
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transformable protein–gold hybrid materials serve as supramolecular vehicles for gene delivery†
Zhenhua
Li
,
Han
Ding
,
Fei
Yan
,
Hongwei
Li
 * and
Zhijun
Chen
*
* and
Zhijun
Chen
*
State Key Laboratory of Supramolecular Structure and Materials, Institute of Theoretical Chemistry, International Joint Research Laboratory of Nano-Micro Architecture Chemistry, Jilin University, No.2699 Qianjin Street, Changchun 130012, China. E-mail: zchen@jlu.edu.cn; lihongwei@jlu.edu.cn
First published on 3rd November 2017
Abstract
Functional DNA cannot pass through plasma membrane of living cells by itself. An efficient non-viral DNA carrier with low cytotoxicity and simple preparation procedure is in high demand. Herein, we describe that protein–gold hybrid nanocubes (PGHNs) could intrinsically recognize DNAs, and transform them into nanoflower-like structures. These supramolecular complexes can be internalized by living yeast cells and allow the coding information of the gene to be transmitted into proteins. PGHN–DNA can be a good model to study DNA–carrier interactions as well as a new carrier for gene delivery research.
Vehicles are the key player in gene delivery system, which help to transport genetic materials toward different cells for various purposes.1–3 These gene vehicles can be divided into two classes, namely viral4–6 and non-viral carriers.7–12 Viral carriers exhibit high DNA transfection efficiency.13 Nevertheless several disadvantages limit their application in biological and clinical research, such as cytotoxicity, carcinogenicity, and immunogenicity.14–16 Non-viral carriers are equally important, which may be partially exempt from the limitations of viral carriers mentioned above. Among them, lipoplexes, cationic lipids, and cationic polymers have been widely explored.17–24 However, these traditional non-viral carriers also exhibit certain weaknesses such as low transfection efficiency, toxicity, or complicated preparation procedure. Hence, new generation non-viral carriers with high transformation efficiency, low cytotoxicity, and simple preparation procedure are highly favored.
In this report, we describe an interesting finding that recently reported protein–gold hybrid nanocubes (PGHNs)25 showed special gene carrier capability as compared to other gold nanoclusters (Au NCs).26,27 PGHNs were cube-like, blue emissive upon UV radiation, and can be readily internalized by living cells.25 Herein we described that PGHN can interact with DNA and transform into higher supramolecular structure. Interestingly, the PGHN–DNAs can be up-taken by classical research model yeast Saccharomyces cerevisiae in an efficient manner. PGHNs can be prepared easily and showed good biocompatibility and low cytotoxicity toward Escherichia coli cells.25 These transformable protein–gold hybrid materials can serve as supramolecular vehicles for gene delivery in fungi (Scheme 1).
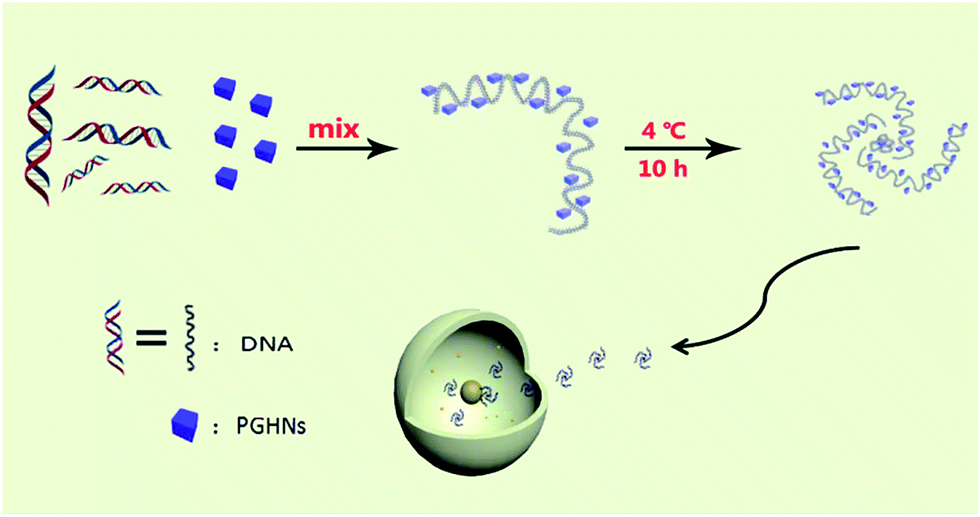 | ||
| Scheme 1 Schematic presentation of PGHN–DNA assembly and its application in gene delivery. The gray wires and blue cubes stand for DNAs and PGHNs, respectively. | ||
