
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of supramolecular organic framework to uranyl-organic coordination complex: a new “matrix-free” strategy for highly efficient capture of uranium†
Bo
Li
a,
Lei
Wang
a,
Yang
Li
a,
Dongqi
Wang
b,
Rui
Wen
a,
Xinghua
Guo
a,
Shoujian
Li
*a,
Lijian
Ma
*a and
Yin
Tian
*c
aCollege of Chemistry, Sichuan University, Key Laboratory of Radiation Physics & Technology, Ministry of Education, No. 29 Wangjiang Road, Chengdu, 610064, P. R. China. E-mail: sjli000616@scu.edu.cn; ma.lj@hotmail.com
bCAS Key Laboratory of Nuclear Radiation and Nuclear Energy Techniques, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China
cSouthwestern Institute of Physics, Chengdu 610041, People's Republic of China. E-mail: tianyin@swip.ac.cn
First published on 30th January 2017
Abstract
Herein, an innovative “matrix-free” strategy has been proposed for highly efficient uranium capture via uranyl-induced disassembly and reassembly of the two functional building blocks of the as-prepared hydrogen-bonded supramolecular organic framework (HSOF), which is composed of N-donor-containing melamine and O-donor-containing trimesic acid self-assembled through hydrogen bonding. The batch experimental results demonstrated that HSOF possesses excellent extraction capacity (qm = 444 mg g−1), >99% removal efficiency in the range of tested U(VI) concentration (20–130 ppm) with a considerably large KUd value of 1.3 × 107 mL g−1 at 130 ppm, and very fast extraction rate (<10 min) for UO22+. Especially, the uranium selectivity (SU = qe-U/qe-tol) of HSOF stays above 80% over the pH range tested in a uranium-containing solution with 11 competing cations, and distinctively, reaches the so far unreported 99% with a great capacity of 309 mg g−1 at pH 2.5. It is worth noting that a clear morphology transformation of HSOF nanowires to nanosheets of the uranyl-organic coordination complex (UOCC) after extraction has been observed only in the presence of uranyl ions. Moreover, according to experimental characterization and DFT studies, a possible mechanism for the efficient capture of uranium is proposed: the stronger coordination interaction among uranyl, TMA and MA could replace the weaker hydrogen-bond interaction originally linking the two building blocks in HSOF in the extraction process.
Introduction
Highly efficient separation and recovery of uranium from various uranium-containing aqueous systems is of great practical and academic significance for the sustainable development of nuclear power, human health, environment protection and resource recycling.1 Over the past few decades, solid-phase extraction (SPE) or adsorption has been considered to be one of the most promising and practical technologies for separation and recovery of uranium and other valuable metals due to its simplicity, convenience, availability, reliability, low-cost, easy to realize large-scale applications etc.2,3 Especially in recent years, a variety of new and powerful SPE techniques have been developed for uranium extraction by functionalization of solid-phase matrix with various chemical components (ligands and small molecules), such as amidoxime-functionalized mesoporous carbon or hydrothermal carbon or nanofibrous or ionic liquid,4–7 organosilica-phosphonate-functionalized SBA-15,8 dihydroimidazole-functionalized SBA-15,9 amino-functionalized MOF,10 coumarin functionalized Zn-MOF-74,11 and benzimidazole-functionalized covalent organic frameworks (COF).12 For most of SPE materials reported, as far as we know, the functional components were usually designed to be grafted or immobilized on and/or in matrix components by strong and irreversible covalent bonds, which is well known as a relatively robust and stable bonding mode in most case. However, this type of bonding mode could usually make it more difficult for the functional components on the SPE materials to adjust themselves to adapt to the coordination geometry of the target ions during the extraction process, and thus limit the efficiency of SPE materials for uranium (and other valuable metals) extraction and separation. Moreover, for those porous SPE materials, the pores and voids in them would usually result in lower selectivity to target metal ion and slower extraction kinetics, due to the unselective physical adsorption and the inevitably intra-particle diffusion. Encouragingly, however, recently emerged supramolecular organic framework (SOF) might provide us a creative approach to ameliorate the problems.SOF, a new type of functional materials, is generally built by using spontaneous self-assembly of appropriate building blocks via intermolecular noncovalent interactions mainly including electrostatic forces, hydrogen bonding, π–π stacking, van der Waals interactions, and so on.13,14 The available important features of SOF materials are as follows: (1) the soft and/or flexible nature based on intermolecular noncovalent interactions, especially weak interactions, endows the SOF materials unique responsiveness to external stimuli (like temperature, pH, light, redox, magnetic, and so on),15–18 (2) the framework structure of the SOF materials can be modified by well-chosen building blocks to meet specific application needs, (3) the morphologies of the materials, such as nanofibers, nanobelts, nanotwists, nanotubes and nanospheres, can be tailored with relatively easy self-assemble process.19 These properties make SOF a “smart”, multipurpose nanomaterials and have attracted increasing interest in the field of chemical storage,20,21 detection,22 drug deliver,15,23 organic electronics,24 and so on.
On the other hand, uranyl ion possesses the unique coordination structure of the two axial oxygen atoms, favoring the accessible coordination of 4 to 6 extra ligands in the equatorial plane, which has strong tendency of forming coordination bond with the adjacent ligands.25,26
In the current study, by combining the features of both hydrogen-bonded SOF and uranyl ion, we develop a new “matrix-free” strategy for highly efficient capture uranium from weak acidic uranium-containing aqueous media. A new hydrogen-bonded SOF (HSOF) material has been prepared via one-step self-assembly under mild condition, using deliberately chosen N-donor-containing melamine (MA) and O-donor-containing trimesic acid (TMA) as the building blocks. The as-synthesized HSOF could be employed directly as “SPE extractant” for the capture of uranium. In this strategy, the functional building blocks, MA and TMA, in HSOF would undergo a disassembly process on the induction of co-existing uranyl ion, and subsequently reassembly with the uranyl ion via coordination bonding, finally forming a new uranyl-organic coordination complex (UOCC).
Batch experiments were performed to investigate the extraction performance of the HSOF towards uranium under various conditions. The results verified that as-prepared HSOF possesses highly specific recognition ability for uranyl ions in both pure uranium solution and multi-ions systems containing 11 competing ions. Finally, possible mechanisms for the extraction of uranium and the structure transformation of HSOF nanowire to UOCC nanosheet were explored detailedly based on experimental characterization and DFT studies.
