
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The rise of organic electrode materials for energy storage
Tyler B.
Schon
,
Bryony T.
McAllister
,
Peng-Fei
Li
and
Dwight S.
Seferos
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario, M5S 3H6 Canada. E-mail: dseferos@chem.utoronto.ca
First published on 7th June 2016
Abstract
Organic electrode materials are very attractive for electrochemical energy storage devices because they can be flexible, lightweight, low cost, benign to the environment, and used in a variety of device architectures. They are not mere alternatives to more traditional energy storage materials, rather, they have the potential to lead to disruptive technologies. Although organic electrode materials for energy storage have progressed in recent years, there are still significant challenges to overcome before reaching large-scale commercialization. This review provides an overview of energy storage systems as a whole, the metrics that are used to quantify the performance of electrodes, recent strategies that have been investigated to overcome the challenges associated with organic electrode materials, and the use of computational chemistry to design and study new materials and their properties. Design strategies are examined to overcome issues with capacity/capacitance, device voltage, rate capability, and cycling stability in order to guide future work in the area. The use of low cost materials is highlighted as a direction towards commercial realization.
 Tyler B. Schon | Tyler Schon received his BSc in chemistry from the University of Western Ontario in 2012. He is currently enrolled in the PhD program in the department of chemistry at the University of Toronto under the supervision of Prof. Seferos. His research is focused on developing polymeric organic electrodes for energy storage devices, both supercapacitors and batteries. In his time outside the lab, Tyler enjoys stereotypical Canadian pastimes such as playing hockey, and camping. |
 Bryony T. McAllister | Bryony McAllister obtained her BSc in chemistry from the University of Victoria in 2015. She is currently pursuing a doctoral degree under the supervision of Prof. Seferos at the University of Toronto. Presently, her research focuses on the development of novel polymers for supercapacitor applications. As a BC native, Bryony is an avid hiker and has hiked the West Coast and Juan de Fuca trails. In her spare time, she enjoys exploring Toronto on bike. |
 Peng-Fei Li | Peng-Fei Li obtained his PhD under the supervision of Prof. Chuan-Feng Chen from the Institute of Chemistry, the Chinese Academy of Sciences in 2013. Afterwards, he carried out postdoctoral research in Prof. Seferos group in University of Toronto. His current research interest focuses on porous materials for optoelectronic and biological applications. |
 Dwight S. Seferos | Dwight Seferos' Research Group develops new organic materials for use in electronics, catalysis, and biomedical applications. Some of our work is focused on energy harvesting and energy storage applications. Dwight has been honored with many national and international awards including a DuPont Young Investigator Award (2011), Canada Research Chair (2012), A.P. Sloan Fellowship (2013), and NSERC Discovery Accelerator Award (2015). Dwight currently serves on the Editorial Advisory Board of ACS journals: Macromolecules and Chemistry of Materials. |
1. Introduction
Energy storage is imperative for the integration of intermittent renewable power sources (solar, wind, tidal) into the grid, the widespread adoption of electric vehicles, and the continued development of portable electronics. Electrochemical energy storage system (EESS) applications are growing enormously on multiple scales, from smart card microbatteries, to large-scale electric vehicle batteries, and warehouse-sized redox flow batteries (RFBs). While much progress has been made, it is clear that higher performing, more versatile, smaller, lighter, and, most importantly, more economically viable energy storage solutions will be required in the future.1The materials used for EESSs are traditionally metal-based inorganic compounds, such as cobalt, iron, tin, or manganese-based materials for lithium-ion battery electrodes and vanadium oxides for redox flow batteries. These inorganic materials rely on changes in metal oxidation state for charge storage and a concomitant balancing of the charged structure with specific counter-ions. In many cases the counter-ion is specific to the crystal structure of the inorganic compound due to size restrictions in the crystal lattice, ionic conductivity, and reversibility of the redox reaction. This inherently restricts the versatility of inorganic compounds, where the same cathode material cannot be used for different series of alkali metal batteries such as lithium and sodium-ion. One of the biggest challenges for inorganic complexes is that they typically require extraction and synthesis techniques that are harmful to the environment. Extraction can release toxic materials that are otherwise trapped underground. Synthesis can create large amounts of heavy metal waste and often requires energy intensive processing. In order to realize the predicted widespread use of EESSs, these challenges must be overcome.
Organic materials provide an excellent opportunity to further improve existing energy storage technologies, and a versatile platform to develop novel EESSs. Organic materials are abundant, relatively inexpensive, and their synthesis can be designed so that it is not energy intensive and produces minimal waste.2 Organic compounds are also structurally diverse, able to be functionalized with relative ease through many synthetic methods. This allows one to tune oxidation and reduction potentials to optimize the operating voltages of EESSs. Modifying the chemical structure is an ideal way to suit the needs of specific applications by changing capacity, solubility, crystal structure, electron transfer rates, ionic conductivity, and mechanical properties. Organic materials are not typically restricted by choice of counter-ion. This means that, to a certain extent, the same organic material can be useful for a wide variety of different energy storage devices such as lithium-ion, sodium-ion, multivalent-ion, and dual-ion batteries.
Organic materials have been studied as electrodes for EESSs since 1969, with the first report of an organic cathode material using dichloroisocyanuric acid.3 After this, multiple research groups tested a variety of organic small molecules such as quinones,4 dianhydrides,5 and phthalocyanines.6 Poly(acetylene)s7 were initially tested as a cathode material, followed by many other conjugated polymers such as polypyrrole, polythiophene, polyaniline, and derivatives thereof.8,9 Research on organic electrode materials for energy storage faded when inorganic transition metal complexes were developed that reversibly intercalate lithium ions at high potentials with high capacity. In the past decade, however, research on organic electrode materials has been reinvigorated due to the increased demand for energy storage that is not only high performing but also inexpensive.
Here we provide an overview on the use of organic electrode materials for EESSs. Our goal is to highlight recent work relating to the development and improvement of organic electrode materials. We have limited our scope to materials that are used to store charge, as there have been a number of excellent reviews and progress reports on organic materials used in other aspects of EESSs including electrolytes, membranes, and binders.10–12 We have also excluded hybrid materials with inorganic compounds including organic–inorganic composites and organometallic compounds where the inherent redox chemistry of the metal is responsible for the charge storage. These types of composites and formulations are undoubtedly important to the field of EESSs, however, in order to examine structure–property relationships and effects of processing, we focus on purely organic materials.12,13 We have separated each EESS based on their architecture and electrochemical characteristics, which will be discussed in the working principles section. We also provide guidelines for future development in the field by reviewing the important metrics associated with individual electrodes and how each metric relates to the performance in devices as a whole.14–17 We will finally consider the use of computational chemistry for the design and understanding of these important materials.
2. Types of EESSs and their working principles
