
Molecular engineering of thiostrepton via single “base”-based mutagenesis to generate side ring-derived variants†
Panpan
Duan
a,
Qingfei
Zheng
a,
Zhi
Lin
a,
Shoufeng
Wang
a,
Dandan
Chen
b and
Wen
Liu
*ab
aState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: wliu@mail.sioc.ac.cn
bHuzhou Center of Bio-Synthetic Innovation, 1366 Hongfeng Road, Huzhou 313000, China
First published on 29th July 2016
Abstract
The quinaldic acid (QA) moiety in the side ring of thiostrepton (TSR), which can be modified regioselectively via precursor-directed mutational biosynthesis, was proven to be biologically relevant but tunable, affecting TSR's outstanding antibacterial activities. In this study, we sought to obtain TSR derivatives with varying amino acid residues connected to the QA moiety. The generation of these TSR derivatives relied on single “base”-based mutagenesis, and six new TSR-type compounds were obtained. Moreover, the simultaneous mutation of Ile1 and Ala2 in the TSR side ring resulted in a naturally occurring compound, siomycin (SIO), together with a new component, SIO-Dha2Ser. The anti-infection assays indicated that all of these new compounds could act as both antimicrobial agents and autophagy inducers, and these two kinds of activities can also be separated via regioselective modifications on the TSR side ring.
Among all the ribosomally synthesized and post-translationally modified peptide (RiPP) natural products, thiostrepton (TSR) possesses the most complex structure and elusive biosynthetic pathway.1 In addition, unlike most RiPPs, TSR has a side ring, of which the key building block, a quinaldic acid (QA) moiety, is biosynthesized independently of the precursor peptide.2 Benefiting from this unique biosynthetic logic, precursor-directed mutational biosynthesis has been conducted to generate side ring-modified TSR derivatives with varying QA moieties via the exogenous chemical feeding of synthetic QA analogs into the corresponding mutant strains.3 The resulting TSR variants show more potent biological activities than the parent compound and exhibit a novel dual mode of action against intracellular pathogens (e.g., Mycobacterium marinum).4 Moreover, other investigations have proven that the side ring of TSR plays significant roles in its antitumor activities and that excessive modification of the QA moiety abolishes