In our previous report, we described that fluorescent PGHNs can be readily internalized by living cells and thus can be used for bioimaging.25 PGHN synthesis was briefly as follows, BSA, tryptophan, and HAuCl4 were mixed and pH of the solution were adjusted to 3.0, followed by incubation at 30 °C for 12 h, where the concentration of these compounds were 5, 20, and 1.6 mg mL−1, respectively. The product was dialyzed, centrifuged, and stored in 4 °C fridge before use. Notably, the concentration of HAuCl4 and PGHNs was referred to the concentration of either Au3+ or Au atoms. The excitation and emission of PGHNs were 395 and 480 nm, respectively. Under UV irradiation (365 nm), PGHN cubes was blue emissive, which was due to the conjugated Au NCs (Fig. S1 and S2†). Although no precise structures of this type of protein–Au NCs available this far, previous researches hinted that proteins were served as capping agents to foster the formation of Au NCs.28–32 During the synthesis process, protein–Au NC complexes can be further assembled into higher nano-structures with large sizes depending on the specific conditions. The photostability of PGHNs was determinated using fluorescence measurement as well as bleaching experiments (Fig. S3 and S4†). Notably, the surface positive charge of PGHNs suggested these materials might interact with negatively charged DNAs. To explore this possibility, plasmid DNA (pGal-sGFP) (Fig. S3 and S5,† 100 ng μL−1) were mixed with PGHNs (1.33 mM L−1) at different volume ratios for a final volume of 100 μL, and rotating incubated at 4 °C for 10 h, and then subjected to agarose gel electrophoresis. The retardation of plasmid DNAs was observed from the gel, indicating that PGHNs interacted with the plasmid DNA (Fig. 1A). This supramolecular interaction can be further optimized via changing the ratio of these two components (Fig. 1A and B). Noteworthy, as PGHN–DNA interaction can be partially damaged in the process due to the harsh condition of electrophoresis, the real number of interacted DNAs toward PGHNs were possibly much higher than that was shown on Fig. 1.
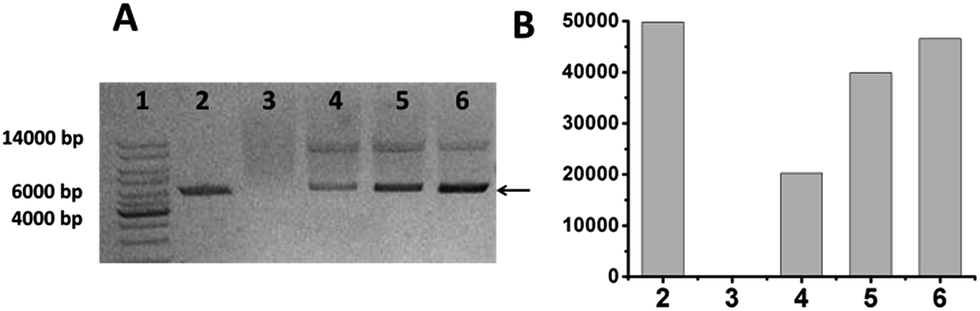 | ||
| Fig. 1 The binding of pGal-sGFP and PGHNs was analyzed using agarose gel electrophoresis. (A) Lane 1, 1 kb DNA ladder; lane 2, pGal-sGFP DNA; lane 3, PGHNs; lane 4–6 represented PGHN–DNA mixture prepared with different volume ratios (the volume of PGHNs was 0, 100, 90, 60, and 30 μL, respectively, where DNA volume was always 10 μL except for lane 3 (0)). (B) The intensity of the lower DNA band (A: arrow) was quantified. | ||
To further investigate the supramolecular interaction between PGHN and DNA, isothermal titration calorimetry (ITC) was applied. PGHNs were titrated towards plasmid DNA of pGal-sGFP with initial value −1.0 kcal mol−1. Following the increased content of PGHNs, the reaction heat was gradually increased, and finally reached −0.3 kcal mol−1 (Fig. 2A). The thermodynamic parameters were calculated and fit by using least square method. The binding affinity turned out to be 1.42 × 104 M−1 with ΔH of −2.79 kcal mol−1 and ΔS = 8.97 cal mol−1 deg−1. In parallel, BSA instead of PGHNs was also titrated toward the plasmid pGal-sGFP (Fig. 2B), suggesting no interaction between BSA and DNA was occurred. These results indicated that AuNCs of PGHNs might be involved in the PGHN–DNA interaction. How was the supramolecular interaction/assembly occurred? The unique DNA binding property of PGHN triggered us to investigate the co-assembly process of PGHN and DNA.