Results and discussion
Preparation of HSOF, UOCC and LaOCC materials
In order to develop a new “matrix-free” strategy for highly selective capture of uranium via reassembly of hydrogen-bonded SOF. Multidentate N- and O-donor ligands, melamine (MA) and trimesic acid (TMA) were deliberately selected to construct flexible hydrogen-bonded SOF via a one-step self-assembly in aqueous solution. The final product, white solid powder (inset in Fig. S1, ESI†), was denoted as HSOF (see Section S1 for details, ESI†). The U(VI)-, La(III)-loaded HSOF samples were denoted as UOCC and LaOCC respectively (see Section S1 for details, ESI†).Quantum chemical calculations
In this work, the electron correlation effects were included by employing density functional theory (DFT)27,28 methods at the CAM-B3LYP29 level of theory which included long range correction by using the Coulomb-attenuating method. In addition, the Grimme's D3-correction with Becke–Johnson damping [D3(BJ)] was added to include London-dispersion.30–32 The Stuttgart quasi-relativistic effective core pseudopotentials (RECPs) were used to describe the uranium atoms.33,34 The small-core RECPs represented 60 core electrons in uranium while the remaining 32 electrons were represented by the corresponding valence basis set. The double-ζ basis set 6-31+G(d) was used to describe oxygen, nitrogen, carbon and hydrogen atoms. The default fine grid (75, 302), having 75 radial shells and 302 angular points per shell, was used to evaluate the numerical integration accuracy. All of the geometric structures were optimized in aqueous solution by using the conductor-like polarized continuum model (CPCM)35 with universal force field (UFF)36 radii. The harmonic vibrational frequencies analysis was carried out after the geometry optimizations to characterize the nature of each stationary point as the true minimum with no imaginary frequency, and provide Gibbs free energies (G). The Wiberg bond indices (WBIs)37 and natural atomic charges were obtained by natural bond orbital (NBO)38,39 analysis at the same level of theory. All calculations have been carried out with Gaussian 09 program.Characterization of materials
Fig. 1a displays dispersed individual HSOF nanowires with uniform width (several micrometers) and length (100–200 micrometers) after ultrasonication in aqueous solution. Interestingly, a dramatic morphological transformation was observed from uniform nanowire to uniform nanosheet (Fig. 1b) after the capture of uranium, which further aggregate together to form flower-like structure. However, under the same extraction conditions, LaOCC shows uniform microrod-like morphology with width less than 1 micrometer and length about 5 micrometers (Fig. 1c). As expected, uniform flower-like nanosheet structure was also observed even in multi-ions system (containing 11 competing cations except UO22+), after uranium capture by HSOF (Fig. S2b, ESI†). The nanosheet morphology can be observed clearly by transmission electron microscopy (Fig. 1d). Energy-dispersive X-ray (EDX) spectroscopy confirm that among the 12 metal ions only uranyl ions was captured and uniformly dispersed into the final structure of UOCC (Fig. S2c–f, ESI†). The HSOF, UOCC and LaOCC showed obvious Tyndall effect, suggesting the small particle size of well-dispersed nanowires, nanosheets and nanorods (insets, Fig. 1a–c). These results strongly verified that HSOF was capable of capturing UO22+ selectively in both pure uranium solution and multi-ions systems. A possible mechanism of the course will be proposed and discussed below. | ||
| Fig. 1 SEM images of (a) HSOF. (b) UOCC, after uranium capture by HSOF in pure uranium solution. (c) LaOCC, after lanthanum capture by HSOF in pure lanthanum solution. The insets show the Tyndall effect of a colloidal suspension. (d) TEM image of UOCC, the inset shows the selected area electron diffraction. | ||
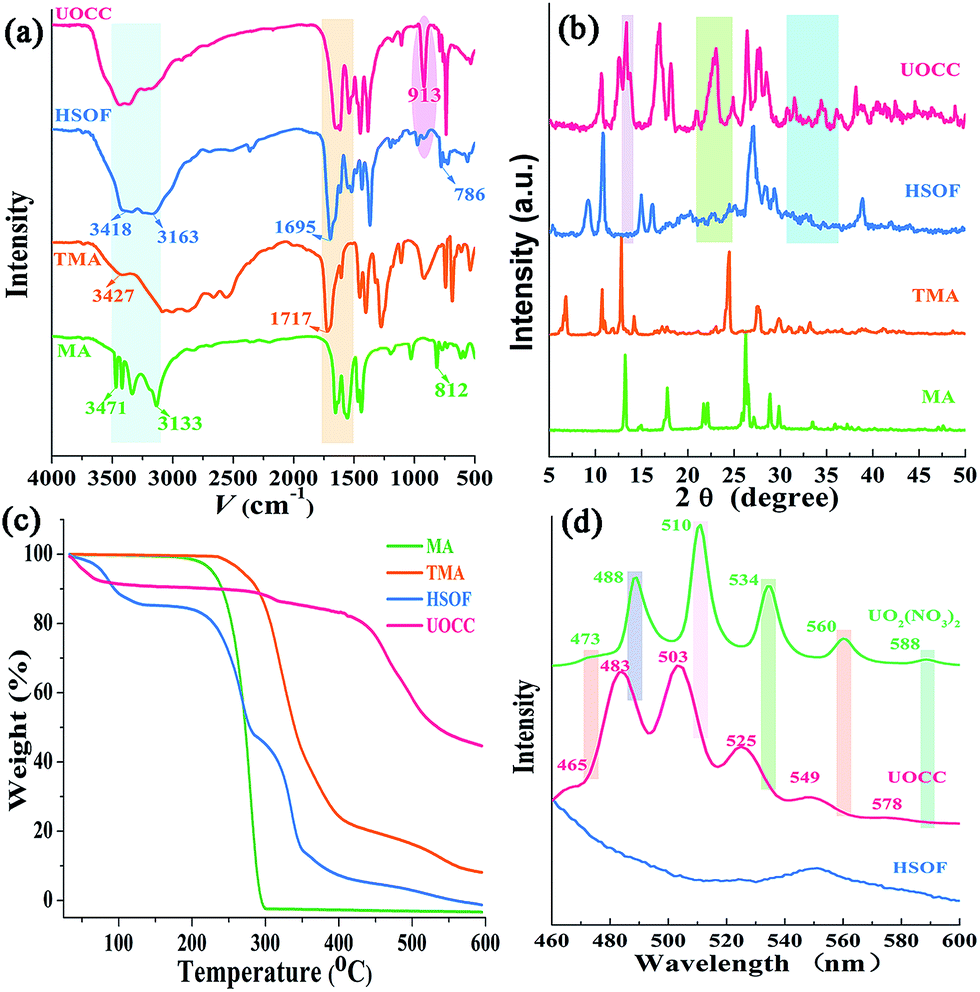
The FT-IR spectra in Fig. 2a shows the –NH2 stretch peak of HSOF is broader and smoother than MA. The triazine ring vibration of MA moves from 812 to 786 cm−1, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak of TMA shifts from 1717 to 1695 cm−1. Furthermore, 1H NMR data (Fig. S3, ESI†) indicated that the amino protons of HSOF shift downfield by 0.5 ppm compared with that of MA. These results imply the formation of hydrogen-bond between MA and TMA. It is noted that the FT-IR spectrum of the UOCC (Fig. 2a) exhibits the existence of a new peak at ∼913 cm−1 assigned to the antisymmetric vibration of the [OUVIO]2+ group,40 which is not present in the spectrum of HSOF (Fig. 2a). Thus it is reasonable to infer that there are interactions between the building blocks and uranyl ions during the extraction process.
O stretching peak of TMA shifts from 1717 to 1695 cm−1. Furthermore, 1H NMR data (Fig. S3, ESI†) indicated that the amino protons of HSOF shift downfield by 0.5 ppm compared with that of MA. These results imply the formation of hydrogen-bond between MA and TMA. It is noted that the FT-IR spectrum of the UOCC (Fig. 2a) exhibits the existence of a new peak at ∼913 cm−1 assigned to the antisymmetric vibration of the [OUVIO]2+ group,40 which is not present in the spectrum of HSOF (Fig. 2a). Thus it is reasonable to infer that there are interactions between the building blocks and uranyl ions during the extraction process.
 | ||
| Fig. 2 (a) FT-IR spectra of MA, TMA, HSOF and UOCC. (b) Powder X-ray diffraction patterns of MA, TMA, HSOF and UOCC. (c) TGA analysis of MA, TMA, HSOF and UOCC. (d) X-ray fluorescence spectra of HSOF, UOCC and UO2(NO3)2·6H2O excited at 428 nm with excitation and emission slit width 3.0 nm. | ||
The PXRD patterns of the prepared materials show well-defined crystalline structure of HSOF and UOCC (Fig. 2b and S4, ESI†). Comparing the PXRD patterns of the as-prepared materials, it is found that four new peaks appear around 2θ = 9.1°, 15.0°, 16.1°, 39° in the patterns of HSOF, which could attribute to the changes of unit cell parameters of the material. So we could infer that a new structure, most likely a self-assembly supramolecular structure, was formed through an intermolecular hydrogen bond between MA and TMA. Several new peaks appear around 2θ = 13.4°, 13.8°, 23°, 24°, 24.8°, 36°, 36.7° in pattern of HSOF, which is consistent with the pattern of UO2(NO3)2·6H2O (Fig. S4, ESI†). Meanwhile, in the pattern of HSOF, some peaks around 2θ = 9.3°, 10.9°, 15°, 16.2° shift to 2θ = 11.7°, 12.5°, 17°, 18.2° respectively, after the extraction of uranium. These results indicate that the accommodation of uranyl ion into the lattice sites of the UOCC and the significant change of band structure. The analysis results are consistent with the FT-IR and SEM analyses. The HSOF exhibits three-stage weight losses at around 100 °C, 260 °C and 340 °C, as indicated in TGA curve (Fig. 2c). According to the weight losses ratio of MA (about 37%) and TMA (about 30%), we can speculate that the molar ratio of MA to TMA is 2.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is basically consistent with the results of elemental analysis (Table S2, ESI†). So, the final ratio of MA to TMA in the HSOF may be 2:1, which self-assemble through hydrogen bonding interactions. Similarly, TGA analysis of UOCC confirms the molar ratio of MA:TMA is 1:2.1 (Fig. 2c).