The working mechanism of any EESS relies on an inherent potential difference between two electrodes known as the operating voltage. The operating voltage of the device is dictated by the differences in redox potential of the positive and negative electrode. The potential difference is used to drive electrochemical reactions on either electrode when they are connected through an external circuit. This creates a flow of electrons from the negative electrode to the positive electrode. The flow of electrons induces oxidation reactions on the negative electrode (anode) and reduction reactions on the positive electrode (cathode) when discharging. The charged electrodes are balanced by a concomitant flow of counter-ions. EESSs are grouped into a number of different categories depending on the composition of the electrodes, the counter-ions, and the nature of the redox reactions (Fig. 1).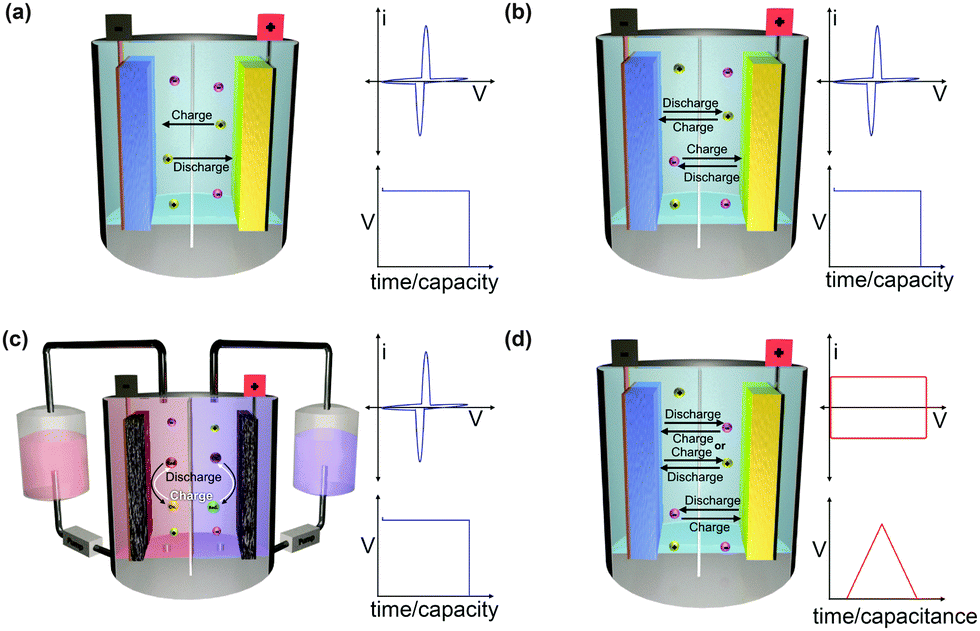 | ||
| Fig. 1 Schematic depicting the working principle of (a) metal-ion batteries, (b) dual-ion batteries, (c) redox flow batteries, and (d) supercapacitors. The insets show the ideal electrochemical behaviour of each type of device when measured using cyclic voltammetry and galvanostatic charging/discharging. Here, V is voltage and i is current. | ||
2.1 Solid electrode batteries
Metal-ion batteries are the most common type of EESSs. They are typically composed of an anode (negative electrode), a cathode (positive electrode), electrolyte (either aqueous, organic, solid-state,18 or polymeric10,19), a separator (to prevent short circuiting), current collectors (to collect charge at each electrode), and a cell casing (to keep the components together and prevent exposure to the external environment). Metal-ion batteries are used for a wide variety of both portable and stationary applications for either primary or back-up power. In metal-ion batteries the charged anodes and cathodes are balanced by the metal ion in a ‘rocking-chair’ type mechanism (Fig. 1a). This is a strict requirement imposed by the definition of metal-ion batteries that should be clearly distinct from dual-ion batteries described below. Metal-ion batteries can be constructed with relatively small amounts of electrolyte because the ions balancing the charge at one electrode are constantly being replenished. Additionally, metal-ion batteries are very attractive candidates for use with solid-state electrolytes because the mobility of only one ion needs to be considered.
Metal–sulfur batteries are a relatively new subset of metal-ion batteries that use (organo)sulfur as an electrode. This is advantageous because sulfur is both inexpensive and has a high theoretical capacity (Ctheor) of 1672 mA h g−1. Metal–sulfur batteries operate by the same mechanism as metal-ion batteries where both electrodes are balanced by metal-ions upon charging (discharging). The electrode that is not composed of sulfur can be composed of a variety of materials, as long as it is balanced by metal-ions in its charged or discharged state. This technology is still in development, but typically the cathode is composed of sulfur impregnated into conductive carbon in order to prevent polysulfide dissolution, which can eventually deplete the capacity of the device.
Metal–air batteries are the newest type of metal-ion battery. Here, the anode can be a number of different materials but the cathode is typically composed of a conductive carbon support with a high surface area that is impregnated with an oxygen reduction/oxidation catalyst. The cathode is exposed to either pure oxygen or ambient air. Oxygen diffuses to the cathode and is reduced to either its alkali metal superoxide or peroxide, the exact species being highly dependent on the metal-ion used.20 The Ctheor of reducing oxygen to peroxide provides a maximum capacity of 1168 mA h g−1 (Li2O2) with a higher voltage than metal–sulfur batteries (2.15 vs. 2.96 V vs. Li/Li+) allowing metal–air batteries to have a much greater energy density (up to ∼3500 W h kg−1) based on the mass of lithium and oxygen alone.21
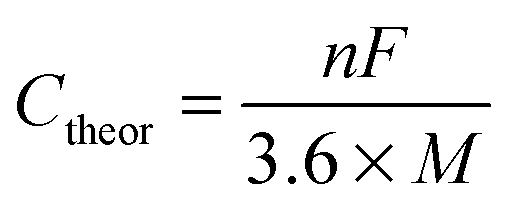 | (1) |
| Structure | # | C theor (mA h g−1) | Electrode composition | Electrolyte | Capacity (mA h g−1), current | Potential: oxidationa/reduction (V), reference | Cycling stability: retention, cycles, current | Ref. |
|---|---|---|---|---|---|---|---|---|
n.r. denotes a value not reported.a If the oxidation potential is not only the reduction potential(s) is listed.b The voltage range specified has a significant sloping voltage profile.c The potential reported is for the first reduction.d The theoretical capacity reported is based on the corresponding molecule accepting 2 electrons.e The theoretical capacity reported is based on the corresponding molecule accepting 4 electrons.f The theoretical capacity reported is determined only for the redox-active group.g The theoretical capacity reported is calculated based on the sulfur content.h The theoretical capacity reported is based on the corresponding repeat unit accepting 6 electrons.i The theoretical capacity reported is based on the corresponding molecule accepting 1 charge per 4 monomer units.j The theoretical capacity reported is based on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid. The abbreviations used in the table are defined as: PVdF = poly(vinylidenefluoride); EC = ethylene carbonate; DEC = diethyl carbonate; DMC = dimethyl carbonate; CB = carbon black; LiTFSI = bis(trifluoromethane)sulfonamide; DOL = dioxolane; EMC = ethyl methyl carbonate; PTFE = poly(tetrafluoroethylene); VGCF = vapor-grown carbon fibers; EiPS = ethyl isopropyl sulfone; CPE = composite polymer electrolyte; PMA = poly(methacrylate); PEO = poly(ethylene oxide); PC = propylene carbonate; TEGDME = tetraethylene glycol dimethyl ether; DEGDME = diethylene glycol dimethyl ether; NaTFSI = sodium bis(trifluoromethane)sulfonamide; BC = butylene carbonate; MWCNT = multiwall carbon nanotubes; EMIC = 1-ethyl-3-methylimidazolium chloride; SBR = styrene-butadiene rubber; CMC = carboxymethyl cellulose. :1 ratio of 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid. The abbreviations used in the table are defined as: PVdF = poly(vinylidenefluoride); EC = ethylene carbonate; DEC = diethyl carbonate; DMC = dimethyl carbonate; CB = carbon black; LiTFSI = bis(trifluoromethane)sulfonamide; DOL = dioxolane; EMC = ethyl methyl carbonate; PTFE = poly(tetrafluoroethylene); VGCF = vapor-grown carbon fibers; EiPS = ethyl isopropyl sulfone; CPE = composite polymer electrolyte; PMA = poly(methacrylate); PEO = poly(ethylene oxide); PC = propylene carbonate; TEGDME = tetraethylene glycol dimethyl ether; DEGDME = diethylene glycol dimethyl ether; NaTFSI = sodium bis(trifluoromethane)sulfonamide; BC = butylene carbonate; MWCNT = multiwall carbon nanotubes; EMIC = 1-ethyl-3-methylimidazolium chloride; SBR = styrene-butadiene rubber; CMC = carboxymethyl cellulose. |
||||||||
| Small molecules | ||||||||
|
1 | 628 | 50:45:5 1:acetylene black:PVdF |
1 M LiPF6 1:1 EC:DEC (v/v) |
340, 200 mA g−1; 90, 800 mA g−1 | 3.5–2.4, Li/Li+b |
58.8%, 40, 200 mA g−1 | 29 |
|
2 | 130 | 70:20:10 2:acetylene black:PVdF |
1 M LiPF6 DMC | 130, 0.2C; 60, 5C | 2.4, Li/Li+ | 120 mA h g−1, 100, 0.1C | 30 |
|
3 | 173 | 70:20:10 3:acetylene black:PVdF |
1 M LiPF6 DMC | ∼150, 0.2C | 2.25, Li/Li+ | ∼75 mA h g−1, 100, 0.1C | 30 |
|
4 | 309.6 | 40:40:10:10 4:CMK-3:CB:PVdF |
1 M LiPF6 EC:DMC (v/v) |
308.6, 0.1C; 245.8, 0.5C | 2.48/2.30, Li/Li+ | 202.6 mA h g−1, 50, 0.1C | 31 |
| 56:24:10:10 4:CMK-3:CB:PVdF |
2 M LiTFSI 1% LiNO3 1:1 DOL:DME (v/v) |
307, 0.2C | ∼2.25, Li/Li+ | ∼80%, 100, 0.2C | 32 | |||
|
5 | 408.9 | 56:24:10:10 5:CMK-3:CB:PVdF |
2 M LiTFSI 1% LiNO3 1:1 DOL:DME (v/v) |
404, 0.2C | ∼2.8, Li/Li+b |
83.9%, 100, 0.2C | 32 |
|
6 | 317.0 | 56:24:10:10 6:CMK-3:CB:PVdF |
2 M LiTFSI 1% LiNO3 1:1 DOL:DME (v/v) |
310, 0.2C | ∼2.4, Li/Li+b |
∼70%, 100, 0.2C | 32 |
|
7 | 326.9 | 56:24:10:10 7:CMK-3:CB:PVdF |
2 M LiTFSI 1% LiNO3 1:1 DOL:DME (v/v) |
322, 0.2C | ∼2.3, Li/Li+ | ∼80%, 100, 0.2C | 32 |
|
8 | 239 | 40:40:20 8:super P:PVdF |
1 M LiPF6 1:1:1 EC:DMC:EMC |
222, 0.1C | 2.6, 2.8, 3.4/2.1, 2.7, Li/Li+ | 33.8%, 40, 0.1C | 33 |
|
9 | 257 | 1.5:4:1 9:acetylene black:PTFE |
1.25 M LiPF6 1:3 EC:EMC (v/v) |
∼225, 0.2C | 2.52, Li/Li+ | Rapid fading due to dissolution | 36 |
|
10 | 253 | 1.5:4:1 10:acetylene black:PTFE |
1 M LiBF4 1:5 EC:DEC (v/v) |
∼205, 0.2C | 2.75, Li/Li+ | Rapid fading due to dissolution | 36 |
|
11 | 255 | 1.5:4:1 11:acetylene black:PTFE |
1.25 M LiPF6 1:3 EC:EMC (v/v) |
∼235, 0.2C | 2.94, Li/Li+ | Rapid fading due to dissolution | 36 |