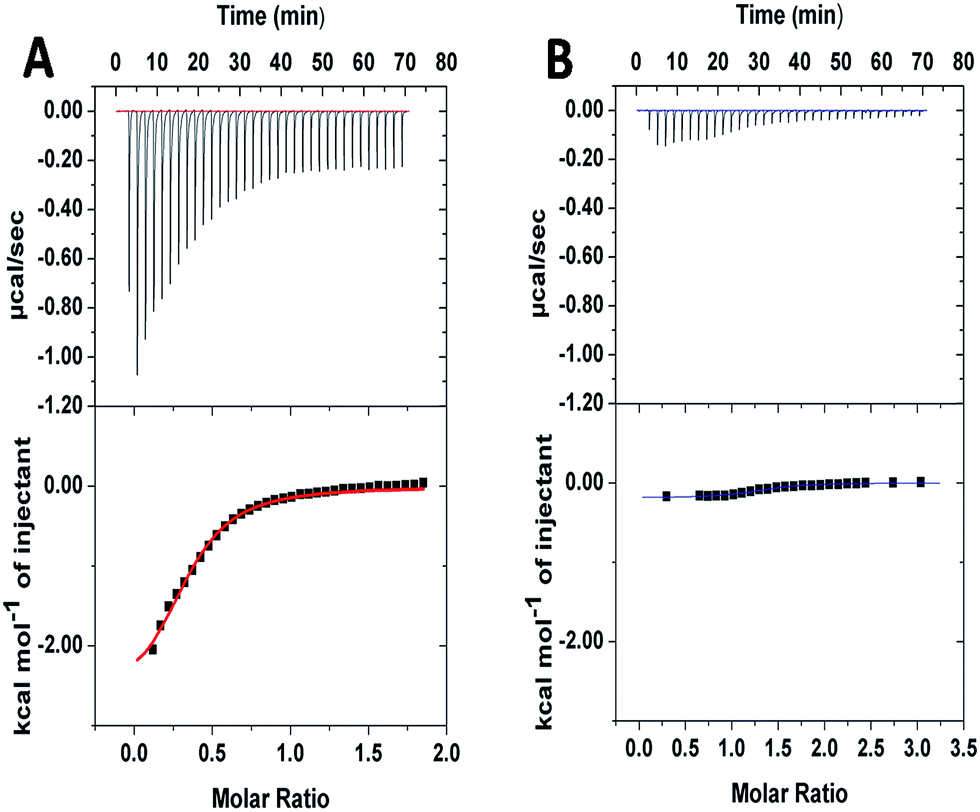 | ||
| Fig. 2 Isothermal titration calorimetry (ITC) showed the energy associated with a reaction triggered by the interaction between pGal-sGFP and PGHNs (A) or pGal-sGFP and BSA (B). | ||
The supramolecular assembly of PGHN–DNA complex was monitored by using scanning electronic microscopy (SEM). This process was initiated by electrostatic interaction between positively charged PGHNs and negatively charged plasmid DNA. Specifically, 90 μL PGHNs (1.33 mM L−1) was mixed with 10 μL plasmid pGal-sGFP (100 ng μL−1), to make a final volume of 100 μL. The mixture was incubated at 4 °C for 10 h. The time and concentration effects were investigated. Interestingly, the recognition and co-assembly of PGHNs and plasmid DNA (pGal-sGFP) turned out to be a slow and sophisticated process. Interestingly, the morphology of PGHN–DNAs gradually shifted from cubic to nanoflower-like structures (Fig. 3). We speculated that DNA may bend and fold into clew-like conformation under the guidance of PGHNs. Notably, during the assembly process, the size of the PGHN–DNAs was gradually increased, shifting from 90 to 500 nm (Fig. 3A–D). This phenomenon was also supported by the data obtained from Dynamic Light Scattering (DLS) (Fig. S4–S7†). The driving force that triggered PGHN–DNAs slowly change the structure may be associated with their supramolecular recognition such as electrostatic interaction.
