
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect electrochemical formation of nanostructured amorphous Co(OH)2 on gold electrodes with enhanced activity for the oxygen evolution reaction†
Md Abu
Sayeed
,
Tenille
Herd
and
Anthony P.
O'Mullane
*
School of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), GPO Box 2434, Brisbane, QLD 4001, Australia. E-mail: anthony.omullane@qut.edu.au
First published on 3rd December 2015
Abstract
The oxides of cobalt have recently been shown to be highly effective electrocatalysts for the oxygen evolution reaction (OER) under alkaline conditions. In general species such as Co3O4 and CoOOH have been investigated that often require an elevated temperature step during their synthesis to create crystalline materials. In this work we investigate the rapid and direct electrochemical formation of amorphous nanostructured Co(OH)2 on gold electrodes under room temperature conditions which is a highly active precursor for the OER. During the OER some conversion to crystalline Co3O4 occurs at the surface, but the bulk of the material remains amorphous. It is found that the underlying gold electrode is crucial to the materials enhanced performance and provides higher current density than can be achieved using carbon, palladium or copper support electrodes. This catalyst exhibits excellent activity with a current density of 10 mA cm−2 at an overpotential of 360 mV with a high turnover frequency of 2.1 s−1 in 1 M NaOH. A Tafel slope of 56 mV dec−1 at low overpotentials and a slope of 122 mV dec−1 at high overpotentials is consistent with the dual barrier model for the electrocatalytic evolution of oxygen. Significantly, the catalyst maintains excellent activity for up to 24 h of continuous operation and this approach offers a facile way to create a highly effective and stable material.
Introduction
The development of electrocatalytically active nanomaterials is a crucial research endeavour given the ever increasing energy demands of our society. This is because electrocatalysis is at the core of many technologies related to the generation, conversion and potential storage of energy in a sustainable and clean manner.1–6 Indeed the most significant hurdle preventing the uptake of renewable energy technology is storage.7 Although there have been many significant developments in the battery space there is still an urgent need to implement large scale storage technologies. The electrochemical splitting of water into oxygen and hydrogen is one such method as it is a very attractive way to store the electricity generated from intermittent renewable energy sources such as wind and solar as a fuel.8,9 However, the generation of hydrogen from water via photochemical, photoelectrochemical or electrochemical methods is still an ongoing challenge due to the inherent chemical stability of water. At the heart of the electrolysis of water are electrocatalytic reactions that generate hydrogen and oxygen where production of the latter is particularly more problematic than the former. There is a large overpotential associated with the evolution of oxygen from water (greater than the theoretical value of 1.23 V) and generally the kinetics of this reaction is sluggish at many materials. There can also be issues associated with deactivation of the catalyst via electrodissolution, in particular at the most active IrO2 and RuO2 which are not stable over prolonged periods in alkaline conditions10–12 due to the potential required to maintain the reaction at a reasonable rate.A particularly interesting material that has shown significant activity for the oxygen evolution reaction (OER) is cobalt oxide, generally with the composition Co3O4.13–24 Among the many alternative metal oxides to iridium and ruthenium, those of iron, nickel and cobalt have gained significant attention due to the balance between cost and activity as well as their tolerance to conditions of high pH. Lyons et al.11 have performed an extensive study on the applicability of oxides of these metals to the OER where they considered dimensionally stable anode type electrodes, hydrous oxide electrodes and bulk oxide/hydroxide electrodes. In that study it was concluded that there is emerging evidence that the acid/base behaviour of the transition metal oxides can be ascribed to the presence of active octahedrally co-ordinated surface groups or surfaquo groups that significantly influence the OER. Taking the cobalt case in particular, many groups have studied the influence of the oxide type and its structure, in particular when at the nanoscale, on the OER.15,17,19,20 Of significant note is the influence on OER activity when Co3O4 is in contact with a metal.18 The presence of electrochemically roughened gold in intimate contact with cobalt oxide has been shown to be a very effective OER catalyst where it was postulated that the underlying gold acts as an electron sink to promote the oxidation of CoII and CoIII centres to CoIV which is regarded as the active site for the evolution of O2.18 The presence of underlying gold was found to be far more effective than platinum, palladium or copper. It has also been demonstrated that Au/Co3O4 core–shell nanomaterials are more active than Co3O4 nanoparticles for the OER13 and recently that Au nanoparticles embedded in mesoporous Co3O4 also improve activity.21 This is consistent with many composite materials in the (electro)-catalysis area where synergistic effects are often seen for supported catalysts and/or bimetallic systems.25–30
It has recently been demonstrated that the formation of amorphous metal oxides is beneficial for the OER which were created via the photochemical reduction of metal–organic species.31 Significantly, Strasser has shown that the outer layers of crystalline Co3O4 is in fact converted into an X-ray amorphous material consisting of CoOx(OH)y which is responsible for increased oxygen evolution activity.32 Therefore, in this work a simple method to produce amorphous and homogeneous cobalt hydroxide films on gold was undertaken by taking advantage of the change in localised pH at an electrode surface during cathodic electrodeposition processes in aqueous solution that induces the complexation of Co2+ ions with electrogenerated OH− ions and precipitation of Co(OH)2 onto the electrode surface. The resulting material was indeed found to be amorphous and far more effective for the OER when deposited on gold compared to glassy carbon, palladium and copper. In addition a highly effective electrocatalyst could be formed in 60 s that showed excellent sustained activity for the OER over a period of 24 h. This approach indicates that a simple electrodeposition protocol to produce metal hydroxides has excellent potential in the area of energy research.
