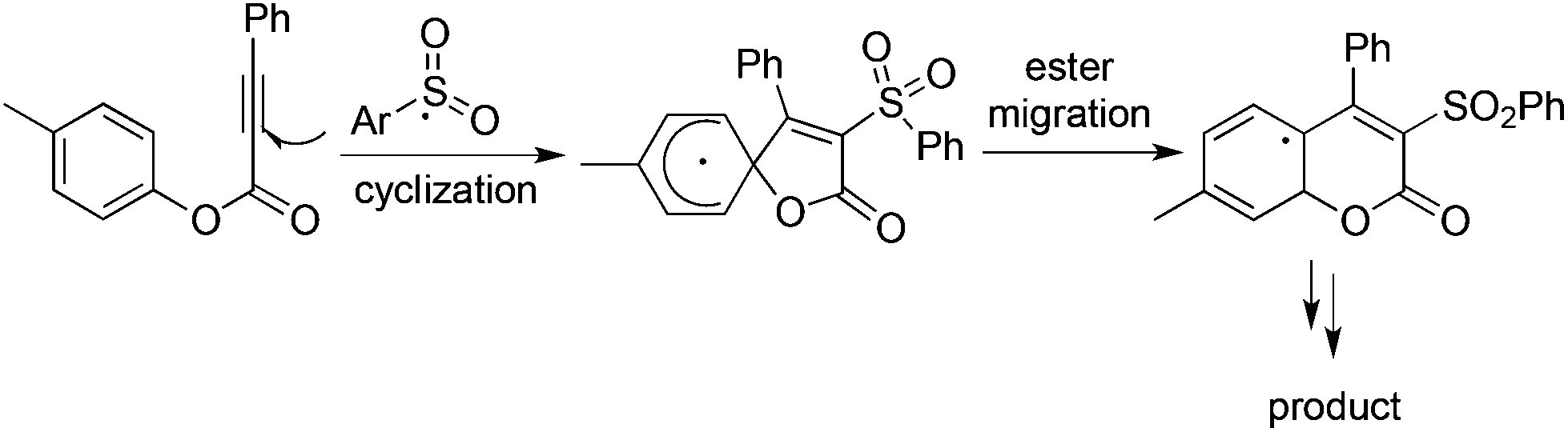 | ||
| Scheme 6 Mechanism studies for the formation of 3-sulfonated coumarins. | ||
Inspired by the above result, various alkyne-based reactions to trap the in situ generated arylsulfonyl radical were then developed. For instance, it was found that propiolic acid was a good reaction partner involving a four-component reaction, which underwent sulfonylation and decarboxylation to provide β-keto sulfones in good yields (Scheme 7).24 Interestingly, a radical α-addition was dominating in the reaction of arylsulfonyl radical with propiolic acid. The use of a copper catalyst facilitated the removal of the carboxylic group in propiolic acid during the reaction process.
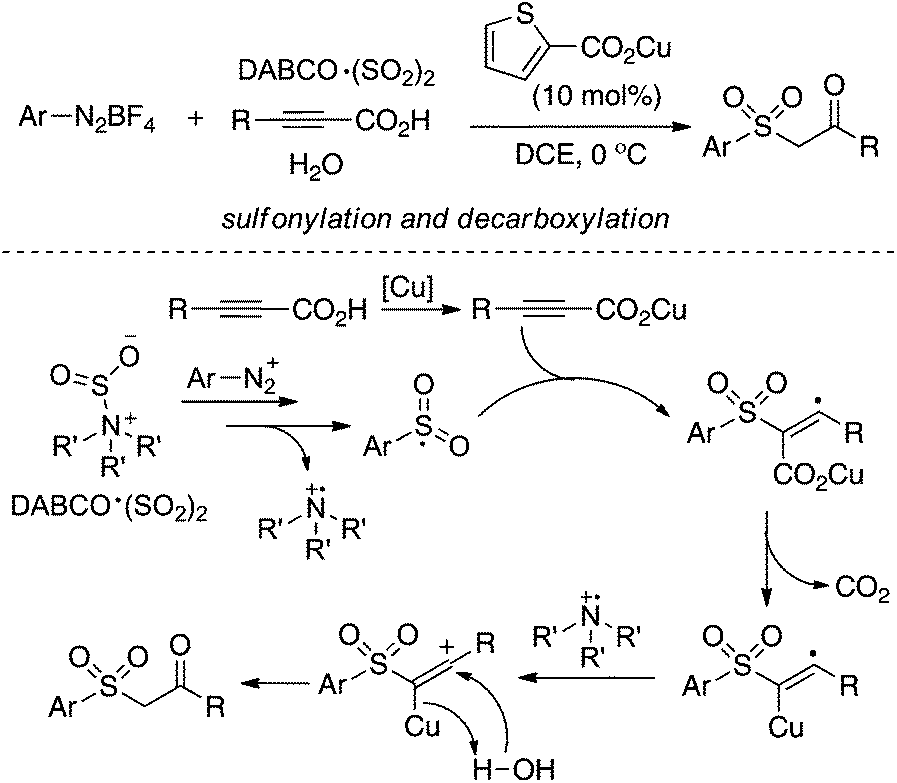 | ||
| Scheme 7 Synthesis of β-keto sulfones via sulfonylation and decarboxylation. | ||
Excellent regioselectivity was observed when treating an arylsulfonyl radical with terminal alkynes. As illustrated in Scheme 5, the reaction of aryldiazonium tetrafluoroborate, DABSO, and 2-functionalized arylacetylene gave rise to the corresponding benzosultam in a 76% yield, and the isomer ratio of E/Z was 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.25 The mechanistic studies showed that the in situ generated arylsulfonyl radical would attack the terminal carbon of arylacetylene to form a vinyl radical (Scheme 8). The arylsulfonyl-containing vinyl radical would be oxidized by the resulting DABCO radical cation into an arylsulfonyl-containing vinyl cation. The arylsulfonyl-containing vinyl cation could be trapped by a nucleophile promoted by the released base of DABCO.25
:1.25 The mechanistic studies showed that the in situ generated arylsulfonyl radical would attack the terminal carbon of arylacetylene to form a vinyl radical (Scheme 8). The arylsulfonyl-containing vinyl radical would be oxidized by the resulting DABCO radical cation into an arylsulfonyl-containing vinyl cation. The arylsulfonyl-containing vinyl cation could be trapped by a nucleophile promoted by the released base of DABCO.25
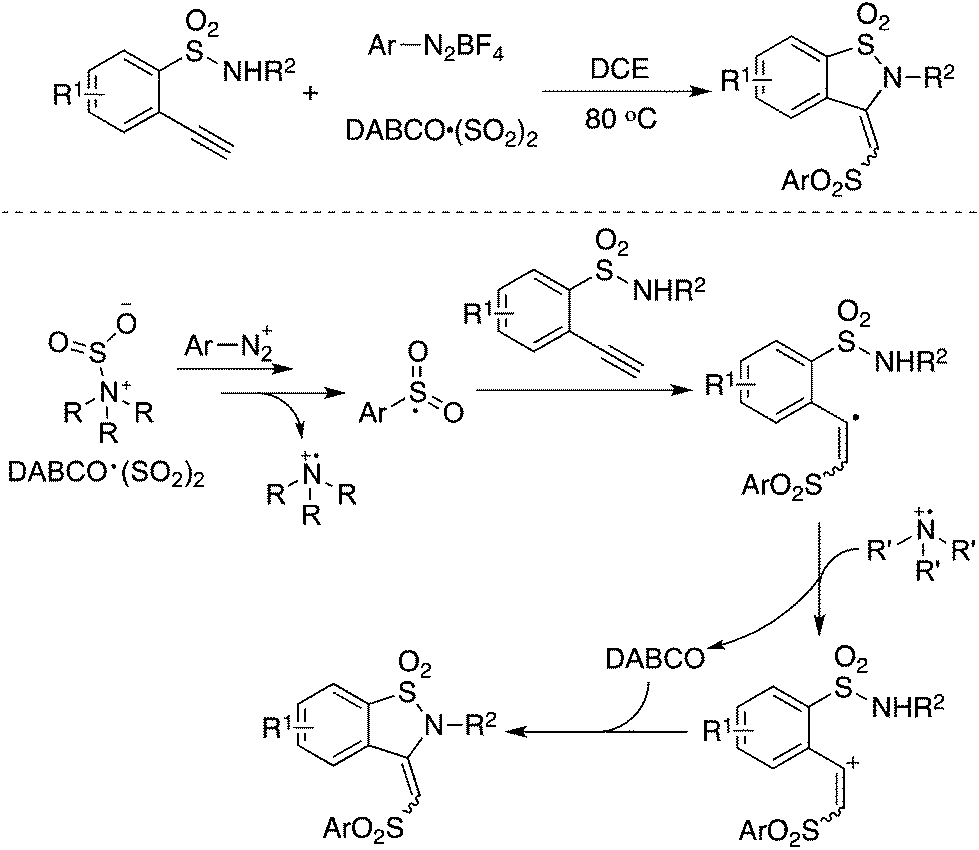 | ||
| Scheme 8 Synthesis of sulfonated benzosultams via the insertion of sulfur dioxide. | ||
Based on the above result, it could be postulated that the generated vinyl cation could be trapped by an additional nucleophile such as a halide anion. Therefore, potassium bromide or iodide was employed in the reaction. As expected, the desired iodinated or brominated vinyl sulfones could be obtained in good yields (Scheme 9).26 In particular, the iodinated vinyl sulfones dominated in producing an E-configuration. The presence of a copper catalyst would facilitate the oxidation of a vinyl radical to a vinyl cation. The resulting vinyl sulfones were reported as a novel class of neuroprotective agents toward Parkinson's disease therapy.27
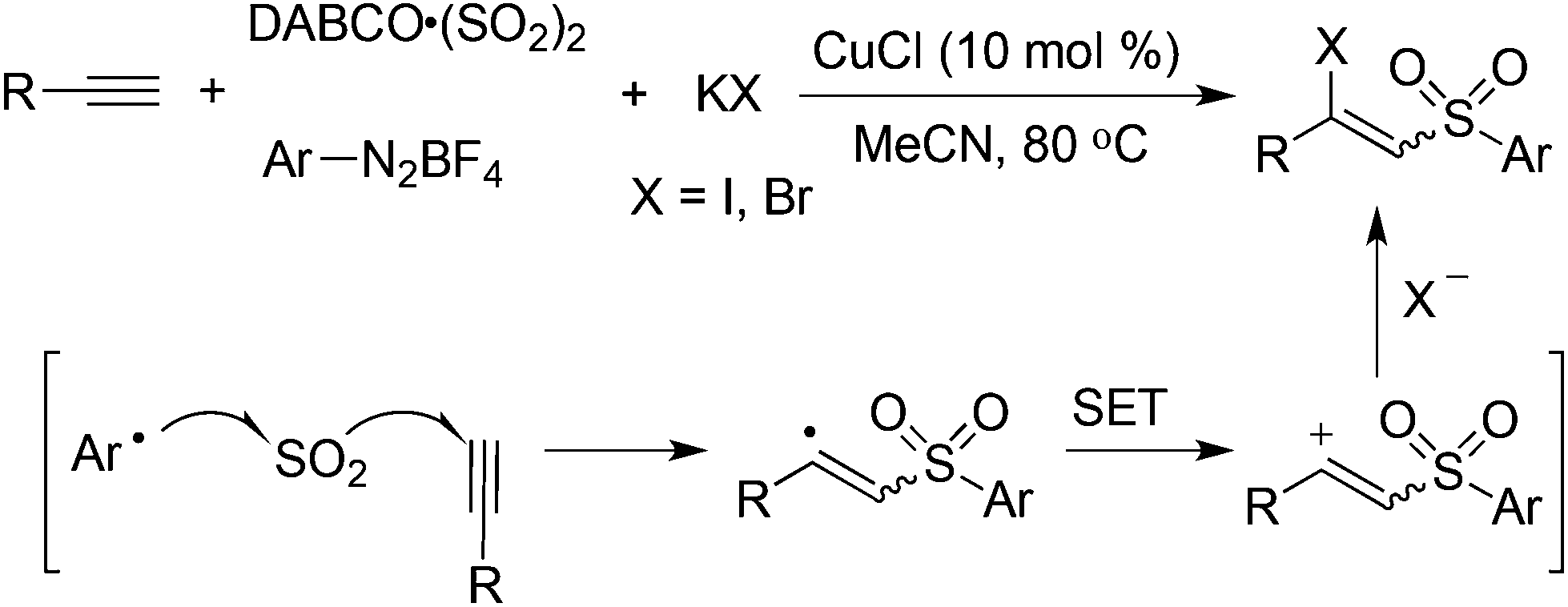 | ||
| Scheme 9 Synthesis of vinyl sulfones via a four-component reaction. | ||
A tandem radical reaction of 1,6-enynes, sulfur dioxide, and aryldiazonium tetrafluoroborates under mild conditions was developed recently (Scheme 10).28 Again, arylsulfonyl radicals from the reaction of aryldiazonium tetrafluoroborates with DABCO·(SO2)2 would attack the triple bond, which initiated the radical cyclization process. Sulfonated tetrahydropyridines and pyrrolidines were obtained depending on the substrates utilized. This reaction proceeds efficiently under mild conditions, and the addition of catalysts or additives was not necessary. It was noteworthy that two molecules of aryldiazonium tetrafluoroborates participated in the reaction.