|
12 | 255 | 1.5:4:1 12:acetylene black:PTFE |
1.25 M LiPF6 1:3 EC:EMC (v/v) |
∼220, 0.2C | 2.73, Li/Li+ | Rapid fading due to dissolution | 36 |
|
13 | 288 | 70:20:10 13:CB:PVdF |
1 M LiPF6 1:1 EC:DEC (v/v) |
200, 0.1C; 100, 6C | 2.0, 2.6/1.6, 2.1, Li/Li+ | 100%, 110, 0.2C | 37 |
|
14 | 241 | 50:50 14:Ketjenblack |
1 M LiPF6 1:1 EC:DMC (v/v) |
105, 0.1C; 63, 2.5C | 2.80, 2.95, Li/Li+b |
100%, 30, various rates | 38 |
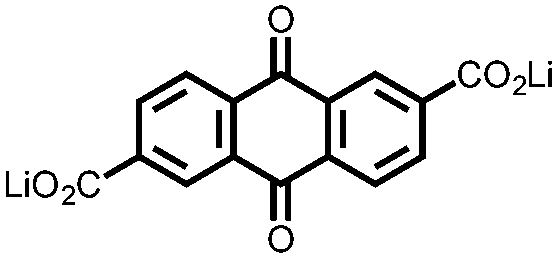
|
15 | 174 | 15:40:10 15:acetylene black:PTFE |
1 M LiPF6 PC | 85, 0.2C | 1.79, Li/Li+b |
82%, 20, 0.2C | 39 |
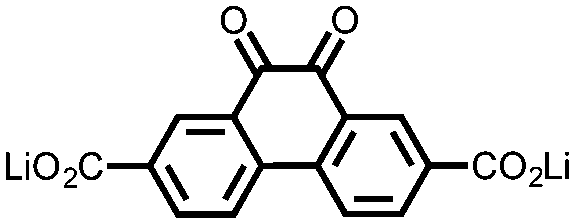
|
16 | 174 | 15:40:10 16:acetylene black:PTFE |
1 M LiPF6 PC | 90, 0.2C | 2.11, Li/Li+b |
105%, 20, 0.2C | 39 |
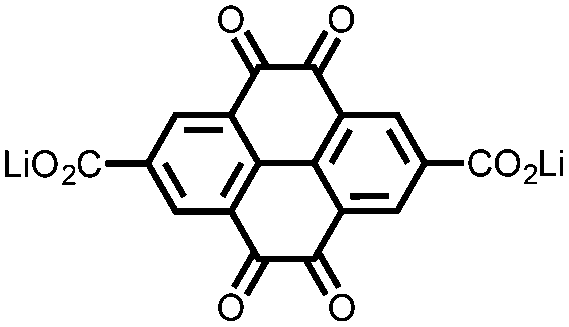
|
17 | 296 | 15:40:10 17:acetylene black:PTFE |
1 M LiPF6 PC | 217, 0.2C; 34.7, 5C | 2.39, Li/Li+b |
86%, 20, 0.2C | 39 |
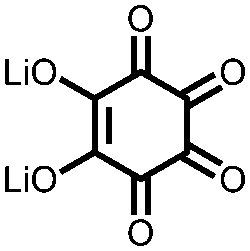
|
18 | 590 | 60:10:30 18:PVdF:Super P |
1 M LiPF6 1:1 EC:DMC |
580, 50 mA g−1 | 2.7, 2.5, 2.0, Li/Li+ | ∼20%, 25, various rates | 40 |
|
19 | 394 | 3:87:10 19:VGCF:PTFE |
1.0 M LiPF6 3:7 EC:DEC (v/v) |
320, n.r. | 2.5, Li/Li+ | 21%, 20, n.r. | 41 |
|
20 | 220 | 3:87:10 20:VGCF:PTFE |
1.0 M LiPF6 3:7 EC:DEC (v/v) |
162, n.r. | 3.0, Li/Li+ | 37%, 20, n.r. | 41 |
| 3:87:10 20:VGCF:PTFE |
1.0 M LiPF6 EiPS | 209, n.r. | 3.0, Li/Li+ | 23%, 20, n.r. | 41 | |||
|
21 | 99 | 3:87:10 21:VGCF:PTFE |
1.0 M LiPF6 3:7 EC:DEC (v/v) |
115, n.r. | 3.1, Li/Li+ | 50%, 20, n.r. | 41 |
|
22 | 66 | 3:87:10 22:VGCF:PTFE |
1.0 M LiPF6 3:7 EC:DEC (v/v) |
177, n.r. | 3.1, Li/Li+ | 55%, 20, n.r. | 41 |
|
23 | 446 | 55:25:3:2:10:5 23:CB:SWCNT:graphene:CPE:PVdF |
PMA/PEO-LiClO4-3 wt% SiO2 CPE | 418, 0.2C; ∼200, 1C | 2.9–2.3, Li/Li+b |
94.7%, 50, 0.2C | 18 |
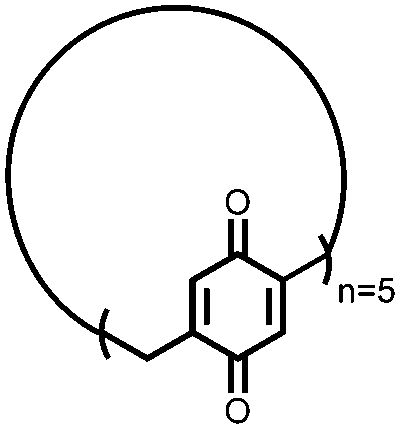
|
24 | 403 | 75:15:10 24:acetylene black:PVdF |
1.0 M LiPF6 1:1:1 DMC:EMC:EC (v/v/v) |
243, n.r. | 2.77, Li/Li+ | Rapid capacity fading due to dissolution | 42 |
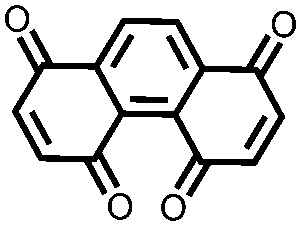
|
25 | 187 | 65:30:5 25:Super P:PVdF |
1 M NaClO4 1:1 EC:DMC (v/v) |
183, 0.1C; ∼80, 5C | 2.35, 2.56/2.10, 2.36, Na/Na+ | 84%, 100, 0.1C | 43 |
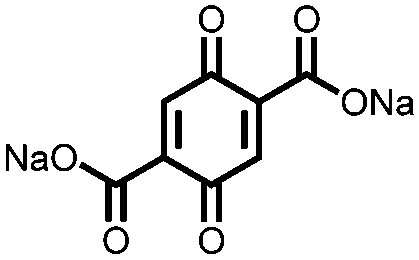
|
26 | 214 | 40:40:20 26:Super-P:PTFE |
1 M NaClO4 45:45:10 EC:PC:DMC |
150, 10 mA g−1 | 2.9, 2.6, Na/Na+ | ∼5%, 20, n.r. | 44 |
| 42:56 26:CMK-3 |
160, 19 mA g−1 | 2.9, 2.6, Na/Na+ | ∼25%, 20, n.r. | |||||
|
27 | 319 | 4:5:1 27:acetylene black:PTFE |
0.5 M Mg(TFSI)2 sulfolane | 100, 10 mA g−1 | 2.9/0.4, Mg/Mg2+ | 20%, 50, 10 mA g−1 | 45 |
| 4:5:1 27:CB:PVdF |
0.5 M Mg(TFSI)2–MgCl2 DME | 226, 0.2C | 2.0, Mg/Mg2+ | 74 mA h g−1, 30, 0.2C | 46 | |||
|
28 | 123 | 45:50:5 28:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
92, 0.2C | 2.5, Li/Li+c |
∼30%, 20, n.r. | 48 |
|
29 | 103 | 45:50:5 29:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
41, 0.2C | 2.3, Li/Li+c |
Rapid capacity fading due to dissolution | 48 |
|
30 | 114 | 45:50:5 30:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
45, 0.2C | 2.6, Li/Li+c |
Rapid capacity fading due to dissolution | 48 |
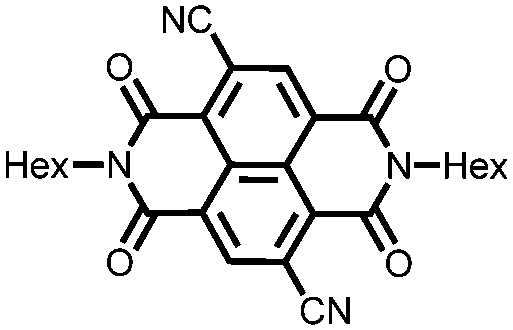
|
31 | 111 | 45:50:5 31:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
100, 0.2C | 2.8, Li/Li+c |
∼30%, 20, n.r. | 48 |
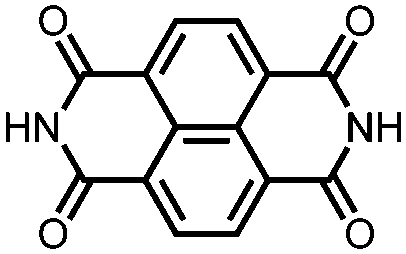
|
32 | 201 | 45:50:5 32:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
121, 0.2C | 2.55, Li/Li+c |
∼90%, 20, n.r. | 48 |
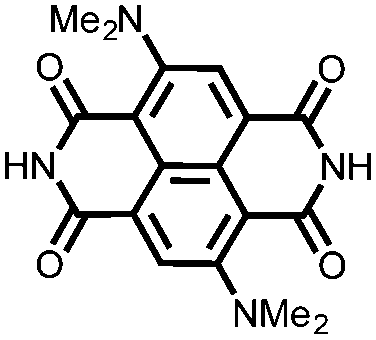
|
33 | 152 | 45:50:5 33:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
30, 0.2C | 2.4, Li/Li+c |
∼75%, 10, n.r. | 48 |
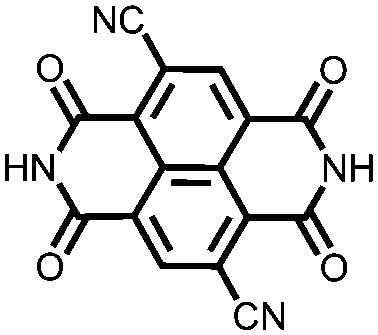
|
34 | 170 | 45:50:5 34:acetylene black:PVdF |
1 M LiClO4 1:1 EC:DMC (v/v) |
34, 0.2C | 2.9, Li/Li+c |
∼50%, 10, n.r. | 48 |
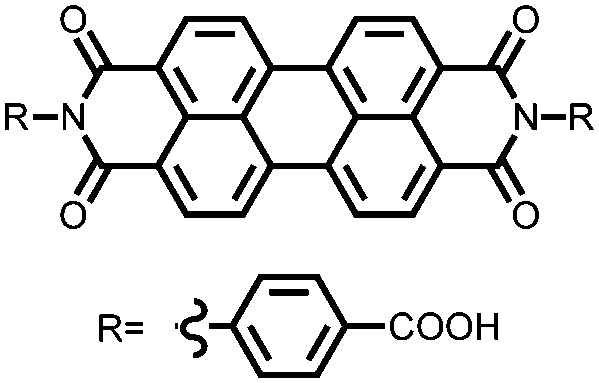
|
35 | 85 | 60:30:10 35:CB:Kynar |
n.r. | 85, 1C; 68, 10C | 2.7/2.0, Li/Li+c |
88%, 200, 5C | 49 |
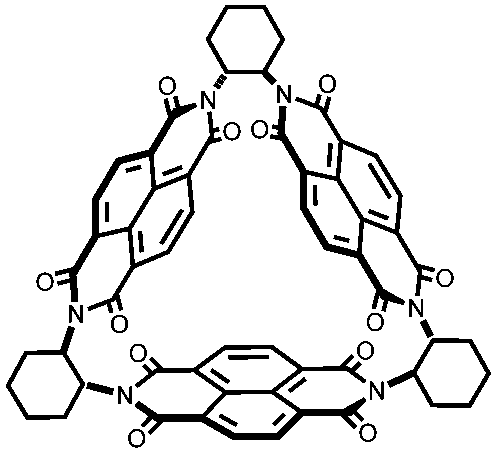
|
36 | 154.8 | 50:40:10 36:Super P:PVdF |
1 M LiTFSI, 0.2 M LiNO3 1:1 DOL:DME (v/v) |
146.4, 0.1C; 58.1, 100C | 2.35, 2.62/2.32, 2.59, Li/Li+ | ∼60%, 300, 10C | 50 |
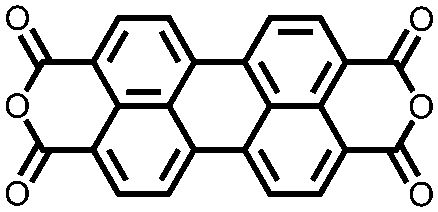
|
37 | 136.6 | 70:20:10 37:Super P:PVdF |
1 M NaPF6 1:1 EC:DEC (v/v) |
145, 10 mA g−1; 91, 1000 mA g−1 | 2.5/2.3, Na/Na+ | 69%, 200, 1.4C | 51 |
| 70:20:10 37:Super P:PVdF |
0.5 M KPF6 1:1 EC:DEC (v/v) |
131, 10 mA g−1; 73, 500 mA g−1 | 2.7, 2.9, 3.2/2.2, 2.4, K/K+ | 90 mA h g−1, 200 50 mA g−1 | 52 | |||
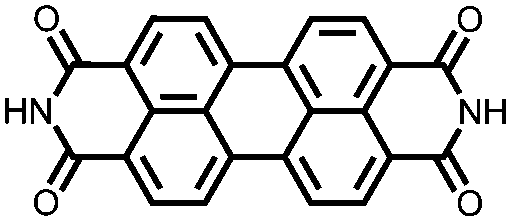
|
38 | 137 | 70:20:10 38:Super P:PTFE |
1 M NaPF6 45:45:10 EC:DEC:PC (v/v/v) |
138.6, 10 mA g−1; 103, 600 mA g−1 | 2.8, 2.5, 2.0/1.7, Na/Na+ | 90%, 300, 200 mA g−1 | 53 |
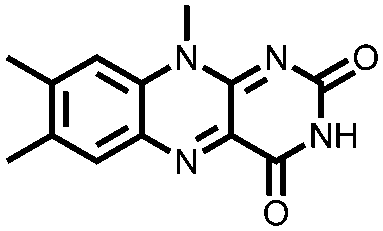
|
39 | 208 | 50:30:20 39:Super P:PTFE |
1 M LiPF6 1:1 EC:DMC (v/v) |
174, 10 mA g−1 | 2.65, 2.3, Li/Li+ | 66.3%, 10, 10 mA g−1 | 54 |
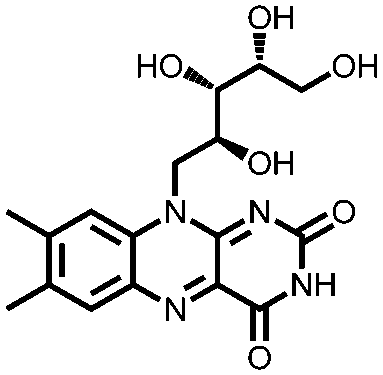
|
40 | 142 | 45:55 39:SWCNTs |
1 M LiPF6 TEGDME | 204, 1C; 125, 78C | 2.67/2.5, Li/Li+ | 99.7%, 100, 0.2 A g−1 | 55 |
| 50:30:20 40:Super:PTFE |
1 M LiPF6 1:1 EC:DMC (v/v) |
106, 10 mA g−1 | 2.65, 2.4, Li/Li+ | 53.6%, 10, 10 mA g−1 | 54 | |||
| 45:55 40:SWCNTs |
1 M LiPF6 TEGDME | ∼150, 0.2 A g−1 | ∼2.5, Li/Li+ | ∼100%, 100, 0.2 A g−1 | 55 | |||
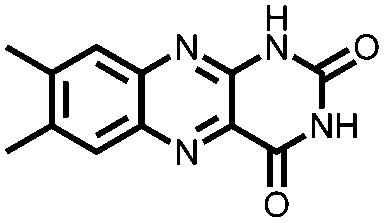
|
41 | 220 | 43:57 41:CNTs |
1 M LiPF6 TEGDME | 215, 1C; 153, 45.5C | 2.50, 2.35, Li/Li+ | 93%, 200, 1.0 A g−1 | 56 |
| 43:57 41:CNTs |
1 M NaPF6 DEGDME | 222, 50 mA g−1 | ∼2.25–1.25, Na/Na+b |
∼50%, 20, n.r. | 56 | |||
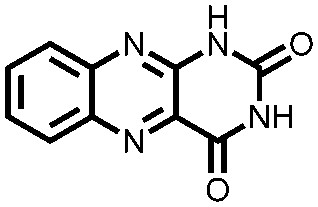
|
42 | 250 | 41:59 42:CNTs |
1 M LiPF6 TEGDME | 236, 1C; 168, 10C | n.r. | 92%, 200, 1.0 A g−1 | 56 |
| 41:59 42:CNTs |
1 M NaPF6 DEGDME | 255, 50 mA g−1 | ∼2.25–1.25, Na/Na+b |
∼50%, 20, n.r. | 56 | |||
|
43 | 357 | 35:65 43:CNTs |
1 M LiPF6 TEGDME | 154, 20 mA g−1 | n.r. | n.r. | 56 |
| 35:65 43:CNTs |
1 M NaPF6 DEGDME | 220, 50 mA g−1 | ∼2.0–1.0, Na/Na+b |
∼50%, 20, n.r. | 56 | |||
|
44 | 115 | 4:5:1 44:acetylene black:PTFE |
n.r. | 110, 10 mA g−1 | 2.2, Li/Li+ | 82.7%, 40, 10 mA g−1 | 57 |
| 1 M NaTFSI BC | 106, 10 mA g−1 | 1.8, Na/Na+ | 81.1%, 40, 10 mA g−1 | 57 | ||||
|
45 | 83 | 50:45:5 45:Ketjen black:PTFE |
1 M LiPF6 1:1 EC:DEC (v/v) |
64.4, 1.2C | 3.9/∼3.7, 2.4, Li/Li+ | ∼90%, 50, 1.2 and 12C | 58 |
|
46 | 77.4 | 50:45:5 46:Ketjen black:PTFE |
1 M LiPF6 1:1 EC:DEC (v/v) |
76.7, 1.2C | 3.7/∼3.0, 2.3, Li/Li+ | ∼90%, 50, 1.2 and 12C | 58 |
|
47 | 74d | 35:35:20:10 47:CB:Ketjen black:PTFE |
0.2 M AlCl3, 0.6 M PhMgCl THF | 50, 19 μA cm−2; 22, 1515 μA cm−2 | 1.3, 1.8/1.1, 1.4 Mg/Mg2+ | ∼10%, 10, 75 μA cm−2; ∼80%, 10, 1515 μA cm−2 | 59 |
|
62 | 256 | 60:30:10 62:Ketjenblack:PTFE |
1 M LiTFSI 1:1 DOL:DME (v/v) |
119, 50 mA g−1 | ∼3.0, 2.0, Li/Li+ | ∼50%, 20, 50 mA g−1 | 70 |
|
63 | 243 | 60:30:10 63:Ketjenblack:PTFE |
1 M LiTFSI 1:1 DOL:DME (v/v) |
193, 50 mA g−1 | 2.3–1.8, Li/Li+b |
∼75%, 20, 50 mA g−1 | 70 |
| Non-conjugated polymers | ||||||||
|
48 | 221.5d |
In situ polymerization, filtered onto SWNT film | 1 M LiTFSI 1:1 DOL:DME (w/w) |
226, 0.1C; 120, 20C | 2.20/2.07, Li/Li+ | 85%, 200, 0.5C | 60 |
| 443e |
60:30:10 48:acetylene black:PVdF |
1 M NaPF6 1:1 EC:DMC (w/w) |
∼124, 25 mA g−1 | 1.73, Na/Na+ | ∼40%, 62, 200 mA g−1 | 62 | ||
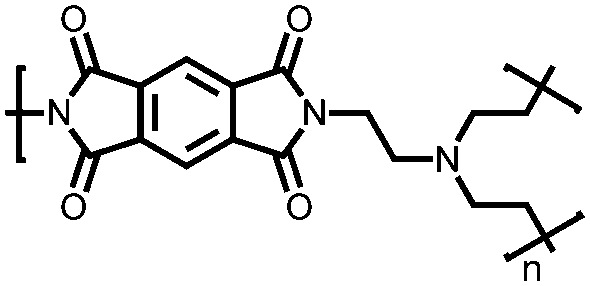
|
49 | 191d |
9:1 49:SWCNT, in situ polymerization |
1 M LiTFSI 1:1 DOL:DME (w/w) |
179, 0.1C; 74, 10C | 2.21/2.09, Li/Li+ | 86.6%, 200, 0.5C | 61 |
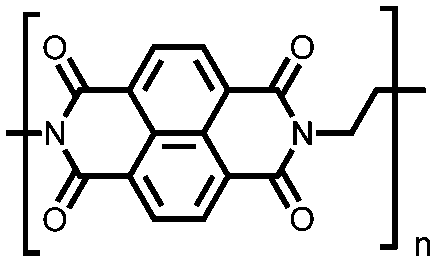
|
50 | n.r. | 60:30:10 50:acetylene black:PVdF |
1 M NaPF6 1:1 EC:DMC (w/w) |
∼132, 25 mA g−1 | 1.89, Na/Na+ | ∼65%, 100, 200 mA g−1 | 62 |
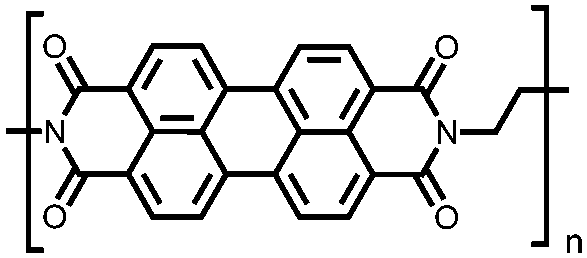
|
51 | 248.6e |
60:30:10 51:acetylene black:PVdF |
1 M NaPF6 1:1 EC:DMC (w/w) |
107.7, 25 mA g−1 | 1.94, Na/Na+ | 83%, 150, 200 mA g−1 | 62 |
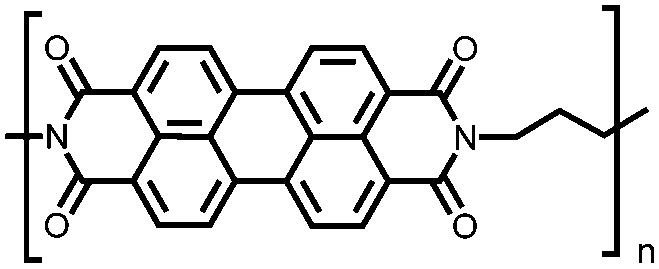
|
52 | 240.8e | 60:30:10 52:acetylene black:PVdF |
1 M NaPF6 1:1 EC:DMC (w/w) |
∼116, 25 mA g−1 | 2.3–2.6, Na/Na+b |
n.r. | 62 |
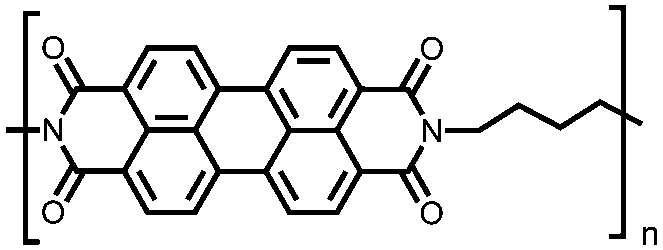
|
53 | 233.5e | 60:30:10 53:acetylene black:PVdF |
1 M NaPF6 1:1 EC:DMC (w/w) |
∼100, 25 mA g−1 | 2.3–2.6, Na/Na+b |
n.r. | 62 |
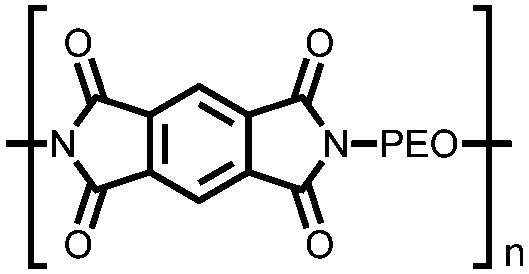
|
54 | 250f | 85:15 54:Ketjen black |
1 M LiTFSI Me-THF | ∼125, 0.1C | 2.7, 2.2/2.2, 1.8, Li/Li+ | 21%, 100, 0.1C | 63 |
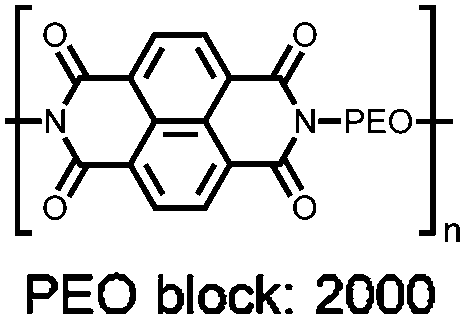
|
55 | 200f | 85:15 55:Ketjen black |
1 M LiTFSI Me-THF | 196, 0.1C | 2.9, 2.8/2.3, 2.1, Li/Li+ | 54%, 100, 0.1C | 63 |
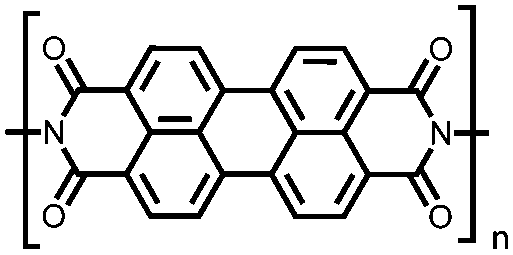
|
56 | 257e | 60:30:10 56:acetylene black:PVdF |
1 M NaPF6 PC | 126, 100 mA g−1; 94.5, 800 mA g−1 | 2.75, 1.97/2.45, 1.86, Na/Na+ | ∼90%, 50, 100 mA g−1 | 64 |
|
58 | 140 | 2:5:0.5 58:CB:PEDOT |