Results and discussion
The electrodeposition of metal and metal oxides is advantageous in that it can be carried out under ambient conditions at room temperature. Using this approach, materials can be produced rapidly in a controlled manner where the applied potential/current and electrolyte composition can be varied to produce films or isolated particles of different sizes and shapes.16,22,33–40 Co(OH)2 can be directly formed on the electrode surface under cathodic conditions using a straightforward protocol. The cyclic voltammogram recorded at a gold electrode in 10 mM Co(NO3)2·6H2O is shown in Fig. 1. The current cross-over seen for the forward and reverse scans is highly indicative of a nucleation and growth phenomenon.41 Interestingly, on the reverse scan it is noteworthy that there is only a minor peak seen at −0.45 V which indicates that the electrodeposited material is not oxidatively removed from the surface of the electrode as would be typical for many metal deposition/stripping systems.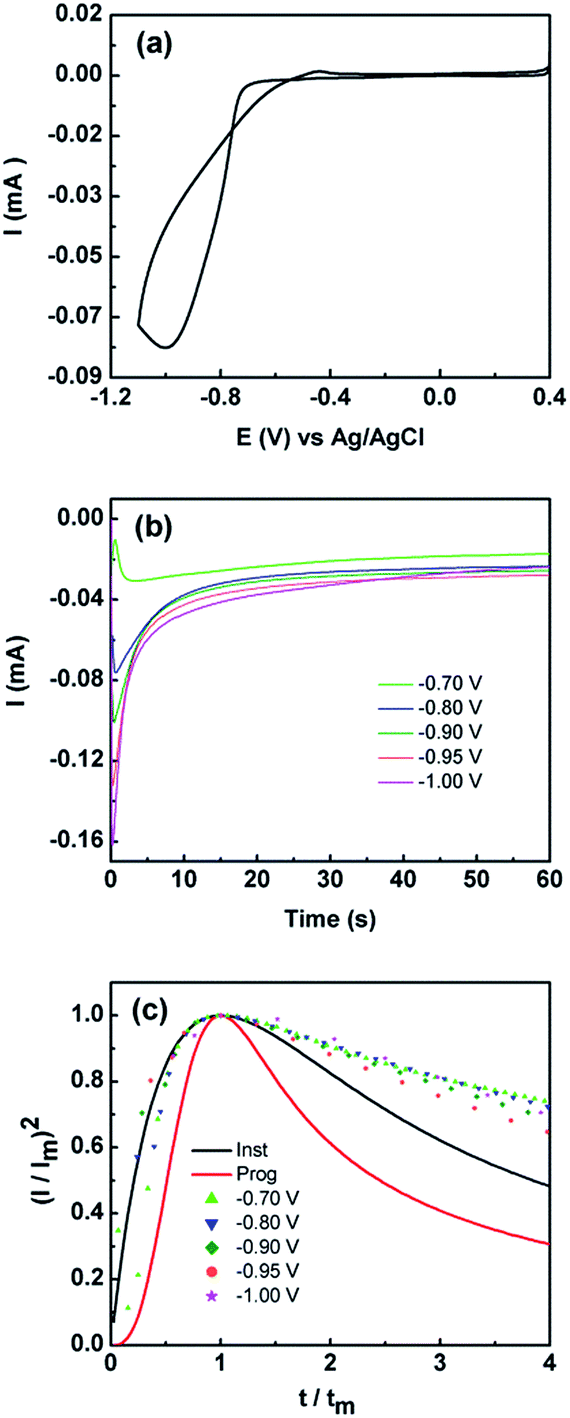 | ||
Fig. 1 (a) Cyclic voltammogram obtained at 50 mV s−1 at a Au electrode in 10 mM Co(NO3)2·6H2O solution (b) current–time curves obtained at different applied potentials and (c) non-dimensional plots of (I/Im)2vs. t/tm for the electrodeposition of Co(OH)2. Overlaid are the theoretical curves calculated using eqn (3) and (4) for progressive ( ) and instantaneous ( ) and instantaneous ( ) nucleation and diffusion limited growth. ) nucleation and diffusion limited growth. | ||
This may originate from two effects, either metallic Co is thermodynamically stable under the applied potentials in Fig. 1, which is highly unlikely, or an oxidised form of Co has been deposited. Given that the pH of the electroplating solution was measured to be 7.4 it is consistent with a mechanism whereby the local pH at the electrode solution interface is increased due to the reduction of water to liberate hydroxide ions which combine with Co2+ ions to precipitate Co(OH)2(s) on to the electrode surface via the following:42
| 2H2O + 2e− → 2OH−(aq) + H2(g) | (1) |
| Co(aq)2+ + 2OH−(aq) → Co(OH)2(s) | (2) |
The formation of Co(OH)2(s) was confirmed by Raman and XPS measurements and discussed below. Co(OH)2 has also been electrochemically produced via other protocols such as the reduction of an alkaline solution of tris(ethylenediamine)cobalt(III),16,17 from CoCl2 in 10% ethanol43 and electrochemical oxidation of a cobalt metal electrode at 100 V for 1.5 h in deionised water.44 However to date Co(OH)2 has been investigated mainly for its applicability in supercapacitor applications43,44 or as a precursor to the formation of CoOOH and Co3O4 materials for the OER.17
Given the current cross-over effect that was observed in Fig. 1 the nucleation-growth mechanism was analysed by performing current–time transients (Fig. 1b) and treating the data by the Hills–Scharifker method.45 This theory describes two limiting cases, i.e. progressive and instantaneous growth mechanisms, for the initial stages of 3D nucleation and 2D growth of deposited nuclei. These processes can be described by the following
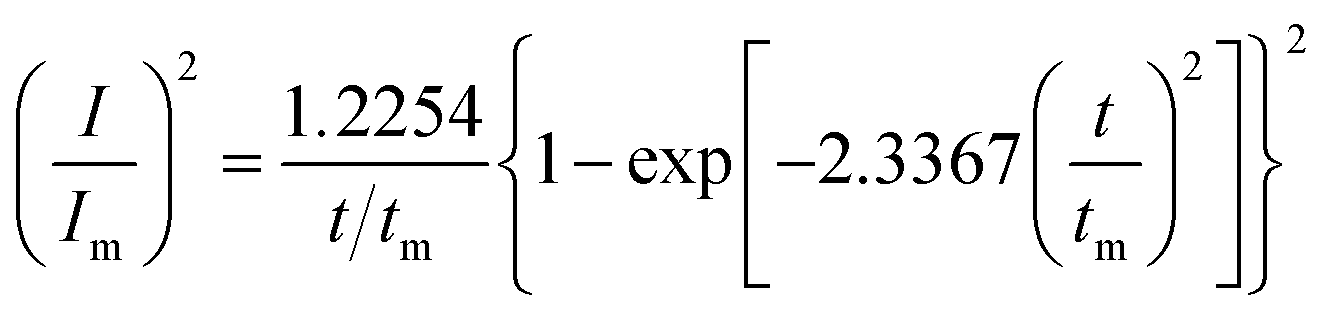 | (3) |
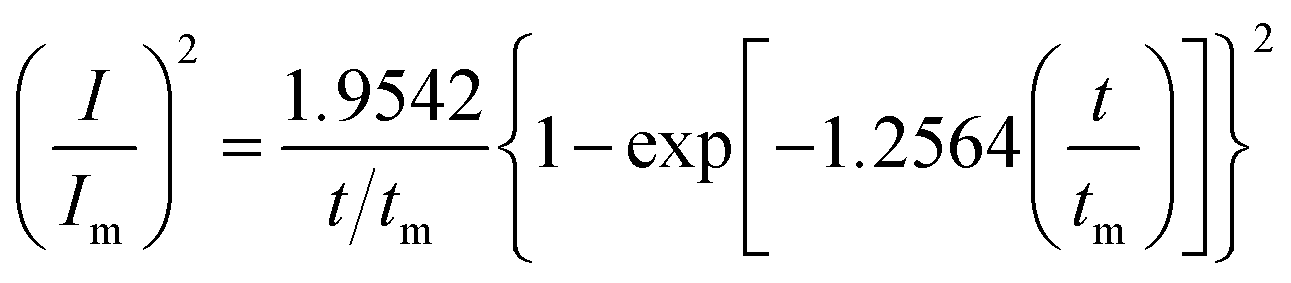 | (4) |
The reason for this becomes evident when the SEM images of the deposit are examined which indicate the formation of a layered sheet like morphology (Fig. 2) where there is some evidence of cracks within a top layer that is covering the underlying lamellar structures. The Hills–Scharifker model assumes the deposition of hemispherical nuclei which grow in a uniform manner which is clearly not the case here and is significantly different to the deposition of metals such as Cu, Au and Pt for instance. The formation of lamellar type structures via electrodeposition at −0.95 V for 60 s, as seen more evidently in Fig. 2b, is highly indicative of the formation of Co(OH)2.43,44