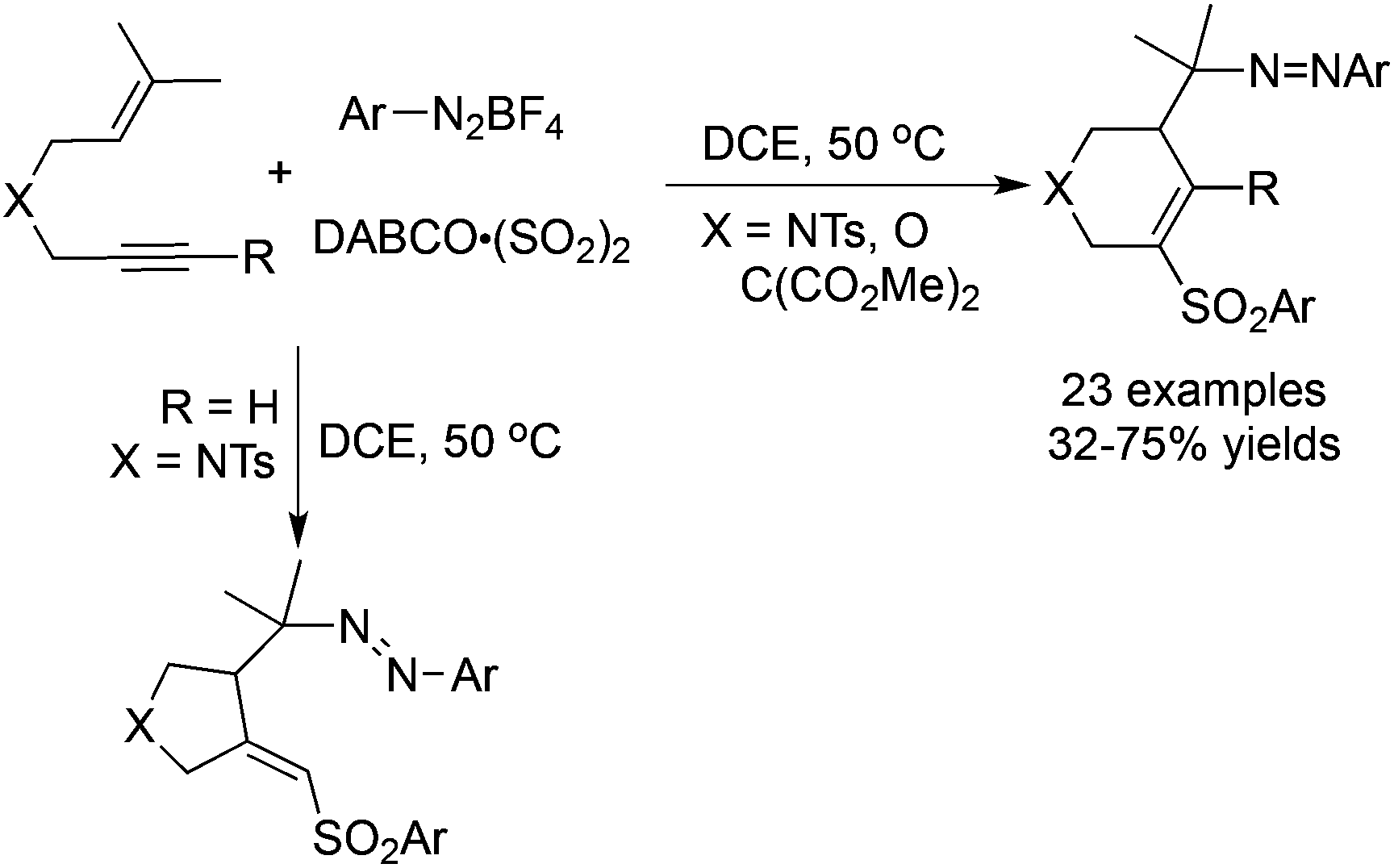 | ||
| Scheme 10 Radical reaction of 1,6-enynes, sulfur dioxide, and aryldiazonium tetrafluoroborates. | ||
2.2 Reactions of aryldiazonium tetrafluoroborates, sulfur dioxide, and alkenes
Based on the positive results of aryldiazonium tetrafluoroborates, DABCO·(SO2)2, and alkynes, the reaction of aryldiazonium tetrafluoroborates, DABCO·(SO2)2, and olefins was explored accordingly. As presented in Scheme 11, various olefins were employed to capture the in situ generated arylsulfonyl radicals.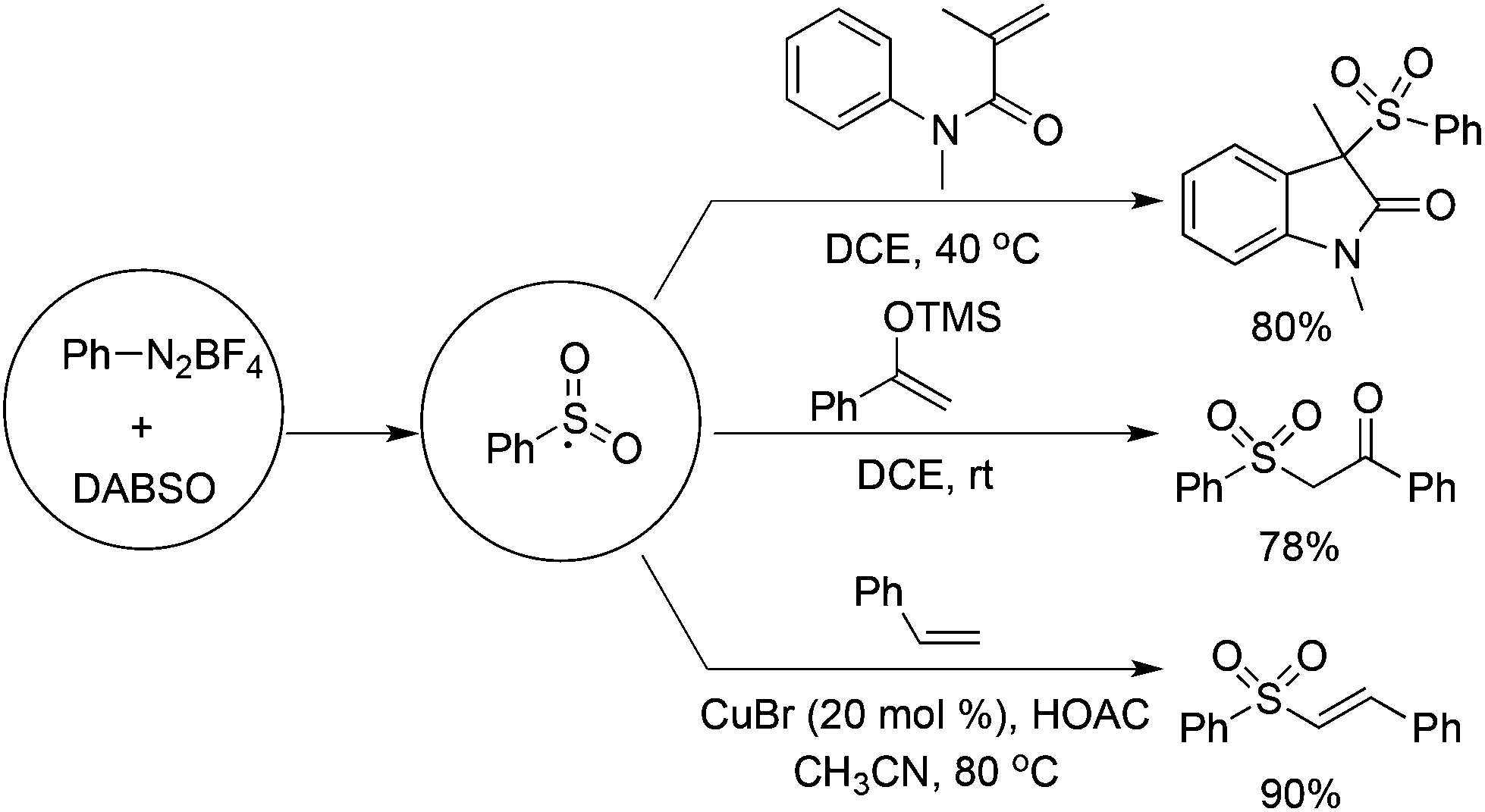 | ||
| Scheme 11 Reactions of aryldiazonium, DABCO·(SO2)2, and olefins. | ||
From Wu's results, the reaction of anilines, N-arylacrylamides, and DABCO·(SO2)2 proceeded efficiently, leading to 3-((arylsulfonyl)methyl)indolin-2-ones in good yields (Scheme 12).29 This one-pot reaction proceeds efficiently via the insertion of sulfur dioxide with a broad reaction scope. It was noteworthy that anilines serve as the aryl source, and the in situ generated sulfonyl radicals from sulfur dioxide as the key intermediates were trapped by N-arylacrylamides to provide the corresponding products under mild conditions. Compared with the reaction of alkynoates shown in Scheme 5, the reaction of N-arylacrylamides proceeded at a lower temperature.
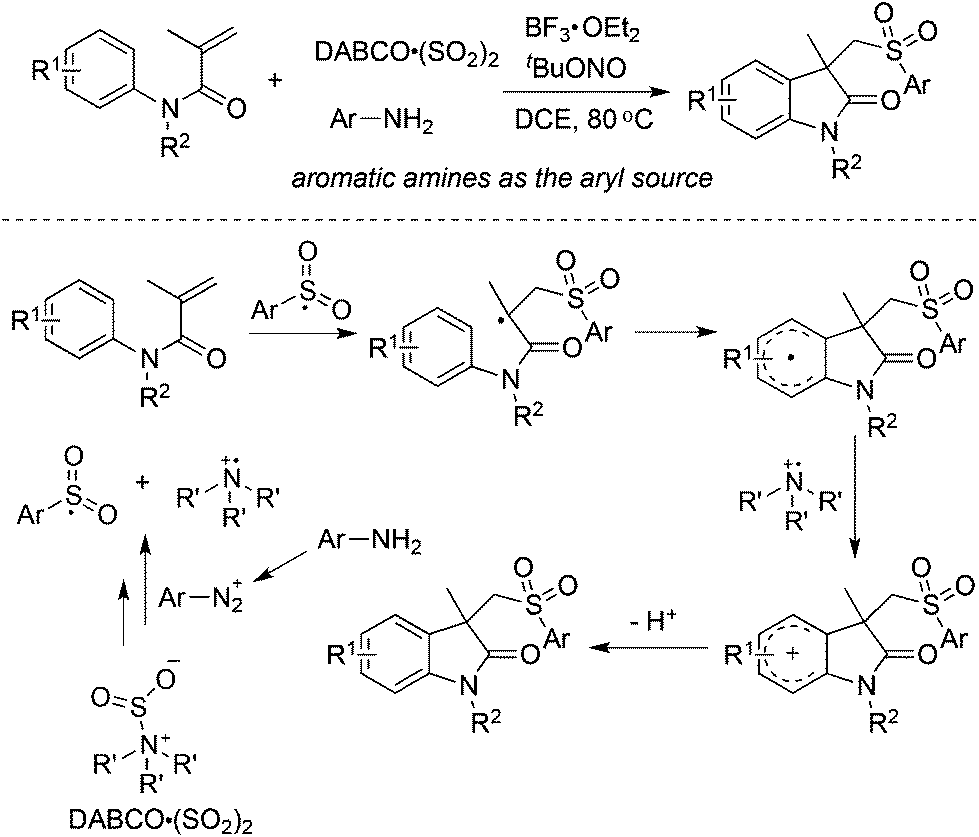 | ||
| Scheme 12 Synthesis of 3-((arylsulfonyl)methyl)indolin-2-ones via the insertion of sulfur dioxide by using anilines as the aryl source. | ||
By altering olefins to silyl enol ethers, β-keto sulfones could be generated through a reaction of aryldiazonium tetrafluoroborates and sulfur dioxide with silyl enol ether under catalyst-free and additive-free conditions (Scheme 13).30 This reaction proceeded efficiently and rapidly at room temperature in the absence of an oxidant and metal catalysis. Again, an arylsulfonyl radical initiated the transformation, and the released DABCO acted as a base in the reaction. Phenyldiazonium tetrafluoroborate with an allylic group attached on the ortho-position was employed in the reaction as well, which proceeded through an alkyl radical intermediate.
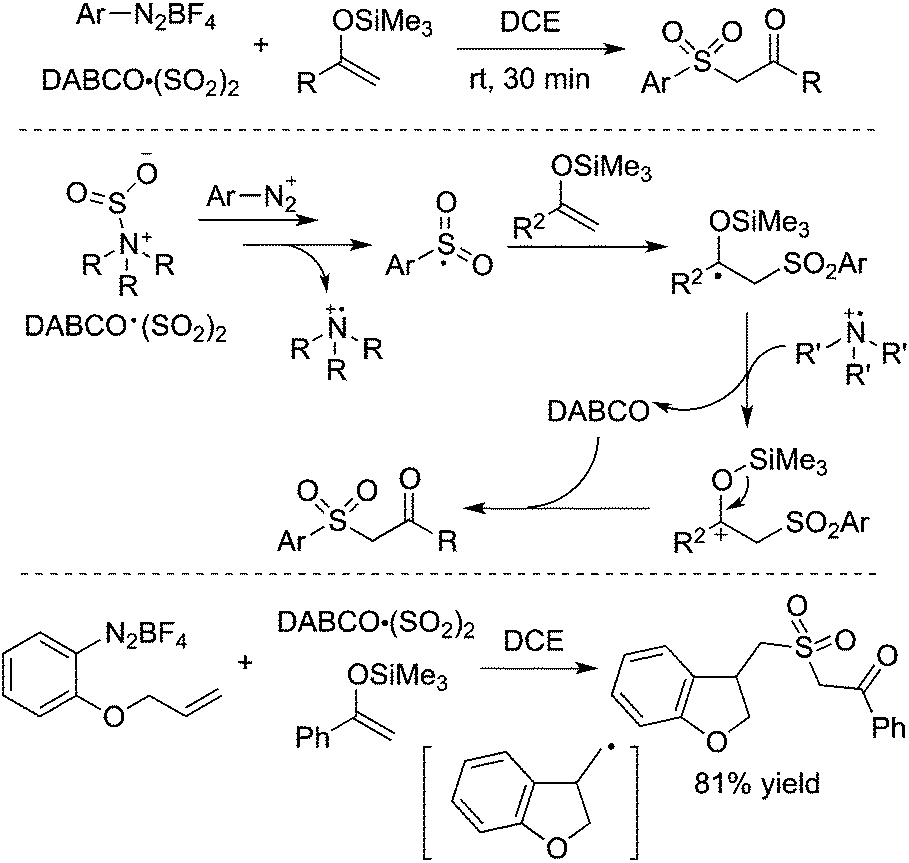 | ||
| Scheme 13 Generation of β-keto sulfones through a reaction of aryldiazonium tetrafluoroborates and sulfur dioxide with silyl enol ether. | ||
Alkenes could be utilized in the reaction of aryldiazonium tetrafluoroborates and sulfur dioxide (Scheme 14).31a Copper(II) bromide acted as an oxidant to promote the transformation of the radical intermediate to the cation. This transformation provides an efficient route to (E)-alkenyl sulfones or allylic sulfones via the insertion of sulfur dioxide, depending on the substrates used in the reactions. Feng and co-workers reported a similar transformation for the synthesis of vinyl sulfones, using aryl/alkyldiazonium tetrafluoroborates, alkenes, and DABCO·(SO2)2 as the starting materials in the presence of tetrabutylammonium iodide (TBAI) and tert-butylhydroperoxide (TBHP).31b However, the result described by Feng was questionable with the reproducibility issue, since extremely unstable alkyldiazonium salts were utilized in the transformation.