1 M LiPF6 1:1 EC:DMC (v/v) |
84, 0.1C; 42, 10C | 2.52, 2.93/2.37, Li/Li+ | ∼95–90%, 100, 0.5C | 66 |

|
59 | 217 | 1:8:1 59:MWCNT:PVdF |
1 M LiClO4 1:1 EC:DMC (m/v) |
219, 1C; 190, 10C | 2.59/2.23, Li/Li+ | 52%, 100, 1C | 67 |
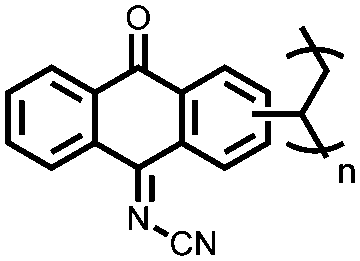
|
60 | 258.5 | 1:1:8 (w/w/w) 60:PVDF:MWCNT |
1 M LiClO4 1:4 EC:DMC (v/v) |
137, 1C | 2.33/2.28, Li/Li+ | 86%, 100, 5C | 68 |
|
61 | 144e | 35:50:15 61:CB:PVdF |
1 M LiPF6 TEGDME | 125, 0.1C; 77, 1C | 2.65, 1.85, Li/Li+ | 28.3%, 200, 1C | 69 |
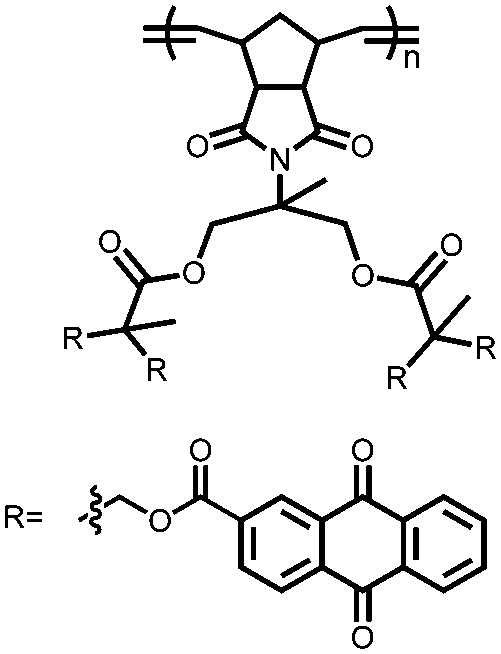
|
118 | 1672g | 75:20:5 118:conductive carbon:polyethylene |
0.38 M LiTFSI, 0.32 M LiNO3 1:1 DOL:DME (v/v) |
1100, 0.1C | 2.4–2.3, 2.1–2.0, Li/Li+ | 74.8%, 100, 0.1C | 118 |
| 1225, 0.1C; 800, 1C | 66.7%, 300, 0.1C | 119 | ||||||
| Conjugated polymers | ||||||||
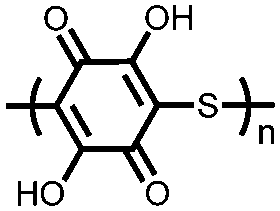
|
64 | 315 | 60:30:10 64:Ketjenblack:PTFE |
1 M LiTFSI 1:1 DOL:DME (v/v) |
214, 50 mA g−1 | ∼3.0, 2.0, Li/Li+ | 47%, 20, 50 mA g−1 | 70 |
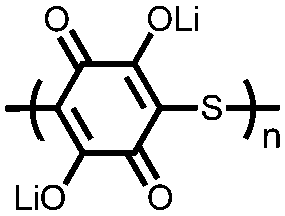
|
65 | 295 | 60:30:10 65:Ketjenblack:PTFE |
1 M LiTFSI 1:1 DOL:DME (v/v) |
247, 50 mA g−1; 124, 10000 mA g−1 |
2.3–1.8, Li/Li+b |
90%, 1500, 500 mA g−1 | 70 |
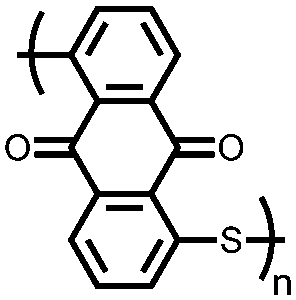
|
66 | 225 | 60:30:10 66:Ketjenblack EC-600JD:PTFE |
1 M LiTFSI 2:1 DOL:DME (v/v) |
213.8, 0.2C | 2.14, Li/Li+ | 98.4%, 100, 0.2C | 23 |
| 50:35:15 66:Printex XE2 carbon:PTFE |
0.37 M MgCl2 0.15 M Mg(TFSI)2 3:2 THF:glyme (v/v) |
225, 50 mA g−1 | 1.5–0.5, Mg/Mg2+b |
∼22%, 100, 50 mA g−1 | 71 | |||
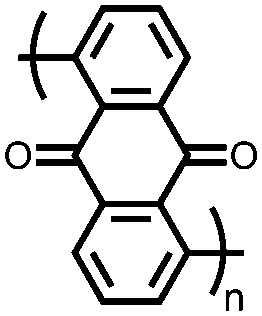
|
67 | 260 | 6:3:1 67:Ketjenblack EC-600JD:PTFE |
1 M LiTFSI 2:1 DOL:DME (v/v) |
240.5, 0.2C | 2.09, Li/Li+ | 67.6%, 100, 0.2C | 23 |
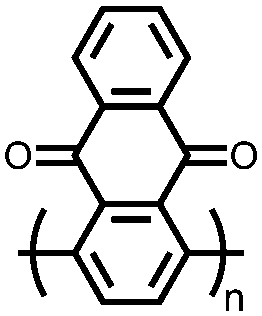
|
68 | 260 | 6:3:1 68:Ketjenblack EC-600JD:PTFE |
1 M LiTFSI 2:1 DOL:DME (v/v) |
263, 0.2C | 2.14, Li/Li+ | 98.3%, 100, 0.2C | 23 |
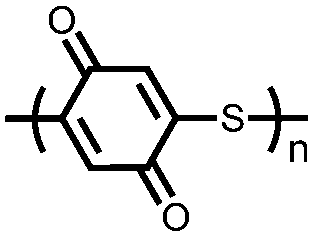
|
69 | 388 | 6:3:1 69:Ketjenblack EC-600JD:PTFE |
1 M LiTFSI 1:1 DOL:DME (v/v) |
275, 50 mA g−1; 198, 5000 mA g−1 | 3.4–2.1, Li/Li+b |
86%, 1000, 500 mA g−1 | 72 |
| 1 M NaTFSI 1:1 DOL:DME (v/v) |
268, 50 mA g−1 | 2.08, Na/Na+b |
68%, 100, 500 mA g−1 | 72 | ||||
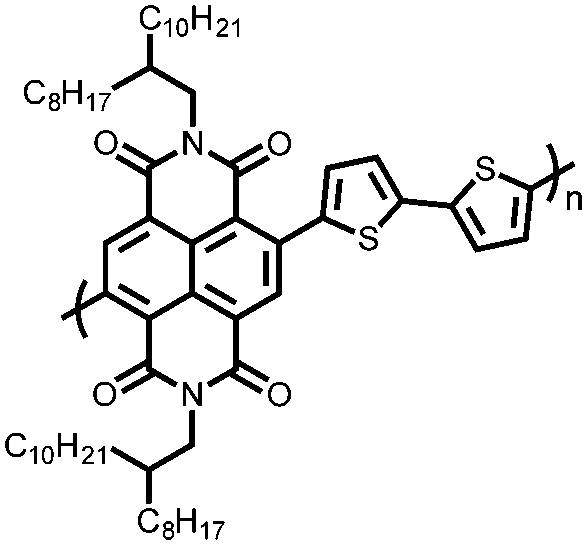
|
70 | 54.2 | 60:40 70:Super P |
1 M LiClO4 1:1 DOL:DME (v/v) |
53.7, 10C; 42.8, 500C | ∼2.55/∼2.45, Li/Li+ | 96%, 3000, 10C | 73 |
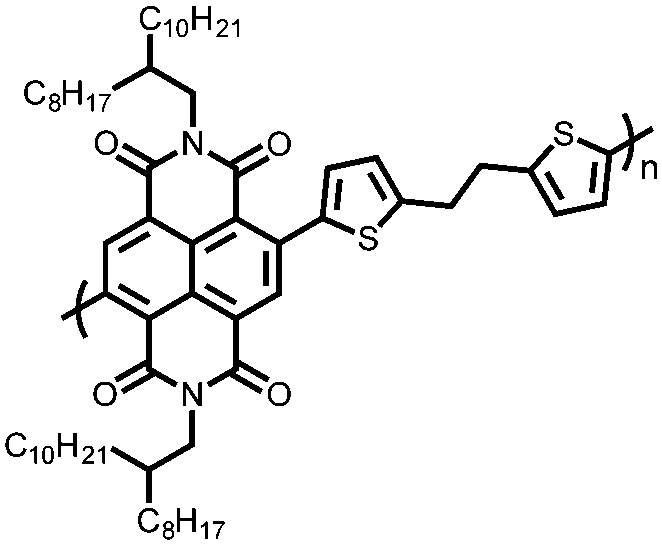
|
71 | 52.7 | 60:40 71:Super P |
1 M LiClO4 1:1 DOL:DME (v/v) |
42.2, 10C; 22.7, 500C | ∼2.5/∼2.4, Li/Li+ | n.r. | 73 |
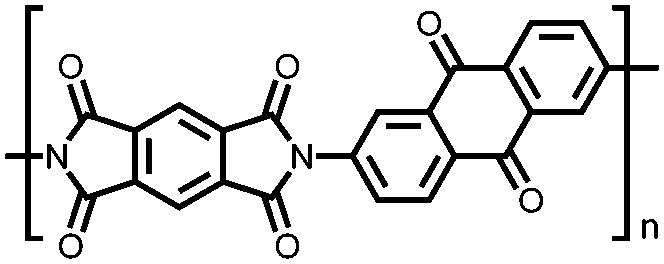
|
72 | 383h | 40:40:20 72:Ketjen black:PVdF |
Saturated NaPF6 1:1 DME:DOL (v/v) |
162, 50 mA g−1 | 1.97, Na/Na+b |
92%, 150, 50 mA g−1 | 74 |
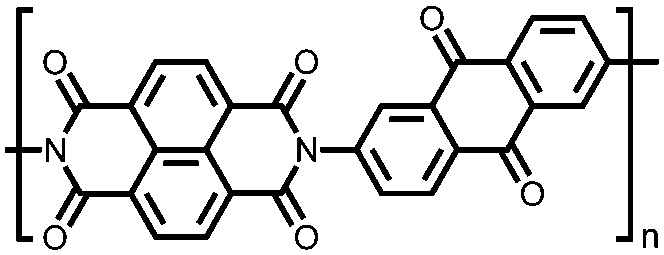
|
73 | 342h | 40:40:20 73:Ketjen black:PVdF |
Saturated NaPF6 1:1 DME:DOL (v/v) |
179, 50 mA g−1 | 2.01, Na/Na+b |
95%, 150, 50 mA g−1 | 74 |
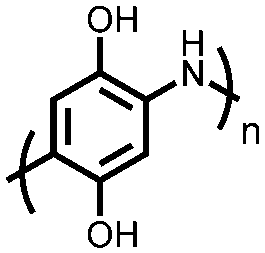
|
74 | 443 | 60:30:10 74:CB:PVdF |
1 M LiPF6 1:1 EC:DEC (v/v) or 1:1:1 EC:DEC:DMC (v/v/v) |
270, 0.1C | ∼2.2, Li/Li+ | ∼7%, 5, n.r. | 75 |
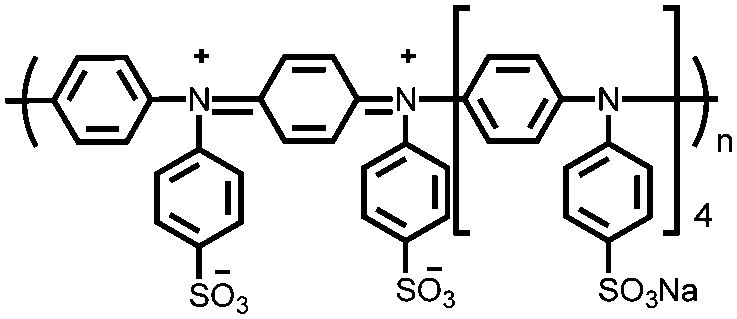
|
75 | 100 | 50:20:20:10 75:carbon fibers:CB:PTFE |
1 M NaPF6 1:1 EC:DEC (v/v) |
100, 50 mA g−1 | 3.5, 3.7/3.3, 3.6, Na/Na+b |
72%, 100, 50 mA g−1 | 76 |
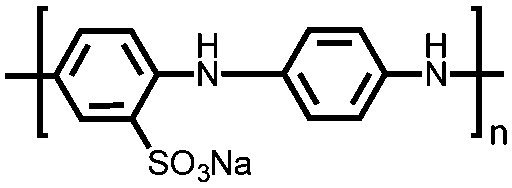
|
76 | 95 | 60:20:10:10 76:Ketjen black:Super P:PTFE |
1 M NaPF6 1:1 EC:DEC (v/v) |
133, 50 mA g−1; 76, 800 mA g−1 | 3.0, 3.6/2.8, 3.4 Na/Na+b |
96.7%, 200, 100 mA g−1 | 77 |
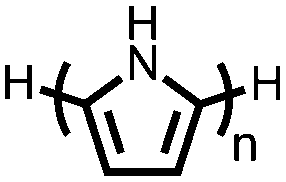
|
77 | 99.9i | 6:3:1:3 77:Kynar Flex 2801:Super P:dibutyl phthalate |
1.5:1 AlCl3:EMIC |
∼50, 0.2C | 0.6–1.8, Al/Al3+b |
∼14–26% loss, 20–100, 0.2C | 78 |
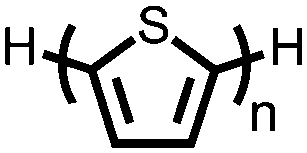
|
78 | 79.6i | 6:3:1:3 78:Kynar Flex 2801:Super P:dibutyl phthalate |
1.5:1 AlCl3:EMIC |
∼80, 0.2C | 1.1–1.9, Al/Al3+b |
13% loss, 20–100, 0.2C | 78 |
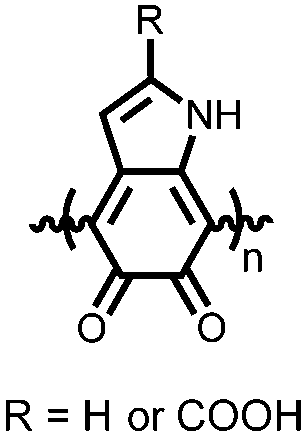
|
79 | 321j | 80:20 79:PTFE |
0.5 M Mg(NO3)2 in water | ∼60, 0.1 A g−1; ∼15, 5.0 A g−1 | ∼0.45/−0.3, SCE | 98–99.5%, 500, 0.1 A g−1 | 79 |
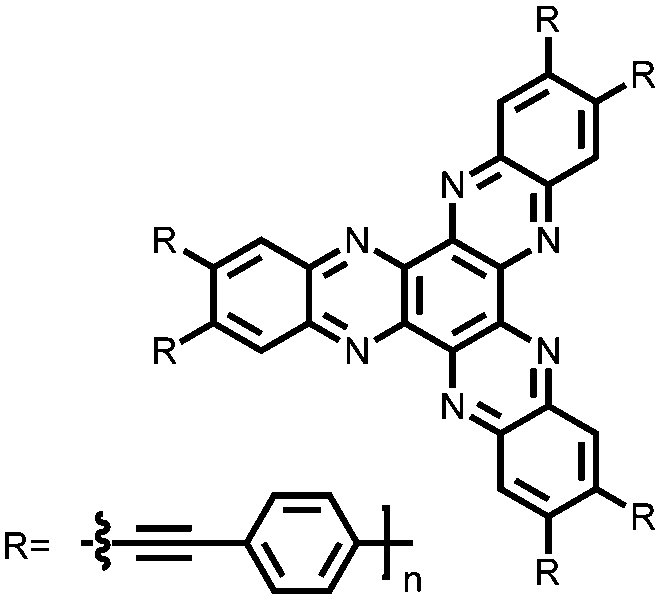
|
80 | 214 | 60:30:20 80:acetylene black:PVdF |
1 M LiPF6 1:1 EC:DMC (w/w) |
147, 100 mA g−1; 50, 1000 mA g−1 | 4.0–1.5, Li/Li+b |
62%, 50, 100 mA g−1 | 80 |
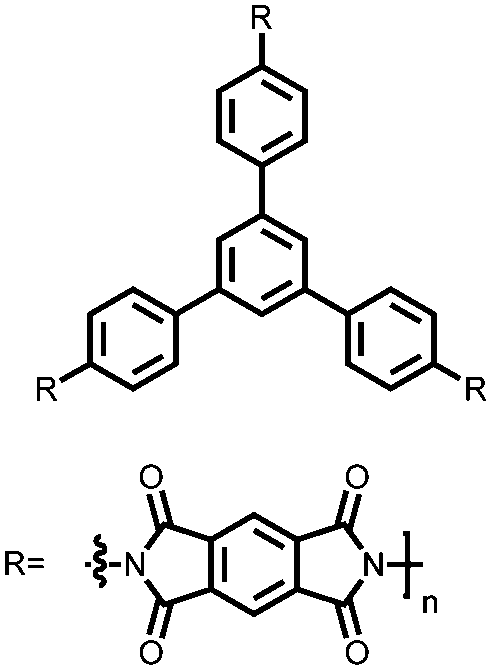
|
81 | 338.2e | 80:15:5 81:acetylene black:PTFE |
1 M LiPF6 1:1:1 EC:EMC:DMC (v/v/v) |
61.7, 25 mA g−1 | 2.35–1.5, Li/Li+b |
∼20%, 35, 25 mA g−1 | 81 |
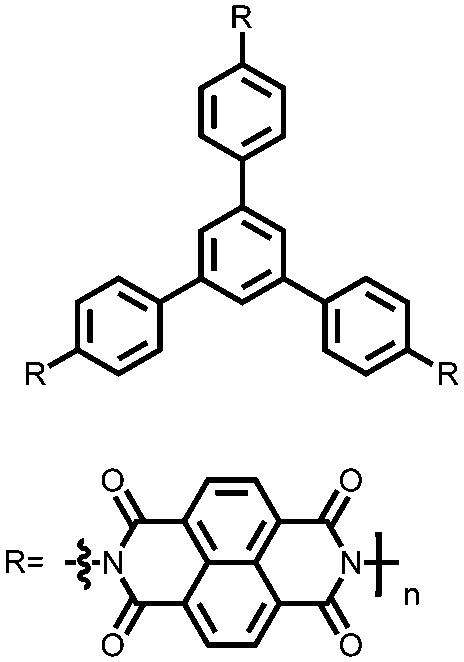
|
82 | 292.1e | 80:15:5 82:acetylene black:PTFE |
1 M LiPF6 1:1:1 EC:EMC:DMC (v/v/v) |
103.4, 25 mA g−1 | ∼2.35, Li/Li+ | 66.2%, 30, 25 mA g−1 | 81 |
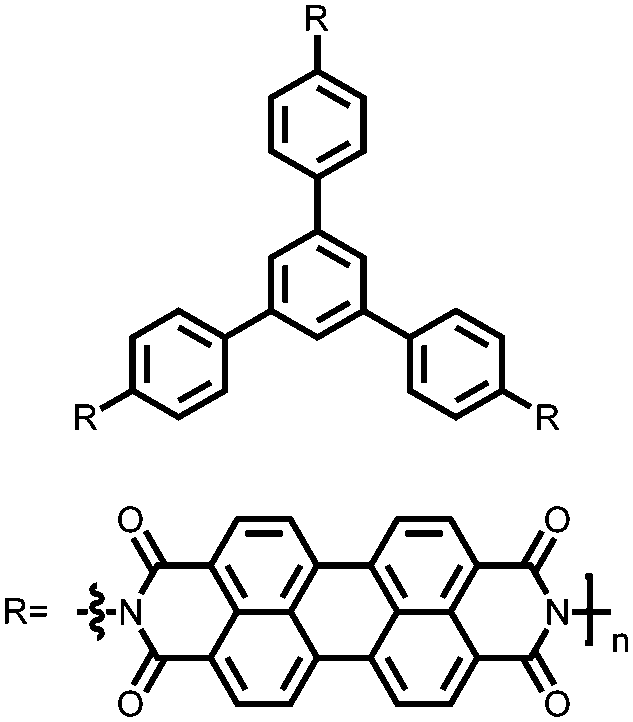
|
83 | 218.3e | 80:15:5 83:acetylene black:PTFE |
1 M LiPF6 1:1:1 EC:EMC:DMC (v/v/v) |
78.1, 25 mA g−1 | ∼2.35, Li/Li+ | 74.1%, 65, 25 mA g−1 | 81 |
|
84 | 82.4 | 48.3:21.7:20:20 84:CNTs:Super P Li:PVdF |
1 M LiPF6 1:1 EC:DMC (w/w) |
69, 2.4C; 58, 12C | 2.5/2.4, Li/Li+ | 100%, 700, 2.4C | 82 |
|
85 | n.r. | In situ polymerization | 0.1 M LiClO4 MeCN | ∼120, 70 μA | −0.83, −0.97, Ag/AgClO4 | n.r. | 83 |
| 0.1 M KClO4 MeCN | ∼95, 70 μA | −0.93, −1.31, Ag/AgClO4 | n.r. | 83 | ||||
| 0.1 M Mg(ClO4)2 MeCN | ∼100, 70 μA | −0.72, −0.72, Ag/AgClO4 | n.r. | 83 | ||||
| 0.1 M TBAClO4 MeCN | ∼110, 70 μA | −0.96, −1.43, Ag/AgClO4 | n.r. | 83 | ||||
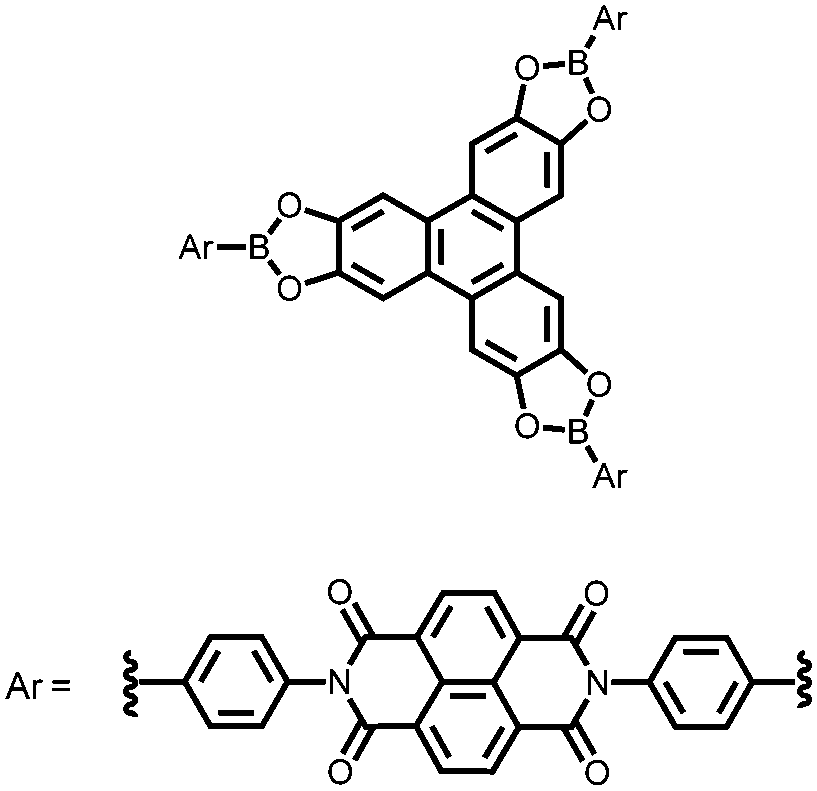
|
119 | 1675g | 70:25:5sulfur:P3HT + Super P :polyethylene |
1 M LiTFSI, 0.2 M LiNO3 1:1 DOL:DME (v/v) |
1212, 0.5C; 739, 1C | ∼2.35, ∼2.1, Li/Li+ | 799 mA h g−1, 100, n.r. | 120 |
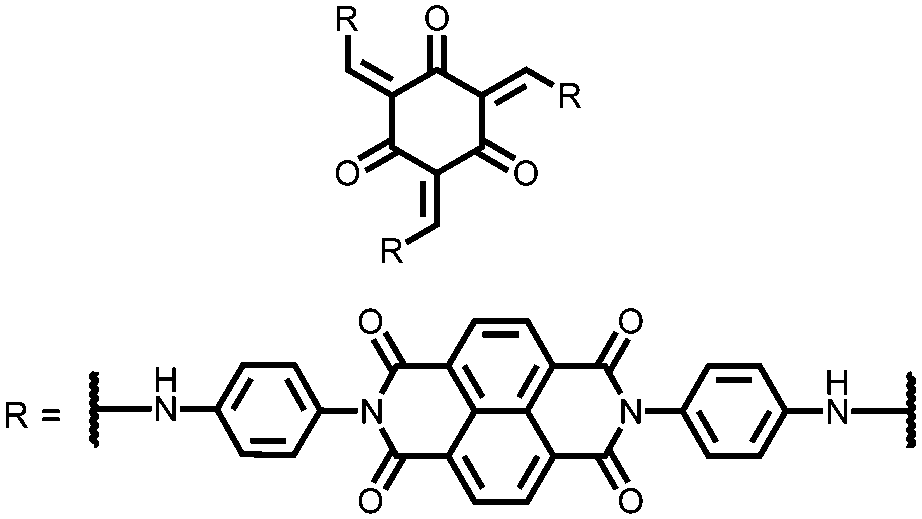
|
120 | 663.6 | 80:10:10 119:denka black:SBR/CMC |
1 M LiTFSI, 0.1 M LiNO3, 0.05 M CsNO3 1:1 DOL:DME (v/v) |
∼9 mA h cm−2, 4.2 mA cm−2 | 2.25–1.75, Li/Li+b |
73.3%, 90, 0.42 mA cm−2 charge, 4.2 mA cm−2 discharge | 121 |
| Structure | # | C theor (mA h g−1) | Electrode composition | Electrolyte | Capacity (mA h g−1), current | Potential: oxidationa/reduction (V), reference | Cycling stability: retention, cycles, current | Ref. |
|---|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a If the oxidation potential is not reported only the reduction potential(s) is listed.b The voltage range specified has a significant sloping voltage profile.c The capacity reported is higher than the theoretical value due to the contribution from CB.d The capacity reported was obtained at an operating temperature of 30 °C.e The theoretical capacity reported is based on the molecule accepting 4 electrons.f The capacity retention was measured in a full cell, based on the mass of both electrodes.g The theoretical capacity reported is based on each atom coordinating to 1 lithium ion.h The measurement was performed in the presence of oxygen.i The measurement was performed in the absence of oxygen.j The measurement was performed at 50 °C. The abbreviations used in the table are defined as: NaFSI = sodium trifluoromethanesulfonimide; SHE = standard hydrogen electrode; SCE = saturated calomel electrode; VC = vinylene carbonate; FEC = fluoroethylene carbonate. | ||||||||
| Small molecules | ||||||||
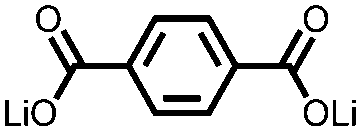
|
86 | 302 | 60:30:10 86:CB:PVdF |
1 M LiPF6 1:1:1 EC:DEC:DMC (v/v/v) |
259, 0.05C; 121, 1C | 0.83, Li/Li+ | 150 mA h g−1, 50, 0.5C | 84 |
| 4:3:1 86:Super P:CMC |
0.8 M LiPF6 1:1 EC:DEC (v/v) |
522, 30 mA g−1 | 0.81, 0.8–0.0, Li/Li+b |
∼75%, 15–50, 30 mA g−1 | 85 | |||
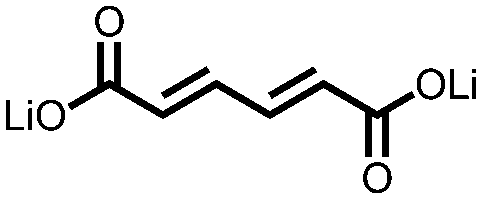
|
87 | 349.1 | 4:3:1 87:Super P:CMC |
0.8 M LiPF6 1:1 EC:DEC (v/v) |
241, 30 mA g−1 | 1.35, Li/Li+ | ∼99%, 50, 30 mA g−1 | 85 |

|
88 | 292 | 4:3:1 88:Super P:CMC |
0.8 M LiPF6 1:1 EC:DEC (v/v) |
850, 30 mA g−1 | ∼1.0, Li/Li+ | n.r. | 85 |
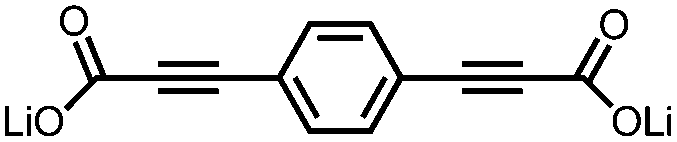
|
89 | 118.6 | 2:1 89:Super P |
1 M LiPF6 1:1 EC:DEC (v/v) |
1363, 1 Li+/50 h | 0.4, 1.1, 2.2, 2.9/0, 0.2, 0.7, Li/Li+ | ∼35%, 100, 1 Li+/h discharge | 87 |
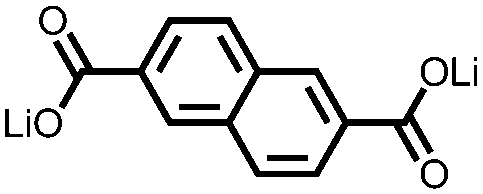
|
90 | 235.3 | 60:40 90:Super P |
1 M LiPF6 1:1 EC:DMC (v/v) |
200, 0.1C; 176, 1C | 0.88, Li/Li+ | 115 mA h g−1, 50, 1C | 88 |
| 77.7:13.7:5.5:3.2 90:CB:CMC:SBR |
1 M LiPF6 30:40:30 EC:DMC:EMC (v/v/v) |
213, 0.1C | 0.8, Li/Li+ | 100%, 10, 0.1C | 89 | |||
| 66.7:11.1:11.1:11.1 90:CB:VGCF:PVdF |
1 M LiPF6 30:40:30 EC:DMC:EMC (v/v/v) |
360, n.r. | 0.8, Li/Li+ | 96%, 100, 0.2C | 90 | |||
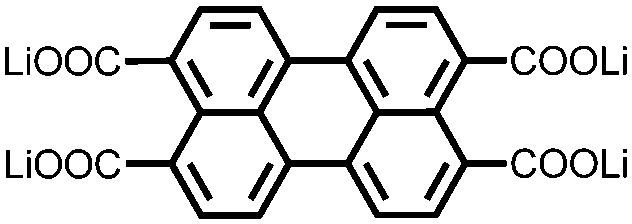
|
91 | 234 | 60:40 91:Super P |
1 M LiPF6 1:1 EC:DMC (v/v) |
222, 1.25C | 1.1, Li/Li+ | 125 mA h g−1, 100, 1.25C | 91 |
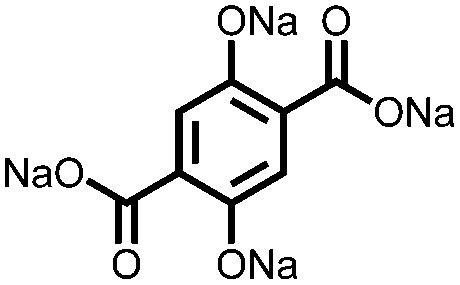
|
92 | 187 | 65:30:5 92:Super P:PVdF |
1 M NaClO4 1:1 EC:DMC (v/v) |
207, 0.1C; 117, 5C | 0.40/0.12, Na/Na+ | 89%, 100, n.r. | 43 |