:1, which is basically consistent with the results of elemental analysis (Table S2, ESI†). So, the final ratio of MA to TMA in the HSOF may be 2:1, which self-assemble through hydrogen bonding interactions. Similarly, TGA analysis of UOCC confirms the molar ratio of MA:TMA is 1:2.1 (Fig. 2c).
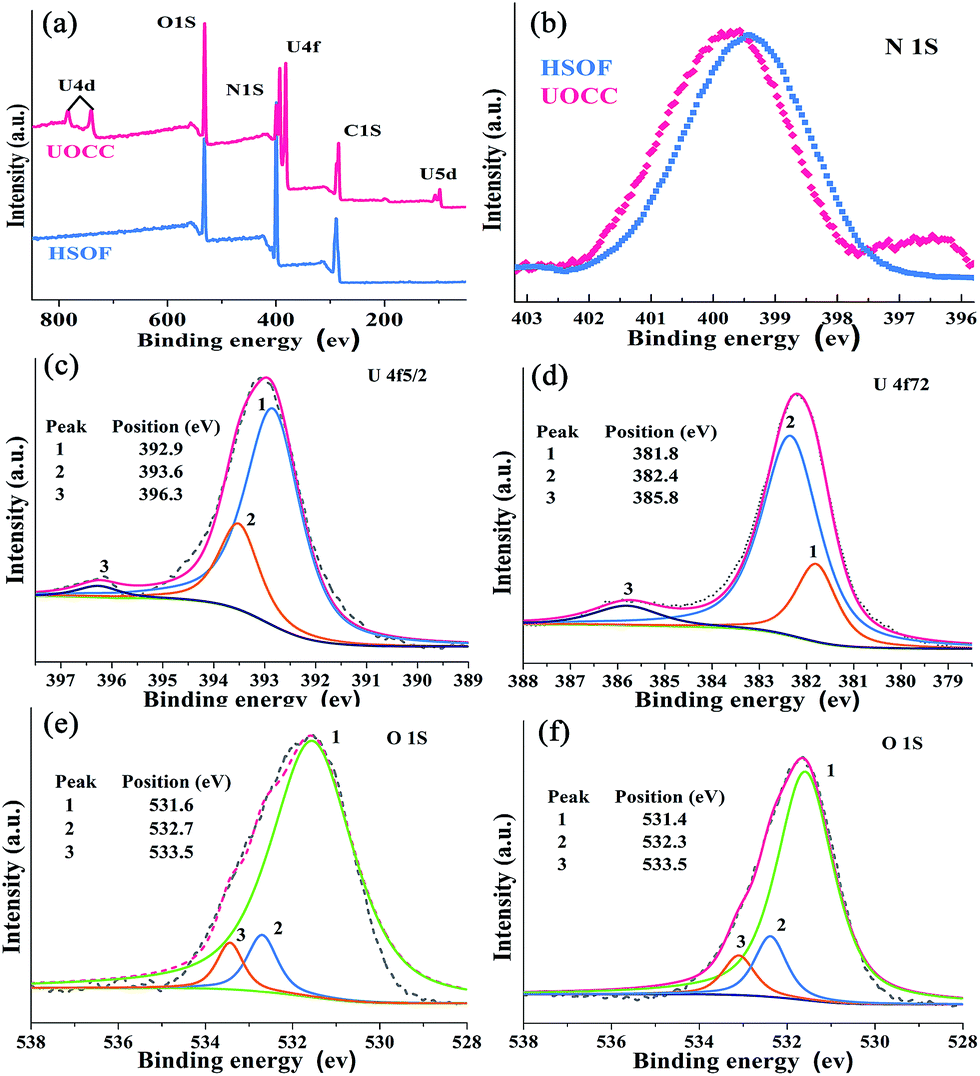
The X-ray fluorescence (XRF) emission spectrum of UOCC (Fig. 2d) shows six emission bands at 465, 483, 503, 525, 549 and 578 nm, quite similar to that of UO2(NO3)2·6H2O with typical bands corresponding to the electronic transitions S11 → S00 and S10 → S0υ (υ = 0–4) of the uranyl ion. The blue-shift effect is attributed to a UO22+–HSOF complexation.41,42 XPS analyses clearly indicate that a new double U4f peaks appears in the UOCC (Fig. 3a), and the corresponding high-resolution U4f5/2 and U4f7/2 core-level spectra (Fig. 3c and d) also reveal the existence of uranium(VI) in the UOCC structure.43 The N 1s core-level peak in Fig. 3b shows about 0.5 eV shift from HSOF (399.4 eV) to UOCC (399.9 eV).44 According to the O 1s core level in Fig. 3e and f, it is found that the peak 1 and peak 2 with binding energies of 531.6 and 532.7 eV (for HSOF) and 531.4 and 532.3 eV (for UOCC) can be assigned to the oxygen atom in the form of CO (carboxyl) and of C–O (hydroxyl),45 respectively. Obviously, the binding energies for peaks 1 and 2 of UOCC have certain degree of shift, which is due to the bonding of uranyl ions onto oxygen atoms and the consequent decrease of the electron density of the oxygen atoms.46 The above results distinctly testify the chemical bonding between uranyl ions and the nitrogen atoms (amino nitrogen and/or heterocyclic nitrogen atoms) and oxygen atoms (carboxyl and hydroxyl atoms) on UOCC. Furthermore, a relative element content (Table S3, ESI†) was also obtained from the integrated XPS peak areas, which is consistent with the theoretical values (MA:TMA:UO22+ = 1:2:1, molar ratio).