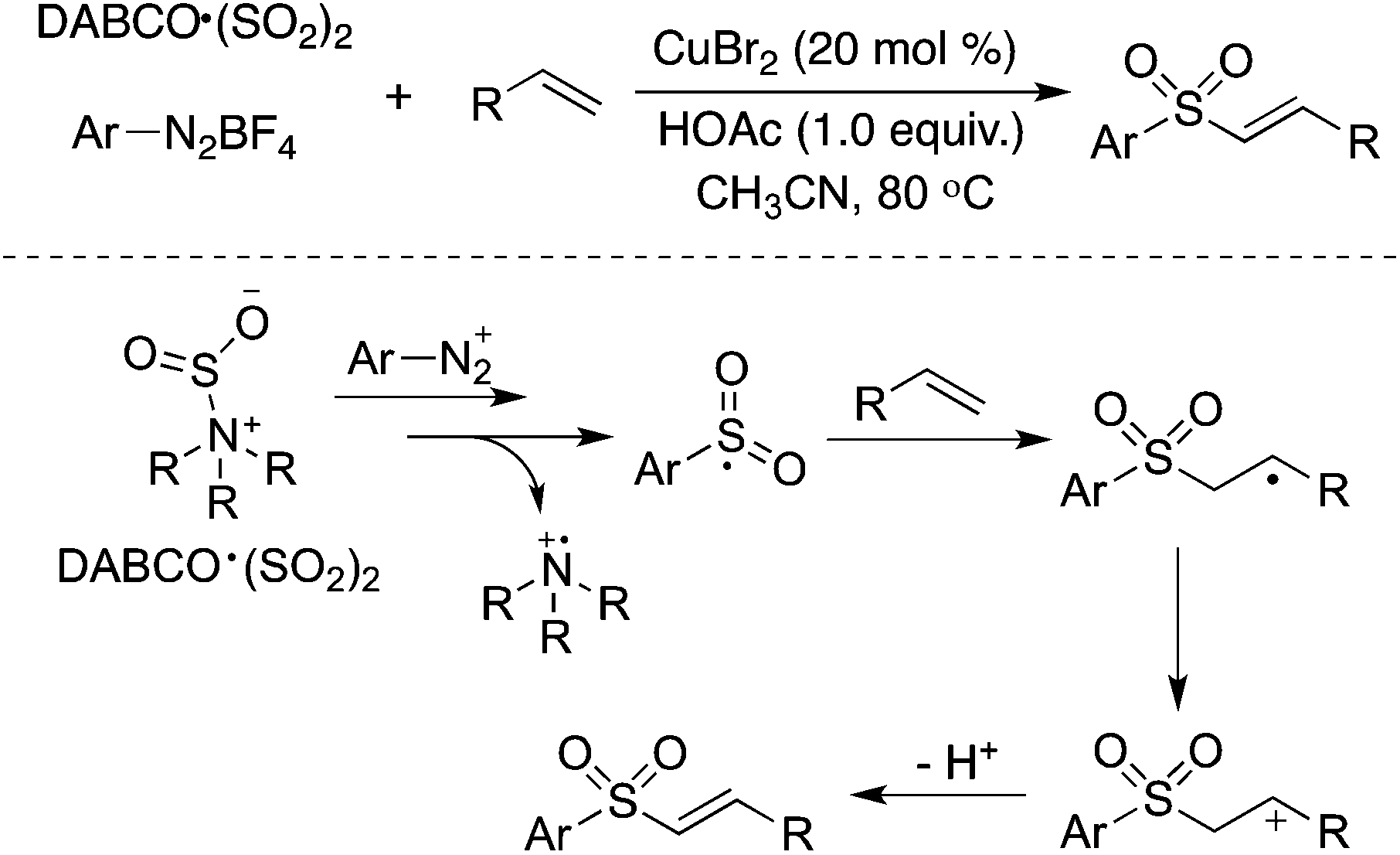 | ||
| Scheme 14 A three-component reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and alkenes. | ||
Prompted by the above result, a three-component reaction of 2-vinylbenzoic acids, aryldiazonium tetrafluoroborates, and sulfur dioxide was designed and developed under photocatalysis in the presence of visible light, which afforded diverse sulfonated isobenzofuran-1(3H)-ones in good yields (Scheme 15, eqn (a)).32a The arylsulfonyl radical generated in situ from aryldiazonium tetrafluoroborate and sulfur dioxide would attack the double bond of 2-vinylbenzoic acids, thus providing the corresponding radical intermediate. A single electron transfer (SET) assisted by a photocatalyst and intramolecular nucleophilic attack would give rise to the desired sulfonated isobenzofuran-1(3H)-ones. Additionally, 4-phenylpent-4-enoic acid and 5-phenylhex-5-enoic acid could be employed in the reactions of aryldiazonium tetrafluoroborates and sulfur dioxide as well under the standard conditions. This approach of using photocatalysis was based on the oxidative ability of the tertiary amine radical cation DABCO˙+ (DABCO, Ered1/2 = +0.69 V vs. SCE),33 which could not facilitate the SET during the reaction process. Thus, a stronger oxidant (e.g., fac-Ir(ppy)3, EIV/III1/2 = +0.77 V vs. SCE) had to be employed for the successful transformation. This strategy was further successfully applied for the synthesis of 4-((arylsulfonyl)methyl)-4H-benzo[d][1,3]oxazines under mild conditions through a photocatalytic reaction of N-(2-vinylphenyl)amides, DABCO·(SO2)2 and aryldiazonium tetrafluoroborates (Scheme 15, eqn (b)).32b
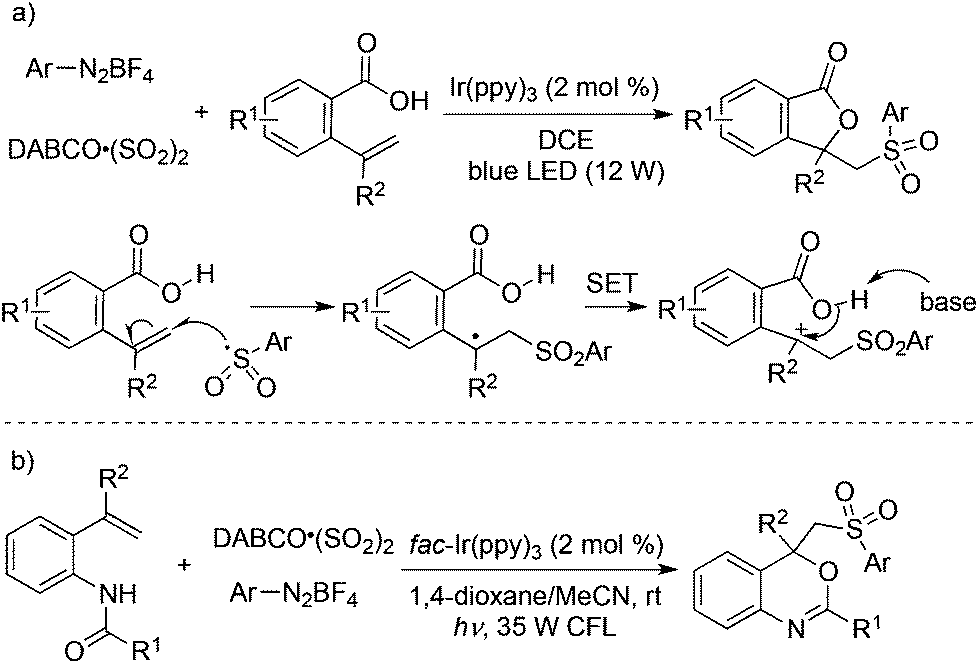 | ||
| Scheme 15 A three-component reaction of 2-vinylbenzoic acids or N-(2-vinylphenyl)amides, aryldiazonium tetrafluoroborates, and sulfur dioxide. | ||
The vicinal difunctionalization of alkenes with the insertion of sulfur dioxide through a four-component reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, alkenes, and hydroxylamines under catalyst-free and additive-free conditions was accomplished (Scheme 16).34 Interestingly, not only DABCO·(SO2)2 but also potassium metabisulfite (K2S2O5) was effective in this radical process with a broad substrate scope. Additionally, the competitive pathway from the three-component reaction of aryldiazonium tetrafluoroborates, sulfur dioxide, and hydroxylamines was not observed, which indicated the high selectivity of this multicomponent reaction.34 The product could be further elaborated. In the presence of Pd/C and hydrogen, the product could be easily transferred to the corresponding alcohol.
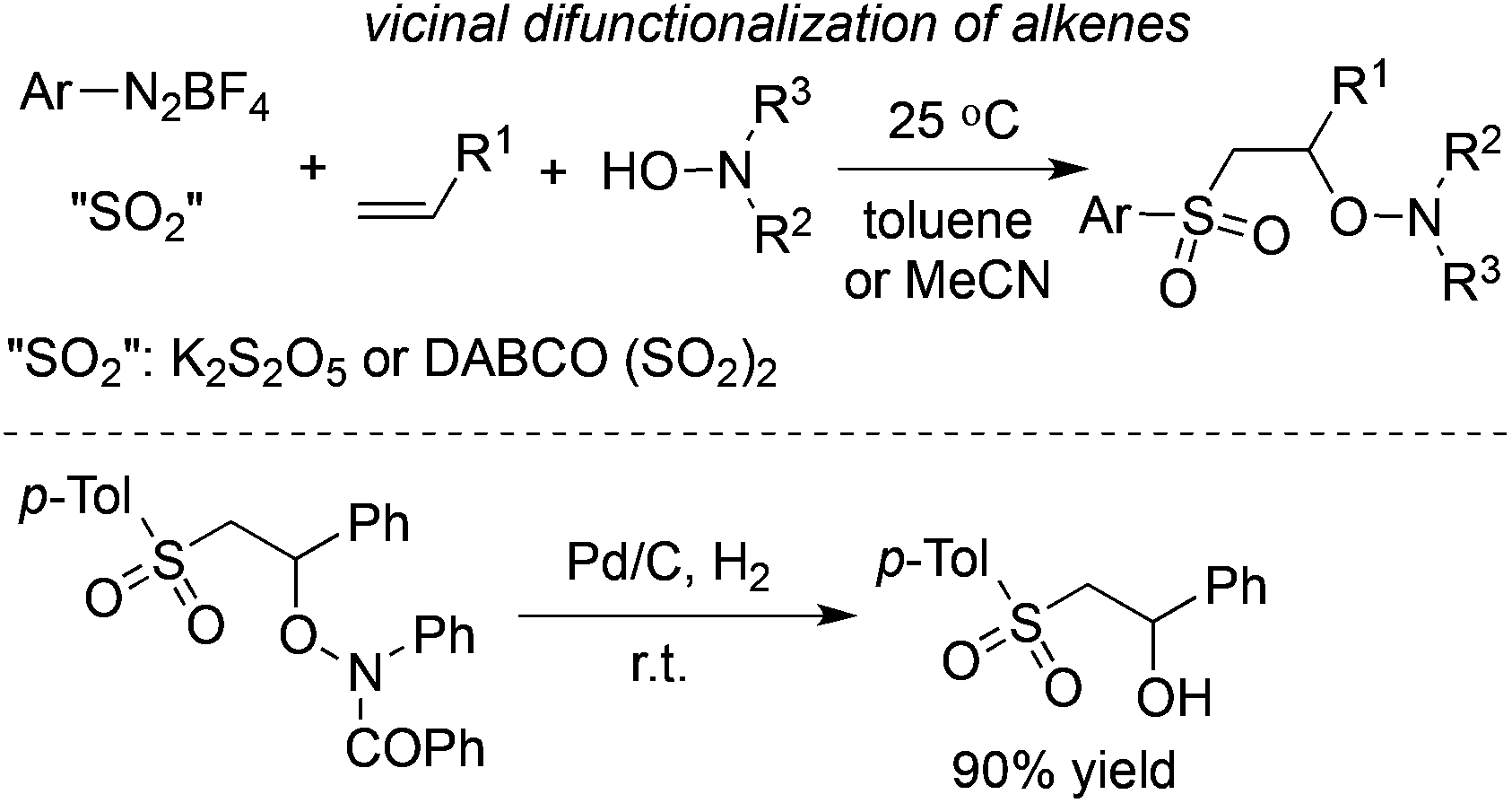 | ||
| Scheme 16 Vicinal difunctionalization of alkenes with the insertion of sulfur dioxide through a four-component reaction. | ||
Jiang and co-workers developed a tandem addition of arylsulfonyl radicals to benzene-linked allene-ynes for the synthesis of functionalized cyclobuta[a]naphthalen-4-ols starting from aryldiazonium tetrafluoroborates, and DABCO·(SO2)2 (Scheme 17, eqn (a)).35a The reaction pathway proceeded through [2 + 2] cycloaddition, insertion of sulfur dioxide via radical addition, 1,4-addition, diazotization, and tautomerization. This approach was further extended to the cyclization of β-alkynyl propenones, leading to sulfonated 1-indenones in good yields with excellent stereoselectivity (Scheme 17, eqn (b)).35b The efficiency of this oxidant-free azosulfonylation of β-alkynyl propenones with aryldiazonium salts and DABCO·(SO2)2 was demonstrated.
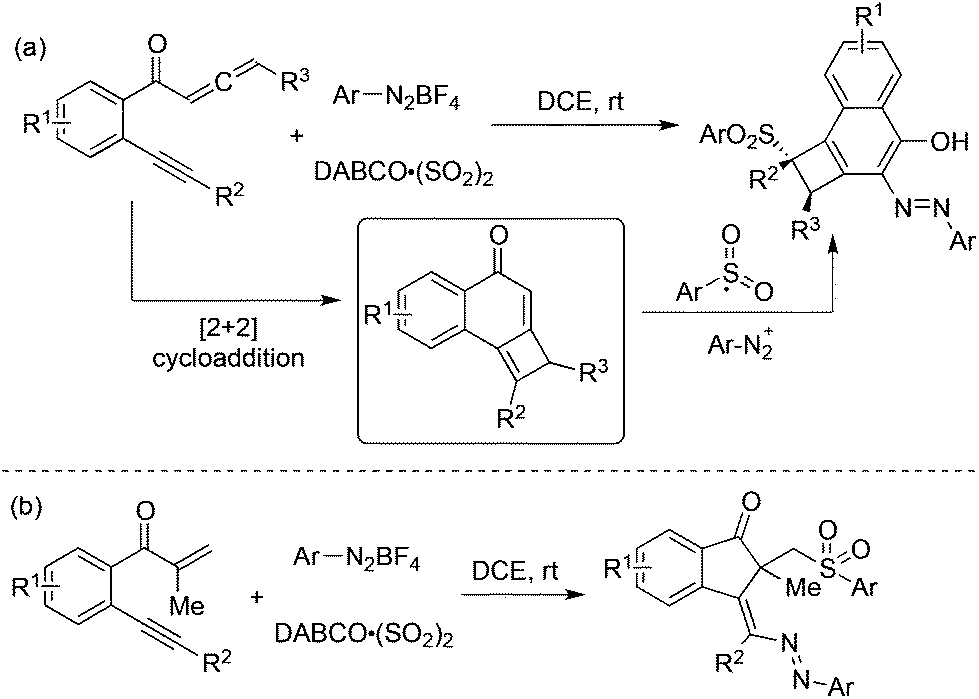 | ||
| Scheme 17 Synthesis of functionalized cyclobuta[a]naphthalen-4-ols and sulfonated 1-indenones with the insertion of sulfur dioxide. | ||
Cheng and co-workers reported a Cu(OAc)2-catalyzed three-component reaction of N-(arylsulfonyl)-acrylamides, aryldiazonium tetrafluoroborates, and DABCO·(SO2)2 (Scheme 18).36 This reaction was also triggered by arylsulfonyl radicals, leading to sulfonated oxindoles in moderate to good yields with good functional group tolerance. The transformation proceeded through a sequential radical addition, radical cyclization, and desulfonylative 1,4-aryl migration to produce the final products.