 | ||
| Fig. 2 (a and b) SEM images of Co(OH)2 electrodeposited on to a Au electrode at a potential of −0.95 V for 60 s in a solution of 10 mM Co(NO3)2·6H2O. | ||
This is verified by XPS data in Fig. 3a which shows the deconvoluted Co 2p3/2 spectrum which is consistent with previously reported spectra for Co(OH)2.44,46 The O 1s spectrum shows a single component at 531.5 eV which is indicative of bound hydroxide groups.44 Further evidence for the presence of the Co2+ oxidation state is seen in the expanded Co 2p spectrum (Fig. S1†) which clearly shows the presence of characteristic satellite peaks at 784.4 and 802.9 eV. The Raman data shown in Fig. 4 is indicative of Co(OH)2 (ref. 46) however there is a distinct peak at 194 cm−1 which could indicate some Co3O4 may have also formed.46 Taking the XPS data into consideration and its low penetration depth suggests that the top layer is Co(OH)2 with the possibility of some Co3O4 beneath this layer. The electrodeposited material is also amorphous as evidenced by the lack of any features in the XRD pattern (Fig. S2†). The preparation of an amorphous material may in fact be quite beneficial as amorphous Co(OH)2 has been reported to have superior properties over the crystalline form for capacitor applications due to an increased number of transportation channels through the material,44 which in principle would also be advantageous for electrocatalytic applications. This material is significantly different to Co(OH)2 generated electrochemically from an alkaline solution of tris(ethylenediamine)cobalt(III) where crystalline β-Co(OH)2 with a distinct disk-like morphology was formed. This material was found to be active for the OER, but no quantitative information with regards to overpotential or Tafel slope was provided.47 In a later publication β-Co(OH)2 was electrochemically converted to CoOOH at elevated temperatures and also to Co3O4via thermal annealing at 300 °C in air and studied for the OER but no direct comparison to the catalytic performance of β-Co(OH)2 was made.17
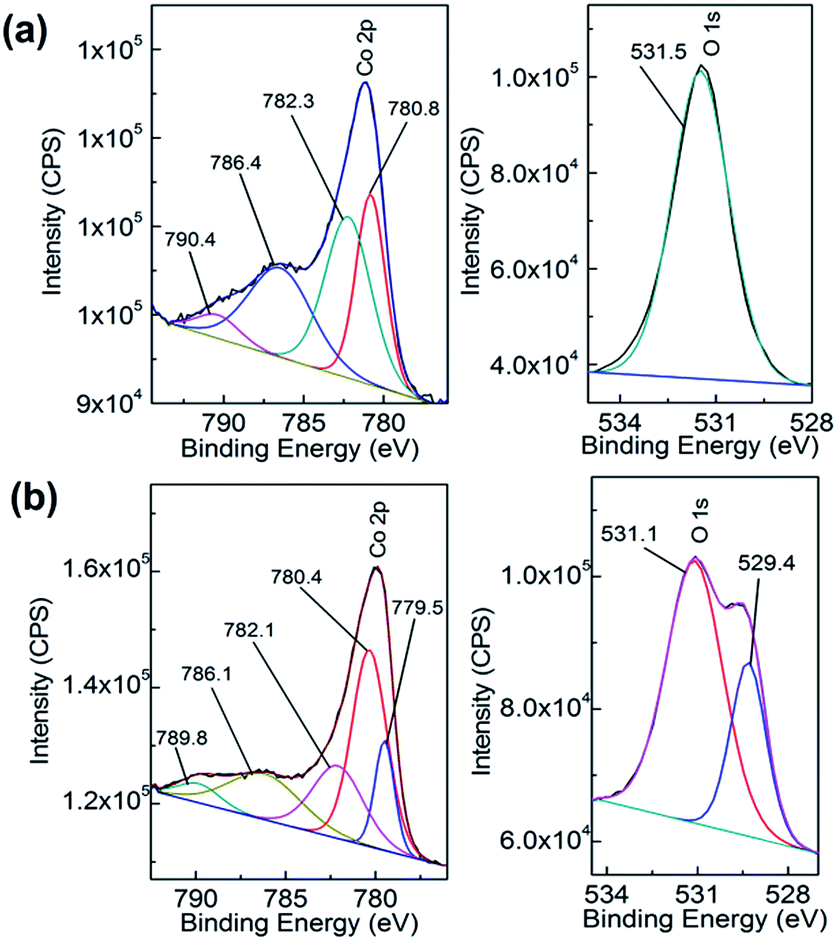 | ||
| Fig. 3 XPS spectra of Co 2p (left panel) and O 1s (right panel) of (a) as deposited Co(OH)2 on Au and (b) after 5 cycles into the OER region. | ||
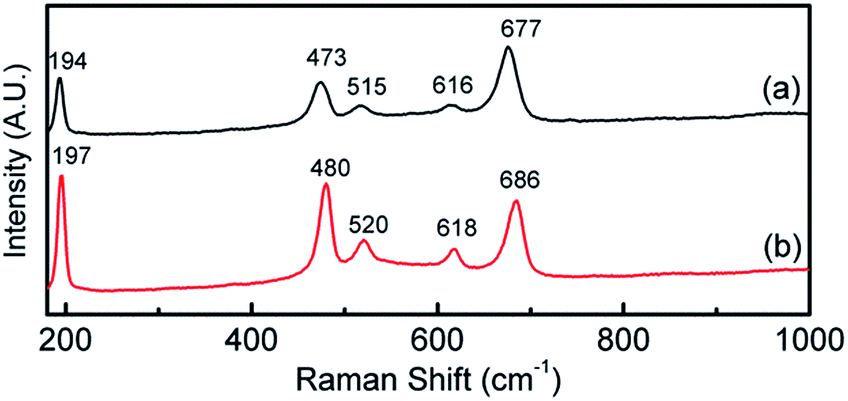 | ||
| Fig. 4 Raman spectra of (a) as deposited Co(OH)2 on Au and (b) after 5 cycles into the OER region. | ||
In this work, the OER was studied in 1 M NaOH to see if the electrodeposited amorphous Co(OH)2 was active. In comparison to Co(OH)2 electrodeposited on GC, Pd and Cu under identical conditions, the current associated with the OER from ca. 0.45 V until the end of the sweep is significantly higher at the Co(OH)2/Au electrode (Fig. 5a). It should be noted that the current has been normalised to the geometric area of the electrodes. On the first cycle it can be seen that there are two peaks at 0.97 and 1.06 V, which are attributed to oxidation of Co(OH)2via the following:
| 3Co(OH)2 + 2OH− → Co3O4 + 4H2O + 2e− | (5) |
| Co(OH)2 + OH− → CoOOH + H2O + e− | (6) |
| CoOOH + OH− → CoO2 + H2O + e− | (7) |
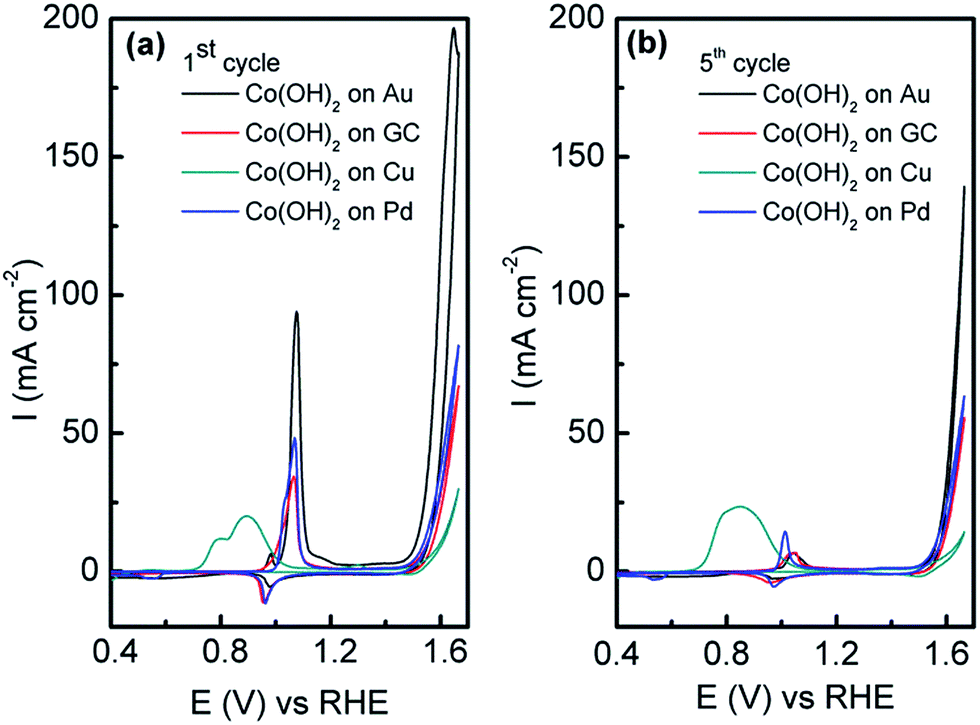 | ||
| Fig. 5 Cyclic voltammograms recorded at Co(OH)2 electrodeposited on Au and GC electrodes and an unmodified Au electrode in 1 M NaOH recorded at 50 mV s−1 showing (a) the first cycle and (b) the fifth cycle. | ||