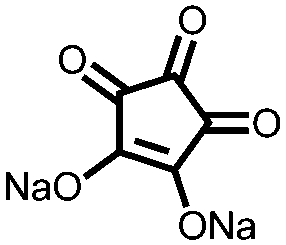
|
13 | 288 | 25.9:44.1:20:10 13:GOshell:CB:PVdF |
1 M NaClO4 1:1 EC:DMC (v/v) |
293, 20c mA g−1 | 1.65, 1.5, 1.15, 1.0, 0.85/1.8, 1.7, 1.25, 1.15, Na/Na+ | ∼40%, 100, 20 mA g−1 | 95 |
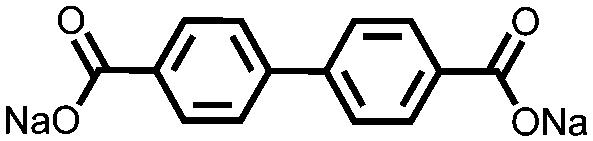
|
93 | 187 | 57.1:28.6:14.3 93:Super P:CMC |
0.8 M NaClO4 1:1 EC:DEC (v/v) |
200, 0.1C; 100, 20d C | ∼0.3, Na/Na+ | ∼100%, 150, 0.1C | 96 |
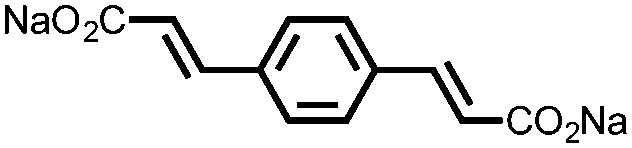
|
94 | 205 | 60:33:7 94:CB:CMC |
1 M NaFSI 1:1.5 EC:DEC |
177.7, 0.025C | 0.9/0.4, Na/Na+ | ∼40 mA h g−1, 40, 0.025C | 97 |
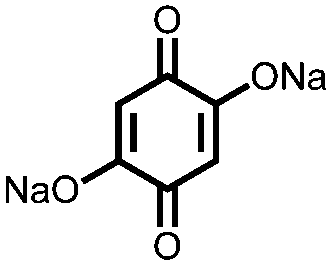
|
95 | 291 | 60:30:10 95:conductive carbon:PVdF |
1 M NaClO4 1:1 EC:DMC (v/v) |
265, 0.1C; 159, 5C | 1.59, 1.28/1.28, 1.20, Na/Na+ | 81%, 300, 1C | 98 |
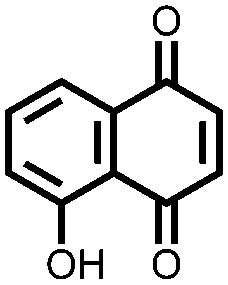
|
96 | 290 | 30.4:69.6 96:reduced GO |
1 M NaClO4 1:1 EC:DMC (v/v) |
398, 0.05 A g−1; 210, 0.4 A g−1 | 1.5/0.6, Na/Na+ | 69.5%, 300, 0.1 A g−1 | 99 |
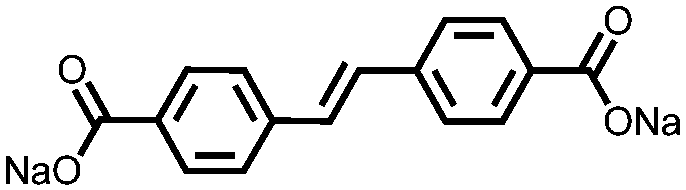
|
97 | 172 | 50:40:10 97:CB:CMC |
1 M NaClO4 PC | 260, 50 mA g−1; 72, 10 A g−1 | 0.95, 0.61/0.45, Na/Na+ | 70%, 400, 1 A g−1 | 100 |
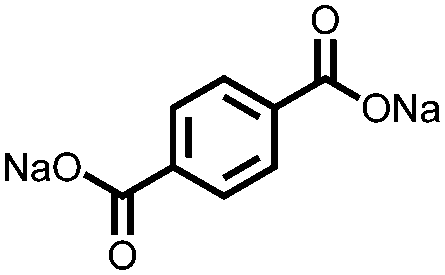
|
57 | 255 | 50:40:10 57:CB:CMC |
1 M NaClO4 PC | 192, 50 mA g−1; 22, 10 A g−1 | ∼0.5/0.18, Na/Na+ | 62 mA h g−1, 400, 1 A g−1 | 100 |
| 40:40:20 57:acetylene black:PVdF |
1 M NaClO4 1:1 EC:DEC (v/v) |
248, 25 mA g−1; 59, 1250 mA g−1 | 0.52/0.18, Na/Na+ | 81%, 100, 250 mA g−1 | 101 | |||
| 50:37.5:12.5 57:acetylene black:PVdF |
1 M NaPF6 PC | 180, 50 mA g−1 | ∼0.25, Na/Na+ | n.r. | 64 | |||
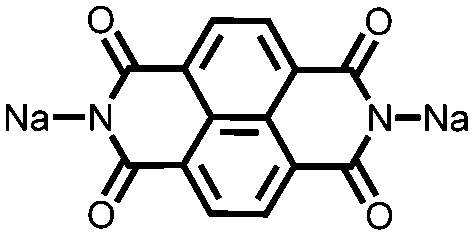
|
98 | 86 | 60:30:10 98:CB:PVdF |
1 M Na2SO4 water | 62, 6C; 40, 24C | −0.25/−0.03, SHE | 74%, 500, 6C | 102 |
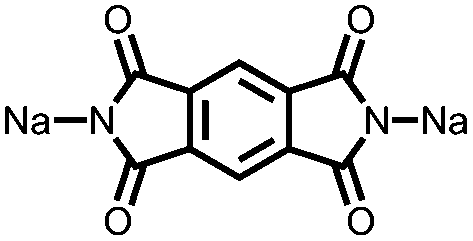
|
99 | 206 | 6:3:1 99:Super P:PVdF |
0.8 M NaPF6 PC | 128.9, 0.025C | 1.6, 1.2/1.3, 1.0, Na/Na+ | 70%, 100, 0.025C | 103 |
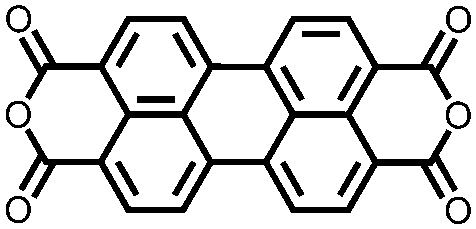
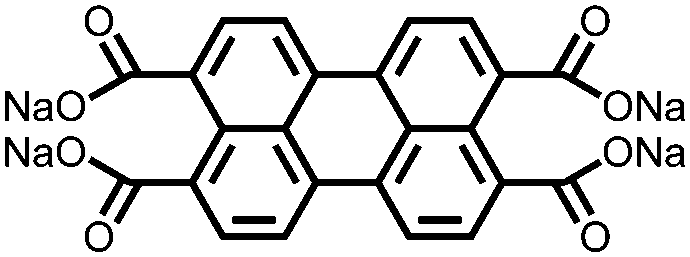
|
37 | 273e | 60:30:10 37:acetylene black:CMC |
1 M NaPF6 1:1 EC:DMC (w/w) |
361, 25 mA g−1; 67.7, 2 A g−1 | 0.5, Na/Na+ | 40.4%, 140, 25 mA g−1 | 104 |
|
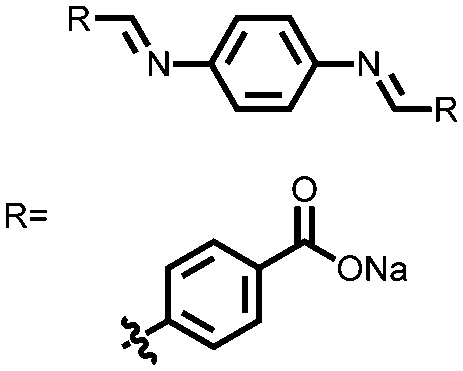
100 | 206 | 60:30:10 100:acetylene black:CMC |
1 M NaPF6 1:1 EC:DMC (w/w) |
350.6, n.r. | ∼0.75, Na/Na+ | 37.4%, 120, 25 mA g−1 | 104 |
|
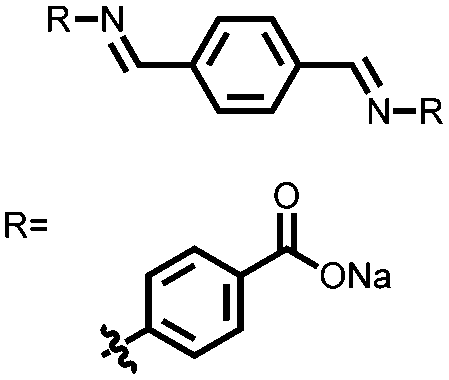
101 | 258 | 80:15:5 101:carbon Super C-65:Ketjen Black |
1 M NaFSI MeTHF | 268, 0.1C | 0.62, 0.90, 1.01/0.53, 0.75, 0.85, Na/Na+ | 97.5%, 25, 0.1C; 92%, 25, 0.2C | 106 |
|
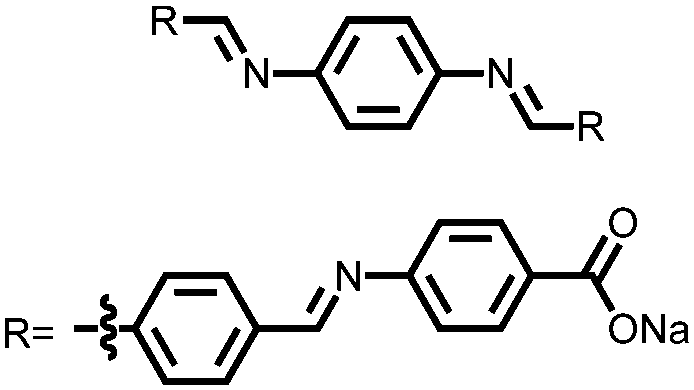
102 | 258 | 80:15:5 102:carbon Super C-65:Ketjen Black |
1 M NaFSI MeTHF | 120, n.r. | 0.87, 1.14/0.31, 0.99, Na/Na+ | ∼80–90%, 25, 0.1C ∼80–90%, 25, 0.2C | 106 |
|
103 | 258 | 80:15:5 103:carbon Super C-65:Ketjen Black |
1 M NaFSI MeTHF | 150, n.r. | 0.81, 1.08/0.79, 0.98, Na/Na+ | ∼80–90%, 25, 0.1C ∼80–90%, 25, 0.2C | 106 |
|
104 | 258 | 80:15:5 104:carbon Super C-65:Ketjen Black |
1 M NaFSI MeTHF | 260, n.r. | 0.65, 0.99, 1.15/0.57, 0.80, 1.02, Na/Na+ | ∼80–90%, 25, 0.1C ∼80–90%, 25, 0.2C | 106 |
| Non-conjugated polymers | ||||||||
|
105 | n.r. | 6:3:1 105:Printex XE2 carbon:PTFE |
5 M LiNO3 in water | 160, 100 mA g−1 | −0.39/−0.50, SCE | 80%, 200, 500f mA g−1 | 107 |
| n.r. | 6:3:1 105:Printex XE2 carbon:PTFE |
5 M NaNO3 in water | 165, 50 mA g−1 | −0.40/−0.55, SCE | 83%, 20, 50 mA g−1 | 107 | ||
|
50 | 120 | 30:60:10 50:acetylene black:PTFE |
5 M LiNO3 in water | 90h or 113,i 1C | 0.2 to −0.8, SCEb | 95%, 1000, 2h C | 108 |
| 120 | 30:50:20 50:CB:PVdF |
1 M NaClO4 1:1 EC:DEC (v/v) |
150, 70 mA g−1; 84, 2520 mA g−1 | 2.4, 2.1/2.25, 1.8, Na/Na+ | ∼95%, 500, 140 mA h g−1 | 109 | ||
|
106 | n.r. | 85:10:5 106:Super P:PTFE |
1 M LiPF6 1:1:1 EC:EMC:DMC (v/v/v) |
484, 20 mA g−1 | 1.06/0.82, Li/Li+ | 70%, 200, 40 mA g−1 | 110 |
| n.r. | 85:10:5 106:Super P:PTFE |
1 M NaPF6 1:1 PC:DMC (v/v) |
208.3, 20 mA g−1 | 0.71/0.48, Na/Na+ | 80%, 200, 40 mA g−1 | 110 | ||
|
121 | 229 | Polymer dropcast, 30 nm thick electrode | 30 wt% NaOH or KOH in water | 217, 3–34 A g−1 | −0.65, Ag/AgCl | ∼90%, 300, 5 A g−1 (for 80 nm thick electrode) | 122 |
|
122 | 212 | Polymer dropcast, 50 nm thick electrode | 10 M NaOH in water | 210, 1–600C | −0.80–0.92, Ag/AgCl | 95%, 500, n.r. | 123 |
| Conjugated polymers | ||||||||
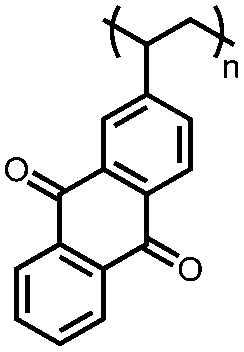
|
107 | n.r. | 60:30:10 107:Super P:PVdF |
1 M LiClO4 PC | 2, 0.05C | 2.0–1.2, Li/Li+b |
98%, 50, n.r. | 111 |
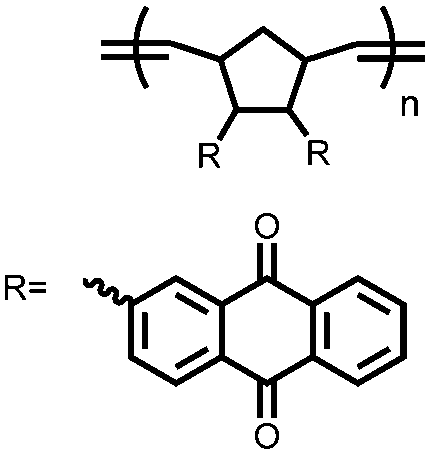
|
108 | n.r. | 60:30:10 108:Super P:PTFE |
1 M LiPF6 1:1:1 EC:DME:DEC (v/v/v) 5% VC |
1042, 20 mA g−1; 117, 2000 mA g−1 | 1.0–0.0, Li/Li+b |
404 mA h g−1, 100, 100 mA g−1 | 112 |
| n.r. | 1 M NaClO4 1:1 EC:DMC (v/v) 5% FEC |
145, 20 mA g−1 | ∼0.75–0.0, Na/Na+b |
100%, 100, 20 mA g−1 | 112 | |||
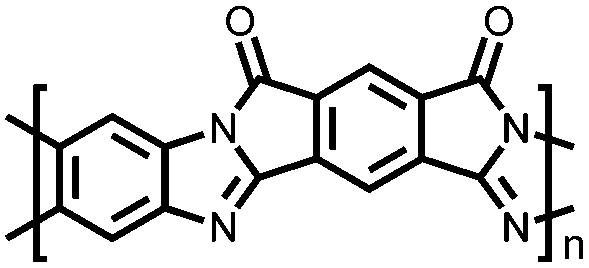
|
109 | 1888g | 70:20:10 109:nanoparticles:CNTs:PVdF |
1 M LiPF6 1:1 EC:DEC (w/w) |
1442, 0.05C; 183, 6C | 1.0–0.0, Li/Li+b |
66.6%, 1000, 3C | 105 |
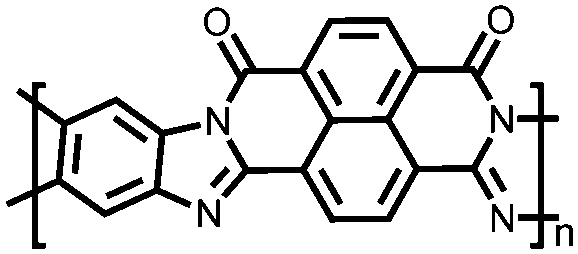
|
110 | 1926g | 70:20:10 110:nanoparticles:CNTs:PVdF |
1 M LiPF6 1:1 EC:DEC (w/w) |
1416, 0.1C; 317, 6C | 1.0–0.0, Li/Li+b |
107%, 1000, 3C | 105 |
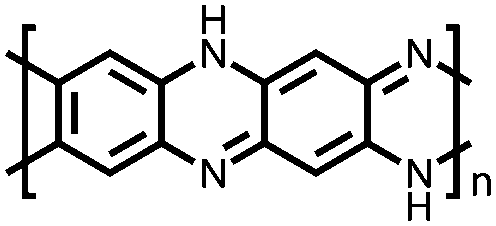
|
111 | 1822g | 70:20:10 111:CNTs:PVdF |
1 M LiPF6 1:1 EC:DEC (w/w) |
1550, 100 mA g−1; 203, 9110 mA g−1 | 1.5–0.0, Li/Li+b |
130% 1000, 2.5Cj | 113 |
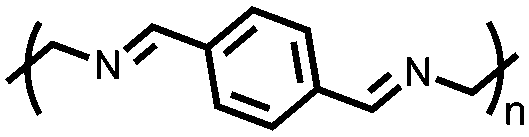
|
112 | 339 | 80:20 112:Carbon Super C-65 |
1 M NaFSI Me-THF | 150, 0.1C | 0.85/0.47, Na/Na+ | 40%, 25, 0.1C | 114 |
|
113 | 312 | 80:20 113:Carbon Super C-65 |
1 M NaFSI Me-THF | ∼50, 0.1C | 0.79/0.37, Na/Na+ | ∼60–80%, 25, 0.1C | 114 |
|
114 | 260 | 80:20 114:Carbon Super C-65 |
1 M NaFSI Me-THF | 180, 0.1C | 0.95/0.75, Na/Na+ | ∼100%, 25, 0.1C | 114 |
|
115 | 231 | 80:20 115:Carbon Super C-65 |
1 M NaFSI Me-THF | ∼138, 0.1C | 0.79/0.65, Na/Na+ | n.r. | 114 |
|
116 | 204 | 80:20 116:Carbon Super C-65 |
1 M NaFSI Me-THF | ∼26, 0.1C | 0.87/0.34, Na/Na+ | n.r. | 114 |
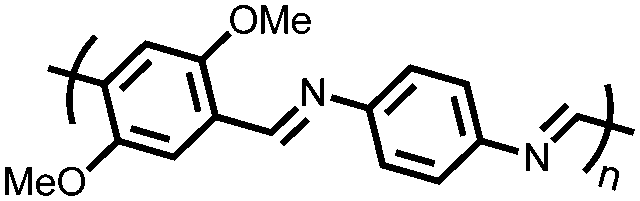
|
117 | 202 | 80:20 117:Carbon Super C-65 |
1 M NaFSI Me-THF | ∼111, 0.1C | 0.87/0.60, Na/Na+ | n.r. | 114 |
| Structure | # | C theor (mA h g−1) | Electrode composition | Electrolyte | Capacity (mA h g−1), current | Potential: oxidationa/reduction (V), reference | Cycling stability: retention, cycles, current | Ref. |
|---|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a If the oxidation potential is not reported only the reduction potential(s) is listed.b The capacity reported is for a symmetric battery.c The voltage range specified has a significant sloping voltage profile.d The capacity was reported at 60 °C. The abbreviations used in the table are defined as: TBAPF6 = tetrabutyl ammonium hexafluorophosphate; TBAClO4 = tetrabutyl ammonium perchlorate; PP13TFSA = N-methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl)amide. | ||||||||
| Small molecules | ||||||||
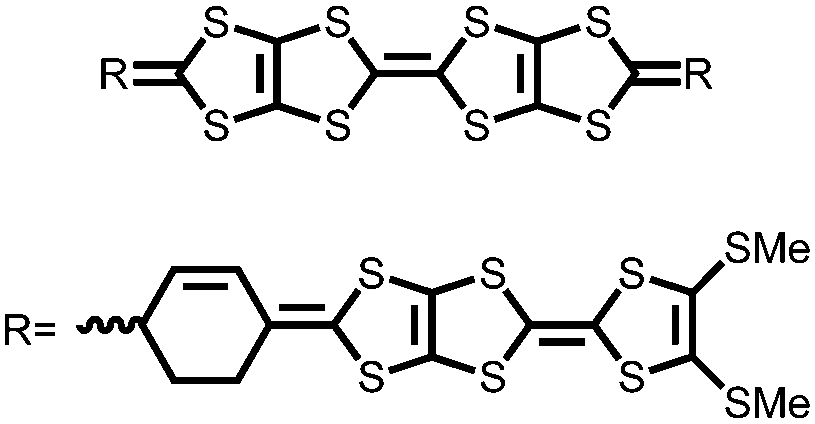
|
123 | 214 | 2:7:1 123:acetylene black:PTFE |
1 M LiPF6 1:1 EC:DEC (v/v) |
196, 0.5C; 125, 100C | 4–3, Li/Li+c |
∼75%, 30, 0.2C charge and 0.5C discharge | 127 |
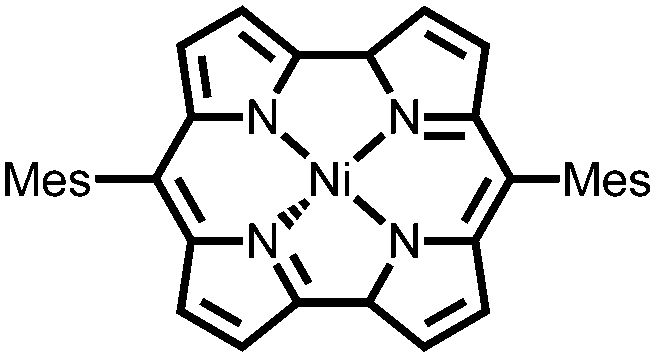
|
124 | 184 | 10:70:20 124:CB:PVdF |
1 M LiPF6 1:1 EC:DEC (v/v) |
200, n.r.; 86, n.r.b | 4.07, 3.43, 2.35, 1.58, Li/Li+c |
90%, 100, n.r.b | 128 |
| Non-conjugated polymers | ||||||||
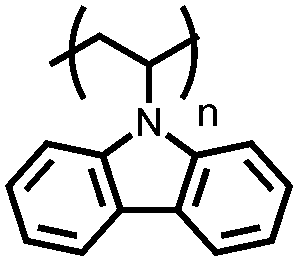
|
125 | 120 | 50:40:1 125:CB:PEDOT-PSS |
1 M LiPF6 1:1 EC:DMC (v/v) |
99.4, 1C; 79.5, 100C | 4.5–3.5, Li/Li+c |
∼100%, 100, 10C | 129 |
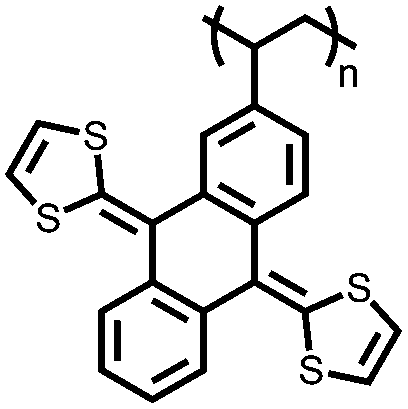
|
126 | 132 | 10:80:10 126:VGCF:PVdF |
0.1 M LiClO4 4:1 DME:PC (v/v) |
108, 1C; 38, 5C | 3.5/3.1, Li/Li+ | 75.9%, 250, 1C | 130 |
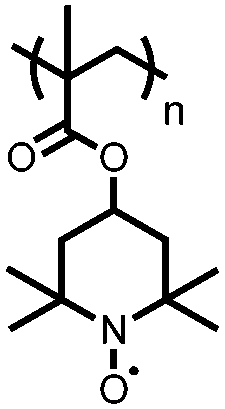
|
127 | 110 | 49:36:15 127:Ketjen black:PTFE |
0.1 M Mg(CF3SO3)2 PP13TFSA | 84.2, n.r.d | ∼2.0/1.7, Mg/Mg2+ | ∼50%, 10, n.r. | 131 |
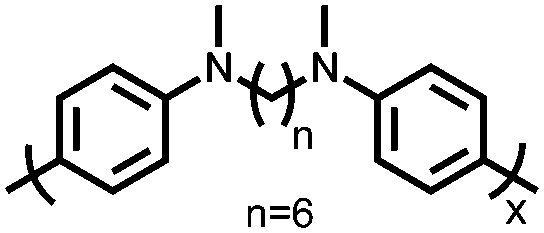
|
128 | 181 | Electro-polymerized film 101 nm thick | 0.5 M TBAPF6 MeCN | 165, 100–1000C | 0.1, 0.4/0.1, 0.3, Ag/Ag+ | 92%, 100, 1000C | 132 |
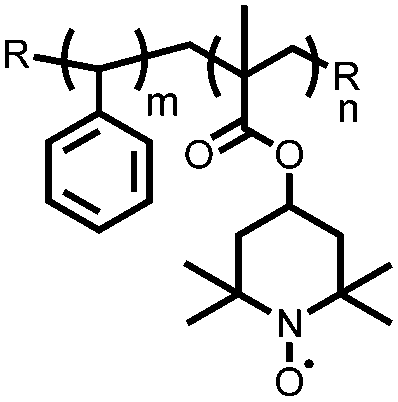
|
129 | n.r. | 80 nm thick spuncast film | 0.1 M TBAClO4 MeCN | ∼27, 23.7 μA cm−2 | 0.46, Ag/Ag+ | ∼95%, 50, 23.7 μA cm−2 | 133 |
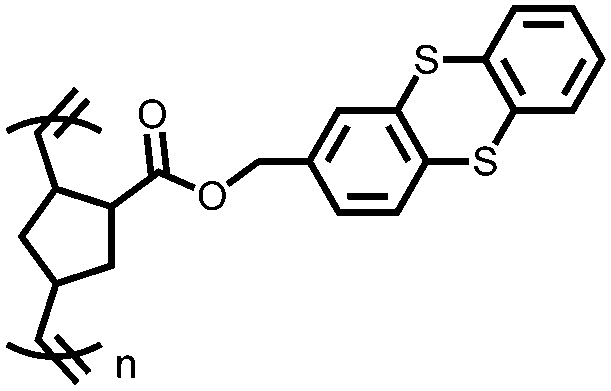
|
130 | 73 | 50:47:3 130:Super P Li:PVdF |
1 M LiPF6 1:1 EC:DMC |
66, n.r. | 4.10/4.05, Li/Li+ | ∼30%, 100, n.r. | 134 |
| Conjugated polymers | ||||||||
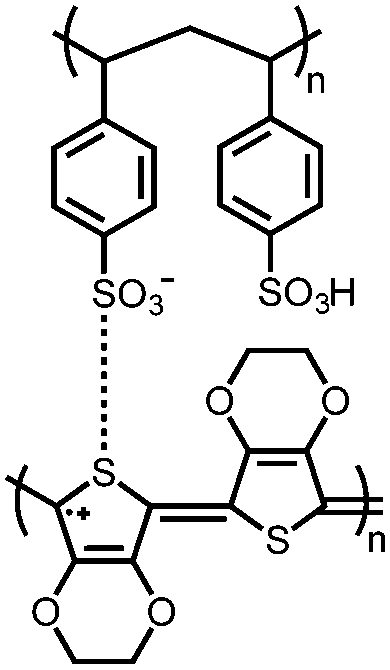
|
131 | n.r. | 200 nm thick 131 film | Solid poly(sodium-4-styrene sulfonate) | ∼0.10, n.r. | ∼0–0.5b | n.r. | 135 |
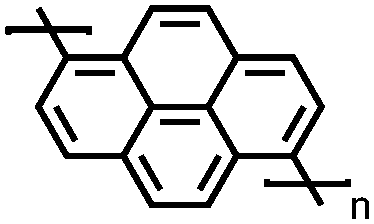
|
132 | 133 | 80:10:10 132:acetylene black:PVdF |