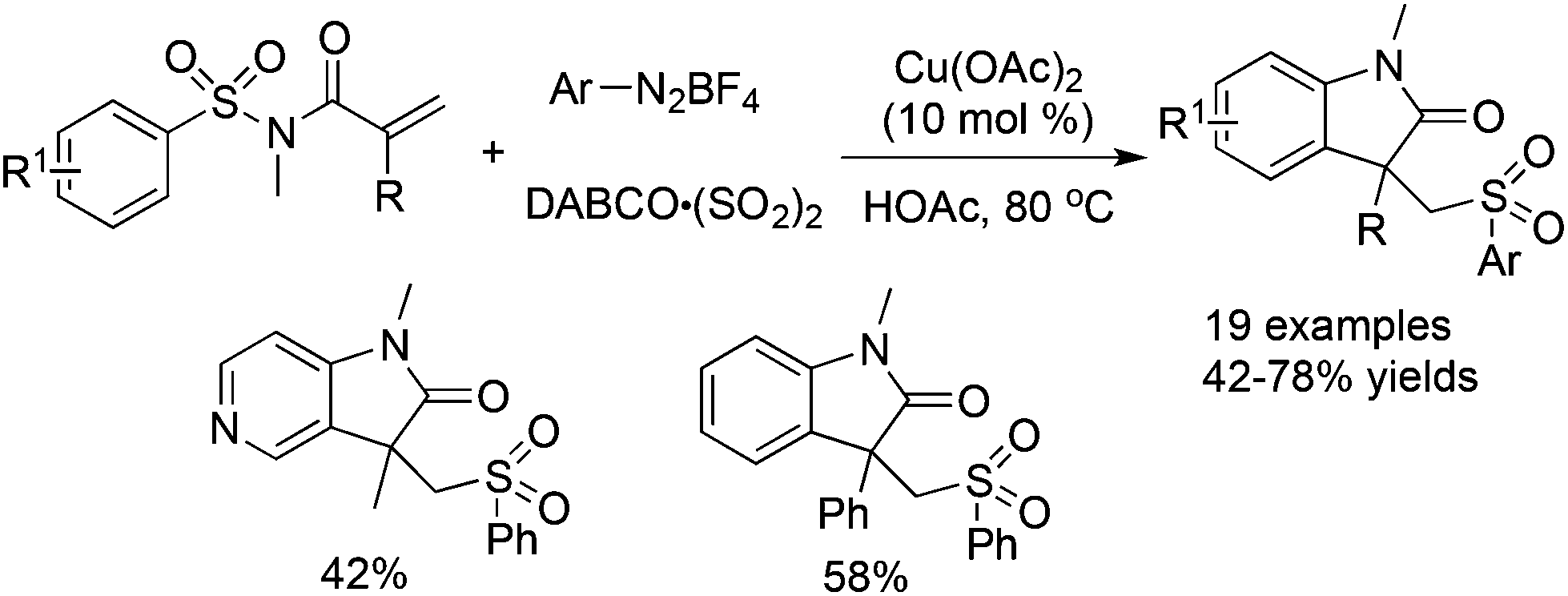 | ||
| Scheme 18 A Cu(OAc)2-catalyzed three-component reaction of N-(arylsulfonyl)acrylamides, aryldiazonium tetrafluoroborates, and DABCO·(SO2)2. | ||
A copper-catalyzed tandem reaction of benzene-tethered 1,7-enynes, aryldiazonium tetrafluoroborates, and DABCO·(SO2)2 for the synthesis of N-polycyclic compounds was developed (Scheme 19).37 In this reaction process, sulfur dioxide was not installed into the products although the arylsulfonyl radical acted as the key intermediate. A blank experiment without the addition of DABCO·(SO2)2 failed to produce the targeted molecules, thus suggesting the importance of DABCO·(SO2)2 for the success of this transformation. In light of the above information, a plausible mechanism including the radical insertion of sulfur dioxide, arylsulfonyl radical-triggered bicyclization, desulfonylation, and intramolecular migration was proposed to account for the formation of N-polycyclic compounds.38 In the process, the arylsulfonyl radical, derived from aryldiazonium tetrafluoroborates and DABCO·(SO2)2, would undergo a cascade attack on the double bond and triple bond of benzene-tethered 1,7-enyne, providing a spiro-intermediate through radical bicyclization and desulfonylation. This followed by a single-electron oxidation would afford a cation intermediate. The subsequent intramolecular vinyl migration and alkyl migration would produce a mixture of N-polycyclic compounds through different pathways. The exploration of the reaction scope showed that the alkyl migration of the cation intermediate was slightly easier in most cases.
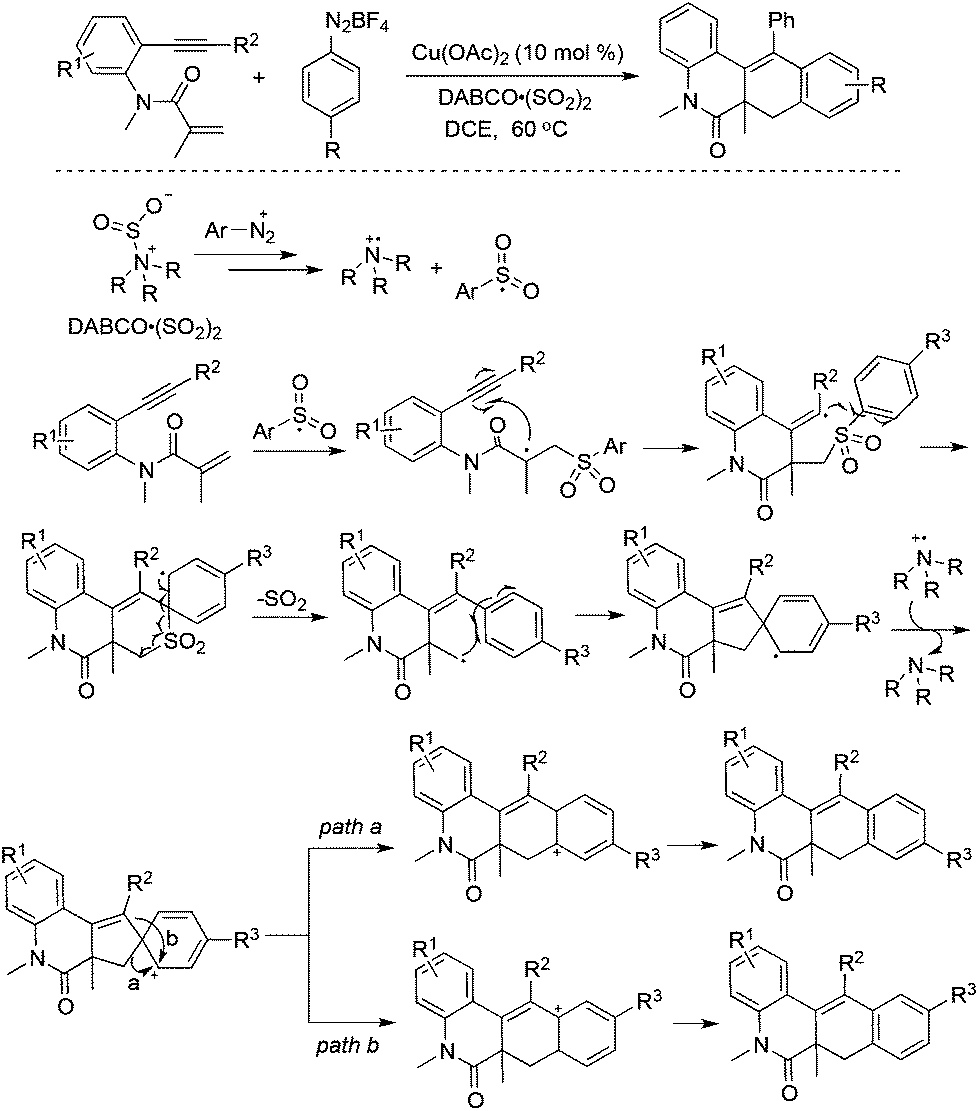 | ||
| Scheme 19 A copper-catalyzed tandem reaction of benzene-tethered 1,7-enynes, aryldiazonium tetrafluoroborates, and DABCO·(SO2)2. | ||
2.3 Insertion of sulfur dioxide with aryldiazonium tetrafluoroborates for C–H bond functionalization
So far, significant progress has been made for the coupling reactions through C–H bond activation under transition metal catalysis in organic synthesis.39 The combination of aryldiazonium tetrafluoroborates and DABCO·(SO2)2 could also be applied to the palladium-catalyzed direct sulfonylation of C–H bonds with the insertion of sulfur dioxide under mild conditions. Wu and co-workers described the sulfonylative couplings with the insertion of sulfur dioxide into C–H bonds by using 8-aminoquinoline amides as the substrates (Scheme 20).40 Interestingly, two classes of sulfonylative products were obtained by the combination of radical chemistry and palladium-catalyzed C–H activation. Additionally, dual C–H functionalization was observed in some cases. As shown in Scheme 20, after introducing the arylsulfonyl group in the 2-position of benzeneamide assisted by palladium catalysis, another aryldiazonium tetrafluoroborate was involved and the aryldiazo group was installed in the para-position of 8-aminoquinoline amide. In the reaction process, the arylsulfonyl radical and palladium-chelated complex via the coordination of the palladium catalyst with the substrate were the key intermediates.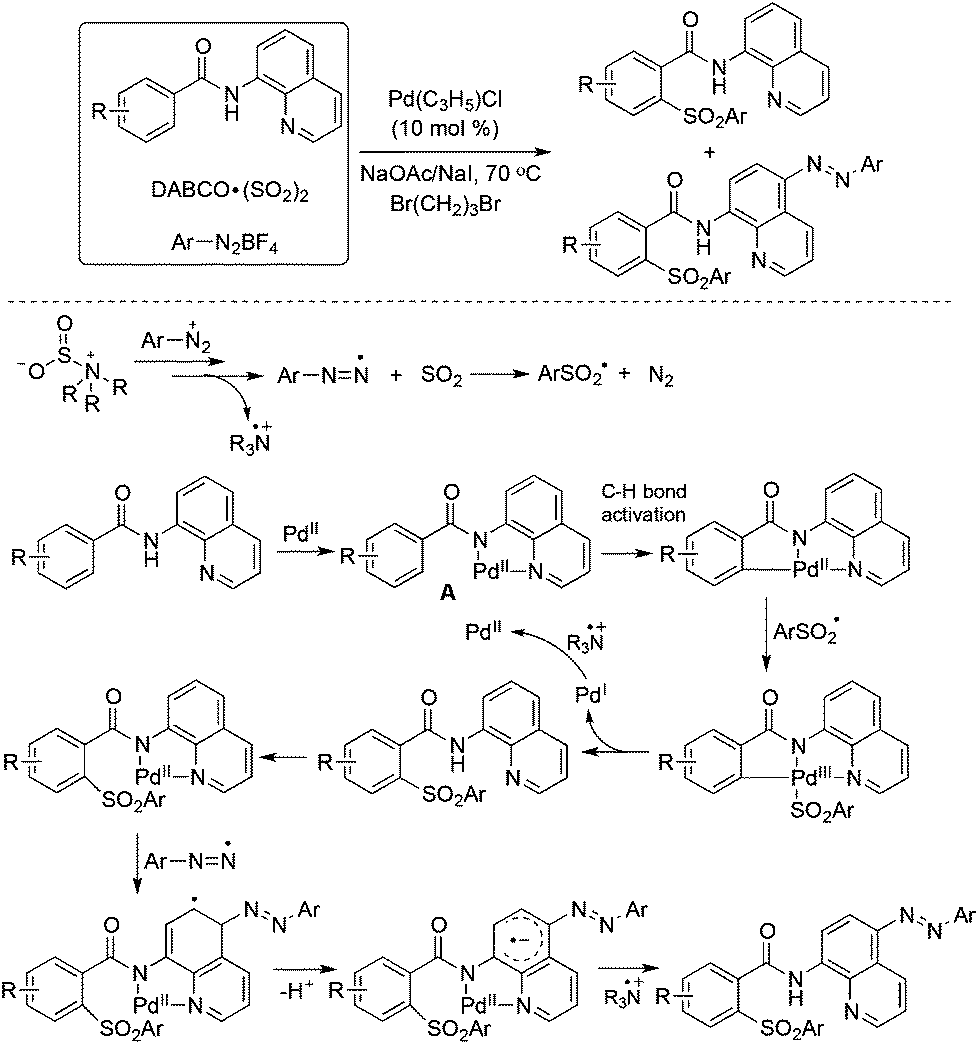 | ||
| Scheme 20 A palladium-catalyzed direct sulfonylation of C–H bonds with the insertion of sulfur dioxide. | ||
To our surprise, the sulfonylative C–H bond functionalization through a three-component reaction of 8-aminoquinoline amides, DABCO·(SO2)2 and aryldiazonium tetrafluoroborates in the presence of a copper catalyst provided a different outcome (Scheme 21).41 The selectivity in the para-position was observed and the arylsulfonyl group was introduced under the copper-catalyzed conditions. Similar to the palladium-catalyzed reaction, this transformation was triggered by a copper-chelated complex via the coordination of the copper catalyst with the substrate. The corresponding 5-sulfonyl-8-aminoquinoline amides were generated in moderate to good yields. Subsequently, Xia and co-workers reported a similar transformation, which utilized a cobalt salt as the catalyst (Scheme 21).42 The same products and selectivity were observed under the conditions. Additionally, anilines were employed successfully as a replacement for aryldiazonium tetrafluoroborates.
 | ||
| Scheme 21 Sulfonylation of C–H bonds with the insertion of sulfur dioxide catalyzed by Cu(OAc)2 or Co(acac)2. | ||
The reaction system of DABCO·(SO2)2 and aryldiazonium tetrafluoroborates was further combined with naphthols. This three-component reaction of naphthols, sulfur dioxide, and aryldiazonium tetrafluoroborates catalyzed by iron(III) chloride worked well, leading to diverse 1-sulfonated 2-naphthols in good yields (Scheme 22).43 The transformation proceeded through a radical process via direct C–H functionalization. The reaction was triggered by iron(III) chloride, giving rise to a naphthol radical through a single electron transfer. The arylsulfonyl radical and the tertiary amine cation radical were involved in the meantime. Sensitive functional group tolerance was observed, and substrates with nitro, halo, cyano, hydroxy, and ester groups were all effective under the conditions.