 | ||
| Fig. 3 (a) Typical XPS survey spectra of HSOF and UOCC. (b) High resolution XPS spectra of N 1s for HSOF and UOCC. (c) U4f7/2 and (d) U4f5/2 for UOCC. O 1s for (e) HSOF and (f) UOCC. | ||
Bath extraction studies of HSOF towards uranium
Extraction capacity (qe, mg g−1) of either U(VI) or other metal ions and distribution coefficient Kd (mL g−1) were calculated by the following eqn (1) and (2):47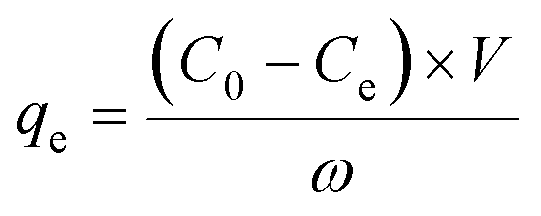 | (1) |
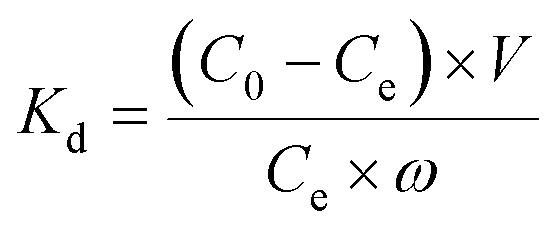 | (2) |
A specific term, uranium-selectivity (SU), was coined to describe the potency and degree of the selectivity of the extractant to uranium:48
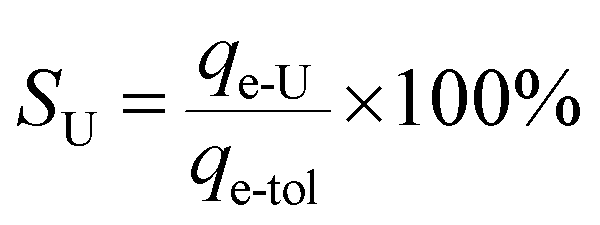 | (3) |
Effect of pH
In order to study the effect of pH on the extraction of U(VI), the stability of HSOF in aqueous solution at different pH values (1.5–8.0) has been investigated. The FT-IR and SEM analysis (Fig. S5 and S6, ESI†) indicate that HSOF is stable enough under all pH conditions tested except pH 1.5. Moreover, according to solubility calculation and modeling of species distribution using a CHEMSPEC (C++) program,7 uranyl ions could be converted into an insoluble specie (UO2(OH)2) at pH value higher than 4.5 in the concentration range used in this study (≤300 mg L−1), (Fig. S7, ESI†). Therefore, the effect of pH on extraction behaviour of HSOF towards U(VI) was investigated from pH 1.0 to 4.5. The results are shown in Fig. 4a. It is clear that the extraction capacity of HSOF towards uranium strongly depends on the solution pH values. At lower pH values (≤1.5), the HSOF basically do not extract of uranium. However, the extraction capacity increases sharply from pH 1.5–2.5, and then slowly increases with the further increase of pH, and reaches 1.74 mmol g−1 (415 mg g−1) at pH 4.5 in pure uranium solution.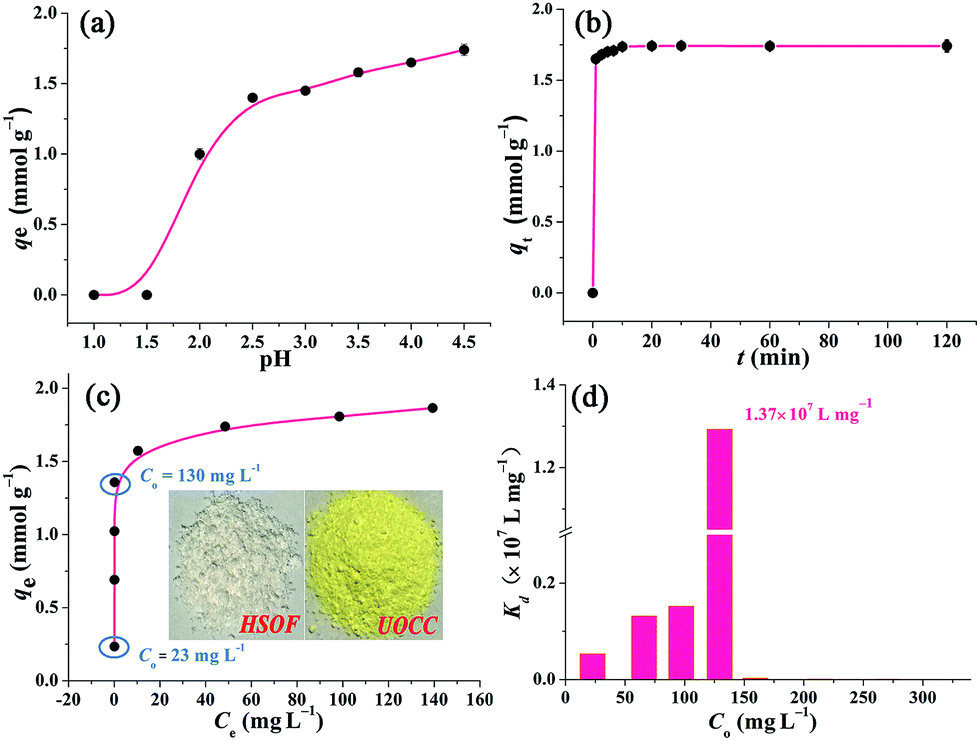 | ||
| Fig. 4 Batch extraction experiments. (a) Effect of pH on the extraction of U(VI) onto HSOF (C0 ≈ 0.9 mmol L−1 in purity uranium solution, t = 120 min, v = 25 mL, T = 298 K, and ω = 10 mg). (b) Effect of contact time on the extraction of U(VI) (C0 ≈ 0.9 mmol L−1, pH ≈ 4.5, v = 25 mL, T = 298 K, and ω = 10 mg). (c) Effect of initial uranium concentration on the extraction of uranium (C0 ≈ 0.08–1.26 mmol L−1 (20–300 mg L−1), pH ≈ 4.5, t = 120 min, v = 25 mL, T = 298 K, and ω = 10 mg), the inset shows the photograph images of HSOF and UOCC. (d) The Kd values of U(VI) on HSOF at different initial uranium concentration (C0 ≈ 20–300 mg L−1, pH ≈ 4.5, t = 120 min, T = 298 K, and ω = 10 mg). | ||
Kinetics studies
Extraction kinetics is of great significance to evaluate the performance of a given extractant. The effect of contact time has been investigated. As shown in Fig. 4b, ten minutes were found to be sufficient for reaching extraction equilibrium (422 mg g−1). The fast equilibrium time in this case outdistances most of the functionalized extractants reported before (Table S6, ESI†). Particularly, over 94% of total extraction capacity occurred during the first minute. These results indicate that the extraction is a very rapid process, and HSOF may be possible to overcome the bottleneck problem of uranium extractant of slow kinetics and low uranium extraction capacity. Three different kinetic models, namely pseudo-first-order, pseudo-second-order model and intraparticle diffusion model, were employed to evaluate the controlling mechanism of the extraction process (see Section S4 for details, ESI†). Results suggest that the pseudo-second-order model could be used for a better description of the extraction process. Meanwhile, the results imply that the current extraction process might be regarded as a chemical process.Isotherm studies
Fig. 4c shows the effect of equilibrium uranium concentration on the extraction of uranium onto HSOF. The extraction capacity exhibits a linear increase with the increase in initial uranium concentration (from about 20 to 130 mg L−1). And the saturation uranium extraction capacity reaches 444 mg g−1. It is noteworthy that the product color changed from nearly colorless to brilliant yellow (insert in Fig. 4c and S1, ESI†) after the capture of uranium. What's more, HSOF shows high removal efficiency (>99% at the range of 20–130 ppm of U(VI) concentration) with considerable large KUd value of 1.3 × 107 mL g−1 at 130 ppm for UO22+, (Fig. 4d, Table S8, ESI†). This value is one of the largest KUd value among the reported extractants (Table 1). To further understand the extraction performance of HSOF towards U(VI), the equilibrium data were fitted to Langmuir, Freundlich and Dubinin and Radushkevich (D–R) isotherm (see Section S5, ESI†). The mean adsorption energy (E) value was calculated to be 31.45 kJ mol−1, indicating the extraction of uranium onto the HSOF was mainly controlled by chemical adsorption.53,54| Extractant | C 0 (mmol L−1) | UREa (%) | K Ud (mL g−1) | Ref. |