 | ||
| Fig. 3 Morphology characterization of PGHNs and PGHNs–DNAs by using SEM and the time effect on the co-assembly of PGHNs–DNAs. (A) Representative SEM image of PGHNs. (B–D) Co-assembly of pGal-sGFP and PGHNs. These two components were mixed for indicated time, 4 h (B), 6 h (C), and 10 h (D). The average size of the nanostructures was approximately 90, 130, 200, and 500 nm, respectively. | ||
Biocompatibility is a critical concern for gene or all the other type of drug carriers. Cytotoxicity of PGHN–DNAs was evaluated by using both prokaryotic and eukaryotic cells via monitoring their growth curve in the presence of PGHN–DNAs. The yeast cells (S. cerevisiae, BY4741) were inoculated and shaking incubated in YPD media at 30 °C. The O/N culture was washed twice by using 1× phosphate-buffered saline (PBS) buffer and transferred into fresh YPD media. The cells were then re-suspended and the cell density was adjusted to OD600 = 0.1. These cells were divided into two groups, where one was mixed with 400 μL PGHNs, the other with equal amount of 1× PBS buffer. These two group of cells were shaking incubated at 30 °C. Samples were taken and subjected to OD600 measurement every 2 h, until reaching static station. The cytotoxicity of PGHNs toward mammalian cells was evaluated using Cell Proliferation and Cytotoxicity Assay Kit (Beijing Seajet Scientific, Beijing, China). The hepatocyte cell line LO2 originating histologically from normal human liver tissue was obtained from Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences. The human cervical cancer cell line Hela was purchased from American Type Culture Collection (ATCC). Two cell lines were cultured in RPMI1640 containing 