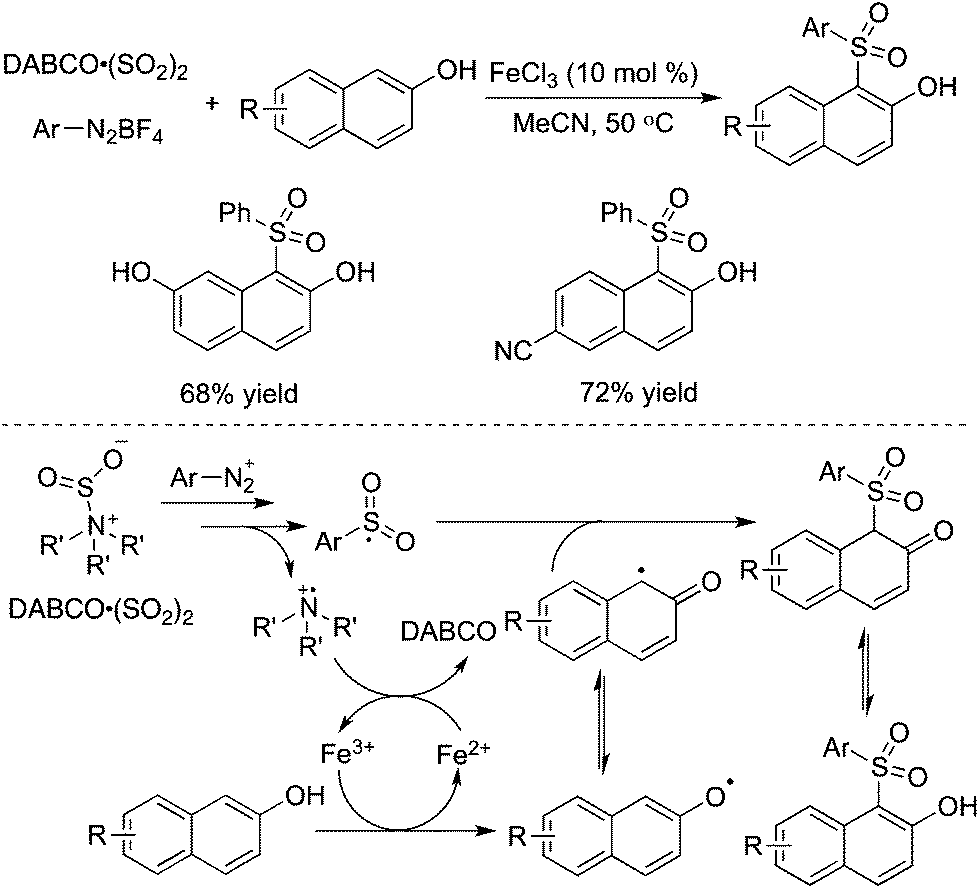 | ||
| Scheme 22 Synthesis of sulfonated naphthols via C–H bond functionalization with the insertion of sulfur dioxide. | ||
Direct C–H bond sulfonylation of indoles was achieved as well in the presence of palladium catalysis with the insertion of sulfur dioxide. A palladium-catalyzed three-component reaction of 1-(pyridin-2-yl)indoles, DABCO·(SO2)2 and aryldiazonium tetrafluoroborates under mild conditions afforded the corresponding 2-sulfonated indoles in good yields (Scheme 23).44 This transformation occurred by using 10 mol% of palladium(II) bromide as the catalyst at room temperature. This radical process by merging palladium catalysis and sulfur dioxide was highly effective. Further investigation revealed that the pyridinyl directing group could be extended to the 2-pyrimidinyl group in the C–H bond sulfonylation. The mechanism was similar to that mentioned in Scheme 20, which combined radical chemistry and palladium-catalyzed C–H activation. A Pd(II)/Pd(IV) catalytic cycle was proposed via a palladium-chelated complex of the substrate and arylsulfonyl radical.
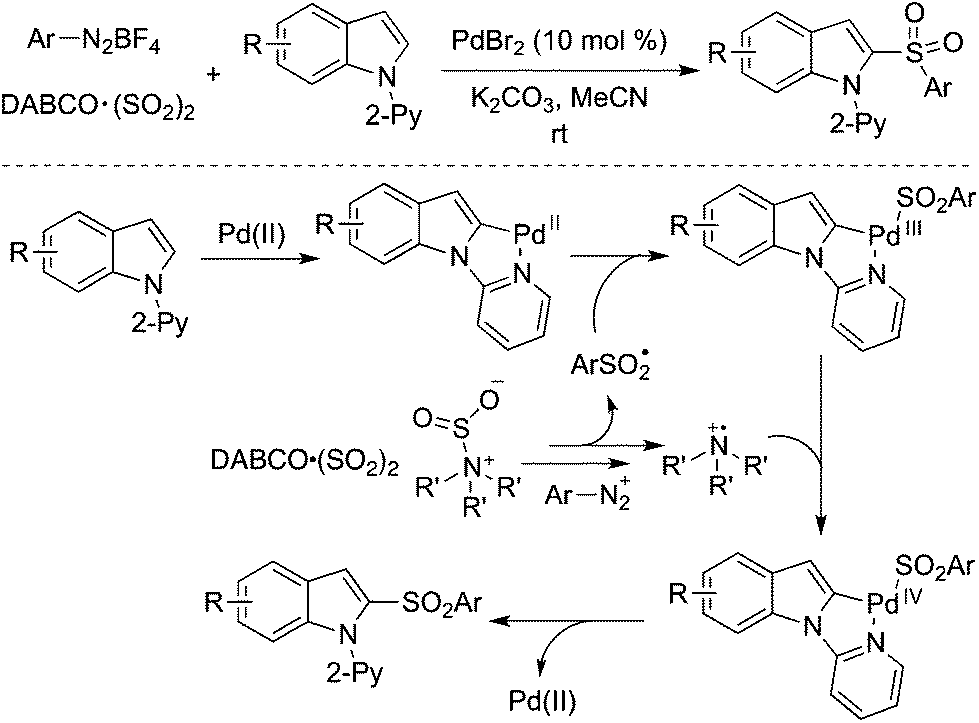 | ||
| Scheme 23 A palladium-catalyzed direct C–H bond sulfonylation of indoles with the insertion of sulfur dioxide. | ||
3. Insertion of sulfur dioxide with aryl halides
As we know, the homolytic cleavage of a carbon–halo bond could happen under UV irradiation.45 The aryl or alkyl radicals could be easily produced from aryl halides or alkyl halides. Consequently, Wu and co-workers realized the UV-irradiated radical insertion of sulfur dioxide into aryl halides and alkyl halides (Scheme 24).46a In the presence of tetra-n-butylammonium iodide (TBAI), aryl halides or alkyl halides could serve as a good source of aryl/alkyl radicals to trap sulfur dioxide, resulting in aryl/alkyl sulfonyl radicals. A halo radical, generated from the homolytic cleavage of a carbon–halo bond, would react with hydrazine to provide a hydrazine radical via hydrogen abstraction. The subsequent coupling of an aryl/alkyl sulfonyl radical with a hydrazine radical would deliver diverse N-aminosulfonamides in 36–94% yields. Not only aryl iodides but also aryl chlorides and aryl bromides were all workable under the conditions. Additionally, alkyl halides were also compatible, giving rise to the corresponding alkyl-connected N-aminosulfonamides in good yields. This result was interesting since alkyl halides were not suitable in the transition metal-catalyzed insertion of sulfur dioxide due to the issue of β-H elimination. The above strategy was further applied to the reaction of N-(2-iodoaryl)acrylamides. N-(2-Iodoaryl)acrylamides reacted with DABCO·(SO2)2 and hydrazines under ultraviolet irradiation, leading to (2-oxoindolin-3-yl) methanesulfonohydrazides in good yields.46b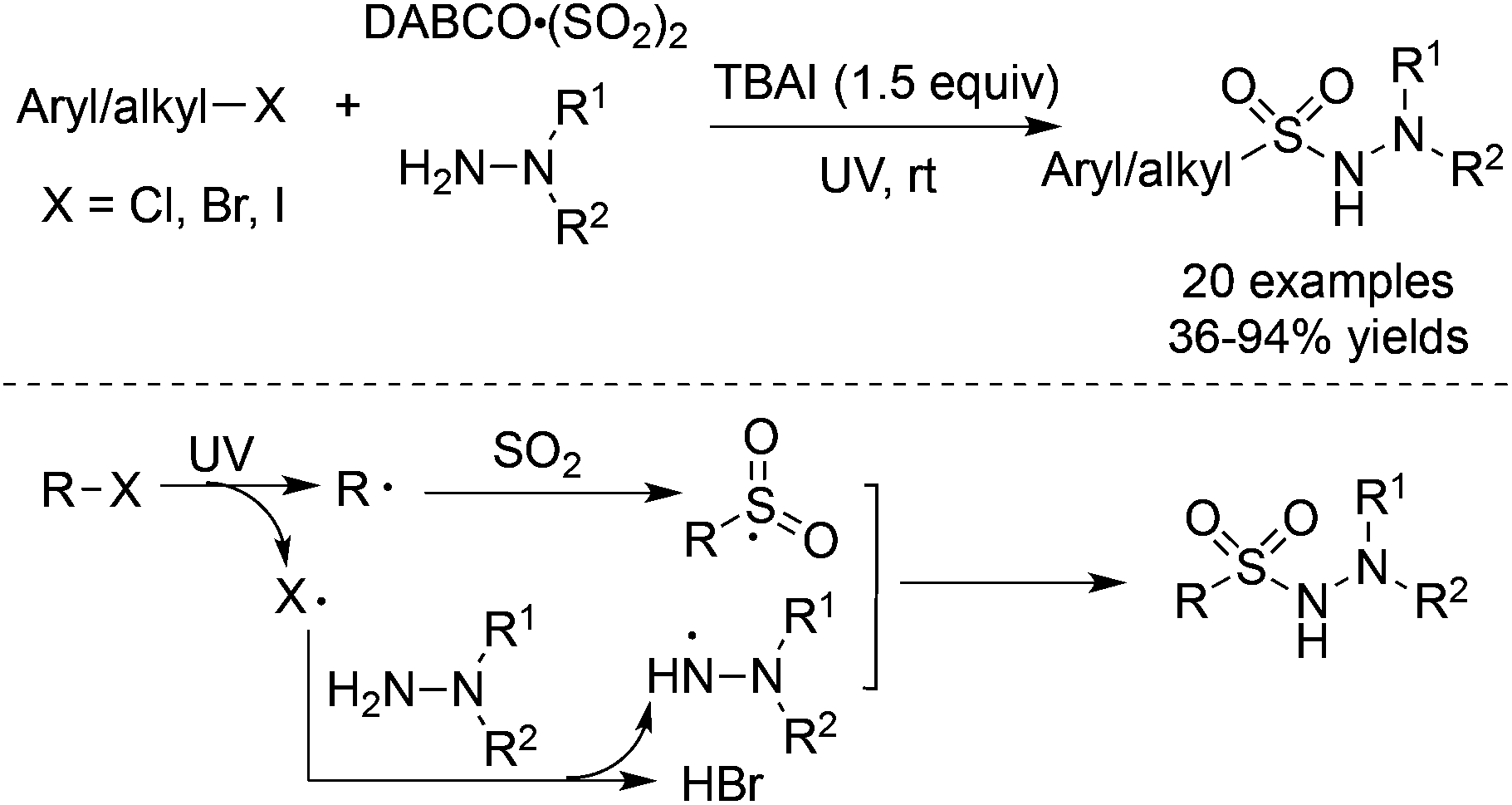 | ||
| Scheme 24 Aminosulfonylation of aryl/alkyl halides, sulfur dioxide, and hydrazines under UV irradiation. | ||
Prompted by the above result, the radical insertion of sulfur dioxide under UV irradiation could be extended to silyl enolate ether. The three-component reaction of aryl/alkyl iodides, DABCO·(SO2)2 and trimethyl((1-arylvinyl)oxy)silanes proceeded smoothly under UV irradiation, providing 1-aryl-2-(arylsulfonyl)ethan-1-ones in 36–81% yields without any additives at room temperature (Scheme 25).47 The exploration of the reaction generality showed that 3-iodopyridine was an efficient reaction partner as well, giving rise to the corresponding product in a moderate yield. Again, not only aryl iodides but also alkyl iodides are suitable substrates in the transformation.
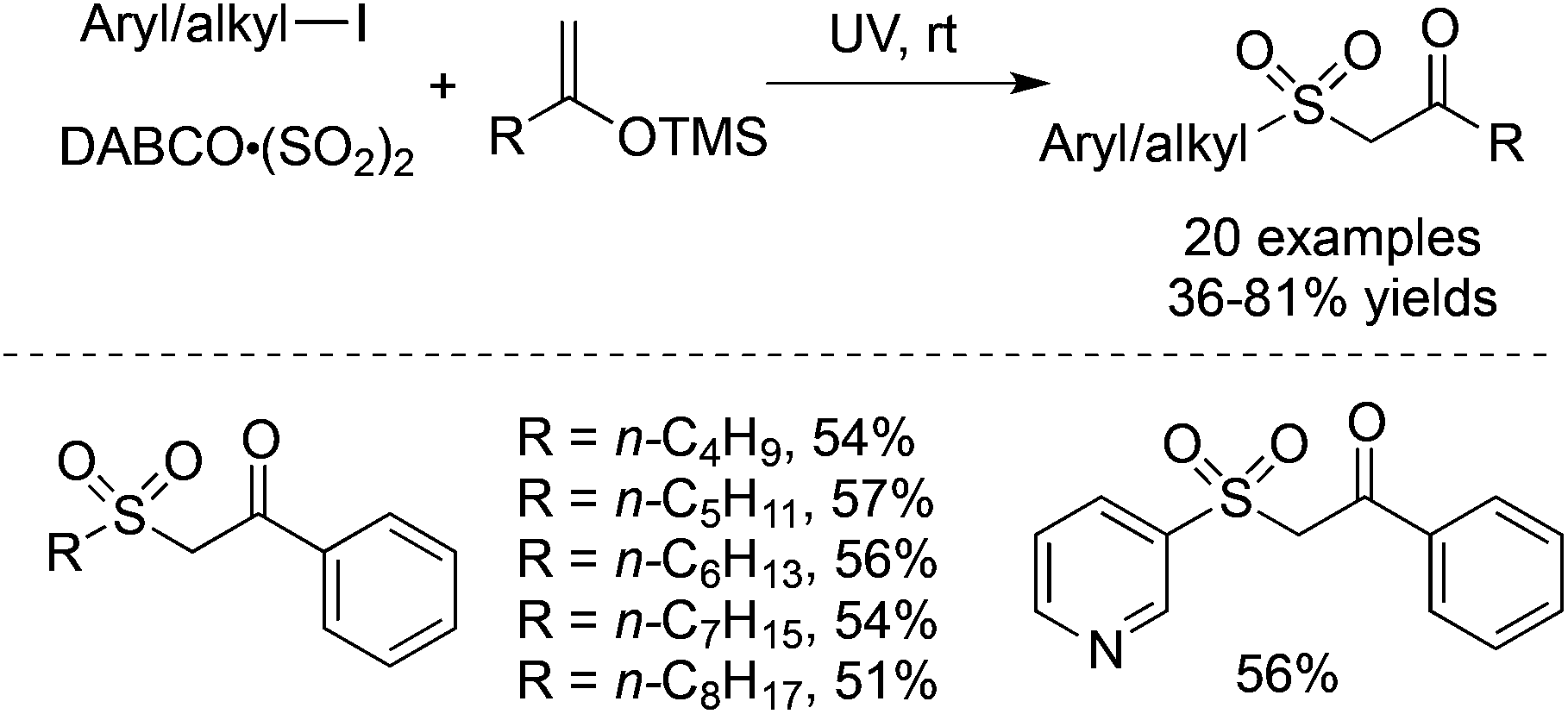 | ||
| Scheme 25 A three-component reaction of aryl/alkyl iodides, DABCO·(SO2)2 and silyl enolate ethers under UV irradiation. | ||
4. Insertion of sulfur dioxide with vinyl radicals
The fixation of sulfur dioxide with aryl radicals and alkyl radicals was achieved by many elegant examples. Therefore, the insertion of sulfur dioxide with vinyl radicals was then explored. A four-component reaction of Togni's reagent, alkynes, sulfur dioxide, and hydrazines at room temperature was designed and developed (Scheme 26).48 This direct vicinal difunctionalization of alkynes under catalyst-free conditions was highly efficient, leading to (E)-3,3,3-trifluoroprop-1-ene-1-sulfonohydrazides in good yields with excellent stereoselectivity and regioselectivity. This one-pot transformation proceeded under mild conditions without any catalysts and additives. Togni's reagent was used as the source of the trifluoromethyl radical. The in situ generated trifluoromethyl