|---|---|---|---|---|
| a Uranium removal efficient. b Salicylideneimine-functionalized hydrothermal carbon. c Phosphonate-functionalized large pore 3-D cubic mesoporous. d Exchangeable potassium ions metal chalcogenide. e Layered sulfide ion exchanger K2MnSn2S6 (KMS-1). f Polysulfide/layered double hydroxide composites. | ||||
| HTC-Salb | 0.5 | 44.4 | 9.50 × 103 | 49 |
| KIT-6c | — | 98 | 1.00 × 104 | 50 |
| K2xSn4−xS8−xd | 0.024 | — | 2.70 × 104 | 51 |
| KMS-1e | 1.68 | 99.9 | 1.80 × 105 | 40 |
| Cs-birnessite | — | ∼100 | 1.70 × 106 | 52 |
| Sx-LDHf | 0.09 | >99.9 | 3.40 × 106 | 1 |
| HSOF | 0.55 | >99.9 | 1.37 × 107 | This work |
Selective extraction of uranium
High efficient separation and recovery of uranium has always been an important research hotspot and difficulty in the field of separation science. The foregoing results verified the high binding affinity, superior extraction capacity and fast kinetics of HSOF towards uranium in pure uranium solution. However, whether the HSOF can selectively recognize and capture uranium in multi-ions aqueous systems? In order to further explore its selectivity recognition ability and binding affinity of HSOF towards uranium in multi-ions solution, a stimulated weak-acid multi-cations competing system has been prepared (see Section S2 and Table S4 for details, ESI†). Some representative transition metal and alkaline earth metal ions were selected as competitive ions, including especially five typical lanthanide nuclides (La3+, Sm3+, Nd3+, Gd3+, Ce3+) which usually have strong coordination capacities to functional ligands and great influence on the selective separation of uranium. Taking into consideration that HSOF could lose its ability to separate uranium at lower pH values (Fig. 4a), batch extraction experiments were carried out at the pH range 2.5–4.5 in multi-ions aqueous systems containing 11 competing cations except UO22+ ion in an attempt to better understand the binding affinity and recognition capability of HSOF towards uranium.Results indicate (Fig. 5) that the total extraction capacity of HSOF towards metal ions increases gradually along with the increasing pH from pH 2.5 (1.38 mmol g−1) to 4.5 (1.89 mmol g−1) and the uranium extraction amount occurs from 329 mg g−1 at pH = 2.5 to 362 mg g−1 at pH = 4.5. Distinctively, it is worth noting that HSOF shows an unprecedented selectivity towards uranium. Throughout the pH range (2.5–4.5) studied, the uranium selectivity, SU, stays above 80% (Fig. 5, inset). Specifically, a so far unreported highest selectivity of 99% with a considerable extraction capacity of 329 mg g−1 is observed at pH 2.5. As we all know, this value is far more than that of all the uranium extractants reported so far (Table 2). To verify the excellent selectivity of HSOF towards uranium, further extraction experiments were performed (Section S3, Fig. S11, ESI†). Results indicated that no Co(II) was extracted even in the pure cobalt solution (C0 ≈ 1.0 mmol L−1, pH = 2.5 and 4.5), and only a little of amount of La(III) (about 0.17 mmol, 23.6 mg) was separated at pH 2.5.
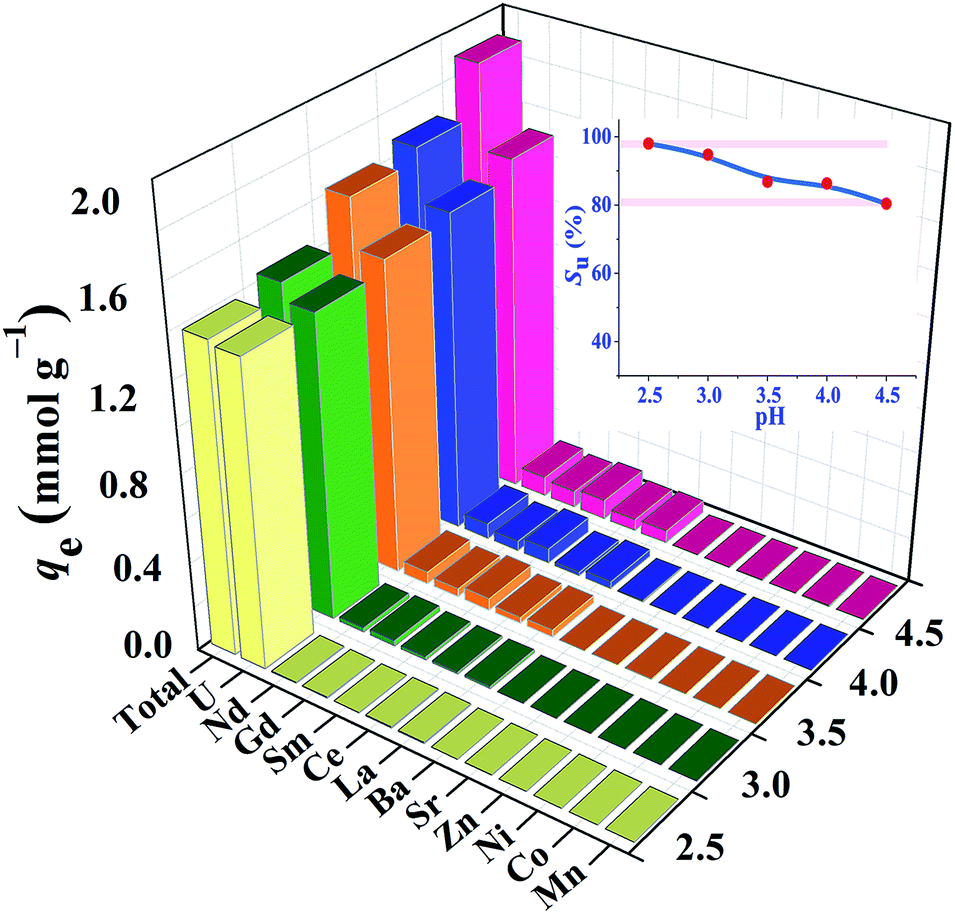 | ||
| Fig. 5 Effect of pH on the extraction of U(VI) in a multi-ion system (C0 ≈ 1.0 mmol L−1 for all cations, t = 120 min, v = 25 mL, T = 298 K, and ω = 10 mg), the inset shows the uranium-selectivity (SU) of HSOF towards uranium at different pH values. | ||
| Extractants | C 0 (mmol L−1) | pH | q U (mg g−1) | Ref. |
|---|---|---|---|---|
| a Diethylenetriamine-functionalized chromium-based MOF. b Dihydroimidazole-functionalized SBA-15. c Phosphine oxide ligands-functionalized mesoporous silicas. d Task specific ionic liquids. e Acylamide- and carboxyl-functionalized metal–organic framework. f Microporous phosphazene-based covalent organic framework. g Amidoxime-functionalized hydrothermal carbon. | ||||
| MIL-detaa | 0.41 | 2.5 | 0 | 10 |
| DIMSb | 0.8 | 2.5 | 0 | 9 |
| SBA-POCc | — | 2.5 | 20 | 55 |
| SSILd | 0.42 | 2.5 | 40 | 56 |
| AC-MOFse | 0.42 | 2.5 | 80 | 57 |
| UiO MOFs | 0.42 | 2.5 | 98 | 58 |
| MPCOFf | 0.45 | 2.5 | 107 | 59 |
| MOF-76 | 0.6 | 2.5 | 200 | 60 |
| HTC-AOg | 1.0 | 2.5 | 269 | 7 |
| HSOF | 0.5 | 2.5 | 282 | This work |
These results significantly demonstrated that HSOF possesses an outstanding binding affinity, specific recognition capability and high extraction capacity towards uranium in a uranium-containing solution with 11 competing cations, which suggests that HSOF possess the potential for highly effective separation and/or recovery of uranium from weakly acidic nuclear industry effluents from such as uranium mining, milling, conversion, enrichment and fuel fabrication processes.