TSR's biological activities.5 Previous studies have shown that changing the amino acid residue connected to the QA moiety affects the conformation of the TSR side ring.6 To regulate the chemical environment of the QA moiety and achieve rational control over TSR's activities, in this work, we sought to replace the Ile1 of TSR with other amino acid residues (Fig. 1).
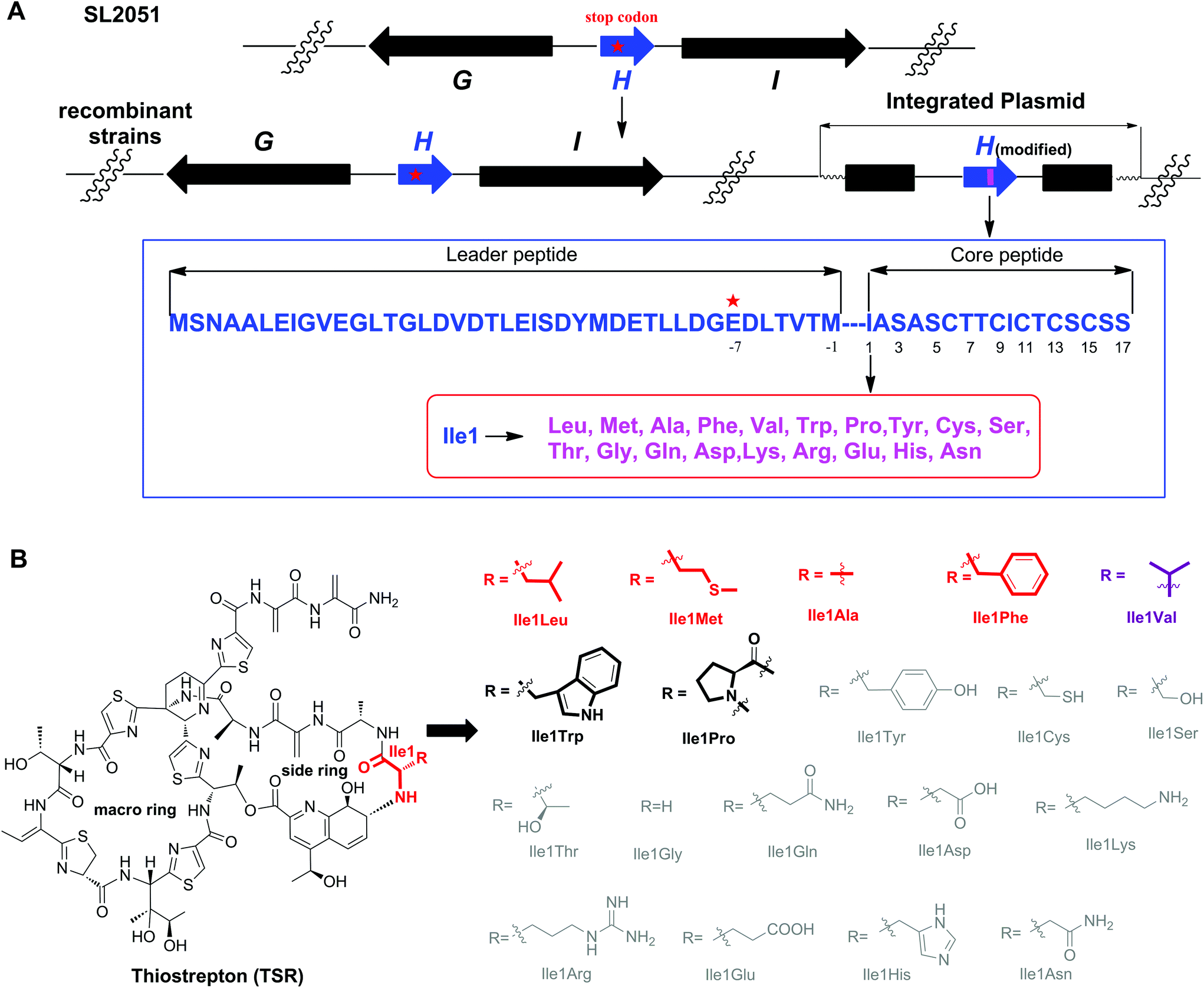 | ||
| Fig. 1 The single “base”-based mutagenesis strategy and the derivatives generated by site-directed mutagenesis of TsrH. (A) Genotypes of the SL2051 (by mutation of GAG for Glu-7 into the stop codon TAG) and recombinant strains with the corresponding encoded precursor peptides. The gene tsrH and post-translationally modified encoding genes are shown in blue and black, respectively. The position where the stop codon was introduced via an in situ single “base”-mutation is indicated by a red star, and the mutated position 1 is indicated by a pink bar. (B) Structures of TSR-Ile1X (X represents natural amino acids other than Ile). The amino acid residue is abbreviated using a three-letter code. The derivatives in gray are not produced. The derivatives with high yields are highlighted in red or purple (TSR-Ile1Val was reported previously), whereas those with low yields are highlighted in black. | ||
The complex structure of TSR poses great challenges for total chemical synthesis,7 however the relatively low genetic complexity associated with TSR biosynthesis makes it an excellent candidate for molecular engineering via synthetic biology strategies.8 In our previous studies, we found that the gene encoding the precursor peptide (tsrH) may serve as an important regulatory element in the gene cluster tsr and that the traditional method of gene deletion-in trans complementation cannot restore TSR production. Accordingly, in a previous study we developed a single “base”-based mutagenesis strategy to modify the TSR precursor peptide.9 Unlike previous methods, in this strategy, the target gene is inactivated by introducing a stop codon into the gene sequence instead of an in frame-gene deletion and, thereby, not disturbing the regulatory function of the open reading frame. Thus, TSR production can be restored, and the TSR derivatives can be generated via in trans complementation with wild-type or modified tsrH genes (Fig. 1).