radical initiated the reaction, which attacked the alkyne regioselectively to form a vinyl radical. Further addition to sulfur dioxide would produce a vinylsulfonyl radical, which would be trapped by hydrazine to provide the final product. A broad substrate scope with good functional group tolerance in the trifluoromethylation of alkynes and aminosulfonylation via the insertion of sulfur dioxide was observed. A plausible mechanism supported by DFT calculations was proposed. This transformation was efficient, since there were several competitive pathways in the reaction process. It was assumed that the trifluoromethylsulfonyl radical generated from sulfur dioxide with the trifluoromethyl radical was not stable in the reaction; thus it underwent reversal to the trifluoromethyl radical. Additionally and more importantly, the reaction speed of the trifluoromethyl radical to the terminal alkyne was faster than that of sulfur dioxide with the trifluoromethyl radical. In the process, hydrazine served as an electron donor to facilitate the formation of the trifluoromethyl radical from Togni's reagent.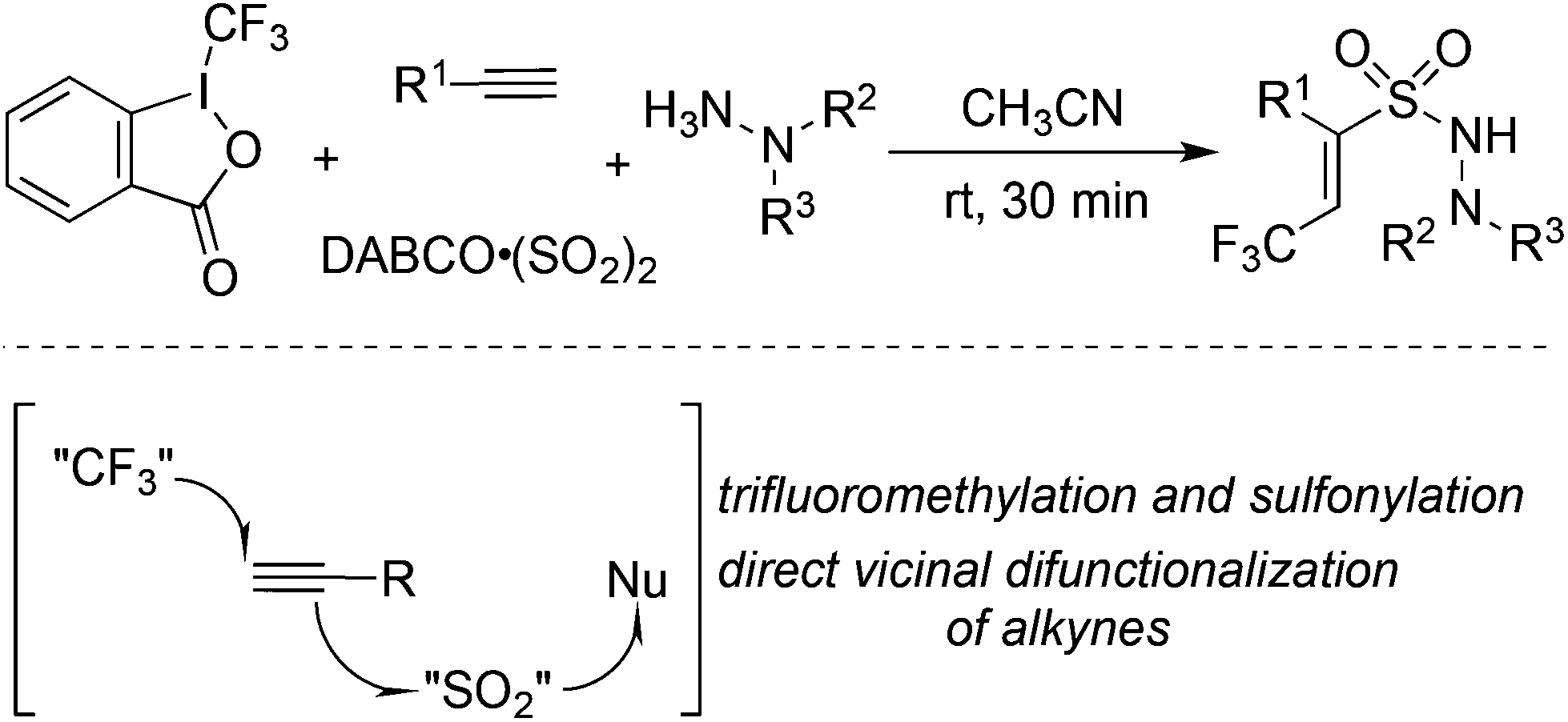 | ||
| Scheme 26 A four-component reaction of Togni's reagent, alkynes, sulfur dioxide, and hydrazines. | ||
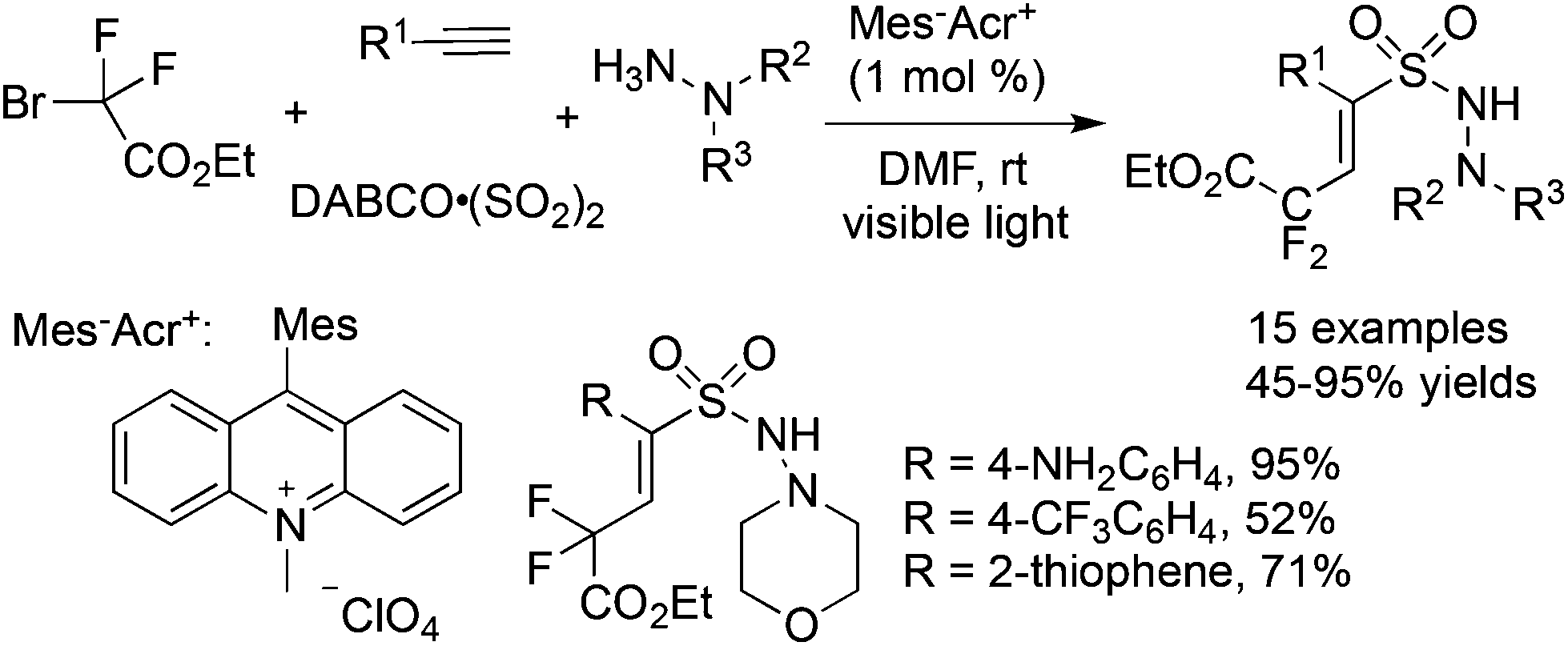
By altering Togni's reagent to the difluoroalkyl radical precursor, a similar pathway took place with the formation of difluoroalkyl-substituted sulfonamides in 45–95% yields (Scheme 27).49 However, the reaction needed to be triggered under photoinduced conditions. The reaction condition optimization suggested that MesAcr+ was the best choice for the photocatalyst. Moreover, terminal alkynes with electron-rich groups were more favorable than alkynes with electron-withdrawing groups. For example, the use of 4-aminophenylacetylene as the substrate under standard conditions provided the corresponding product in a 95% yield, while the reaction of 4-trifluoromethylphenylacetylene afforded the desired product in a 52% yield. It was worth noting that many highly reactive groups such as amino groups and heterocycles were compatible in the reaction.
 | ||
| Scheme 27 A four-component reaction of difluoroalkyl bromides, alkynes, sulfur dioxide, and hydrazines. | ||
5. Insertion of sulfur dioxide with nitrogen-centered radicals
In addition to carbon-centered radicals, nitrogen-centered radical species under visible light photocatalysis have attracted continuous attention.50 Therefore, a nitrogen-centered radical was also employed in the insertion of sulfur dioxide. As presented in Scheme 28, the homolytic cleavage of the N–O bond in N-oxime would provide an imine radical in the presence of blue LED.51 From the established experimental results, the imine radical was not captured by sulfur dioxide, but converted into a carbon-centered radical through 1,5-H abstraction. The resulting carbon radical was then trapped by sulfur dioxide. This procedure represented an initiative study on the N-radical triggered aminosulfonylation of unactivated C(sp3)–H bonds. The use of 2,4-dinitrophenyl-connected N-oximes facilitated the homolytic cleavage of the N–O bond to produce the N-radical intermediate. Further exploration and radical trapping experiment provided solid experimental evidence that the reaction underwent a radical pathway. Reaction scope investigation showed that heterocycle-connected substrates were workable as well in the reaction. For example, the reaction of a thiophene-substituted substrate provided the desired product in a reasonable yield.51 The relative low reaction efficiency might be ascribed to a side reaction of the resulting C(sp3)-center radical with a thiophene core.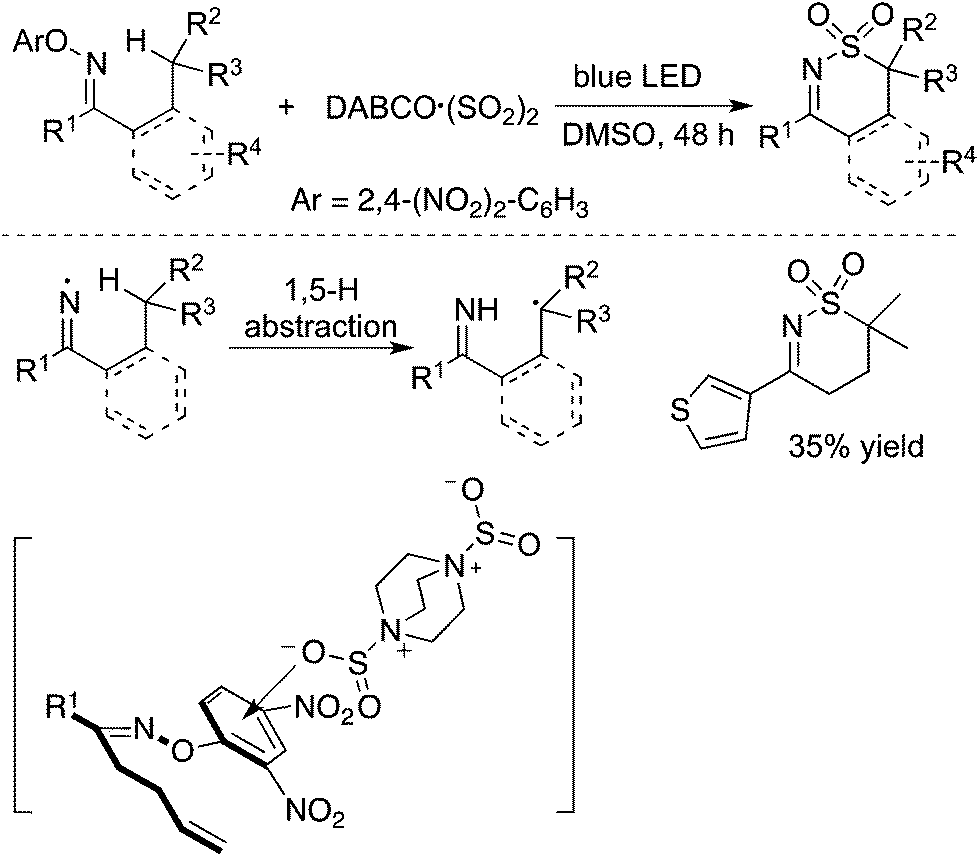 | ||
| Scheme 28 Reaction of N-oxime with the insertion of sulfur dioxide through 1,5-H abstraction. | ||
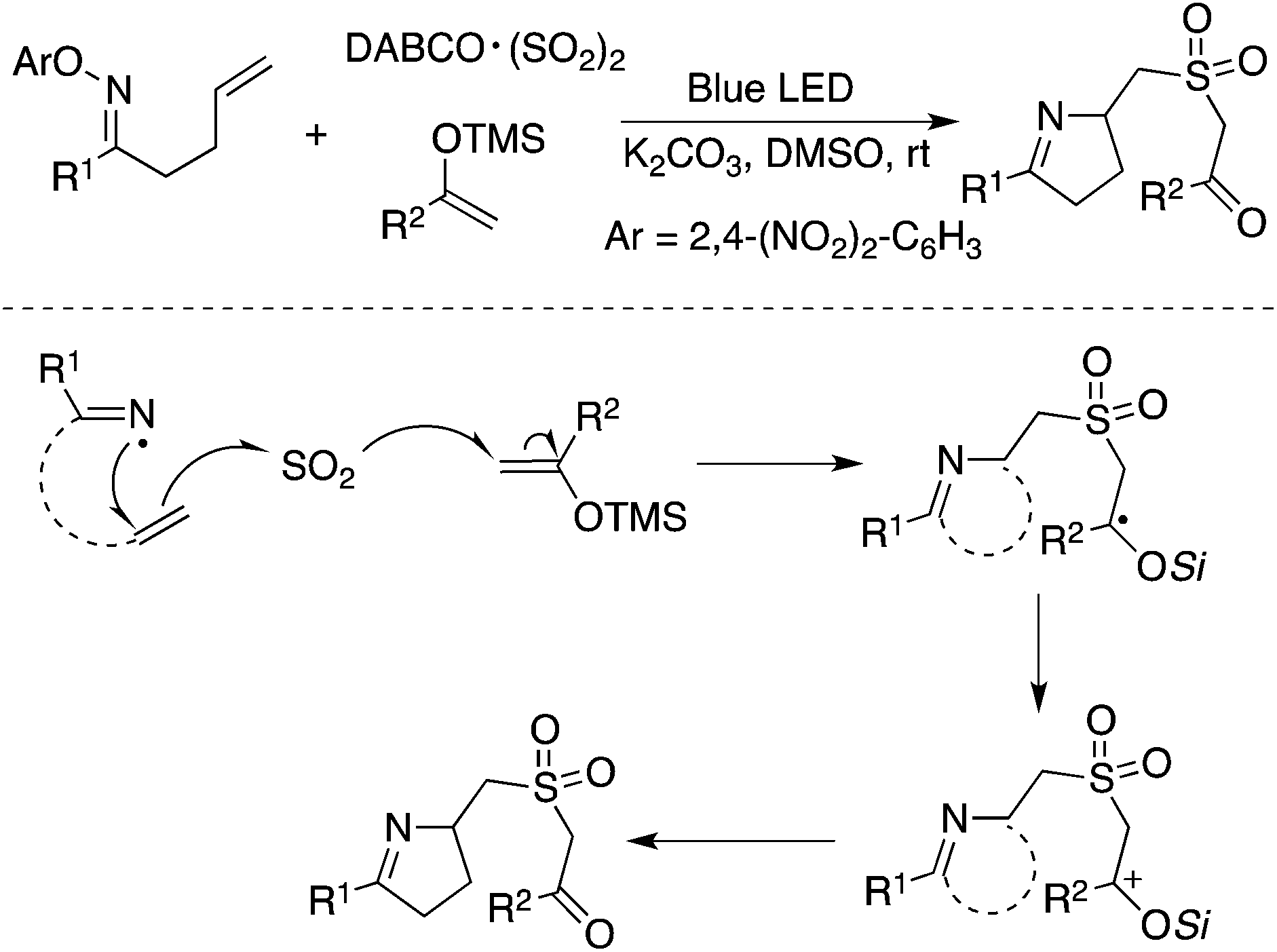
The synthesis of sulfonated 3,4-dihydro-2H-pyrroles was developed under photoinduced sulfonylative conditions (Scheme 29).52 This transformation proceeded through a N-radical initiated cyclization with the sulfonylation of unactivated alkenes through the insertion of sulfur dioxide in the presence of visible light under catalyst-free conditions. The products of 2-(((3,4-dihydro-2H-pyrrol-2-yl)methyl)sulfonyl)-1-arylethanones could be easily converted to 3,4-dihydro-2H-1,4-thiazine 1,1-dioxides. High efficiency and good selectivity were observed for this photoinduced transformation at room temperature under catalyst-free conditions. This approach provided another example of the iminyl radical-mediated cyclization with the insertion of sulfur dioxide.