DFT calculations of uranyl-HSOF interactions
To gain deeper insights into the extraction process, we further studied this conversion reaction by DFT. Recently, theoretical and experimental studies indicate that uranyl ions could form a stable hydrated ion ([UO2(H2O)5]2+) that contains five water molecules in its equatorial plane in aqueous solution, or form the neutral uranyl nitrate hydrates [UO2(NO3)2(H2O)2] that contains two nitrates as bidentate ligands also in the equatorial plane in high concentration of nitrate solution. In this work, the optimized structures of the [UO2(H2O)5]2+ and UO2(NO3)2(H2O)2 are depicted in Fig. S12, ESI.† So, the UOCC formation reaction in this study is in essence an exchange process between the ligands (MA and TMA) from the disassembly of HSOF and the coordinated water or nitrates in the first coordination sphere of the uranyl ion.61The optimized structures of two possible products UOCC-1 and UOCC-2 were shown in Fig. 6a and b, respectively. For the first possible hexa-coordination product UOCC-1 (Fig. 6a), the carboxyl in TMA coordinated to the uranyl ion as bidentate chelating ligand through oxygen donor atoms which replace the coordinated water molecules or nitrates in the first coordination sphere of the uranyl ion. In addition, the MA also interacts with the TMA through hydrogen bond. As described in Table 3, the changes in the Gibbs free energy (ΔG) of this process were −56.82 kJ mol−1 and −23.73 kJ mol−1 in aqueous solution and in nitrate solution, respectively. But, the MA played the bidentate ligand roles in the second possible product UOCC-2 (Fig. 6b), which coordinated with uranyl through nitrogen atoms. The ΔG values of this process were −21.08 kJ mol−1 and 13.46 kJ mol−1 in aqueous solution and in nitrate solution, respectively, which indicates that UOCC-1 are the main product because of the weak coordination ability of nitrogen atom in MA with uranyl ions. These results can also be qualitatively explained through the analysis of the Wiberg bond indices (Table S10, ESI†). The order of WBIs of the U–L bonds in these two produces is U–NL < U–Owater < U–Onitrate < U–Ocarboxyl, which corresponds to the coordination ability with uranyl ions.
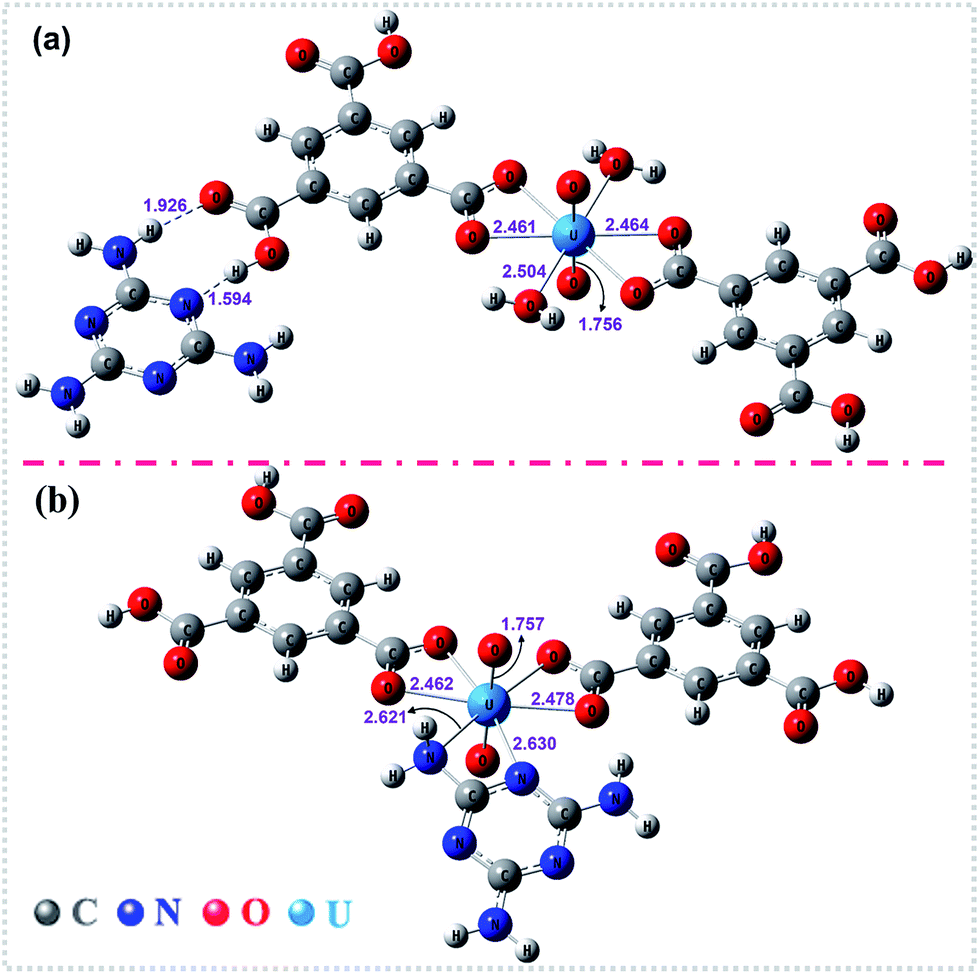 | ||
| Fig. 6 The optimized structures of the stationary points for (a) UOCC-1 and (b) UOCC-2 by DFT calculations. The important bond lengths (angstrom) are also shown in this figure. | ||
| Reaction | ΔG |
|---|---|
| [UO2(H2O)5]2+ + HSOF → UOCC-1 | −56.82 |
| UO2(NO3)2(H2O)2 + HSOF → UOCC-1 | −23.73 |
| [UO2(H2O)5]2+ + HSOF → UOCC-2 | −21.08 |
| UO2(NO3)2(H2O)2 + HSOF → UOCC-2 | 13.46 |
Proposed mechanism
Various characterization techniques and the DFT method were employed to gain deeper insight into the essence of the uranium separation process. Main characterization results can be summarized below: (1) the significant changes in color from nearly colorless to brilliant yellow (Fig. 4c, insert) and in morphology from nanowire (Fig. 1a) to nanosheet (Fig. 1b and S2b, ESI†) for the extractant used after loading of uranium were observed, which imply some structural changes in the extractant occurring during extraction. (2) The analyses of FT-IR, PXRD and XRF in Fig. 2a, b and d respectively for both HSOF and UOCC indicated that uranyl ion was incorporated into the structure of UOCC via chemical bonds. (3) XPS measurements (Fig. 3) confirmed the metal–ligand coordination between uranyl ions and oxygen atoms (hydroxyl and carbonyl) of TMA and nitrogen atoms (amino and triazine) of MA in the structure of UOCC. (4) Anticipatively, before extraction, the molar ratio of MA to TMA in HSOF could be inferred as 2:1 according to the results of elemental analyses (Table S2, ESI†) and TGA analyses (Fig. 2c), but after extraction, the molar ratio MA:TMA:UO22+ in UOCC is significantly found to be 1:2:1 obtained from integration of the XPS peak areas (Table S3, ESI†) and TGA analysis (Fig. 2c), and furthermore, based on these outcomes, DFT calculations demonstrated that the reaction processes for the disassembly of HSOF and the reassembly of UOCC could be spontaneous (ΔG < 0, Fig. 6 and Table 3). According to above analyses, the possible mechanism for the uranium extraction in this study is proposed as illustrated in Scheme 1. Under the inducing of the coexisting uranyl ion, the hydrogen bonds (O–H⋯N, O⋯H–N and N–H⋯N) between the MA and TMA in SOF would be disassembled, and two kinds of uranyl-organic