We first conducted saturation mutagenesis to transform Ile1 into the other 19 natural amino acids (Fig. 1). In addition to the previously reported variant TSR-Ile1Val,9 high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS) and ultraviolet (UV) absorption analyses revealed that six new TSR-type compounds were produced when Ile1 was replaced with Leu, Phe, Ala, Met, Trp and Pro (Fig. 2 and ESI†). The structures of TSR-Ile1Leu, TSR-Ile1Phe, TSR-Ile1Ala and TSR-Ile1Met were determined by 1H, 13C and two-dimensional (2D) nuclear magnetic resonance (NMR) analyses (ESI†). Overall, the spectra of TSR-Ile1Leu, TSR-Ile1Phe, TSR-Ile1Ala and TSR-Ile1Met are similar to that of the parent compound TSR (ESI†). The analysis of their 1D and 2D NMR spectra revealed that the only difference existed in residue 1. The four new TSR analogs all clearly lacked the 1H NMR and 13C NMR signals of Ile1 in TSR. In TSR-Ile1Leu, the establishment of correlation spectroscopy (COSY; C6, C5, C4, C3 and C2) and heteronuclear multiple bond correlation (HMBC; H3-6CH3 to C4 and C3; H3-5CH3 to C4 and C3; and H-2CH to C3) further supports the substitution of a tert-butyl group at C2 in residue 1 in place of the isobutyl group in TSR (Fig. 2). This result is consistent with residue 1 in TSR-Ile1Leu being derived from Leu (rather than Ile in TSR). The structures of TSR-Ile1Phe, TSR-Ile1Ala and TSR-Ile1Met were further verified in the same way (Fig. 2), whereas the structures of TSR-Ile1Trp and TSR-Ile1Pro were determined by UV absorption and high-resolution mass spectrometry (HRMS) because of their low yields (ESI†).
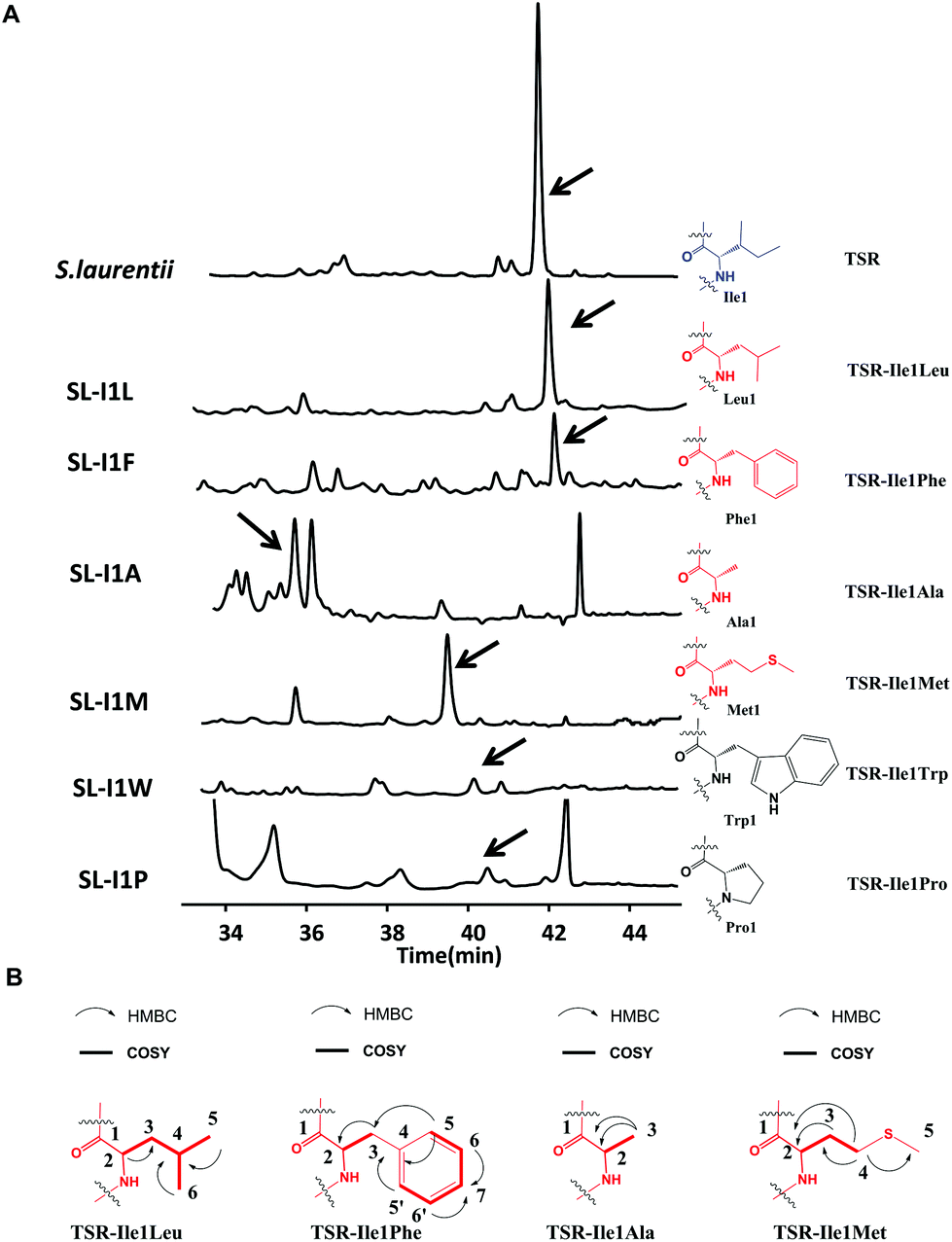 | ||
| Fig. 2 Production of new Ile1-derived TSR derivatives and their structural elucidation. (A) HPLC analyses of the fermentation cultures of the wild-type TSR-producing S. laurentii strain and the mutant strains SL-I1L, SL-I1F, SL-I1A, SL-I1 M, SL-I1 W, and SL-I1P. The resulting TSR and its analogs are indicated by arrows. TSR is shown in blue. The derivatives with high yields are highlighted in red, whereas those with low yields are highlighted in black. (B) Selected HMBC and COSY correlations for residue 1 in the derivatives with high yields. | ||