1 M NaClO4 PC | 120, 20 mA g−1; 60, 100 mA g−1 | 3.78/3.54, Na/Na+ | ∼70%, 50, 20 mA g−1 | 136 |
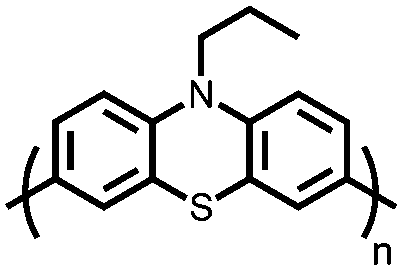
|
133 | 112 | 65:20:5:10 133:Super P:VGCF:PVdF |
1 M LiPF6 1:1 EC:DMC (w/w) |
80, 0.1C | 4.0–3.2, Li/Li+c |
99.75%, 9–10, 0.1C | 137 |
|
134 | n.r. | 70:20:10 134:CB:CMC |
1 M LiPF6 1:1 EC:DMC (v/v) |
65, 0.05 A g−1; 17, 2.0 A g−1 | 0–3.5b,c | ∼100%, 1000, 1.0 A g−1 | 138 |
|
135 | 130 | 50:40:10 135:acetylene black:PVdF |
1 M LiPF6 1:1 EC:DMC (v/v) |
129.1, 20 mA g−1; 92.8, 500 mA g−1 | 3.8, 3.3, Li/Li+ | 85.6%, 50, 20 mA g−1 | 139 |
| Miscellaneous polymers | ||||||||
|
136 | 132 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 46, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
137 | 196 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 44, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
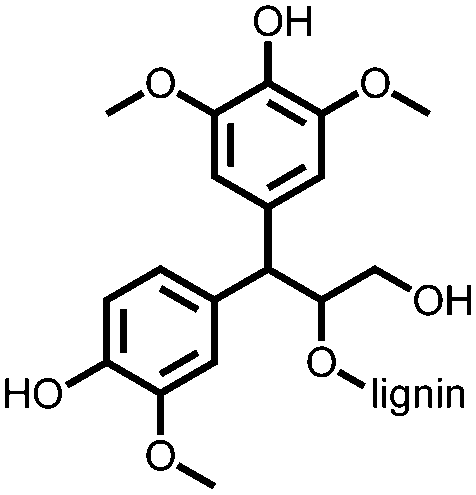
|
138 | 127 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 51, A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
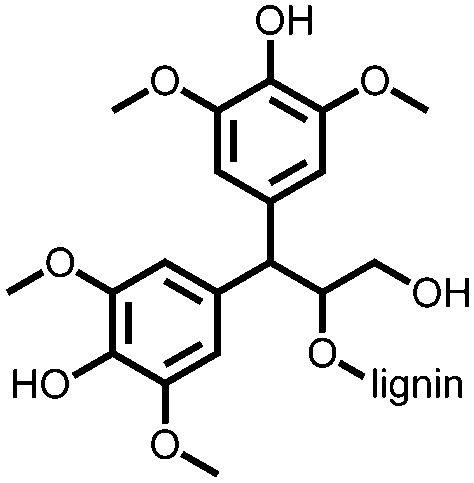
139 | 127 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 49, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
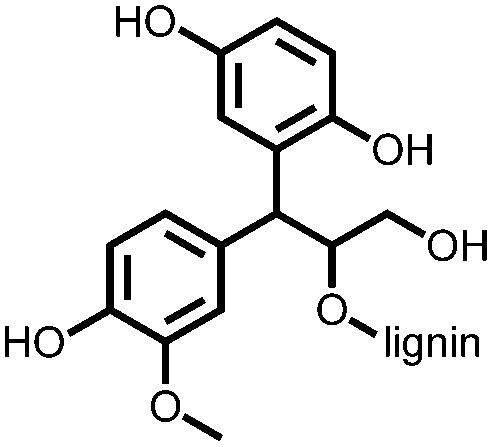
140 | 185 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 69, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
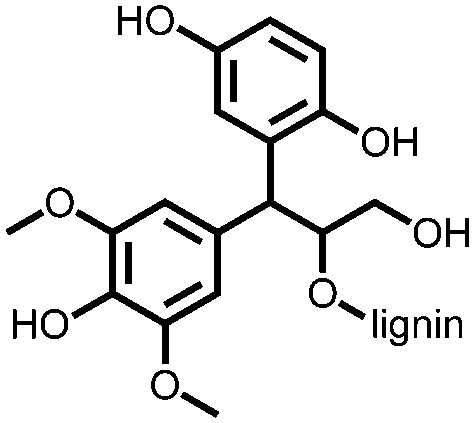
141 | 148 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 54, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
142 | 95 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 52, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
|
143 | 106 | Electro-polymerized with pyrrole | 0.1 M HClO4 in water | 47, 1 A g−1 | 0.1–0.6, Ag/AgClc | n.r. | 140 |
| Structure | # | C theor (mA h g−1) | Electrode composition | Electrolyte | Capacity (mA h g−1), current | Potential: oxidationa/reduction (V), reference | Cycling stability: retention, cycles, current | Ref. |
|---|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a If the oxidation potential is reported, if not only the reduction potential(s) is listed.b The voltage range specified has a sloping voltage profile.c The voltage range specified is for a symmetric device. The abbreviations used in the table are defined as: TBAOH = tetrabutyl ammonium hydroxide. | ||||||||
| Non-conjugated polymers | ||||||||
|
144 | 92 | 10:56:24:10 144:graphite:VGCF:PVdF |
0.1 M TBAOH in water | 51, 0.5C | −0.55, Ag/AgCl | 35%, 40, 0.5C | 141 |
|
145 | 90 | 10:56:24:10 145:graphite:VGCF:PVdF |
0.1 M TBAOH in water | 40, 1C | −0.55, Ag/AgCl | 100%, 50, 1C | 141 |
|
146 | 92 | 10:56:24:10 146:graphite:VGCF:PVdF |
0.1 M TBAOH in water | 60, 1C | −0.60, Ag/AgCl | 100%, 100, n.r. | 141 |
| Conjugated polymers | ||||||||
|
134 | n.r. | 70:20:10 133:CB:CMC |
1 M LiPF6 1:1 EC:DMC (v/v) |
65, 0.05 A g−1; 17, 2.0 A g−1 | 0–3.5 Vb,c | ∼100%, 1000, 1.0 A g−1 | 138 |
| Catholyte, # | Anolyte, # | Electrolyte | Operating potentialb (V) | Capacity (Cv), current, CE, VE, EE | Cycling stability (retention cycles, current) | Energy density (ED), power density (PD), diffusion coefficient (Do, cm2 s−1), charge transfer rate constant (k, cm s−1) | Ref. |
|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a The power density was obtained at an operating temperature of 45 °C.b Based on an average of the discharge voltage. | |||||||
| Br2/Br− |
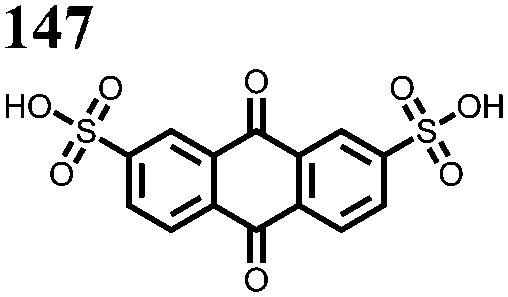
|
1 M 147 in 1 M H2SO4, 3 M HBr for catholyte | 0.69–0.92, 10−90% SOC | n.r. | 99%, 15, 0.5 A cm−2 | ED: >50 W h L−1 >50 W h kg−1 | 142 |
| PD: 0.6 W cm−2 at 1.3 A cm−2 (90% SOC), 0.246 W cm−2 (10% SOC) | |||||||
| D o: 3.8 × 10−6 | |||||||
| k: 7.2 × 10−3 | |||||||
| 147 in 1 M H2SO4, Br2 in 3 or 3.5 M HBr | 0.8 | n.r. | n.r. | PD: 1 W cm−2 | 143 | ||
|
|
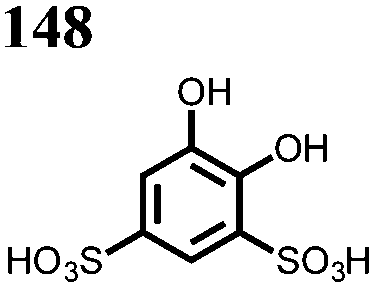
1 M 148 sulfuric acid; 0.2 M 149, 0.5 M 150 | 0.6, 100% SOC | n.r. | 100%, 12, n.r. | D o: 148: 3.8 × 10−6149: 3.40 × 10−6150: 3.71 × 10−6 | 144 |
|
k: 148: 1.55 × 10−4149: 1.52 × 10−4150: 2.25 × 10−4 | 144 | |||||
|
|
2 M NaCl | 1.1, 100% SOC | C v: 8.2 A h L−1 | 80%, 10000, 20 mA cm−2 |
D o: 151: 7.0 × 10−8152: 7.6 × 10−7 | 146 |
| CE: ∼99% | ED: 8.0 W h L−1 | ||||||
| EE: ∼75–80% | |||||||
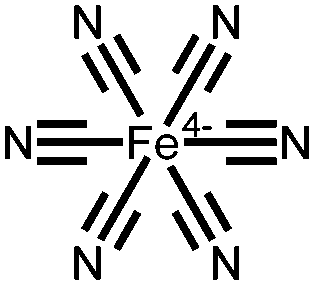
|
|
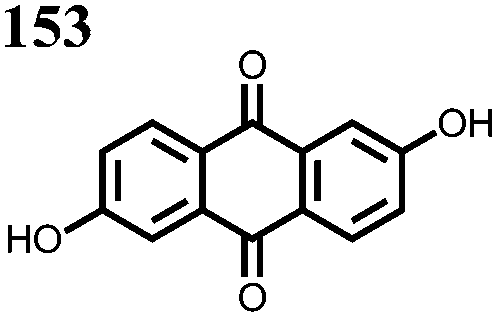
0.5 M 153 1 M KOH | 1.2, 50% SOC | C v: 27 A h L−1 | 84%, 100, 0.1 A cm−2 | ED: 6.8 W h L−1 | 147 |
| CE: >99% | PD: >0.45 W cm−2, 0.7a W cm−2 | ||||||
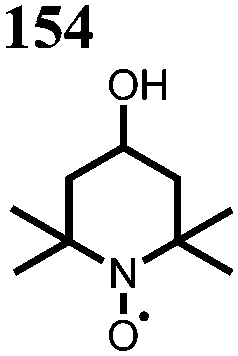
|
|
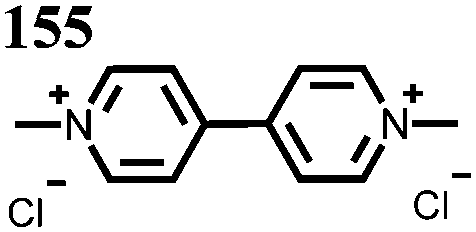
0.5 M 155, 1.5 M NaCl | 0.9 | C v: 9.58 A h L−1 | 89%, over 100 cycles, 60 mA cm−2 | D o: 154: 2.95 × 10−5155: 2.57 × 10−5 | 148 |
| CE: >99% | k: 154: 2.6 × 10−4155: 2.8 × 10−4 | ||||||
| VE: 62.1% | |||||||
| EE: 62.5% | |||||||
| Catholyte, # | Anolyte, # | Electrolyte | Operating potentiald (V) | Capacity (Cv or Csp), current, CE, VE, EE | Cycling stability (retention cycles, current,) | Energy density (ED), power density (PD), diffusion coefficient (Do, cm2 s−1), charge transfer rate constant (k, cm s−1) | Ref. |
|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a Microspheres of 78 were used in this study.b The capacitance value reported is based on the mass of the limiting solution.c These values were obtained at operation temperatures of 60 °C.d Based on an average of the discharging voltage. The abbreviations used in the table are defined as: TEABF4 = tetraethyl ammonium tetrafluoroborate; TEATFSI = tetraethylammonium bis(trifluoromethylsulfonyl)imide. | |||||||
|
|
8.4 g L−1 polythiophene, 2 g L−1 Ketjen black in 1.0 M TEABF4 PC | 2.5 | C sp: 110 mA h g−1, 0.5 mA cm−2 | 100.2 ± 2.4%, per cycle, 0.5 mA cm−2 | n.r. | 149 |
| CE: 77.5% | |||||||
| VE: 78.6% | |||||||
| EE: 60.9% | |||||||
|
|
0.1 M 156 or 157, 1.0 M TEATFSI DME | 2.37 | C v: 1.04 A h L−1, n.r. | 90%, 50, 10 mA cm−2 | n.r. | 150 |
| CE: ∼94% | |||||||
| VE: ∼86% | |||||||
| EE: ∼82% | |||||||
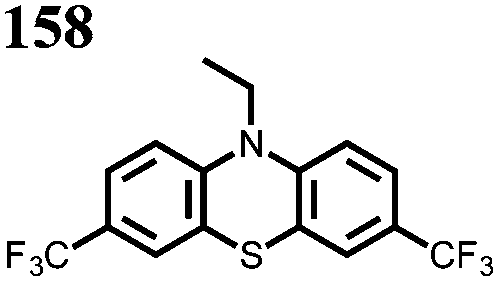
|
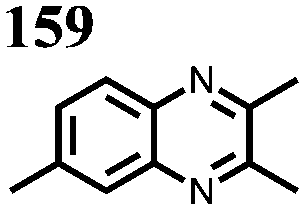
|
0.05 M 158 or 159, 0.2 M LiBF4 PC | 1.4 | C sp: 0.62b mA h g−1 | 0%, 100 cycles, n.r. | n.r. | 151 |
| CE: 92% | |||||||
| n/a |
|
Neutral 1.6 M in MeCN, fully reduced 62 ± 7 mM in MeCN | −1.1, −1.48 vs. Ag/Ag+ | n.r. | n.r. |
D
o: 1st reduction: 1.1 × 10−5
2nd reduction: 1.8 × 10−5 |
152 |
|
k: 1st reduction: 6.0 × 10−3
2nd reduction: 4.7 × 10−3 |
|||||||
|
|
50 mM compound 3:2 MeCN:PhMe 100 mM TBAPF6 |
∼2.35/∼1.20 | CE: 81–61% | n.r. | D o: 10−7 to 10−6 for all redox states | 153 |
| VE: 53–44% | |||||||
| EE: 43–28% | k: ∼10−2 for all redox states | ||||||
|
Lithiated graphite felt | 2.0 M TEMPO, 2.3 M LiPF6 EC:PC:EMC 4:1:5 (w/w/w) |
3.5 vs. Li/Li+ | CE: 84% | ∼80% over 100 cycles, 5.0 mA cm−2 | ED: 126 W h L−1 | 154 |
| VE: 82% | |||||||
| EE: 69% | |||||||
|
Li metal | 1 mM active material 0.5 M LiBF4 PC | 3.9 vs. Li/Li+ | EE: 80.9% | ∼75% over 30 cycles, 0.4 mA | D o: 1.8 × 10−6 | 155 |
|
Li metal | 0.05 M 164 1.3 M LiTFSI TEGDME | ∼2.20 vs. Li/Li+ | C sp: 172 mA h g−1, 24 mA g−1 | ∼70% over 100 cycles, 24c mA g−1 | n.r. | 156 |
|
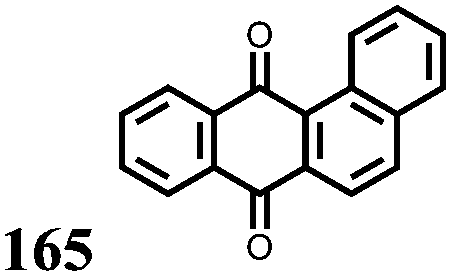
Li metal | 0.05 M 165 1.3 M LiTFSI TEGDME | 2.24, 2.43 vs. Li/Li+ | C sp: 169 mA h g−1, 24 mA g−1 | 93.5%, 100, 24c mA g−1 | ED: 55 W h L−1 | 156 |
| CE: ∼100% | |||||||
| EE: ∼80% | |||||||
| Structure | # | Electrode composition | Electrolyte | Redox potential (V), reference | Counter electrode, operating potential (V) | Capacitance (F g−1), current | Cycling stability (%, cycles, current, CE) | Ref. |
|---|---|---|---|---|---|---|---|---|
| n.r. denotes a value not reported.a Capacitance retention was reported for a device cycled at a 150° bending angle.b Capacitance loss occurred only in the first few cycles, followed by slow current decay.c Thermal cyclodebromination was performed at 500 °C.d The capacitance values reported are for films of thicknesses 62, 98 and 250 nm.e The capacitance retention reported is for films of thickness 98 nm and 250 nm.f The values reported are in electrolyte TBASbF6.g The values reported are in electrolyte TEABF4. The abbreviations used in the table are defined as: GNP = graphene nanoplatelet; FWNT = few-walled carbon nanotubes; PVA = poly(vinyl alcohol); EMIMBF4 = 1-ethyl-3-methylimidazolium tetrafluoroborate; PVdF-co-HFP = poly(vinylidenefluoride-co-hexafluoropropylene); PMMA = poly(methyl methacrylate); DCM = dichloromethane; BMIMPF6 = 1-butyl-3-methylimidazolium hexafluorophosphate; Fc = ferrocene; TBASbF6 = tetrabutyl ammonium hexafluoroantimonate. | ||||||||
| Positive charge-accepting | ||||||||
|
166 | GO and 166 on Pt foil | 1 M H2SO4 | n.r. | 166, 1 | Device: 441, 1 A g−1; 353, 20 A g−1 | Device: 86%, 10000, n.r., n.r. |