coordination complex (UOCC-1 and UOCC-2, Scheme 1) would subsequently be reorganized via coordination-driven self-assembly. During the novel extraction process, the stronger coordination interaction among uranyl and MA and TMA of UOCC would replace the weaker hydrogen-bond interaction originally existing between MA and TMA in HSOF. In essence, the conversion process from the smart HSOF to the UOCC, which could result from the unique coordination structure and strong electropositive nature of uranyl ions with an effective charge of 3.2+ (ref. 62) combining with the flexible nature of the soft HSOF with the distinctive responsiveness to external stimuli, and especially, the significant difference in bond energies between weaker hydrogen bond (12–25 kJ mol−1)63,64 and coordinate bond (250–800 kJ mol−1, 701 kJ mol−1 for U–O bond).65
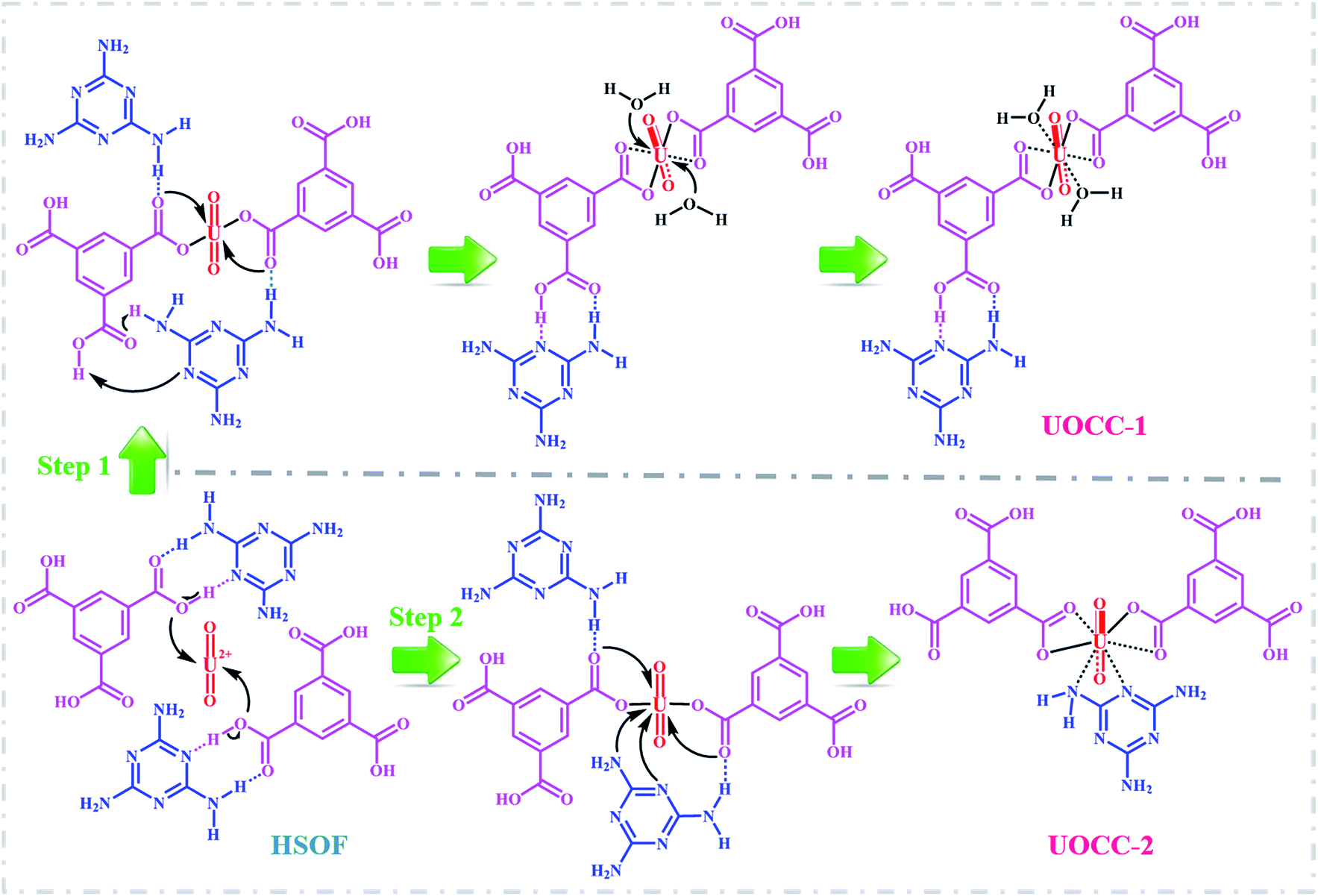 | ||
| Scheme 1 Two possible mechanisms for uranium extraction. | ||
Conclusion
In summary, we proposed a novel “matrix-free” strategy for highly efficient capture of target uranyl ion by solid–liquid extraction. The essence of the strategy is that under the inducing of the coexisting uranyl ion, the hydrogen bonds between TMA and MA in HSOF would be disassembled, and uranyl-organic coordination complex, UOCC would subsequently be reorganized via coordination-driven self-assembly. DFT calculations indicate that the disassembly and reassembly process is spontaneous (ΔG < 0).Based on this strategy, we successfully prepared a new hydrogen-bonded SOF (HSOF) material, which shows highly efficient recognition capability, excellent extraction capacity and very fast extraction rate for capture of UO22+ in both pure uranium solution and multi-ions systems containing 11 competing cations.
Compared with conventional SPE extractants mostly composed of solid matrix and functional groups, such as amidoxime (functional group)-grafted polymer or carbon4–6 (matrix) and dihydroimidazole-functionalized SBA-15,9 the as-prepared HSOF contains no matrix component, and does not need extra functionalization process, which actually changed the basic way for SPE from the ordinary extraction/sorption relying on functionalized fixed-sites on the conventional SPE extractants to a much more flexible extraction via uranyl-induced spontaneous disassembling and reassembling based on this kind of HSOF materials. Consequently, we achieved the following expected results: (1) actually simplifying the structure of SPE material and the synthesis procedure; (2) avoiding undesired non-selective physical adsorption and intra-particle diffusion caused by pores and voids in porous sorbents; (3) averting some negative factors such as possible steric hindrance during coordination reaction between target metal ions and functional groups grafted on the surface of matrix component, and greatly enhancing the utilization rate of functional components in SPE materials.
The significant findings suggest that the as-synthesized HSOF is among the most attractive and highly efficient uranium extractant, and has great potential in the uranium separation from weakly acidic nuclear industry effluents. The new strategy also may provide a practical yet meaningful idea to rationally design of new SPE materials for high efficient separation of uranium or other interested metal ions.
Acknowledgements
The financial support from the National Natural Science Foundation of China (Grants 21671140) and the Science Challenge Project JCKY2016212A504 are gratefully acknowledged. Calculations were done on the computational grids in the Supercomputing Center of Chinese Academy of Sciences (SCCAS).Notes and references
- S. Ma, L. Huang, L. Ma, Y. Shim, S. M. Islam, P. Wang, L.-D. Zhao, S. Wang, G. Sun, X. Yang and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 3670–3677 CrossRef CAS.
- T.-F. Lin and J.-K. Wu, Water Res., 2001, 35, 2049–2057 CrossRef CAS.
- Y. Shen, L. Li and Z. Zhang, J. Phys. Chem. C, 2016, 120, 6659–6668 CrossRef CAS.
- J. Górka, R. T. Mayes, L. Baggetto, G. M. Veith and S. Dai, J. Mater. Chem. A, 2013, 1, 3016–3026 RSC.
- P. S. Barber, S. P. Kelley, C. S. Griggs, S. Wallace and R. D. Rogers, Green Chem., 2014, 16, 1828–1836 RSC.
- S. Y. Xie, X. Y. Liu, B. W. Zhang, H. J. Ma, C. J. Ling, M. Yu, L. F. Li and J. Y. Li, J. Mater. Chem. A, 2015, 3, 2552–2558 RSC.
- X. D. Yang, J. Li, J. Liu, Y. Tian, B. Li, K. C. Cao, S. B. Liu, M. Hou, S. J. Li and L. J. Ma, J. Mater. Chem. A, 2014, 2, 1550–1559 RSC.