However in reality the process is far more complicated and as discussed by Burke likely to involve the formation of hydrous oxides48 as well as species such as [Co(CoO)(CoOOH)·xH2O] as proposed by Gomez Meier.49 It is clear that there is only a minor reduction process on the reverse sweep indicating that irreversible oxidation of Co(OH)2 occurs. This is even more apparent on the 5th cycle (Fig. 5b) where the oxidation of Co(OH)2 is significantly reduced. For the Cu electrode there are additional processes in the anodic sweep from 0.70 to 1.00 V which are attributed to the oxidation of Cu to Cu2O and the oxidation of both Cu and Cu2O to a mixture of Cu(OH)2 and CuO.50 It is likely that such extensive oxidation of the surface with oxides of Cu inhibit the activity of the surface for the OER as it is known that only appreciable current densities can be achieved at copper oxides at overpotentials of ca. 0.60 V or greater.51 It should be pointed out that the charge associated with Co(OH)2 oxidation on Au is slightly greater than that observed on GC, Pd and Cu (Fig. 5a). This is quite surprising given that the charge passed during the electrodeposition process was 2.4, 4.2, 5.2 and 7.6 mC for Au, GC, Pd and Cu electrodes respectively (Fig. S3†). However the charge passed for the oxidation of Co(OH)2 on the 5th cycle is comparable (Fig. 5b), yet the OER current is significantly higher when deposited on Au compared to all other electrodes. This clearly demonstrates the benefit of electrodepositing this material on the surface of Au rather than carbon, Pd or Cu. The influence of morphology can be ruled out as Co(OH)2 electrodeposited on GC (Fig. S4†) under the same conditions has the same lamellar type structure as shown in Fig. 2 on Au. Therefore this is further evidence that there is a strong synergism between Co(OH)2 and Au that is facilitating the OER.
There is still much speculation on the active site involved in the OER but there is significant evidence for the participation of CoIV cations during the reaction which can be enhanced by the presence of Au (ref. 18) and is consistent with the data presented here in that the activity at the carbon, palladium and copper electrodes are lower in terms of current density. After 5 cycles of the potential the Au/Co(OH)2 electrode, as well as the other systems, maintains activity for the OER while there is a significant decrease in the processes associated with the oxidation of Co(OH)2 which is expected given the irreversible nature of this process as discussed above. The Au/Co(OH)2 electrode was then investigated via XPS (Fig. 3b) and Raman spectroscopy (Fig. 4) after 5 cycles of the potential into the OER region, where it was found that the composition had changed slightly. The XPS data indicated the likely formation of Co3O4 (Fig. 3b)46 where it can also be seen in Fig. S1† that the satellite peaks are more suppressed compared to the as-deposited material. The O 1S spectrum is also split into two components which is consistent with some Co3O4 formation. This would be expected under the conditions employed of cycling the electrode 5 times to an upper limit of 0.70 V.17 The Raman data is less convincing as there are only minor shifts in the peak positions which could indicate the presence of both Co(OH)2 and Co3O4 materials. Given that the penetration depth of XPS is ca. 10 nm indicates that the Co3O4 material that is formed is confined largely to the surface. SEM images of the surface after the OER (Fig. 6) showed that the surface morphology doesn't change significantly compared to the as-deposited material (Fig. 2). The bulk of the material also remains amorphous when the XRD data is considered as no distinct peaks were observed (Fig. S2b†). These data suggests therefore that there is not a large scale conversion of the as-deposited Co(OH)2 material into Co3O4, and that it is essentially limited to the surface. This is consistent with previous reports where it was found that for full conversion of Co(OH)2 into higher oxidation states required electrochemical oxidation at 95 °C via slow potential cycling at 1 mV s−1 over the region encompassing Co(OH)2 oxidation.17 However, a consequence of the OER performed here is that the lamellar structure becomes more defined and the overall structure is significantly more open and porous which can be attributed to the vigorous evolution of oxygen from the surface which is very visible by eye at a potential of 1.67 V.