159 |
| on GO | Free-standing film | H2SO4, PVA (∼10:10 wt%) |
n.r. | 166, 1 | Device: 412, 1 A g−1; 304, 20 A g−1 | Device: 87%, 10000, 10 A g−1, n.r.a |
159 | |
|
167 | 73:12:10:5 167:GNP:acetylene black:PTFE |
1 M H2SO4 | 0–1, SCE | n.r. | Single electrode: 206, 1 A g−1; 186, 3 A g−1 | Single electrode: 78%, 1000, 1 A g−1, n.r. | 160 |
|
168a | 168a electro-polymerized onto oxidized FWNTs | 1 M LiPF6 3:7 EC:DMC (v/v) |
1.5–4.5, Li/Li+ | Li, 4.5 | Device: ∼60, 0.05 A g−1 | n.r. | 161 |
|
168b | 168b electro-polymerized onto oxidized FWNTs | 1 M LiPF6 3:7 EC:DMC (v/v) |
1.5–4.5, Li/Li+ | Li, 4.5 | Device: ∼210, 0.05 A g−1; 147, 10 A g−1 | Device: ∼100%, 1000, 10 A g−1, ∼100%; 85%, 10000, 10 A g−1, n.r. |
161 |
|
168c | 168c electro-polymerized onto oxidized FWNTs | 1 M LiPF6 3:7 EC:DMC (v/v) |
1.5–4.5, Li/Li+ | Li, 4.5 | Device: ∼113, 0.05 A g−1; 79.1, 10 A g−1 | n.r. | 161 |
|
169 | 85:10:5 169:CB:PTFE |
EMIMBF4 | n.r. | 169, 3.0 or 3.5 | Device: 151.3, 0.1 A g−1 (3 V) | Device: 85%, 10000, 10 A g−1, n.r. (3 V) |
162 |
|
170 | 20:70:10 170:CB:PTFE |
0.1 M TBAClO4 | 0–0.8, Ag/Ag+ | n.r. | Single electrode: 170–100%: 167, 100 mA g−1; 113, 2000 mA g−1 170–50%: 124, 100 mA g−1; 101, 2000 mA g−1 | Single electrode: 170–50%: 100%, 100, 500 mA g−1, n.r. | 163 |
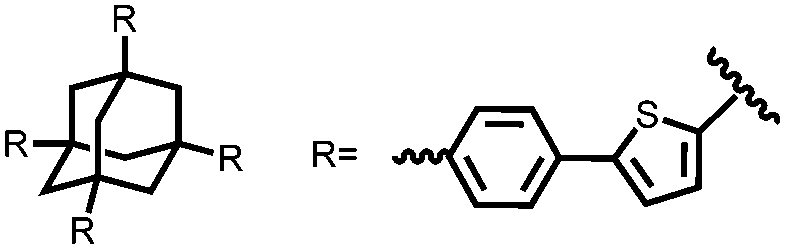
|
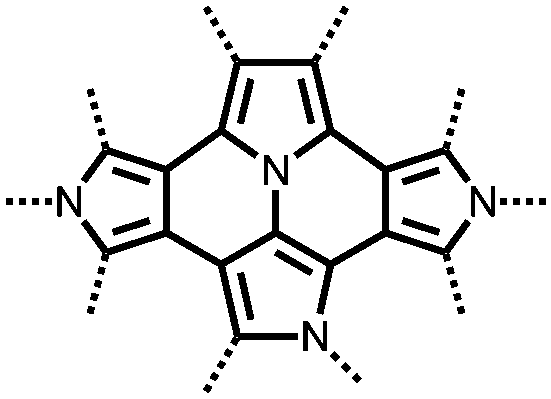
171 | 50:40:10 CB:171:PVdF-co-HFP |
0.1 M TBAPF6 in MeCN, 15 wt% PMMA | n.r. | CB, 2.25 | Device: 4.01 ± 0.05 mF cm−3, 0.1 A g−1 | Device: 80%, 500, n.r., n.r.b | 164 |
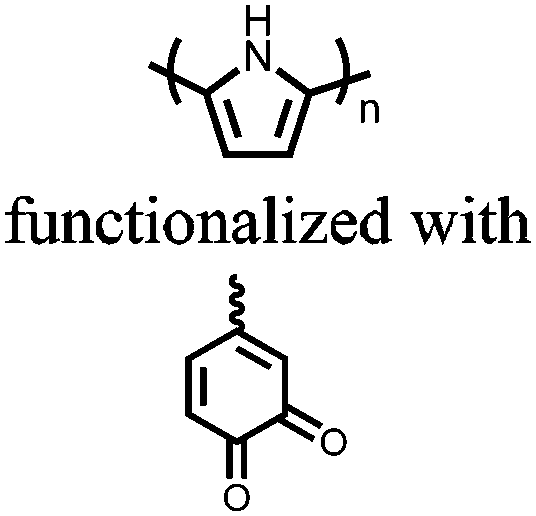
|
172 | 80:10:10 172:cSuper-P:PVDF on nickel foam |
1 M KCl | −1.0–0.2, SCE | 172, 0.8 | Single electrode: 423, 0.1 A g−1; 143 F cm−3, 1 A g−1; 1.94 mF cm−2, 1 A g−1 | Single electrode: 88.2%, 2100, 1 A g−1, n.r. | 165 |
| Device: 216, 0.5 A g−1;101 F cm−3, 0.5 A g−1; 1.37 mF cm−2, 0.5 A g−1 | Device: 78.3%, 2000, 0.5 A g−1, n.r.; 77.4%, 4000, 0.5 A g−1, n.r. | 165 | ||||||
|
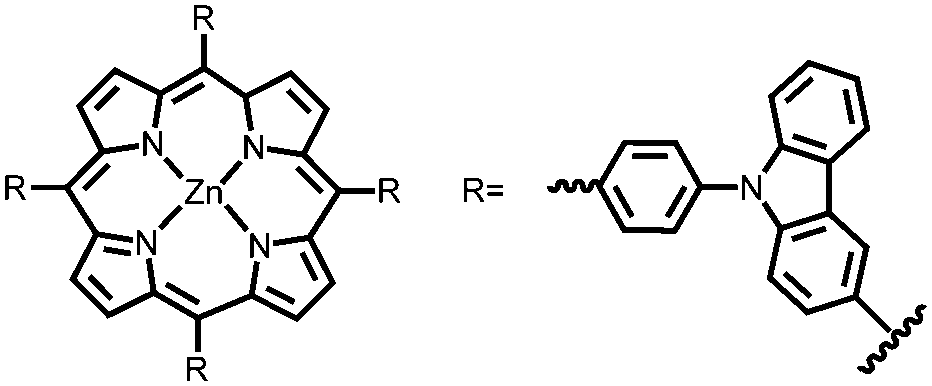
173 | Polystyrene opal template on Au/Cr coated Si substrate | 0.1 M aqueous HClO4 | −0.1–0.5, Ag/AgCl | n.r. | Single electrode: 385, 0.4 A cm−3; 288.8, 5.6 A cm−3 | Single electrode: 75%, 10000, 2 A cm−3, 97% |
166 |
|
174 | p-Doped 174 films | 0.1 M TBAPF6 in DCM | 0.4–1.4, Ag/Ag+ | 174, 1.4 | Device: 142, 5 A g−1, 99.4, 50 A g−1 | n.r. | 167 |
|
175 | 75:20:5 175:acetylene black:PTFE |
1 M H2SO4 | 0.2–0.9, Ag/AgCl | 175, 0.9 | Single electrode: 406.3, 0.2 A g−1; 256, 50 A g−1 | Device: 90.7%, 20000, 1 A g−1, n.r. |
168 |
|
176 | Electro-polymerized onto Pt wire | 0.1 M LiClO4 in MeCN | ∼0.1–1.1, Ag/AgCl | n.r. | Single electrode: 249.4, 5 A g−1; 226, 30 A g−1 | Single electrode: 25.5%, 500, 10 A g−1, n.r. | 169 |
| Electro-polymerized onto Pt wire | 0.1 M LiClO4 in MeCN and BF3OEt2 | ∼0.1–1.1, Ag/AgCl | n.r. | Single electrode: 392, 5 A g−1 | Single electrode: 67%, 500, 10 A g−1, n.r. | 169 | ||
| Electro-polymerized onto Pt wire | BMIMPF6 | ∼0.1–1.1, Ag/AgCl | n.r. | Single electrode: 209.4, 5 A g−1 | Single electrode: 32.4%, 600, 10 A g−1, n.r. | 169 | ||
|
177 | Electro-polymerized onto Pt | Electrode: 0.1 M TBAPF6 in DCM Device: 0.1 M | −0.5 to 1.1, Ag/AgCl | 177, ∼1.1 | Single electrode: 132.5, 1 A g−1; 123.6, 35 A g−1 | Single electrode: 53.1%, 1000, 10 A g−1, n.r. | 170 |
| Device: 31.2, 0.5 A g−1; 23.8, 17.5 A g−1 | ||||||||
|
178 | Electro-polymerized onto Pt | TBAPF6 in MeCN | −0.5 to 1.1, Ag/AgCl | 178, ∼1.1 | Single electrode: 135.4, 1 A g−1; 112.4, 35 A g−1 | Single electrode: 84.6%, 1000, n.r., n.r. | 170 |
| Device: 31.8, 0.5 A g−1; 26.9, 17.5 A g−1 | ||||||||
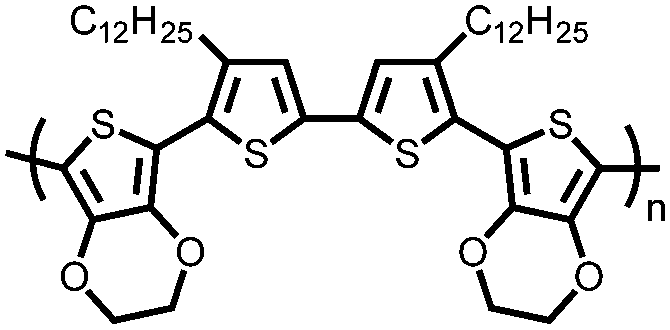
|
179 | Electro-polymerized onto Pt | Electrode: 0.1 M TBAPF6 in DCM Device: 0.1 M TBAPF6 in MeCN | −0.5 to 1.1, Ag/AgCl | 179, ∼1.1 | Single electrode: 129.3, 1 A g−1; 108.9, 35 A g−1 | Single electrode: 78.4%, 1000, n.r., n.r. | 170 |
| Device: 30.2, 0.5 A g−1; 25.5, 17.5 A g−1 | ||||||||
| Negative charge-accepting | ||||||||
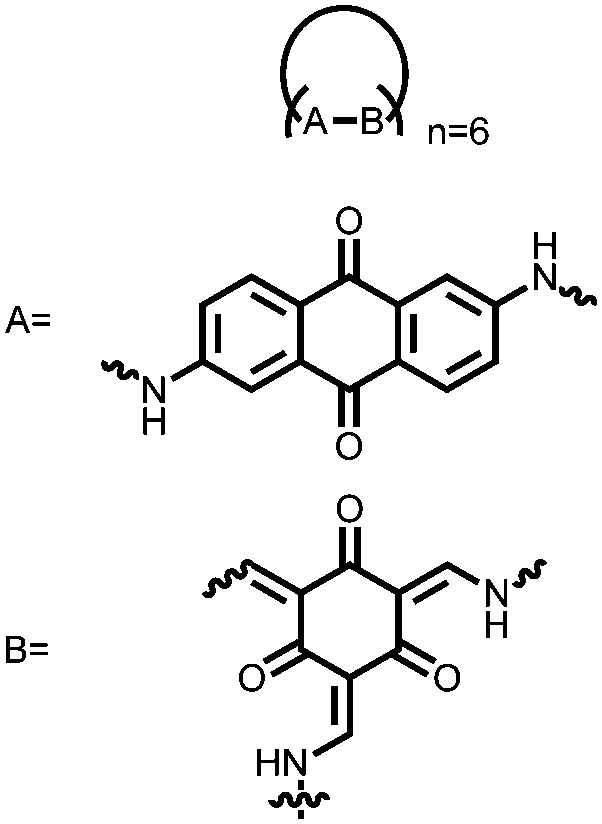
|
180 | 35:60:5 180:CB:PVDF |
1 M H2SO4 | −0.5 to 0.3, Ag/AgCl | n.r. | Single electrode: 48 ± 10, 0.1 A g−1 | Single electrode: 40 ± 9 F g−1, 5000 cycles, 0.1 A g−1, n.r. | 171 |
| Solvo-thermal growth onto Au | 0.1 M TBAPF6 in MeCN | −2.0 to 0.5, Fc/Fc+ | n.r. | Single electrode: 1.2–3.0 mF cm−2, 10–150 μAd cm−2 | Single electrode: 7% loss, 5000, 150 μA cm−2, n.r.e | 172 | ||
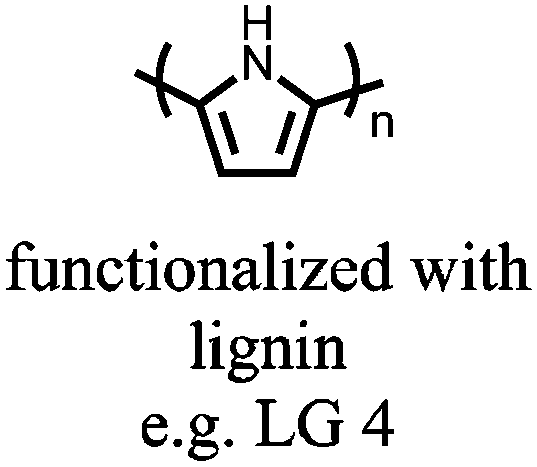
|
181 | 0.025 M pyrrole and 5 mg mL−1 lignin electro-polymerized onto Au | 1.0 M HClO4/water:MeCN (1:1) |
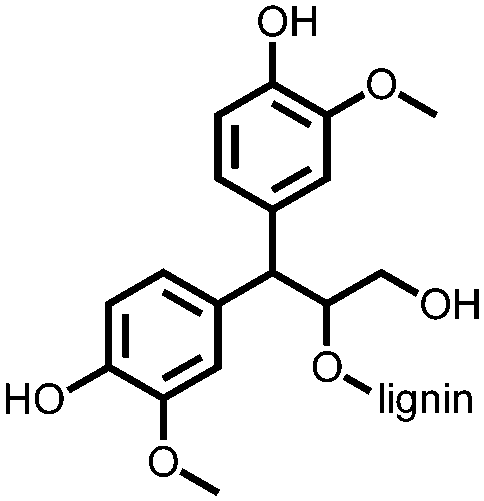
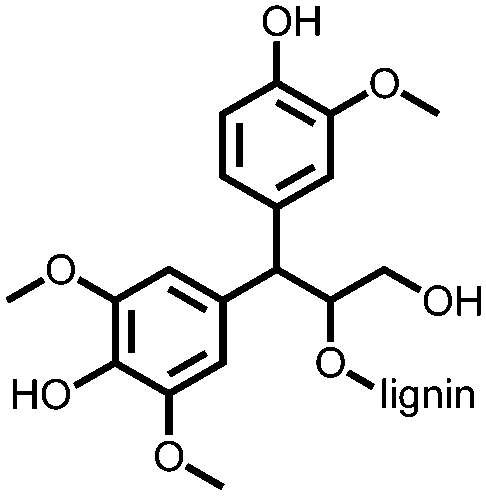
−0.2 to 0.8, Ag/AgCl/KCl (3.0 M NaCl) | n.r. | Single electrode: LG 1: 206 LG 2: 220 LG 3: 239 LG 4: 282 1 A/g LG 1-4, S/G ratio increased from 0.3–2.6 | n.r. | 173 |
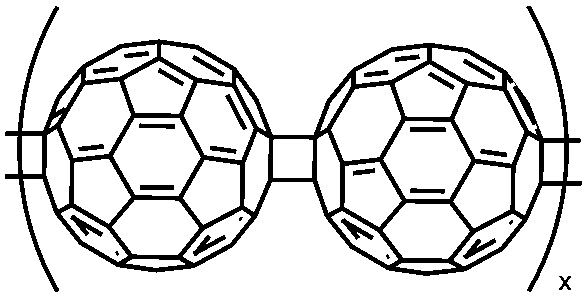
|
182 | Electro-polymerized onto Au-Kapton | 0.1 M TBASbF6 or 0.1 M TEABF4 in MeCN | −0.9 to −1.5, Fc/Fc+ | PEDOT, 2.2 | Single electrode: 110−220 F cm−3, 100–10 A cm−3; 87 F cm−3, 10 A cm−3 | Single electrode: n.r., n.r., 12 A cm−3, 60%;f n.r., n.r., 100 A cm−3, 39%;g n.r., n.r., 10 A cm−3, 25% | 174 |
| Device: 7.9 ± 1.1 F cm−3, 0.11 mA | Device: 50, n.r.; ∼0%, 250, n.r | 174 | ||||||
|
183 | 183, 3 wt% PTFE | 1 M TEABF4 1:1 PC:DMC |
−2.0 to 0.0, Ag/AgNO3 | 183, 2.0 | Device: 0.5, 0.1 mA | Device: ∼90%, 500, n.r., n.r. | 175 |
| Activated carbon, 2.0 | Device: 22.0, 0.1 mA | Device: ∼90%, 500, n.r., n.r. | 175 | |||||

|
184 | 184, 3 wt% PTFE | 1 M TEABF4 1:1 PC:DMC |
−2.0 to 0.0, Ag/AgNO3 | Activated carbon, 2.0 | Device: 4.92, 0.1 mA | n.r. | 175 |
|
185 | 185, 3 wt% PTFE | 1 M TEABF4 1:1 PC:DMC |
−2.0 to 0.0, Ag/AgNO3 | Activated carbon, 2.0 | Device: 4.94, 0.1 mA | n.r. | 175 |
| Donor–acceptor materials | ||||||||
|
186 | Electro-polymerized onto Au-Kapton | 1 M LiTFSI in PMMA 7% (m/v) in PC | −1.4 to 0.8, Ag/Ag+ | Device 1: 186, 0.5 Device 2: 186, 2.25 | Single electrode: 1.7 mF cm−2, 50 mV s−1 | Single electrode: n.r. | 176 |
| Device (0.5 V): 14, 50 mV s−1 | Device (0.5 V): 80%, 10000, 200 mV s−1, n.r. |
|||||||
| Device (2.25 V): n.r. | Device (2.25 V): ∼0%, 200, 50 mV s−1, n.r. | |||||||
|
187 | Electro-polymerized on Pt disk | 1 M TBAPF6, 10 wt% PMMA in MeCN | 2.5, Ag/Ag+ | 187, 2.5 | Device: 201, 100 mV s−1 | Device: n.r., n.r., 1 A g−1, 38% | 177 |
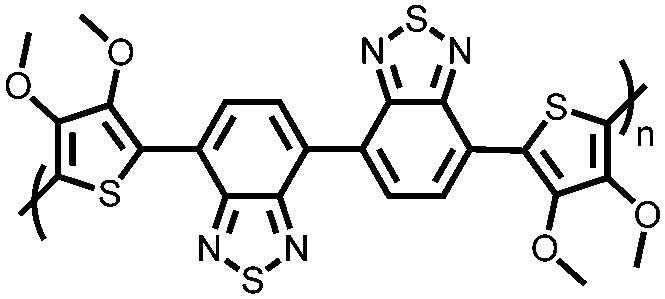
|
188 | Electro-polymerized on Pt disk | 1 M TBAPF6, 10 wt% PMMA in MeCN | 2.5, Ag/Ag+ | 188, 2.5 | Device: 91, 50 mV s−1 | Device: 75%, 100, 1 A g−1; 30%, 1000, 1 A g−1, 60% | 177 |
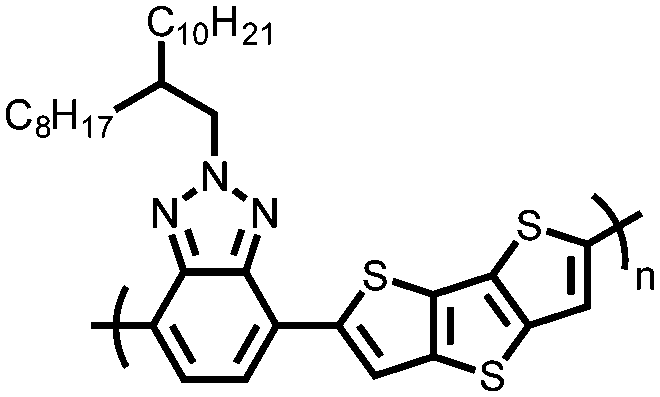
|
189 | Spray-coated onto SWCNT thin film | 0.1 M LiClO4 in PC | −0.3–1.5, Ag/AgCl | n.r. | Single electrode: 112.4, 1.0 A g−1; 59.8, 16.0 A g−1 | Single electrode: 82%, 12500, 1.0 A g−1, ∼100% |
178 |
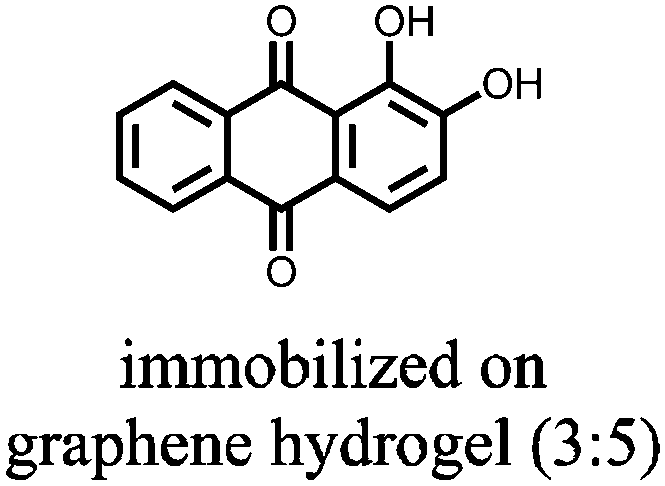
|
190 | 85:15 190:acetylene black |
1 M H2SO4 | −0.4–1.0, SCE | 190, 1.4 | Single electrode: 350, 1 A g−1; 213.5, 200 A g−1 | Device: 88%, 1000, 5 A g−1 | 179 |
| Device: 285.6, 1 A g−1; 180.5, 50 A g−1 | ||||||||
The specific capacity (Csp) is the measured capacity of the electrode at a specific current density for either charging or discharging. The Csp is reported in mA h g−1 and by measuring Csp at different rates (usually reported as a C-rate, where 1C is the amount of current it would take to collect the total charge of the Ctheor in 1 hour) the rate capabilities of the electrode can be determined. The Csp is typically calculated from galvanostatic charge/discharge curves using eqn (2):
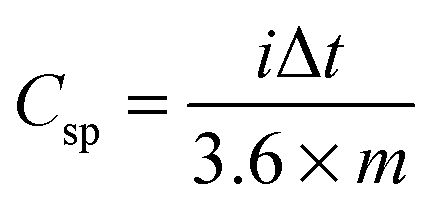 | (2) |
The coulombic efficiency (CE) is measured by dividing the Csp for discharging by the Csp for charging. This provides insight into the reversibility of the redox reactions and indicates whether any side reactions occur with the electrode and electrolyte. The CE is a good indicator of whether a stable solid electrolyte interface (SEI) is formed in the charging cycles and if the material itself will be stable upon extended cycling. If the CE is low in the first charging cycles but increases to ∼100% afterwards, it is typically attributed to SEI formation.
The cycling stability is an important parameter that quantifies the retention of capacity upon charging and discharging the electrode multiple times. Usually this measurement is performed under galvanostatic conditions and is reported as a percentage of the initial capacity after a specified number of cycles. The current density (or C-rate) must be specified for these measurements because the rate can have a significant effect on the cycling stability. This effect is especially pronounced if capacity fading is due to electrode dissolution, which is a common problem with organic electrode materials.