The successful engineering of TSR's Ile1 residue into six alternative amino acids suggested that this position is highly variable and encouraged us to extend the applications of the single “base”-based mutagenesis strategy. Siomycin (SIO), isolated from Streptomyces sioyaensis, is a naturally occurring TSR analog in which the 1st and 2nd residues in the side ring are Val1 and Dha2, respectively (rather than Ile1 and Ala2 in TSR; Fig. 3).10 SIO shows more potent activities against tumor cells and plasmodia than TSR.11 However, the fermentation yield of SIO is much lower than that of TSR, and the genetic manipulation and fermentation processes are more challenging for S. sioyaensis than for the TSR-producing strain S. laurentii.12 Thus, the simultaneous mutagenesis of TSR's Ile1 and Ala2 into Val1 and Dha2 was performed to produce SIO in the TSR-producing strain. Because the Dha2 in SIO is generated from Ser by a glutamate-dependent dehydratase during its biosynthesis,13 the codons for Ser and Val were introduced into the complementation gene to replace those for Ala2 and Ile1 in tsrH. HPLC and LC-MS analyses revealed that SIO can thus be produced by the engineered strain SL-I1V-A2S (Fig. 3), and an unexpected new component with a molecular weight of 1665.4 (18 Da larger than that of SIO) was also found (Fig. 3). 1H, 13C, 2D NMR and HRMS analyses confirmed the structure of SIO and revealed that this new component is a hydroxyl SIO analog, SIO-Dha2Ser (Fig. 3 and ESI†). The unexpected production of SIO-Dha2Ser most likely occurred because the glutamate-dependent dehydratase in the TSR biosynthetic system cannot accurately recognize or completely transform Ser2 in the core peptide. Meanwhile, the remarkable improvement of the SIO yield (up to 1.3 g L−1) showed that our engineering strategy may have great potential for applications in future synthetic biology research.