- P. J. Lebed, J.-D. Savoie, J. Florek, F. O. Bilodeau, D. Lariviére and F. Kleitz, Chem. Mater., 2012, 24, 4166–4176 CrossRef CAS.
- L. Y. Yuan, Y. L. Liu, W. Q. Shi, Z. J. Li, J. H. Lan, Y.-X. Feng, Y. L. Zhao, Y. L. Yuan and Z. F. Chai, J. Mater. Chem., 2012, 22, 17019–17026 RSC.
- Z. Q. Bai, L. Y. Yuan, L. Zhu, Z. R. Liu, S. Q. Chu, L. R. Zheng, J. Zhang, Z. F. Chai and W. Q. Shi, J. Mater. Chem. A, 2015, 3, 525–534 RSC.
- L. Zhang, L. L. Wang, L. L. Gong, X. F. Feng, M. B. Luo and F. Luo, J. Hazard. Mater., 2016, 311, 30–36 CrossRef CAS PubMed.
- J. Li, X. D. Yang, C. Y. Bai, Y. Tian, B. Li, S. Zhang, X. Y. Yang, S. D. Ding, C. Q. Xia and X. Y. Tan, J. Colloid Interface Sci., 2015, 437, 211–218 CrossRef CAS PubMed.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- Q. P. Duan, Y. Cao, Y. Li, X. Y. Hu, T. X. Xiao, C. Lin, Y. Pan and L. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10542–10549 CrossRef CAS.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11, 1025–1038 CrossRef CAS.
- Y. B. He, S. C. Xiang and B. L. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS.
- W. B. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa and N. R. Champness, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS.
- S. Ishihara, J. M. Azzarelli, M. Krikorian and T. M. Swager, J. Am. Chem. Soc., 2016, 138, 8221–8227 CrossRef CAS.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 115, 4788–4791 CrossRef.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- G. N. Greaves, N. T. Barrett, G. M. Antonini, F. R. Thornley, B. T. M. Willis and A. Steel, J. Am. Chem. Soc., 1989, 111, 4313–4324 CrossRef CAS.
- S. Shinkai, H. Koreishi, K. Ueda, T. Arimura and O. Manabe, J. Am. Chem. Soc., 1987, 109, 6371–6376 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- L. Goerigk and J. R. Reimers, J. Chem. Theory Comput., 2013, 9, 3240–3251 CrossRef CAS.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- S. Marque, V. Razafimahaleo, A. Dinut, G. Grach, D. Prim, X. Moreau and R. Gil, New J. Chem., 2013, 37, 2683–2690 RSC.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- M. J. Manos and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 16441–16446 CrossRef CAS.
- S. Fery-Forgues, M. T. Le Bris, J. P. Guette and B. Valeur, J. Phys. Chem., 1988, 92, 6233–6237 CrossRef CAS.
- I. Mihalcea, N. Henry, C. Volkringer and T. Loiseau, Cryst. Growth Des., 2011, 12, 526–535 CrossRef.
- S. Amayri, T. Arnold, T. Reich, H. Foerstendorf, G. Geipel, G. Bernhard and A. Massanek, Environ. Sci. Technol., 2004, 38, 6032–6036 CrossRef CAS.
- N. Xiao, D. Lau, W. Shi, J. Zhu, X. Dong, H. H. Hng and Q. Yan, Carbon, 2013, 57, 184–190 CrossRef CAS.
- F. Tao, S. Dag, L.-W. Wang, Z. Liu, D. R. Butcher, H. Bluhm, M. Salmeron and G. A. Somorjai, Science, 2010, 327, 850–853 CrossRef CAS.
- T. Classen, M. Lingenfelder, Y. L. Wang, R. Chopra, C. Virojanadara, U. Starke, G. Costantini, G. Fratesi, S. Fabris and S. de Gironcoli, J. Phys. Chem. A, 2007, 111, 12589–12603 CrossRef CAS.
- B. Li, L. Ma, Y. Tian, X. D. Yang, J. Li, C. Y. Bai, X. Y. Yang, S. Zhang, S. J. Li and Y. D. Jin, J. Hazard. Mater., 2014, 271, 41–49 CrossRef CAS.
- B. Li, C. Bai, S. Zhang, X. Zhao, Y. Li, L. Wang, K. Ding, X. Shu, S. Li and L. Ma, J. Mater. Chem. A, 2015, 3, 23788–23798 RSC.
- H. Wang, L. Ma, K. Cao, J. Geng, J. Liu, Q. Song, X. Yang and S. Li, J. Hazard. Mater., 2012, 229–230, 321–330 CrossRef CAS.
- P. J. Lebed, K. de Souza, F. Bilodeau, D. Lariviere and F. Kleitz, Chem. Commun., 2011, 47, 11525–11527 RSC.
- D. Sarma, C. D. Malliakas, K. S. Subrahmanyam, S. M. Islam and M. G. Kanatzidis, Chem. Sci., 2016, 7, 1121–1132 RSC.
- L. Al-Attar and A. Dyer, Sorption behaviour of uranium on birnessite, a layered manganese oxide, J. Mater. Chem., 2002, 12, 1381–1386 RSC.
- C. A. Başar, J. Hazard. Mater., 2006, 135, 232–241 CrossRef.
- E. Oguz, Colloids Surf., A, 2005, 252, 121–128 CrossRef CAS.
- W. Zhang, G. Ye and J. Chen, J. Mater. Chem. A, 2013, 1, 12706–12709 RSC.
- R. Ruhela, N. Iyer, M. Yadav, A. K. Singh, R. Hubli and J. K. Chakravartty, Green Chem., 2015, 17, 827–830 RSC.
- F. Luo, L. L. Dang, J. Q. Li, S. Juan Liu, M. B. Luo and L. L. Wang, J. Mater. Chem. A, 2015, 3, 13724–13730 RSC.
- M. Carboni, C. W. Abney, S. B. Liu and W. B. Lin, Chem. Sci., 2013, 4, 2396–2402 RSC.
- S. Zhang, X. Zhao, B. Li, C. Bai, Y. Li, L. Wang, R. Wen, M. Zhang, L. Ma and S. Li, J. Hazard. Mater., 2016, 314, 95–104 CrossRef CAS.
- W. T. Yang, Z. Q. Bai, W. Q. Shi, L. Y. Yuan, T. Tian, Z. F. Chai, H. Wang and Z. M. Sun, Chem. Commun., 2013, 49, 10415–10417 RSC.
- Y. Tian, J. Fu, Y. Zhang, K. Cao, C. Bai, D. Wang, S. Li, Y. Xue, L. Ma and C. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 7214–7223 RSC.
- A. F. Lucena, J. M. Carretas, J. Marçalo, M. D. C. Michelini, P. X. Rutkowski and J. K. Gibson, J. Phys. Chem. A, 2014, 118, 2159–2166 CrossRef CAS.
- M. S. Searle, D. H. Williams and U. Gerhard, J. Am. Chem. Soc., 1992, 114, 10697–10704 CrossRef CAS.
- D. A. Rodham, S. Suzuki, R. D. Suenram, F. J. Lovas, S. Dasgupta, W. A. Goddard and G. A. Blake, Nature, 1993, 362, 735–737 CrossRef CAS.
- R. G. Denning, in Complexes, Clusters and Crystal Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992, pp. 215–276, DOI:10.1007/bfb0036502.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of HSOF materials, preparation of U(VI), La(III) and Co(II) stock solution, test solution and multi-ion solution, characterization of HSOF materials including additional photograph and SEM images, FT-IR spectra, EA analysis, PXRD patterns, TG and DTG curve, XPS spectra of C 1s, extraction experimental including kinetic studies and isotherm studies, uranium(VI) species distribution, and other experimental results including figures and tables. See DOI: 10.1039/c6ra28356j |
| This journal is © The Royal Society of Chemistry 2017 |