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 10% fetal bovine serum. Hela (cancer cells) or LO2 (normal cells) cells were treated with different doses of nanoparticles for 48 and 72 hours, and subjected to CCK-8 assay for cytotoxicity analysis. The results revealed that PGHNs were noncytotoxic to all the cell lines tested so far (Fig. 4).
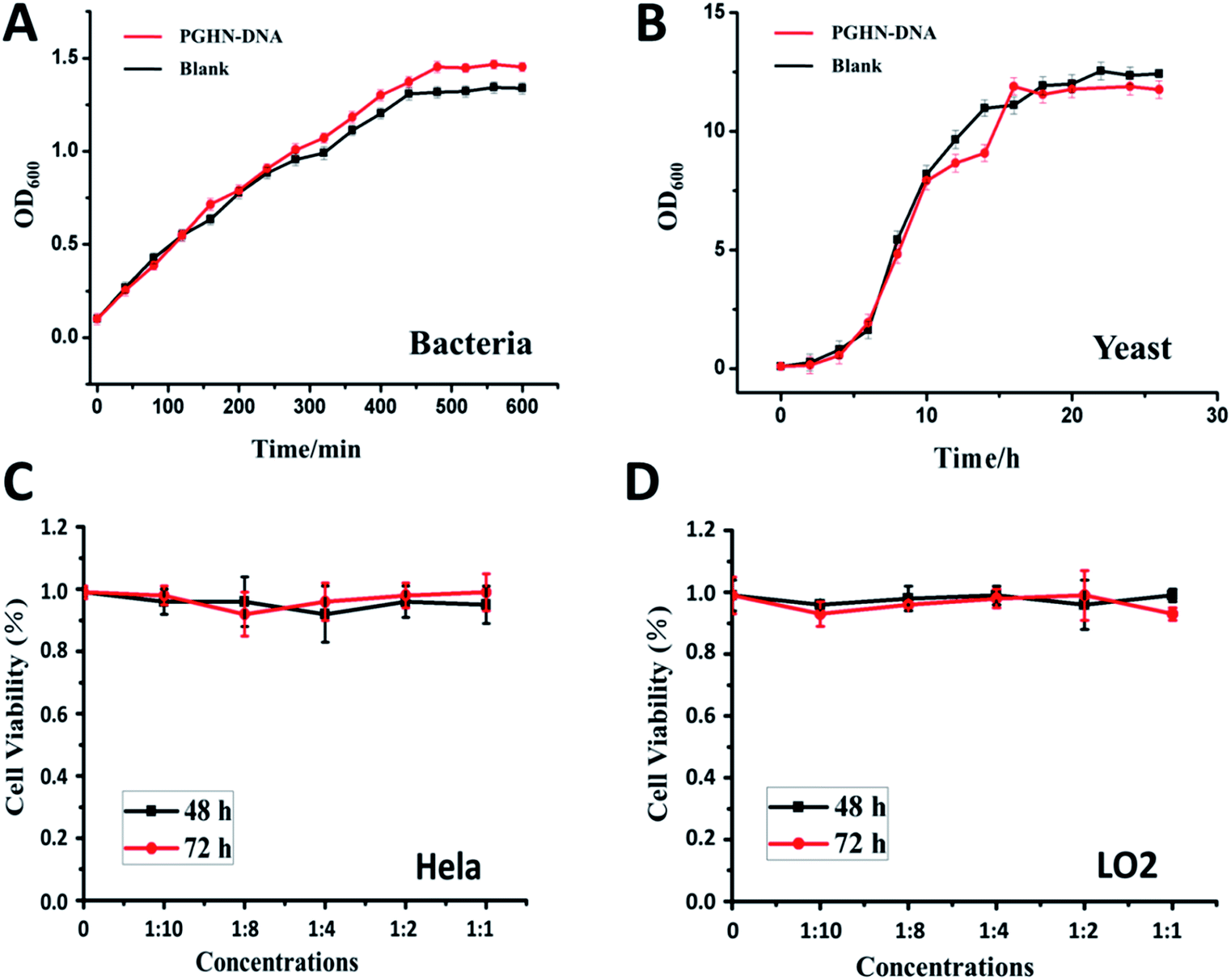 | ||
| Fig. 4 The cytotoxicity experiments of PGHN–DNAs with different biological research models. The optical density (OD) measurement of the PGHN–DNAs incubated with bacteria (A) and yeast (B) cells. Cell viability measurement of the PGHN–DNAs incubated with cancer (Hela) (C) and normal liver (LO2) cells (D). As shown on the growth curve, PGHN–DNAs did not show a clear observable toxic effect. The black lines represent test group, the red lines represent control group. The value are plotted as mean ± SD (n = 3). | ||
The size, morphology, and surface charge of vehicles are important measures to assess the plausibility if they can be internalized by living cells. DNA is strong negatively charged biopolymer, which hinders their crossing of plasma membrane of cells. Notably, during the supramolecular assembly of PGHN–DNAs, the strong negatively charged DNAs were largely neutralized as shown on the zeta potential measurement with final value approaching nearly zero (Fig. S8†). The change of size and surface charge of PGHNs after interacting with DNA suggested a gene carrier possibility of these materials. Altogether, the suitable size, low cytotoxicity, DNA binding capability, and non-viral origin indicated PGHNs might serve as a new type of DNA carrier for gene transformation research.