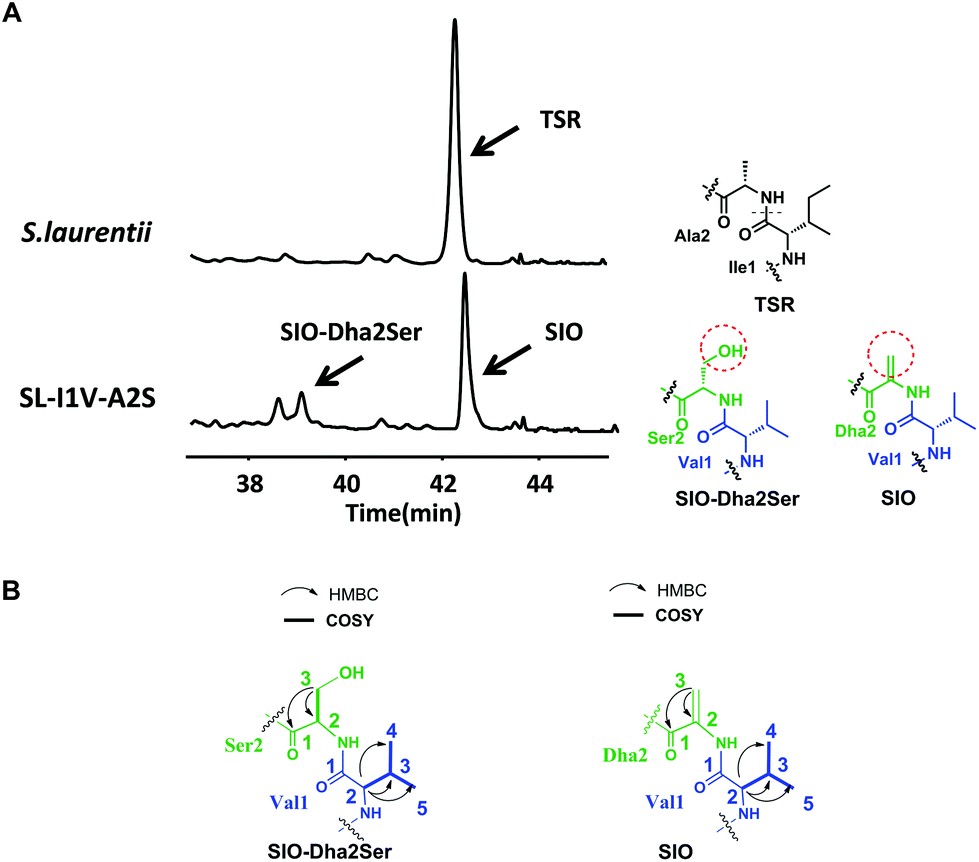 | ||
| Fig. 3 Production of SIO derivatives and their structural elucidation. (A) HPLC analyses of the fermentation cultures of the wild-type TSR-producing strain S. laurentii and the mutant strain SL-I1V-A2S. The resulting TSR and TSR analogs are indicated by arrows. Ile1 and Ala2 in TSR are indicated in black. Val1 in SIO and SIO-Dha2Ser is shown in blue, whereas Ser2 in SIO and Dha2 in SIO-Dha2Ser are presented in green. The differences between SIO and SIO-Dha2Ser are shown by a red dashed circle. (B) Selected HMBC and COSY correlations for residues 1 and 2 in the derivatives. | ||
To examine the antibacterial activities of these newly generated TSR-type antibiotics, the corresponding bioassays were performed to determine their minimum inhibitory concentrations (MICs). The results showed that all the Ile1-mutated derivatives possess equal or reduced in vitro bioactivities compared with TSR, whereas the derivative SIO-Dha2Ser lost the remarkable antibacterial activity of its parent compound SIO (Table 1). Thus, the naturally selected amino acid patterns of TSR and SIO appear to be the most advantageous in terms of antibacterial effects, and the replacement of the first and second residues in their side ring with the other naturally occurring amino acids may reduce their in vitro antibacterial activities. In our previous work, we discovered that TSR-type antibiotics can serve as both prokaryotic ribosome inhibitors and autophagy inducers.4a Thus, we detected the in vivo antibacterial activities of the aforementioned engineered compounds (Fig. 4A) and their autophagy-induction properties by detecting their capacities of activating the overexpression of LC3 and the transformation of LC3-I into LC3-II (Fig. 4B). Intriguingly, the results showed that although SIO-Dha2Ser does not possess outstanding in vitro antibacterial activities, it is a good autophagy inducer. Separating the bioactivities of SIO (i.e., ribosome inhibition and autophagy induction) by introducing a hydroxyl group onto Dha2 provides new insights into the complex structure–activity relationship (SAR) of TSR-type antibiotics.
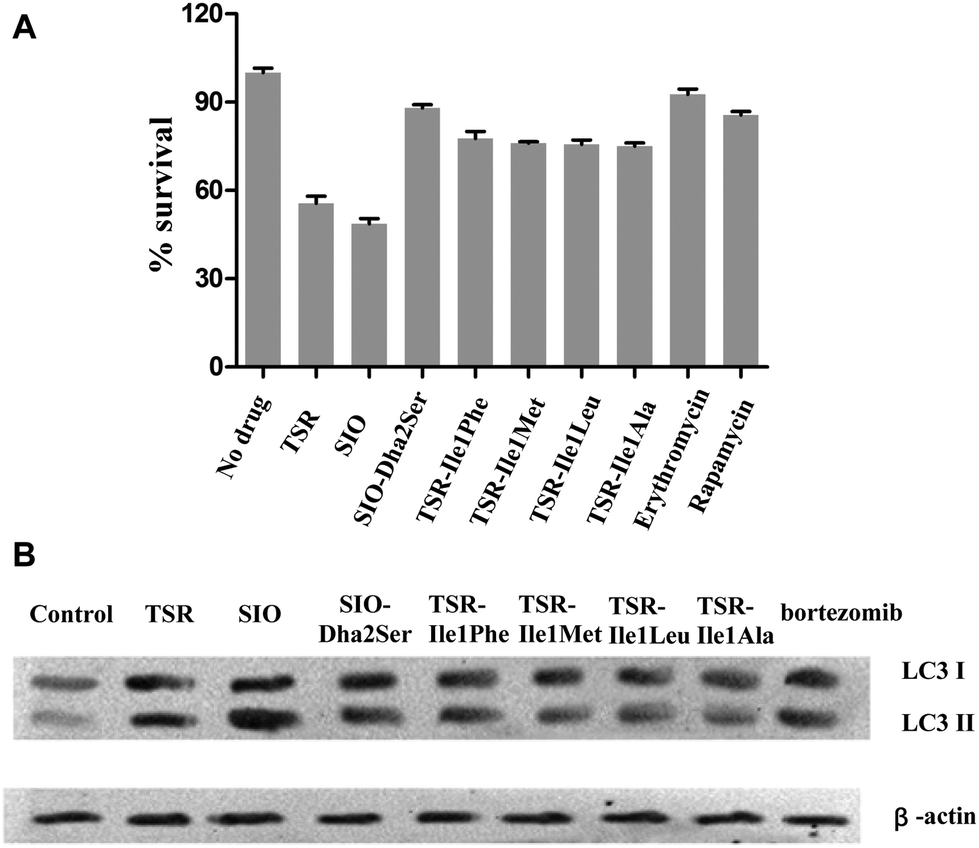 | ||
| Fig. 4 In vivo antibacterial activities of TSR and its analogs and their autophagy-induction properties. (A) Survival rates of pathogen colony-forming units (CFU) when the M. marinum-infected RAW 264.7 cells were treated with different compounds. Erythromycin (a typical ribosome inhibitor) and rapamycin (a typical autophagy inducer) were chosen as control drugs. The error bars represent the standard deviation from three different experiments. (B) LC3 conversion in the M. marinum-infected RAW 264.7 macrophage cell line treated with each drug (1 mM). β-Actin was chosen as an internal control biomarker, and a clinically used proteasome inhibitor and autophagy inducer, bortezomib, was chosen as a control drug. | ||
| TSR | SIO | SIO-Dha2Ser | TSR-Ile1Phe | TSR-Ile1Met | TSR-Ile1Leu | TSR-Ile1Ala | VAN | INH | |
|---|---|---|---|---|---|---|---|---|---|
| Streptococcus pneumoniae PRSP1063a | 0.001 | 0.001 | 0.5 | 0.125 | 0.25 | 0.125 | 0.5 | 0.25 | — |
| Streptococcus pneumoniae PRSP2831a | 0.001 | 0.002 | 1 | 0.125 | 0.25 | 0.25 | 0.5 | 0.25 | — |
| Streptococcus pneumoniae PRSP224588a | 0.008 | 0.004 | 1 | 0.25 | 1 | 0.5 | 0.25 | 0.25 | — |
| Staphylococcus aureus MRSA-S1a | 0.032 | 0.032 | >1 | 0.25 | 0.125 | 0.25 | 0.5 | 0.5 | — |
| Staphylococcus aureus MRSA-SAU3a | 0.064 | 0.064 | >1 | 0.25 | 0.125 | 0.25 | 0.25 | 0.5 | — |