 | ||
| Fig. 6 SEM images of electrodeposited Co(OH)2 on the Au electrode after the OER performed for 5 cycles under the conditions of Fig. 5. | ||
The Au/Co(OH)2 system was then optimised to attain the best possible OER performance and therefore the effect of deposition potential (Fig. S5†) was carried out where it was found that a potential of −0.95 V was the optimum value. When the SEM images are considered (Fig. S6†); films that are deposited at a less negative potential of −0.75 V show the distinctive lamellar type structures, however they are slightly patchy and the outer continuous type layer is absent as seen in Fig. 2. However after potential cycling experiments like those in Fig. 5, there is clear degradation of the film which explains the reduced activity. At more negative deposition potentials of −1.40 V, there is more extensive coverage on the electrode surface but the morphology is much less uniform than seen for the case at −0.95 V. This is also reflected in data obtained via linear sweep voltammetry for Co(OH)2 electrodeposited at various times which covers the potential range for Co(OH)2 oxidation only (Fig. S7†). There is an increase in the magnitude of the oxidation process when Co(OH)2 was electrodeposited at −0.75 to −0.95 V but this dramatically decreases at more negative potentials. This indicates that the amount of electrochemically active material is greatest when an applied electrodeposition potential of −0.95 V is used and hence gives the highest OER response. When a constant deposition potential of −0.95 V was used and the time was varied it was found that 60 s was the optimum time for OER activity (Fig. 7). The SEM images (Fig. S8†) show that at a lower time of 10 s that an open porous lamellar structured film is obtained that maintains its morphology after the OER. For 90 s deposition there is clear evidence of a continuous film covering the lamellar structures which is also cracked. After OER experiments the film is quite dense with less evidence of a porous morphology or lamellar structures seen for the optimum case of 60 s deposition. Therefore it appears that an open porous structure consisting of lamellar Co(OH)2 where the underlying Au electrode is accessible to a certain extent is the optimum condition for this electrode system. There is clearly a minimum coverage of Co(OH)2 however that is required as the much lower deposition times from 1–10 s were not as active. However it is quite significant that Co(OH)2 deposited for only 1 s shows quite good activity for the OER. This is consistent with previous work where multilayers of Co3O4 deposited on gold were less effective for the OER,18i.e. for gold to promote CoIV formation, the overlying active material should not be too thick to allow for effective electron transfer. Therefore maximising the active material/Au contact interface should be beneficial for the greatest activity. It should be noted that the Au electrode is not active for OER over the potential range of interest (Fig. S9†).
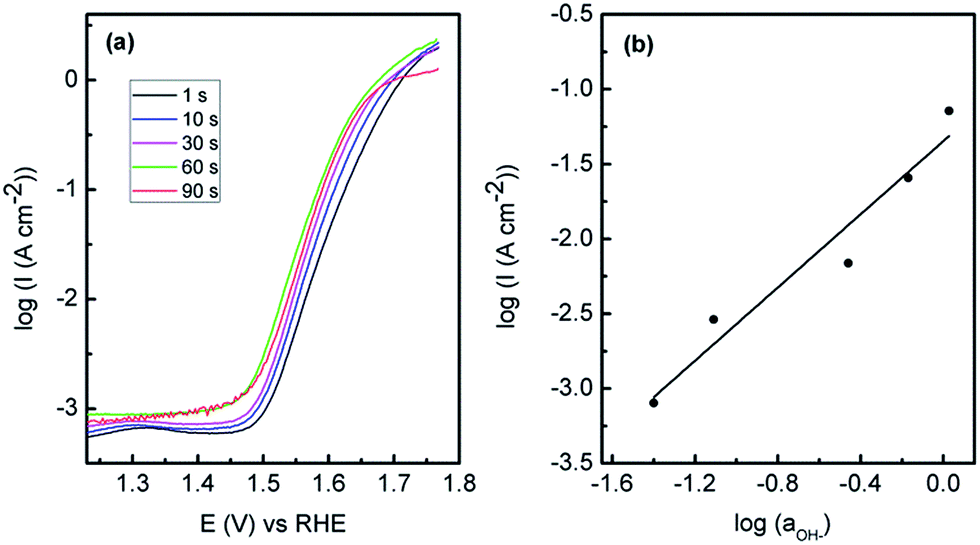 | ||
| Fig. 7 (a) Tafel plots showing OER performance for Au/Co(OH)2 electrodeposited from 10 mM Co(NO3)2·6H2O at different deposition time, (b) effect of hydroxide ion activity on OER. The cyclic voltammetric experiments used to obtain the data was recorded at a sweep rate of 1 mV s−1 in 1 M NaOH. | ||
This result is also consistent with the trend in OER activity for Co3O4 deposited on other electrodes such as Cu, Pt and Pd.18 The same trend is observed here in that the activity of Co(OH)2 follows the order of Au > Pd > Cu. This was explained in terms of the electronegativity of the underlying electrode in that Au being the most electronegative would facilitate the oxidation of CoII and CoIII into the CoIV active species to a greater extent than the less electronegative metals such as Pd and Cu. Previous density functional theory calculations have showed that the d-band centre of Co on Au shifts positively by 0.74 eV relative to pure Co. This higher d-band centre results in a stronger Co–O bond thereby making Co easier to oxidise.13 Evidence for this is shown in Fig. S10† where the oxidation of CoII to CoIII occurs as a distinct peak at the Au electrode at a lower potential compared to the other electrodes where broad shoulders are seen for this process prior to the main peak at ca. 1.06 V. Also the broad peak for oxidation into CoIV at ca. 1.37 V is more readily observable at Au compared to the other electrodes. The latter observation has also been reported by Zhao et al. for gold nanoparticles incorporated into mesoporous Co3O4 compared to mesoporous Co3O4 only.21 The rate limiting step for the OER has been postulated to be the reaction of OH− with adsorbed O atoms on the surface to form adsorbed OOH species. Once this species is formed it reacts with more OH− ions to give adsorbed O2 and H2O where finally O2 desorbs from the surface. The CoIV species enhances the electrophilicity of the adsorbed O atoms which via nucleophilic attack with OH− ions forms O–OH species. In addition CoIV has been postulated to aid in the deprotonation of these OOH species to form O2.18
Recently a significant insight into the OER on cobalt based materials has been reported by Strasser.32 In that study they found that crystalline Co3O4 is converted into X-ray amorphous CoOx(OH)y containing di-μ-oxo bridged Co3+/4+ ions during high rates of oxygen evolution. Once the material returns to non-catalytic conditions it recrystallizes to Co3O4 and this process was found to be reversible. Therefore we electrodeposited Co(OH)2 onto a gold coated TEM grid (2–3 nm thick) and investigated it for the OER. The TEM grid consists of gold islands dispersed over the carbon coated grid (Fig. 8a). Comparable OER activity to that of the Au electrode was found for Co(OH)2 electrodeposited on the Au TEM grid (Fig. 8b). When the surface was analysed it was found that Co(OH)2 was deposited to an equal extent over all areas of the grid and not exclusively to the areas with a high coverage of gold (Fig. S11†). Significantly it was found after the OER that some of the cobalt species was in fact crystalline in nature as shown in Fig. 8c and d. This crystalline material was found on both the gold and carbon rich areas of the grid. The lattice fringe with an interplanar spacing of 0.23 nm (Fig. 8d) is ascribed to the (111) plane of Au (ref. 52) but there are also lattice fringes with an interplanar spacing of 0.275 nm evident which can be indexed to Co3O4 species.24 This is in agreement with the XPS data recorded after the OER on the Au electrode (Fig. 3b) which showed the formation of Co3O4. Therefore in this case it appears that once Co(OH)2 partakes in the OER and CoIV species are created as the active material, they are subsequently converted to the more stable crystalline form upon return to non-catalytic conditions. As indicated by the GIXRD data (Fig. S2b†) this conversion is limited to the surface of the material which is in agreement with Strasser's observations.