To investigate the plasma membrane crossing capability of PGHN–DNAs, yeast S. cerevisiae BY4741 were used. Yeast was a classical eukaryotic model for biological study, which was very much similar to mammalian cells in terms of subcellular structure and key signaling and metabolic pathways.33,34S. cerevisiae cells were shaking incubated overnight (O/N) at 30 °C. 1 mL of the O/N culture was centrifuged and washed 2 times using 1× PBS buffer. The collected cells were mixed with 400 μL PGHN–DNA nanoflowers, transferred to 5 mL fresh yeast extract peptone dextrose (YPD) media containing 2% galactose, and then incubated by shaking for 1 h, followed by further shaking incubation at 30 °C for additional 2 h. Notably, the presence of galactose induced the expression of green fluorescent protein (GFP) which was under the control of Gal promoter (Fig. S5†). Yeast cells in the absence of PGHN–DNA (replaced by 400 μL dH2O) were followed the same procedure to serve as negative control. The classical yeast transformation approach (trimix-heat-shock) was applied as a positively control. This trimix-heat-shock approach can be described briefly as follows, the collected cells from 1 mL O/N culture were mixed with 80 μL trimix (0.2 M LiAc, 40% PEG3350, 100 μM DTT), followed by sequently adding 4 μL ssDNA (10 mg mL−1) and plasmid DNA (4 μg). The mixture was incubated at 45 °C for 45 min, and then transferred to 5 mL fresh YPD media, followed by shaking incubation at 30 °C for 2 h. These yeast cells were collected by using centrifugation and then washed twice using 1× PBS buffer, and then transferred into YPD media containing galactose (2%) for additional 2 h for galactose induction. For fixation, 37% paraformaldehyde (PFA) were added into cell culture to make a final concentration of 3.7%, followed by shaking incubation at 30 °C for 30 min. The cells were then washed twice using solution P (0.1 M phosphate buffer, pH 6.5, 1.5 M sorbitol), followed by re-suspending of cells using solution P containing additional 25 mM DTT and incubation at room temperature for 10 min. The cell samples were mounted on cover slips and subjected to fluorescent microscopic analysis.
Fluorescent microscopic analysis showed that the pGal-sGFP DNA had been successfully uptaken by S. cerevisiae cells using this new method. Importantly, the plasmid DNA that entered the cells remained functional as shown by the green fluorescent signals, indicating the induction and expression of GFP (Fig. 5A–D). Significantly, compared to well-established classical trimix-heat-shock approach, this new method showed even higher transformation efficiency (Fig. 5E–H and S9†). In contrast, the negative control group did not show any positive fluorescent signal (Fig. 5I–L), confirming that naked DNA cannot enter the yeast cells under this condition. Moreover, in the absence of DNA, PGHNs can be internalized by yeast cells (Fig. 5M–P), exhibiting blue emission, whereas no green emission was detected in the green channel. Notably, the fluorescent pattern of GFP (Fig. 5A–D) and PGHNs (Fig. 5M–P) were slightly different, indicating their different subcellular localization.