| Staphylococcus aureus MRSA-SAU5a | 0.064 | 0.064 | >1 | 0.5 | 0.5 | 1 | 0.5 | 1.0 | — |
| Enterococcus faecium VRE3a | 0.032 | 0.016 | >1 | 0.25 | 1 | 0.25 | 0.5 | >256 | — |
| Enterococcus faecium VRE73a | 0.064 | 0.032 | >1 | 0.125 | 0.5 | 0.5 | 0.25 | 256 | — |
| Enterococcus faecium VRE83a | 0.064 | 0.064 | >1 | 0.25 | 0.25 | 0.25 | 1 | >256 | — |
| Mycobacterium. marinum b | 0.5 | 0.5 | >8 | 8 | 4 | 2 | 2 | — | 25 |
Conclusions
In conclusion, by utilizing a single “base”-based mutagenesis strategy, we engineered the Ile1 residue of TSR into other amino acids. The replacement of Ile1 with six different amino acids was tolerated by the TSR biosynthetic system and resulted in six new side ring-derived TSR variants. Moreover, the simultaneous mutagenesis of TSR's Ile1 and Ala2 residues allowed SIO to be produced in the original TSR-producing strain. An unpredicted SIO derivative, SIO-Dha2Ser, was also found in this engineered system. The detection of the in vivo and in vitro antibacterial activities of the compounds studied here facilitates the understanding of the complex SAR of TSR-type antibiotics and provides insights into their bioactivity-separation.Acknowledgements
This work was supported in part by grants from the NSFC (31430005 and 91413101), STCSM (13XD1404500 and 14JC1407700), “973 program” (2012CB721100), and MST (2012AA02A706) of China to Prof. Wen Liu. This research was also funded in part by NSFC (81402831) to Dr Dandan Chen. We also thank Prof. Heinz G. Floss, University of Washington, for providing the TSR-producing strain S. laurentii ATCC 31255.Notes and references
- (a) P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108 RSC; (b) Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218 RSC.