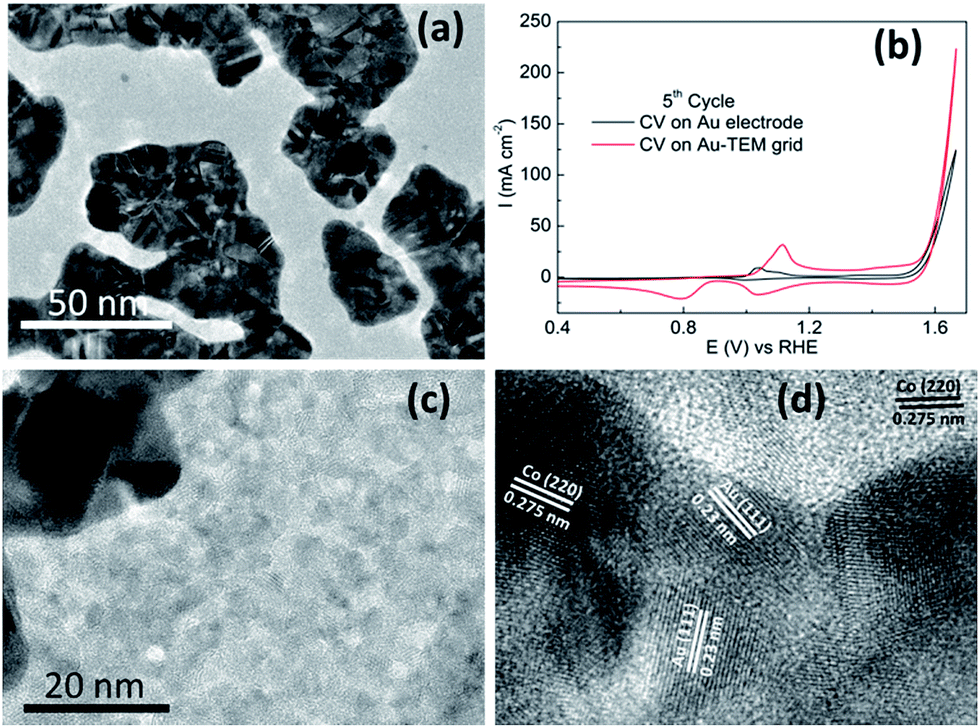 | ||
| Fig. 8 HR-TEM images of (a) pristine Au grid, (b) OER performance comparison; CV were recorded at 50 mV s−1 in 1 M NaOH, (c) Co(OH)2 deposited on Au support, (d) magnified HR-TEM images showing lattice spacing of Co3O4 and Au. | ||
For the optimised Au/Co(OH)2 system, the onset potential for the OER is 1.48 V. Significantly a current density of 10 mA cm−2 can be achieved at an overpotential (η) of 360 mV greater than the theoretical value of 1.23 V. From a Tafel plot analysis (Fig. 7a) the Tafel slope was found to be 56 mV dec−1 with an exchange current density (j0) of 1.8 × 10−10 A cm−2. It was also found that the Tafel slope (56 mV ± 4 mV) is independent of the electrodeposition time (calculated from Fig. 7a). The lower activity achieved for Co(OH)2 on the different electrodes is also reflected in the Tafel slopes of 59, 61 and 74 mV dec−1 recorded for C, Pd and Cu respectively. The Tafel slope value for the gold supported material is lower than that reported recently for Co3O4 supported on reduced graphene oxide (68 mV dec−1),14 where a lower Tafel slope is indicative of a more active electrocatalyst. The performance of this material is also comparable to a graphene–Co3O4 sandwich structure (10 mA cm−2 at η = 313 mV in 1 M NaOH with a Tafel slope of 56 mV dec−1),24 however the method of synthesis here is more facile compared to a multistep chemical synthesis including a calcination step followed by immobilisation onto an electrode surface secured via a Nafion coating. It is also comparable to the OER activity in 1 M NaOH for Co3O4 (Tafel slope of 61 mV dec−1, j0 = 6.0 × 10−9 A cm−2) and CoOOH (Tafel slope of 54 mV dec−1 and j0 = 1.2 × 10−10 A cm−2) formed via conversion of electrodeposited β-Co(OH)2,17 but again the approach taken in this study is advantageous in that only a simple one step room temperature deposition process is required. In comparison to a commercial catalyst such as IrOx, a current density of 10 mA cm−2 in 1 M NaOH can be achieved at an overpotential of 0.32 V (ref. 53) which shows the applicability of the Au/Co(OH)2 material. As discussed above, the Tafel slope at low overpotentials is 56 mV dec−1 and in this region a reaction order mOH− = 1.2 was determined (Fig. 7b). The Tafel slope changes to 122 mV dec−1 at higher overpotentials and taking the pH dependence into account is consistent with the analysis undertaken by Lyons et al. for an aged Co substrate that had been subjected to extensive potential cycling where an oxide layer had built up on the electrode surface.54 They proposed a dual barrier model to explain the change in the OER kinetic data whereby an inner anhydrous oxide layer is covered by a more dispersed hydrous oxide layer. Ionic transport through the inner layer is more difficult but charge percolation through the outer hydrous oxide layer is facile. The active site for the OER was proposed to be [Co(IV)Om(OH)n]p− (p = 2m + n − 4) rather than discrete CoO2 species described in eqn (7).