 | ||
| Scheme 29 A N-radical initiated cyclization with the sulfonylation of unactivated alkenes through the insertion of sulfur dioxide. | ||
6. Other SO2-based transformations
As mentioned in Scheme 27, a four-component reaction of terminal alkynes, ethyl α-bromo-α,α-difluoroacetate, DABCO·(SO2)2, and hydrazines gave rise to difluoroalkyl-substituted sulfonamides.49 We reasoned that the resulting difluoroalkylsulfonyl radical was not stable due to the electron-deficient nature of the difluoroacetate part. In the process, the electron-withdrawing sulfonyl radical would be reversed to the difluoroalkyl radical. In other words, sulfur dioxide just served as a stabilizer to prolong the lifetime of the difluoroalkyl radical in the reaction. As we know, many metal salts such as copper salt and nickel salt exhibited similar properties to stabilize the radical intermediate. To verify the hypothesis, a reaction of prop-2-yn-1-ones and α-bromo-α,α-difluoroacetate was carried out in the presence of 0.2 equiv. DABCO·(SO2)2 under photocatalysis.53 As expected, a series of 3-fluorinated indenones was achieved in 52–97% yields (Scheme 30). A blank experiment without the addition of DABCO·(SO2)2 did not provide the corresponding indenones, proving the necessity of using DABCO·(SO2)2 as an additive. Interestingly, the DFT theoretical calculation on the free-energy reaction profile presented that the sulfur dioxide surrogate of DABCO·(SO2)2 also acted as an efficient catalyst for the cleavage of the difluoroalkyl–bromo bond under light irradiation.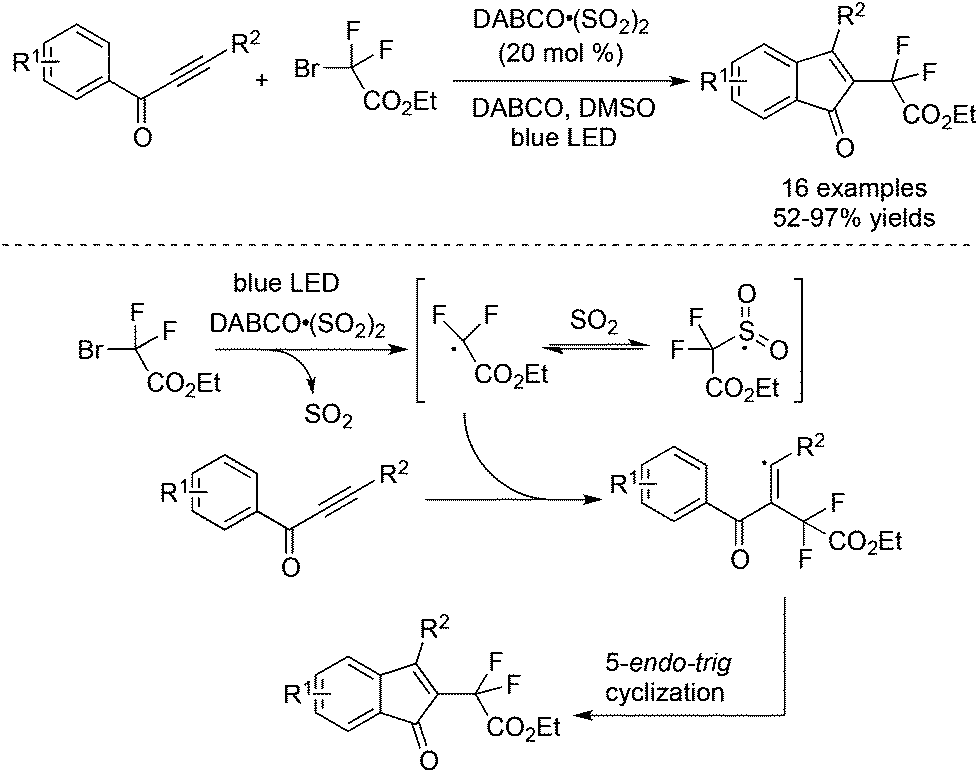 | ||
| Scheme 30 Reaction of prop-2-yn-1-ones and α-bromo-α,α-difluoroacetate in the presence of DABCO·(SO2)2. | ||
Thiophosphates could be produced through a three-component reaction of diaryliodonium tetrafluoroborates, DABCO·(SO2)2, and diarylphosphine oxides under visible light irradiation (Scheme 31).54 Interestingly, the formation of sulfonyl-substituted diarylphosphine oxide was not observed in this transformation. On treating with visible light irradiation (36 w) and Ir(ppy)3 in MeCN as a solvent, the reaction provided S-aryl diphenylphosphinothioate in 46–91% yields. In the reaction process, sulfur dioxide behaved as the sulfur source. Mechanistic studies revealed that the generation of aryl radicals from diaryliodonium salts would happen under photocatalysis, and the resulting arylsulfonyl radical would be reduced by an organophosphorus reagent in the presence of Ir(IV) to produce an arylsulfur cation. The nucleophilic capture of the arylsulfur cation by diarylphosphine oxide would release the desired thiophosphates.
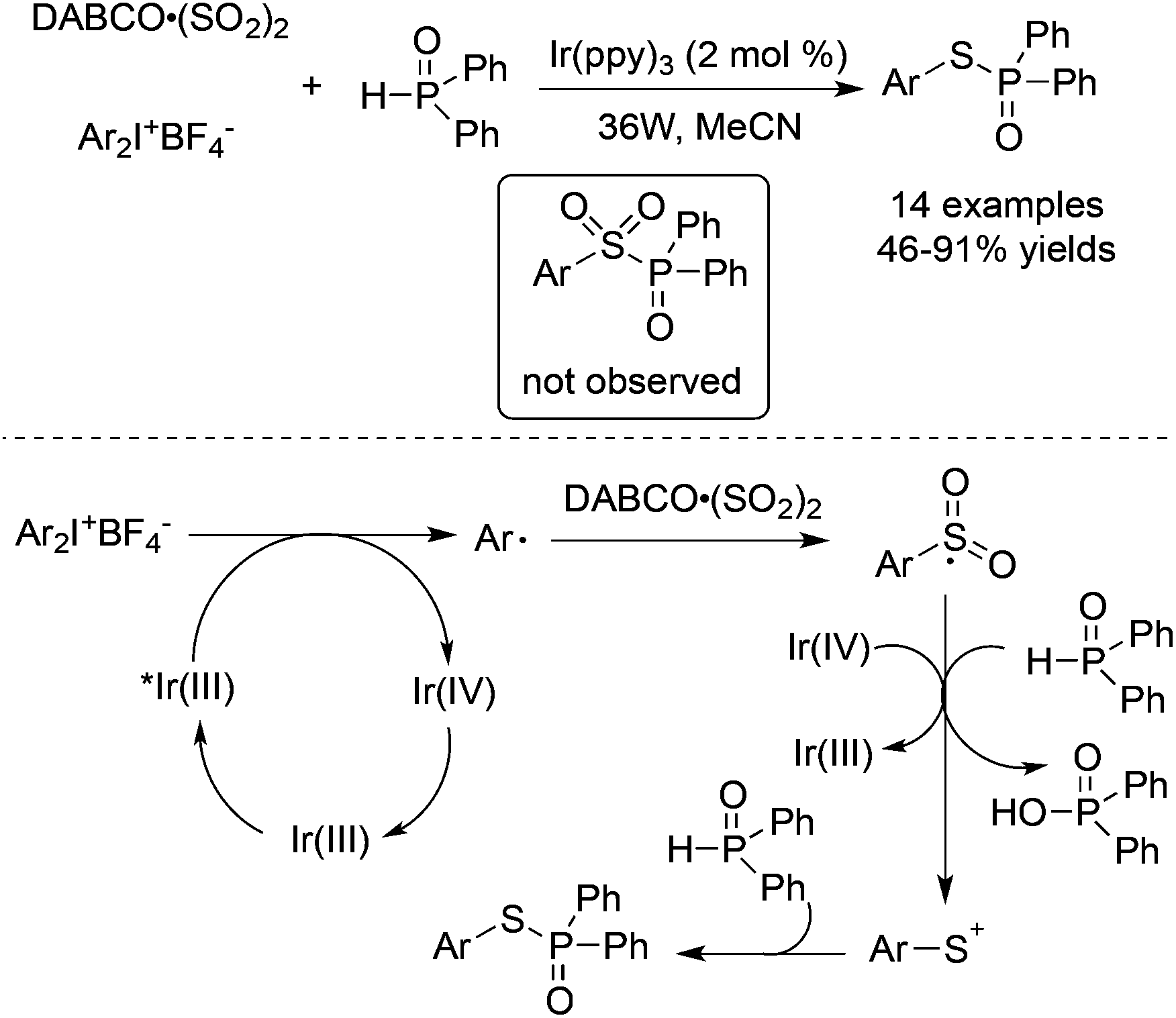 | ||
| Scheme 31 A three-component reaction of diaryliodonium tetrafluoroborates, DABCO·(SO2)2, and diarylphosphine oxides under visible light irradiation. | ||
Manolikakes and co-workers reported a visible-light photoredox-catalyzed aminosulfonylation of diaryliodonium salts, sulfur dioxide, and hydrazines (Scheme 32).55 In the reaction process, perylene dye PDI served as the photoredox catalyst. Either DABCO·(SO2)2 or potassium/sodium metabisulfite (K2S2O5/Na2S2O5) with acid could be used as the source of sulfur dioxide. A plausible mechanism was shown in Scheme 32. The generation of the aryl radical and nitrogen radical of hydrazine would lead to the formation of N-aminosulfonamides.
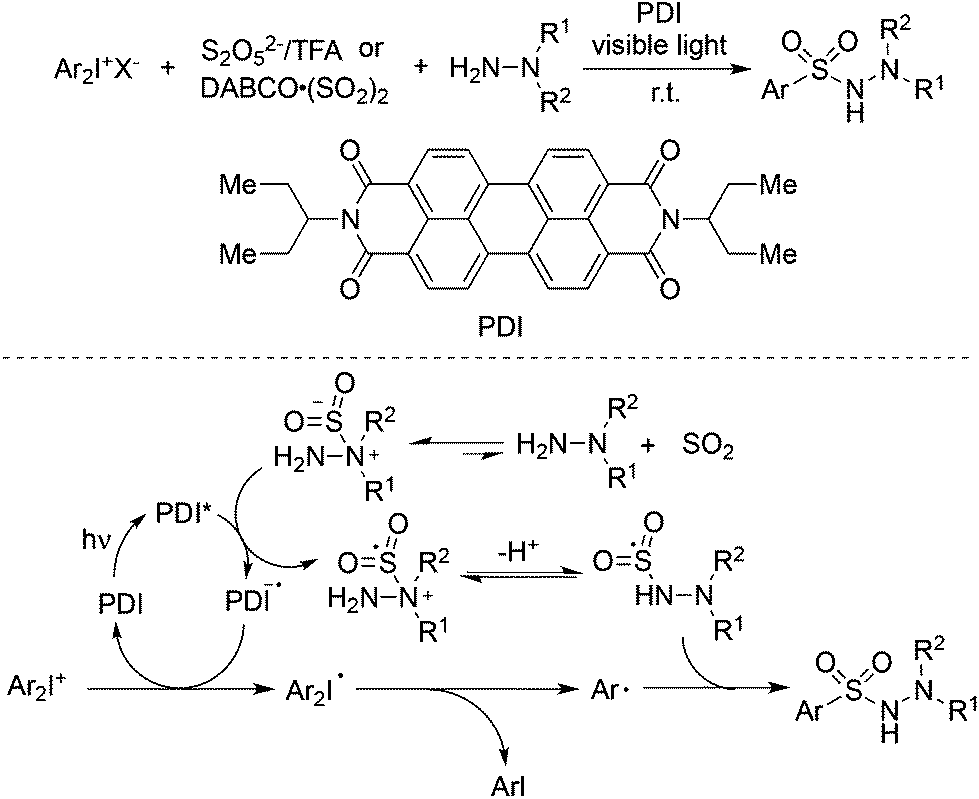 | ||
| Scheme 32 A three-component reaction of diaryliodonium salts, sulfur dioxide, and hydrazines under visible light irradiation. | ||
Jiang and co-workers described an example for the generation of diarylannulated sulfones via the SO2/I exchange of iodonium(III) salts catalyzed by copper(II) trifluoromethanesulfonate (Scheme 33).56 In this report, the readily available sodium metabisulfite (Na2S2O5) was used as the source of sulfur dioxide. Additionally, this method was successfully applied for the synthesis of organic light emitting diode (OLED) material molecules in the gram scale.