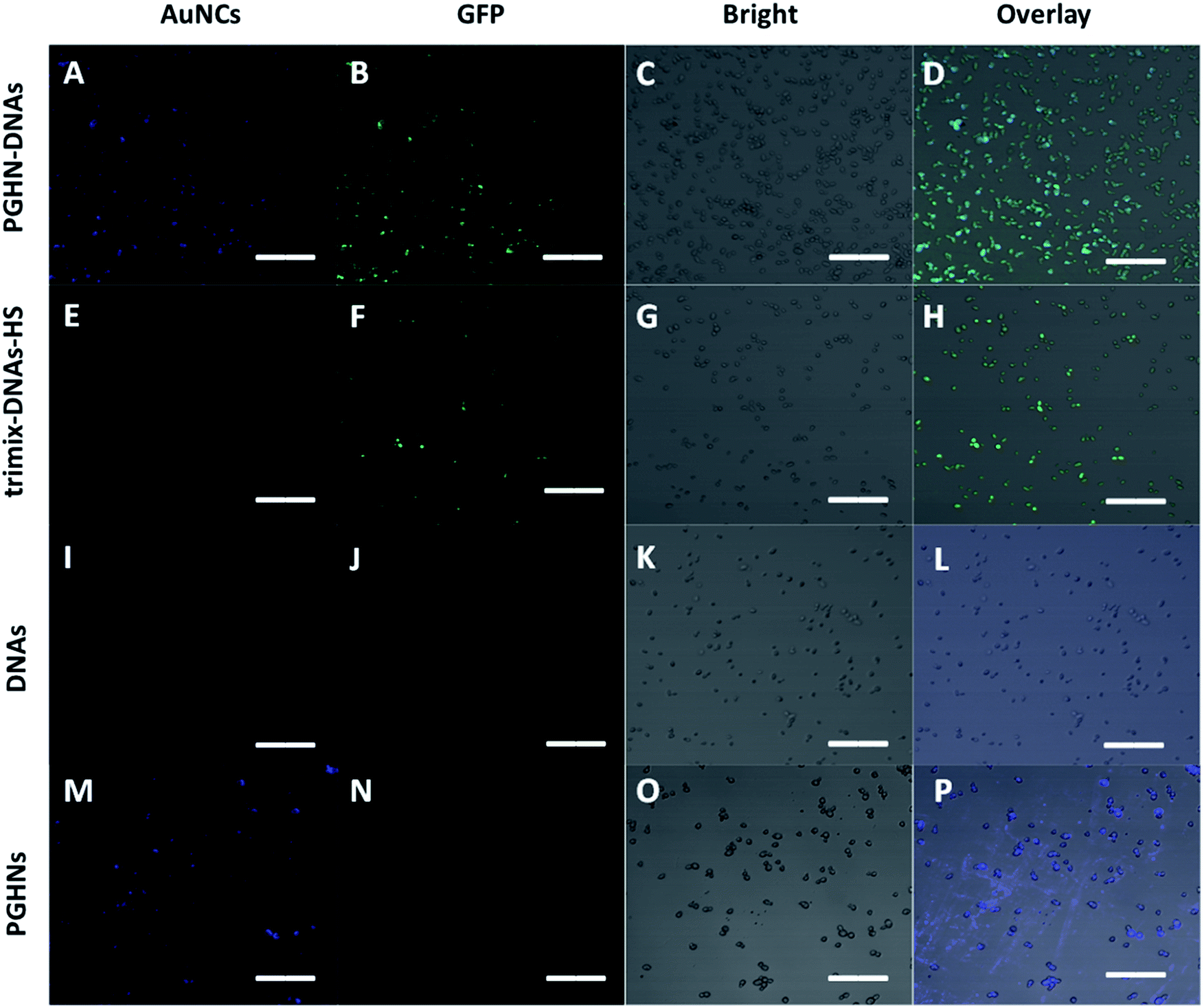 | ||
| Fig. 5 Fluorescence microscopic images of EGFP expression in yeast cells transfected using different methods, (A–D) referred to DNA transformation by using this new method. (E–H) Referred to DNA transformation by using traditional approach (trimix-heatshock). (I–L) Referred to negative control 1 where pGal-sGFP DNAs were present while PGHNs were absent. (M–P) Referred to negative control 2 where PGHNs was present while pGal-sGFP DNAs were absent. All scale bars were 20 μm. | ||
Conclusions
Taken together, as recently emerged new protein–gold hybrid materials, PGHNs can interact with plasmid DNA likely through electrostatic interaction. This process has been investigated by using both agarose gel electrophoresis, ITC, and SEM. The supramolecular co-assembly of PGHNs and plasmid DNA was slow and dynamic. During this course, the size, surface charge, and morphology of the PGHN–DNA complexes were significantly changed. Hence, PGHNs can serve as shape-transformable gene vehicles, which is special and worthy to explore further. Moreover, these complexes showed hardly any toxicity toward both bacteria and yeast cells, indicating a good biocompatibility. Finally, PGHN–DNAs can be readily internalized by yeast cells with high efficiency as compared to the traditional yeast transformation approach. We expect that PGHN–DNA can be a good model to study DNA–carrier interaction as well as new carriers for gene delivery research.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We would like to acknowledge the financial support from the National Natural Science Foundation of China (NSF 21372097).Notes and references
- Y. Tao, E. Ju, J. Ren and X. G. Qu, Chem. Commun., 2013, 49, 9791 RSC.
- F. McCormick, Nat. Rev. Cancer, 2001, 1, 130–141 CrossRef CAS.
- L. Brannon-Peppas, B. Ghosn, K. Roy and K. Cornetta, Pharm. Res., 2007, 24, 618–627 CrossRef CAS.
- M. A. Kay, J. C. Glorioso and L. Naldini, Nat. Med., 2001, 7, 33–40 CrossRef CAS.
- M. A. Zanta, P. Belguise-Valladier and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 91–96 CrossRef CAS.
- M. Rafalski, A. Ortiz, A. Rockwell, L. C. Van Ginkel, J. D. Lear, W. F. DeGrado and J. Wilschut, Biochemistry, 1991, 30, 10211–10220 CrossRef CAS.
- M. D. Brown, A. G. Schätzlein and I. F. Uchegbu, Int. J. Pharm., 2001, 229, 1–21 CrossRef CAS.