The turnover frequency (TOF), which is used extensively in the molecular catalysis area, can also be utilised for electrocatalytic reactions. The TOF is defined as the number of molecules reacting for a given reaction per unit time. As outlined by Lyons et al. the electrochemical equivalent of the TOF can be written as:55
| TOF = 1NA/4FNatoms = J/4Q | (8) |
The stability of the Au/Co(OH)2 material was then investigated over a period of 24 h where an overpotential of 440 mV was applied (Fig. 9). After the initial drop in the current density over the first hour, from 45 to 35 mA cm−2, there is only a gradual decrease in activity over the next 23 hours where a current density of 25 mA cm−2 was maintained. The noise in the trace is due to the evolution of oxygen bubbles from the surface of the electrode. This clearly indicates that a film deposited from a relatively dilute precursor metal salt for a period of only 60 s results in a highly robust material that is tolerant to quite robust oxygen evolution conditions.
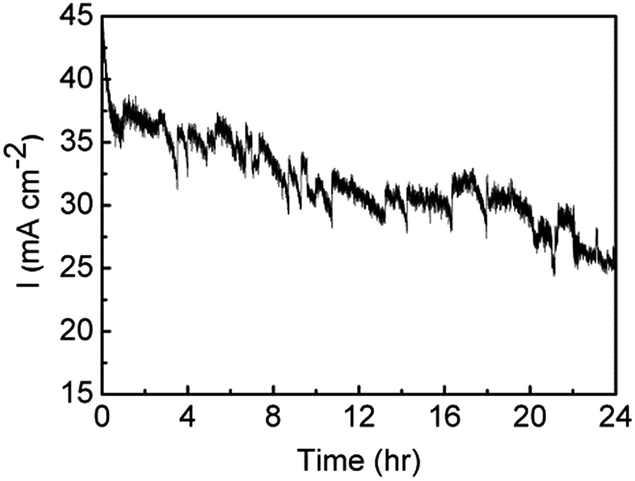 | ||
| Fig. 9 Stability measurements of Au/Co(OH)2 held at 1.67 V for 24 h in 1 M NaOH. | ||
Conclusions
The direct electrochemical formation of an active amorphous Co(OH)2 material has been reported that is highly active for the OER. The presence of an underlying gold electrode is critical to the performance of the catalyst and is consistent with previous work where the formation of the CoIV active site is benefited by the underlying Au electrode when compared to other metals such as Pd or Cu and follows the trend whereby increased electronegativity of the underlying metal increases OER activity. The morphology of the material can be controlled by the applied deposition potential and time where the optimum conditions was found to produce lamellar type structures that became open and porous during the course of the OER and exposed the underlying Au electrode, thereby maximising the cobalt oxide/Au interface. In addition there was evidence that the surface of the amorphous material was converted to crystalline Co3O4 once the OER was completed. The mechanism of catalysis was found to be consistent with a dual barrier model and gave excellent current densities at low overpotential with a TOF as high as 2.1 s−1. In addition this activity could be maintained for up to 24 h of continuous operation. This simple approach may be of benefit for other transition metal oxides that are also active for the OER.Experimental
Materials
Cobalt(II) nitrate hexahydrate (Chem-Supply), and sodium hydroxide (98%) (Sigma-Aldrich) were used as received from the suppliers and made up with deionised water (resistivity of 18.2 MΩ cm) purified by use of a Milli-Q reagent deioniser (Millipore).Electrochemical measurements
Electrochemical measurements were undertaken at (20 ± 2) °C with a BioLogic VSP workstation and a standard three-electrode cell configuration, consisting of a working electrode, a reference electrode and a counter electrode. The reference electrode was Ag/AgCl (3 M KCl) in all experiments. For voltammetric experiments, gold (1.6 mm diameter), glassy carbon (3 mm diameter), Pd (3 mm diameter) or copper (1.6 mm diameter) from Bioanalytical Systems were used as the working electrode, and a platinum wire as an auxiliary electrode. Prior to each deposition, the surface of each electrode was mechanically polished with 0.3 μm-sized alumina powder on a Microcloth pad and rinsed in Milli-Q water. These procedures were repeated several times. For all electrochemical experiments the electrolyte solution was purged for 10 min with nitrogen prior to performing any experiments to remove dissolved oxygen from the solution. The plating solution for the electrodeposition of Co oxide nanostructures consisted of 10 mM Co(NO3)2·6H2O. The electrodeposition of Co hydroxide nanostructures onto the electrodes was optimized by varying the potential and deposition time using the chronoamperometric technique. The electrodeposited films were washed with Milli-Q water several times ensuring no residual cobalt nitrate salt remained on the surface. The deposited film was then dried under a stream of nitrogen gas.For the OER the reproducibility of the measurements was achieved by carrying out five replicates for each experiment. In all cases iR correction was applied to all cyclic voltammograms and the solution was stirred at ca. 300 rpm using a magnetic stirrer at the bottom of the electrochemical cell. For the OER data the potential has been converted to the RHE scale via ERHE = EAg/AgCl + 0.059 × pH + 0.197 V. The current density reported in this work was normalized to the geometric surface area of the electrodes.
Physical measurements
Raman spectra were collected using an Olympus BHSM microscope, equipped with 10 and 50× objectives and part of a Renishaw 1000 Raman microscope system that is also equipped with a monochromator, a filler system, and a charge coupled device (CCD). Raman spectra were excited by a HeNe laser (633 nm) in the range between 100 and 1000 cm−1. Several acquisitions were used to improve the signal-to-noise ratio. X-ray photoelectron spectroscopy data were collected using an Omicron Multiscan Lab Ultra-high Vacuum Scanning Tunnelling Microscope (UHV-STM) incorporating a 125 mm hemispherical electron energy analyser. XPS measurements were performed using non-monochromatic Mg Kα (1253.6 eV) X-ray source (DAR 400, Omicron Nanotechnology), 300 W incident angle at 65° to the sample surface. Wide scans were observed at an analyser pass energy of 50 eV with 0.5 eV steps and 200 ms dwell time. Narrow high-resolution scans for Co 2p, O 1s, Au 4f, and C 1s were taken at 20 eV pass energy, 0.2 eV steps, 200 ms dwell time. The base pressure in the analysis chamber was 1.0 × 10−9 torr and 1.0 × 10−8 torr when the sample was analysed. Atomic compositions of cobalt oxide surface were calculated using the CasaXPS version 2.3.15 software and a linear baseline with Kratos library Relative Sensitivity Factors (RSFs). Sample preparations for XPS measurements were same with SEM sample preparations.SEM and EDX were performed on JEOL 7001F at an operating voltage of 5 KV and 15 KV, respectively. Samples were prepared by electrodeposition onto 100 nm thick Au coated silicon substrates following the same parameters used for the Au electrode (BAS). The surface area of the electrodeposited cobalt hydroxide film on the Au coated silicon substrates was carefully controlled using a mask (∼17 mm diameter). HRTEM images were taken using a JEOL 2100 instrument at 200 KV. A high-sensitivity silicon drift X-ray detector for more accurate compositional analysis and a Gatan Orius SC1000 CCD camera is equipped for better image acquisition. Ultrathin gold supported films on 3 mm standard TEM grids (Substratek™, TED PELLA) were used as the working electrode for the electrodeposition of Co(OH)2.