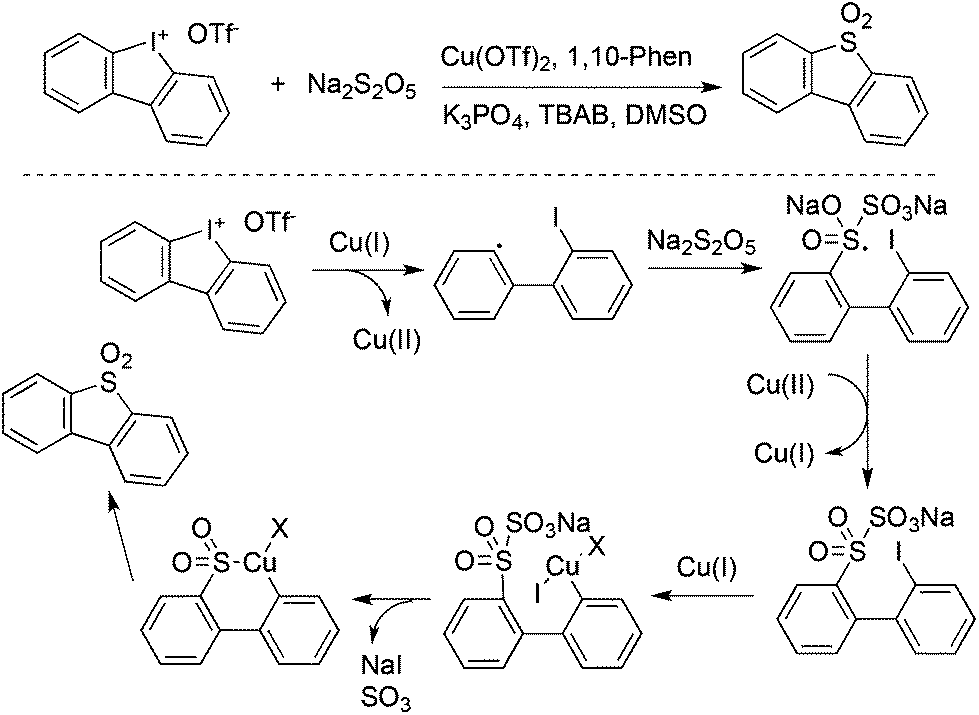 | ||
| Scheme 33 Generation of diarylannulated sulfones via the SO2/I exchange of iodonium(III) salts. | ||
7. Conclusions and Outlook
This review is focused on the recent advances in the chemistry of radical sulfur dioxide fixation. The high efficiency, broad reaction scope, and mild reaction conditions made this type of transformation attractive in the field of organic synthesis. In general, aryl radicals, vinyl radicals, alkyl radicals, and N-centered radicals succeeded in the reactions of sulfur dioxide with the formation of aryl/alkylsulfonyl radicals. The resulting aryl/alkylsulfonyl radical could be captured by nucleophiles, olefins and alkynes leading to diverse sulfones and other sulfonyl compounds. Various sources of aryl radicals from aryldiazonium tetrafluoroborates, aryl/alkyl halides, and diaryliodonium salts were demonstrated as well. Interestingly, the established achievements showed that the sulfur dioxide surrogate of DABCO·(SO2)2 could serve as a catalyst to facilitate the cleavage of the C–Br bond to form a fluoro-containing carbon-centered radical. Additionally, the application of sulfur dioxide was also highlighted as a source of sulfur.Although with unprecedented achievements in hand, radical chemistry involving sulfur dioxide still remains in an infancy stage. There are a number of unsettled scientific issues that need to be explored. Considering its great potential of synthetic applications, it is believed that the insertion of sulfur dioxide through radical processes will appear continuously in the near future. Moreover, the synthetic utilities of sulfonyl radicals generated from sulfur dioxide will be witnessed in organic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21502069 and 21772067) and the Science and Technology Project of Jiangxi Provincial Education Department (No. 151357) is gratefully acknowledged.Notes and references
- (a) M. Bartholow, Top 200 Drugs of 2011. Pharmacy Times. http://www.pharmacytimes.com/publications/issue/2012/July2012/Top-200-Drugs-of-2011, accessed on Jan 9, 2013; (b) For a list of the top drugs by year, see: http://cbc.arizona.edu/njardarson/group/toppharmaceuticals-poster, accessed on Jan 9, 2013; (c) J. Drews, Science, 2000, 287, 1960 CrossRef CAS PubMed.
- For selected examples, see: (a) A. S. Deeming, C. J. Rusell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150 CrossRef CAS PubMed; (b) B. N. Rocke, K. B. Bahnck, M. Herr, S. Lavergne, V. Mascitti, C. Perreault, J. Polivkova and A. Shavnya, Org. Lett., 2014, 16, 154 CrossRef CAS PubMed; (c) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679 CrossRef CAS PubMed.
- For selected reviews on CO2-based carboxylation, see: (a) K. Dong and X.-F. Wu, Angew. Chem., Int. Ed., 2017, 56, 5399 CrossRef CAS PubMed; (b) Y.-G. Zhang and S. Riduan, Angew. Chem., Int. Ed., 2011, 50, 6210 CrossRef CAS PubMed; (c) X. Li, X. He, X. Liu and L.-N. He, Sci. China: Chem., 2017, 60, 841 CrossRef CAS; (d) L. Zhang, Z. Han, L. Zhang, M. Li and K. Ding, Chin. J. Org. Chem., 2016, 36, 1824 CrossRef CAS.
- B. Nguyen, E. J. Emmet and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372 CrossRef CAS PubMed.
- (a) S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753 RSC; (b) S. Ye, H. Wang, Q. Xiao, Q. Ding and J. Wu, Adv. Synth. Catal., 2014, 356, 3225 CrossRef CAS; (c) Y. An, H. Xia and J. Wu, Org. Biomol. Chem., 2016, 14, 1665 RSC.
- (a) M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., Int. Ed., 2014, 53, 4404 CrossRef CAS PubMed; (b) R. Mao, D. Zheng, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 693 RSC; (c) X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056 CrossRef CAS PubMed; (d) D. Zheng, R. Mao, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 359 RSC; (e) D. Zheng, M. Chen, L. Yao and J. Wu, Org. Chem. Front., 2016, 3, 985 RSC.
- (a) S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037 RSC; (b) W. Li, H. Li, P. Langer, M. Beller and X.-F. Wu, Eur. J. Org. Chem., 2014, 3101 CrossRef.
- For reviews: (a) G. Liu, C. Fan and J. Wu, Org. Biomol. Chem., 2015, 13, 1592 RSC; (b) P. Bisseret and N. Blanchard, Org. Biomol. Chem., 2013, 11, 5393 RSC; (c) A. S. Deeming, E. J. Emmett, C. S. Richards-Taylor and M. C. Willis, Synthesis, 2014, 2701 Search PubMed; (d) E. J. Emmett and M. C. Willis, Asian J. Org. Chem., 2015, 4, 602 CrossRef CAS; (e) D. Zheng and J. Wu, Sulfur Dioxide Insertion Reactions for Organic Synthesis, Nature Springer, Berlin, 2017 Search PubMed.
- Synthetic application of N-aminosulfonamides, see: (a) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222 RSC; (b) W. Zhang and M. Luo, Chem. Commun., 2016, 52, 2980 RSC; (c) B. Du, Y. Wang, W. Sha, P. Qian, H. Mei, J. Han and Y. Pan, Asian J. Org. Chem., 2017, 6, 153 CrossRef CAS.
- (a) P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2001 Search PubMed; (b) J.-Y. Tsai and P. B. Shevlin, J. Org. Chem., 1998, 63, 3230 CrossRef CAS; (c) J. S. Hwang and C. P. Tsonis, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 1417 CrossRef CAS; (d) M. S. Kharasch and H. N. Friedlander, J. Org. Chem., 1948, 13, 882 CrossRef CAS PubMed; (e) A. Good and J. C. J. Thynne, Trans. Faraday Soc., 1967, 63, 2708 RSC; (f) A. Good and J. C. J. Thynne, Trans. Faraday Soc., 1967, 63, 2720 RSC; (g) S. W. Benson, Chem. Rev., 1978, 78, 23 CrossRef CAS; (h) M. P. Bertrand, Org. Prep. Proced. Int., 1994, 26, 257 CrossRef CAS.
- (a) C. F. Reed, US2046090A, 1936 Search PubMed; (b) J. Texter, Reactions and Synthesis in Surfactant Systems, Marcel Dekker, New York, 2001 Search PubMed; (c) M. A. Smook, E. T. Pieski and C. F. Hammer, Ind. Eng. Chem., 1953, 45, 2731 CrossRef CAS.
- (a) H. Meerwein, G. Dittmar, R. Göllner, K. Hafner, F. Mensch and O. Steinfort, Chem. Ber., 1957, 90, 841 CrossRef CAS; (b) P. J. Hogan and B. G. Cox, Org. Process Res. Dev., 2009, 13, 875 CrossRef CAS.
- D. H. R. Barton, B. Lacher, B. Misterkiewicz and S. Z. Zard, Tetrahedron, 1988, 44, 1153 CrossRef CAS.
- R. M. Wilson and S. W. Wunderly, J. Am. Chem. Soc., 1974, 96, 7350 CrossRef CAS.
- L. Malet-Sanz, J. Madrzak, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2010, 8, 5324 CAS.
- D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed., 2014, 53, 2451 CrossRef CAS PubMed.
- D. Zheng, Y. Li, Y. An and J. Wu, Chem. Commun., 2014, 50, 8886 RSC.
- (a) Y. An, D. Zheng and J. Wu, Chem. Commun., 2014, 50, 11746 RSC; (b) Y. Luo, X. Pan, C. Chen, L. Yao and J. Wu, Chem. Commun., 2015, 51, 180 RSC; (c) J. Sheng, Y. Li and G. Qiu, Org. Chem. Front., 2017, 4, 95 RSC; (d) D. Zheng, Y. Kuang and J. Wu, Org. Biomol. Chem., 2015, 13, 10370 RSC; (e) X. Liu, W. Wang, D. Zheng, X. Fan and J. Wu, Tetrahedron, 2015, 71, 3359 CrossRef CAS.
- W. Li, M. Beller and X.-F. Wu, Chem. Commun., 2014, 50, 9513 RSC.
- Y. Wang, B. Du, W. Sha, H. Mei, J. Han and Y. Pan, Org. Chem. Front., 2017, 4, 1313 RSC.
- T. Liu, D. Zheng, Z. Li and J. Wu, Adv. Synth. Catal., 2017, 359, 2653 CrossRef CAS.
- D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925 CrossRef CAS PubMed.
- X. Wang, T. Liu, D. Zheng, Q. Zhang and J. Wu, Org. Chem. Front., 2017, 4, 2455 RSC.
- J. Yu, R. Mao, Q. Wang and J. Wu, Org. Chem. Front., 2017, 4, 617 RSC.
- K. Zhou, H. Xia and J. Wu, Org. Chem. Front., 2017, 4, 1121 RSC.
- Y. Xiang, Y. Kuang and J. Wu, Chem. – Eur. J., 2017, 23, 699 Search PubMed.
- S. Y. Woo, J. H. Kim, M. K. Moon, S.-H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473 CrossRef CAS PubMed.
- Y. An and J. Wu, Org. Lett., 2017, 19, 6028 CrossRef CAS PubMed.
- T. Liu, D. Zheng and J. Wu, Org. Chem. Front., 2017, 4, 1079 RSC.
- T. Liu, D. Zheng, Y. Ding, X. Fan and J. Wu, Chem. – Asian J., 2017, 12, 465 CrossRef CAS PubMed.
- (a) R. Mao, Z. Yuan, R. Zhang, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2016, 3, 1498 RSC; (b) W. Fan, J. Su, D. Shi and B. Feng, Tetrahedron, 2015, 71, 6740 CrossRef CAS.
- (a) J. Zhang, K. Zhou and J. Wu, Org. Chem. Front., 2017 10.1039/C7QO00987A; (b) T. Liu, D. Zheng, Z. Li and J. Wu, Adv. Synth. Catal., 2018 DOI:10.1002/adsc.201701187.
- J. P. Barham, M. P. John and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 15482 CrossRef CAS PubMed.
- J. Zhang, Y. An and J. Wu, Chem. – Eur. J., 2017, 23, 9477 CrossRef CAS PubMed.
- (a) F. Liu, J.-Y. Wang, P. Zhou, G. Li, W.-J. Hao, S.-J. Tu and B. Jiang, Angew. Chem., Int. Ed., 2017, 56, 15570 CrossRef CAS PubMed; (b) Z.-J. Shen, Y.-N. Wu, C.-L. He, L. He, W.-J. Hao, A.-F. Wang, S.-J. Tu and B. Jiang, Chem. Commun., 2017 10.1039/c7cc08516h.
- H. Wang, S. Sun and J. Cheng, Org. Lett., 2017, 19, 5844 CrossRef CAS PubMed.
- Y. An, J. Zhang, H. Xia and J. Wu, Org. Chem. Front., 2017, 4, 1318 RSC.
- Y.-L. Zhu, B. Jiang, W.-J. Hao, J.-K. Qiu, J. Sun, D.-C. Wang, P. Wei, A.-F. Wang, G. Li and S.-T. Tu, Org. Lett., 2015, 17, 6078 CrossRef CAS PubMed.
- For selected examples, see: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) L. McMurray, F. O. Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (d) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (e) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (f) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (g) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (h) Q.-F. Wu, P.-X. Shen, J. He, X.-B. Wang, F. Zhang, Q. Shao, R.-Y. Zhu, C. Mapelli, J. X. Qiao, M. A. Poss and J.-Q. Yu, Science, 2017, 355, 499 CrossRef CAS PubMed; (i) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538 CrossRef CAS PubMed; (j) P. Jain, P. Verma, G. Xia and J.-Q. Yu, Nat. Chem., 2017, 9, 140 CrossRef CAS PubMed; (k) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (l) G. Chen, W. Gong, Z. Zhuang, M. S. Andrä, Y.-Q. Chen, X. Hong, Y.-F. Yang, T. Liu, K. N. Houk and J.-Q. Yu, Science, 2016, 353, 1023 CrossRef CAS PubMed; (m) A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80 CrossRef CAS PubMed; (n) G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Nat. Chem., 2016, 8, 810 CrossRef CAS PubMed.
- H. Xia, Y. An, X. Zeng and J. Wu, Chem. Commun., 2017, 53, 12548 RSC.
- H. Xia, Y. An, X. Zeng and J. Wu, Org. Chem. Front., 2017, 4 10.1039/C7QO00866J.
- K. Wang, G. Wang, G. Duan and C. Xia, RSC Adv., 2017, 7, 51313 RSC.
- K. Zhou, M. Chen, L. Yao and J. Wu, Org. Chem. Front., 2017 10.1039/C7QO00811B.
- T. Liu, W. Zhou and J. Wu, Org. Lett., 2017, 19, 6638 CrossRef CAS PubMed.
- (a) W. Liu, L. Li and C.-J. Li, Nat. Commun., 2015, 6, 6526 CrossRef CAS PubMed; (b) L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed.
- (a) Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574 RSC; (b) K. Zhou, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 865 RSC.
- X. Gong, Y. Ding, X. Fan and J. Wu, Adv. Synth. Catal., 2017, 359, 2999 CrossRef CAS.
- Y. Li, Y. Xiang, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 1493 RSC.
- Y. Xiang, Y. Li, Y. Kuang and J. Wu, Chem. – Eur. J., 2017, 23, 1032 CrossRef CAS PubMed.
- For recent reviews, see: (a) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC; (b) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069 RSC; (c) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC.
- Y. Li, R. Mao and J. Wu, Org. Lett., 2017, 19, 4472 CrossRef CAS PubMed.
- R. Mao, Z. Yuan, Y. Li and J. Wu, Chem. – Eur. J., 2017, 23, 8176 CrossRef CAS PubMed.
- Y. Li, Y. Lu, R. Mao, Z. Li and J. Wu, Org. Chem. Front., 2017, 4, 1745 RSC.
- X. Gong, J. Chen, J. Liu and J. Wu, Org. Chem. Front., 2017, 4, 2221 RSC.
- N.-W. Liu, S. Liang and G. Manolikakes, Adv. Synth. Catal., 2017, 359, 1308 CrossRef CAS.
- M. Wang, S. Chen and X. Jiang, Org. Lett., 2017, 19, 4916 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2018 |