- (a) R. Liao, L. Duan, C. Lei, H. Pan, Y. Ding, Q. Zhang, D. Chen, B. Shen, Y. Yu and W. Liu, Chem. Biol., 2009, 16, 141 CrossRef CAS PubMed; (b) L. Duan, S. Wang, R. Liao and W. Liu, Chem. Biol., 2012, 19, 443 CrossRef CAS PubMed.
- (a) S. Wang, Q. Zheng, J. Wang, Z. Zhao, Q. Li, Y. Yu, R. Wang and W. Liu, Org. Chem. Front., 2015, 2, 106 RSC; (b) S. Wang, Q. Zheng, J. Wang, D. Chen, Y. Yu and W. Liu, Org. Chem. Front., 2016, 3, 496 RSC.
- (a) Q. Zheng, Q. Wang, S. Wang, J. Wu, Q. Gao and W. Liu, Chem. Biol., 2015, 22, 1002 CrossRef CAS PubMed; (b) Q. Zheng and W. Liu, Mycobact. Dis., 2016, 6, 203 Search PubMed.
- N. S. Hegde, D. A. Sanders, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2011, 3, 725 CrossRef CAS PubMed.
- F. Zhang, C. Li and W. L. Kelly, ACS Chem. Biol., 2016, 11, 415 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, B. S. Safina, M. Zak, A. A. Estrada and S. H. Lee, Angew. Chem., Int. Ed., 2004, 43, 5087 CrossRef CAS PubMed; (b) K. C. Nicolaou, M. Zak, B. S. Safina, S. H. Lee and A. A. Estrada, Angew. Chem., Int. Ed., 2004, 43, 5092 CrossRef CAS PubMed.
- M. A. Ortega and W. A. van der Donk, Cell Chem. Biol., 2016, 23, 31 CrossRef CAS PubMed.
- H. Guo, J. Wang, Y. Li, Y. Yu, Q. Zheng, J. Wu and W. Liu, Chem. Sci., 2014, 5, 240 RSC.
- (a) H. Nishimura, S. Okamoto, M. Mayama, H. Ohtsuka, K. Nakajima, K. Tawara, M. Shimohira and N. Shimaoka, J. Antibiot., 1961, 14, 255 CAS; (b) K. Tori, K. Tokura, Y. Yoshimura, K. Okabe, H. Otsuka, F. Inagski and T. Miyazawa, J. Antibiot., 1979, 32, 1072 CrossRef CAS PubMed; (c) N. J. Clayden, F. Inagaki, R. J. Williams, G. A. Morris, K. Tori, K. Tokura and T. Miyazawa, Eur. J. Biochem., 1982, 123, 127 CrossRef CAS PubMed.
- (a) U. G. Bhat, P. A. Zipfel, D. S. Tyler and A. L. Gartel, Cell Cycle, 2008, 7, 1851 CrossRef CAS PubMed; (b) U. G. Bhat, M. Halasi and A. L. Gartel, PLoS One, 2009, 4, e6593 Search PubMed; (c) U. G. Bhat, M. Halasi and A. L. Gartel, PLoS One, 2009, 4, e5592 Search PubMed; (d) M. Halasi, H. Zhao, H. Dahari, U. G. Bhat, E. B. Gonzalez, A. V. Lyubimo, D. A. Tonetti and A. L. Gartel, Cell Cycle, 2010, 9, 1214 CrossRef CAS PubMed.
- S. Yagi, S. Kitai and T. Kimura, Appl. Microbiol., 1971, 22, 153 CAS.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 16012 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00320f |
| This journal is © the Partner Organisations 2016 |