Acknowledgements
AOM gratefully acknowledges funding through a Future Fellowship from the Australian Research Council (FT110100760). The XPS and SEM data reported in this paper were obtained at the Central Analytical Research Facility operated by the Institute for Future Environments (QUT). Access to CARF is supported by generous funding from the Science and Engineering Faculty (QUT).Notes and references
- A. P. O'Mullane, Nanoscale, 2014, 6, 4012–4026 RSC.
- N. M. Markovic, Nat. Mater., 2013, 12, 101–102 CrossRef CAS PubMed.
- A. S. Bandarenka and M. T. M. Koper, J. Catal., 2013, 308, 11–24 CrossRef CAS.
- Electrocatalysis in Fuel Cells, ed. M. Shao, Springer, 2013 Search PubMed.
- A. Rabis, P. Rodriguez and T. J. Schmidt, ACS Catal., 2012, 2, 864–890 CrossRef CAS.
- S. E. F. Kleijn, S. C. S. Lai, M. T. M. Koper and P. R. Unwin, Angew. Chem., Int. Ed., 2014, 53, 3558–3586 CrossRef CAS PubMed.
- S. Weitemeyer, D. Kleinhans, T. Vogt and C. Agert, Renewable Energy, 2015, 75, 14–20 CrossRef.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- M. E. G. Lyons and R. L. Doyle, Int. J. Electrochem. Sci., 2012, 7, 9488–9501 CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737–13783 RSC.
- K. Kinoshita, Electrochemical Oxygen Technology, Wiley, 1992 Search PubMed.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- R. Ramsundar, J. Debgupta, V. Pillai and P. Joy, Electrocatalysis, 2015, 6, 331–340 CrossRef CAS.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- Y.-C. Liu, J. A. Koza and J. A. Switzer, Electrochim. Acta, 2014, 140, 359–365 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587–5593 CrossRef CAS PubMed.
- Y. Li, J. Zhao, Y. Dan, D. Ma, Y. Zhao, S. Hou, H. Lin and Z. Wang, Chem. Eng. J., 2011, 166, 428–434 CrossRef CAS.
- X. Deng, W. N. Schmidt and H. Tüysüz, Chem. Mater., 2014, 26, 6127–6134 CrossRef CAS.
- X. Lu, Y. H. Ng and C. Zhao, ChemSusChem, 2014, 7, 82–86 CrossRef CAS PubMed.
- B. H. R. Suryanto, X. Lu, H. M. Chan and C. Zhao, RSC Adv., 2013, 3, 20936–20942 RSC.
- X. Zhou, Z. Xia, Z. Tian, Y. Ma and Y. Qu, J. Mater. Chem. A, 2015, 3, 8107–8114 CAS.
- Y. Zhao, S. Chen, B. Sun, D. Su, X. Huang, H. Liu, Y. Yan, K. Sun and G. Wang, Sci. Rep., 2015, 5, 7629 CrossRef CAS PubMed.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55–68 CAS.
- B. J. Plowman, I. Najdovski, A. Pearson and A. P. O'Mullane, Faraday Discuss., 2013, 164, 199 RSC.
- W. Yu, M. D. Porosoff and J. G. Chen, Chem. Rev., 2012, 112, 5780–5817 CrossRef CAS PubMed.
- H. Wang and X. Ge, Electroanalysis, 2012, 24, 911–916 CrossRef CAS.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099–8139 RSC.
- A. Pearson and A. P. O'Mullane, Chem. Commun., 2015, 51, 11297–11300 RSC.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araujo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- Y. D. Gamburg and G. Zangari, Theory and Practice of Metal Electrodeposition, Springer, New York, 2011 Search PubMed.
- B. J. Plowman, S. K. Bhargava and A. P. O'Mullane, Analyst, 2011, 136, 5107–5119 RSC.
- A. Pearson and A. P. O'Mullane, Chem. Commun., 2015, 51, 5410–5413 RSC.
- R. Sivasubramanian and M. V. Sangaranarayanan, CrystEngComm, 2013, 15, 2052–2056 RSC.
- J. Elias, M. Gizowska, P. Brodard, R. Widmer, Y. deHazan, T. Graule, J. Michler and L. Philippe, Nanotechnology, 2012, 23, 255705 CrossRef PubMed.
- W. Simka, D. Puszczyk and G. Nawrat, Electrochim. Acta, 2009, 54, 5307–5319 CrossRef CAS.
- L. P. Bicelli, B. Bozzini, C. Mele and L. D'Urzo, Int. J. Electrochem. Sci., 2008, 4, 356–527 Search PubMed.
- P. M. Vereecken, R. A. Binstead, H. Deligianni and P. C. Andricacos, IBM J. Res. Dev., 2005, 49, 3–18 CrossRef CAS.
- A. R. Harris, A. K. Neufeld, A. P. O'Mullane, A. M. Bond and R. J. S. Morrison, J. Electrochem. Soc., 2005, 152, C577–C583 CrossRef CAS.
- E. M. Garcia, J. S. Santos, E. C. Pereira and M. B. J. G. Freitas, J. Power Sources, 2008, 185, 549–553 CrossRef CAS.
- C. Qian, T. Jie, S. Norio and Q. Lu-Chang, Sci. Technol. Adv. Mater., 2014, 15, 014206 CrossRef.
- H. B. Li, M. H. Yu, X. H. Lu, P. Liu, Y. Liang, J. Xiao, Y. X. Tong and G. W. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 745–749 CAS.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CAS.
- J. A. Koza, C. M. Hull, Y.-C. Liu and J. A. Switzer, Chem. Mater., 2013, 25, 1922–1926 CrossRef CAS.
- L. D. Burke, M. E. Lyons and O. J. Murphy, J. Electroanal. Chem. Interfacial Electrochem., 1982, 132, 247–261 CrossRef CAS.
- H. G. Meier, J. R. Vilche and A. J. Arvía, J. Electroanal. Chem. Interfacial Electrochem., 1982, 138, 367–379 CrossRef.
- A. Balkis and A. P. O'Mullane, Aus. J. Chem., 2015, 68, 1213–1220 CAS.
- X. Liu, H. Jia, Z. Sun, H. Chen, P. Xu and P. Du, Electrochem. Commun., 2014, 46, 1–4 CrossRef CAS.
- S. Singh, R. Pasricha, U. M. Bhatta, P. V. Satyam, M. Sastry and B. L. V. Prasad, J. Mater. Chem., 2007, 17, 1614–1619 RSC.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2010, 641, 119–130 CrossRef CAS.
- I. J. Godwin and M. E. G. Lyons, Electrochem. Commun., 2013, 32, 39–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS data for as-deposited films and films after OER, XRD data, chronoamperometry data recorded at Au and GC electrodes and SEM images showing the effect of deposition time and potential. See DOI: 10.1039/c5ta09125j |
| This journal is © The Royal Society of Chemistry 2016 |