- K. Kodama, Y. Katayama, Y. Shoji and H. Nakashina, Curr. Med. Chem., 2006, 13, 2155–2161 CrossRef CAS.
- B. Zhang, X. Ma, W. Murdoch, M. Radosz and Y. Q. Shen, Biotechnol. Bioeng., 2013, 110, 990–998 CrossRef CAS.
- T. B. Ren, L. Li, X. J. Cai, H. Q. Dong, S. M. Liu and Y. Y. Li, Polym. Chem., 2012, 3, 2561–2569 RSC.
- S. T. Guo, Y. Y. Huang, W. D. Zhang, W. W. Wang, T. Wei, D. S. Lin, J. F. Xing, L. D. Deng, Q. Du, Z. C. Liang, X. J. Liang and A. J. Dong, Biomaterials, 2011, 32, 4283–4292 CrossRef CAS.
- X. J. Cai, H. Q. Dong, W. J. Xia, H. Y. Wen, X. Q. Li, J. H. Yu, Y. Y. Li and D. L. Shi, J. Mater. Chem., 2011, 21, 14639–14645 RSC.
- S. Munier, I. Messai, T. Delair, B. Verrier and Y. Ataman-Onal, Colloids Surf., B, 2005, 43, 163–173 CrossRef CAS.
- N. S. Templeton, Biosci. Rep., 2002, 22, 283–295 CrossRef CAS.
- T. Nagasaki and S. Shinkai, J. Inclusion Phenom. Macrocyclic Chem., 2007, 58, 205–219 CrossRef CAS.
- T. A. Ratko, J. P. Cummings, J. Blebea, J. Blebea and K. A. Matuszewski, Am. J. Med., 2003, 115, 560–569 CrossRef CAS.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS.
- M. C. P. de Lima, S. Simões, P. Pires, P. Pires, H. Faneca and N. Duzgunes, Adv. Drug Delivery Rev., 2001, 47, 277–294 CrossRef.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- S. B. Zhang, Y. M. Xu, B. Wang, W. H. Qiao, D. L. Liu and Z. S. Li, J. Controlled Release, 2004, 100, 165–180 CrossRef CAS.
- D. Schaffert and E. Wagner, Gene Ther., 2008, 15, 1131–1138 CrossRef CAS.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- C. Rudolph, J. Lausier, S. Naundorf, R. H. Müller and J. Rosenecker, J. Gene Med., 2000, 2, 269–278 CrossRef CAS.
- S. Y. Wong and J. M. D. Pelet, Proc. Natl. Acad. Sci. U. S. A., 2007, 32, 799–837 CAS.
- H. Ding, D. Y. Yang, C. Zhao, Z. k. Song, P. C. Liu, Y. Wang, Z. J. Chen and J. C. Shen, ACS Appl. Mater. Interfaces, 2015, 7, 4713–4719 CrossRef CAS.
- L. Shang and G. U. Nienhaus, Acc. Chem. Res., 2017, 50, 387–395 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- G. A. Simms, J. D. Padmos and P. Zhang, J. Chem. Phys., 2009, 131, 214703 CrossRef.
- J. S. Gebauer, M. Malissek, S. Simon, S. K. Knauer, M. Maskos, R. H. Stauber, W. Peukert and L. Trrurl, Langmuir, 2012, 28, 9673–9679 CrossRef CAS.
- R. Khandelia, S. Bhandari, U. N. Pan and S. S. Ghosh, Small, 2015, 11, 4075–4081 CrossRef CAS.
- W. Gu, Q. Zhang, T. Zhang, Y. Li, J. Xiang, R. Peng and J. Liu, J. Mater. Chem. B, 2016, 4, 910–919 RSC.
- J. L. Burt, C. Gutiérrez-Wing, M. Miki-Yoshida and M. José-Yacamán, Langmuir, 2004, 20, 11778–11783 CrossRef CAS.
- D. Botstein and G. R. Fink, Science, 1988, 240, 1439–1443 CrossRef CAS.
- D. Botstein and G. R. Fink, Genetics, 2011, 189, 695–704 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional the experimental section and Fig. S1–S9. See DOI: 10.1039/c7ra10141d |
| This journal is © The Royal Society of Chemistry 2017 |