The potential at which the redox process(es) occur(s) is also a very important parameter. Combined with the capacity, the redox potential can be used to predict the energy density of the device when paired with an anode/cathode of known redox potential. To have a high energy density, the potential of cathode material should be as high as possible while that of anode material should be as low as possible within the electrochemical window of the electrolyte, or within the electrolytes' ability to form a stable SEI. Although an ideal battery maintains a constant voltage while it discharges, real batteries tend to have a decreased voltage with decreasing state-of-charge (SOC). This creates a sloping voltage plateau that is especially apparent in polymeric electrodes or in electrodes with multiple redox events.23 The reduction and oxidation peak splitting is also important to provide insight into electron-transfer kinetics, and to predict the energy efficiency of the device.
While energy and power density are important parameters to gauge the performance of energy storage, we chose to exclude them from our evaluation of solid organic electrode materials since they pertain to fully assembled devices and relate to the combined performance of all aspects of the device including both the anode and cathode, the electrolyte, membrane, and resistances associated with various aspects of the device. Additionally, it is important to report the electrode formulations and procedure for electrode manufacturing, electrode morphologies, electrode thicknesses, electrolyte, and the conditions under which the experiments are being performed. All of these factors can have an enormous effect on device performance. For example, in our lab we have observed that changes in the electrolyte solvent can influence the electrochemical properties, such as the capacity, by as much as an order of magnitude. Therefore, we encourage others to report the details of electrode preparation and testing in full.
2.2 Redox flow batteries
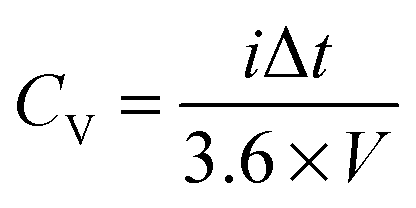 | (3) |
In terms of overall device performance, the energy density dictates the amount of energy the device can deliver. It is usually reported in W h L−1 and is calculated using eqn (4):
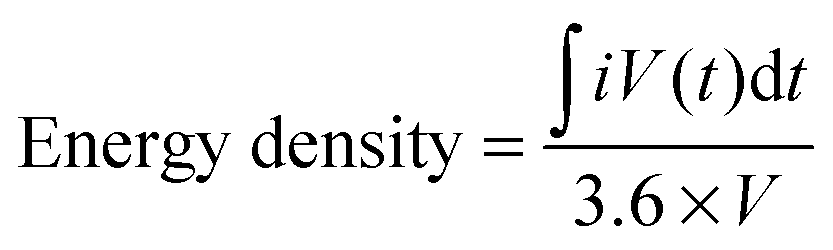 | (4) |
 | (5) |
2.3 Supercapacitors
SCs have power densities orders of magnitude higher than batteries and can store a significant amount of energy, although typically less than batteries. SCs are intermediate in terms of their power and energy densities compared to conventional capacitors and batteries (Fig. 2). As such, they are ideal for complementing or replacing batteries in applications that require rapid charge/discharge, such as load-leveling, storage of energy generated from intermittent renewable power sources, and for acceleration and braking in electric vehicles. SCs are classified into two categories based on their mechanism of charge storage: electric double layer capacitors (EDLC); and pseudocapacitors. EDLCs are the more traditional type of SCs, and store charge electrostatically between the electrode and electrolyte interface. This type of capacitance relies on a surface charging of conductive materials, usually HSACs, therefore capacitance is limited by surface area.27 Pseudocapacitors store charge through fast, reversible surface (or near surface) formal redox reactions. As Conway describes it, “pseudocapacitance arises when the extent of reaction, Q, is some continuous function of potential, V, so that a derivative, dQ/dV, arises that has properties of capacitance”.28 Pseudocapacitance arises from many different mechanisms and the reader is referred to the work of Conway for further information.28
 | ||
| Fig. 2 Ragone plot comparing the performance of various EESSs. | ||
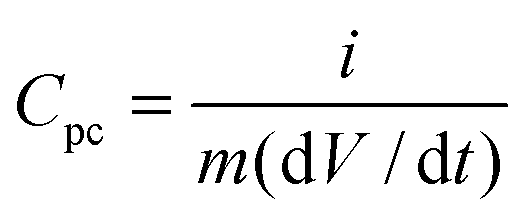 | (6) |
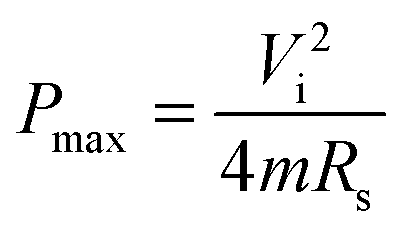 | (7) |
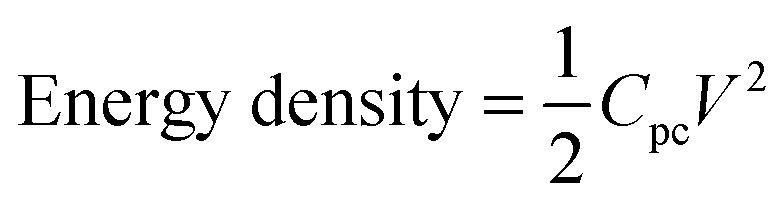 | (8) |
Importantly, SC electrode materials must have high cycling stability. Materials suitable for commercial devices must experience little capacity fade over thousands of cycles.
3. Solid electrode batteries
3.1 Metal-ion batteries
Organic materials can be used as the anode and/or cathode in metal-ion batteries. Typically, organic materials are synthesized in the neutral state without charge-balancing ions incorporated into their structure. In order for metal-ion batteries to function, the opposite electrode must contain the charge-balancing metal ions. For example, if a cathode material does not contain metal ions in its structure, the anode must contain metal ions and vice versa. In order to satisfy this requirement, the counter electrode is usually a reduced metal (e.g. magnesium, sodium, lithium) regardless of whether the organic electrode is the anode or cathode material. Using reduced metals as the counter electrode maximizes the operating potential of the battery when different cathode materials are tested. Recent work in organic electrodes for metal-ion batteries has focussed on a number of aspects in order to improve performance and the overall cost of the entire device. The relatively low potential at which organic electrodes operate limits the overall energy and power density when incorporated into a full device. By adding electron withdrawing groups (EWGs), the potential at which the organic cathode accepts an electron is increased. Conversely, adding electron-donating groups (EDGs) to anode materials lowers the reduction potential and also increases the operating potential of the device. These increase the overall operating voltage of the device, but also have the effect of decreasing the Ctheor since the EWG/EDGs typically add mass to the compound without affecting the number of electrons it can accept. Other popular strategies to tune the redox potential include substituting heteroatoms into the aromatic core, and developing different isomers without significantly changing the mass:charge ratio of the compound.
Another popular strategy to overcome the low voltages of organic compounds is to use compounds that inherently have a high mass:charge ratio. These have high a Ctheor and can exhibit high energy densities without necessarily having a high voltage. Obviously, the best solution would be to combine the two strategies of increasing voltage and Ctheor, however more work is needed to find an optimal trade-off between Ctheor and voltage.
Decreasing the cost of the electrode, especially the cathode, is a major motivation for the investigation of organic electrode materials. This is because the cathode in lithium-ion batteries can account for ∼30% of the cost of the device. Although the investigation of organic electrode materials with high performance is important, it is equally important to develop low cost materials for applications that do not necessarily require a high energy or power density. The use of electrolytes other than lithium can also greatly decrease the cost of the device. Although lithium has the lowest reduction potential and highest Ctheor out of all the alkali metals, it is also the most expensive. It is expected that the cost of lithium will continue to increase due to depletion of resources.2
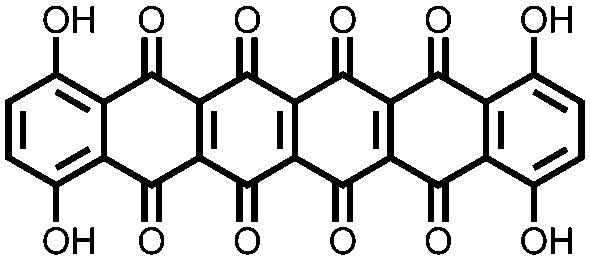
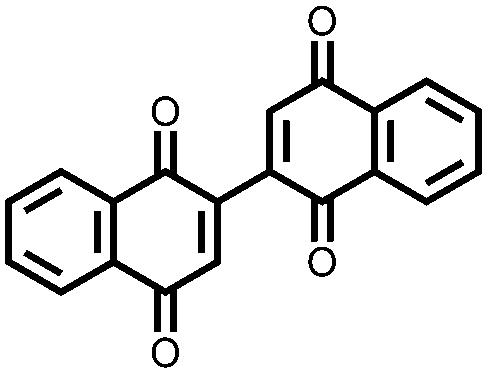
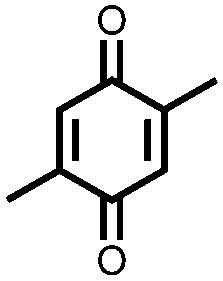
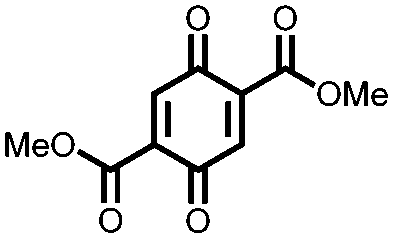
In an effort to both decrease the solubility and increase the capacity of quinones, Zou et al. synthesized a tetrahydro-hexaquinone, 1, which has a high Ctheor of 628 mA h g−1.29 When measured at 200 mA g−1 it reaches 54.1% of its Ctheor, retaining 26.5% of that value when the current is increased to 800 mA g−1. The electrode has a sloping voltage plateau between 3.5 and 2.4 V vs. Li/Li+ due to the complex redox chemistry of the multiple carbonyl groups on the compound. The large aromatic structure along with a lack of solubilizing alkyl groups make it less soluble than its parent anthraquinone. Nevertheless, slight solubility still causes a decrease in cycling stability to only 58.8% of the initial capacity after 40 cycles. Although increasing the size of the aromatic structure decreases solubility and can yield large Ctheor's, the proximity of the redox groups can impose electrostatic repulsion upon reduction. This will limit the Csp and may also disrupt the crystal packing leading to dissolution and capacity fading.
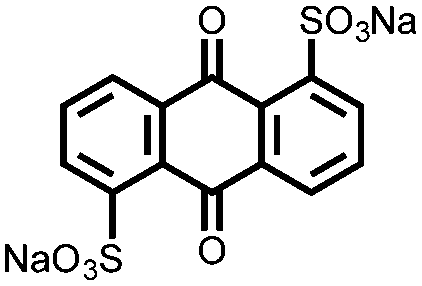
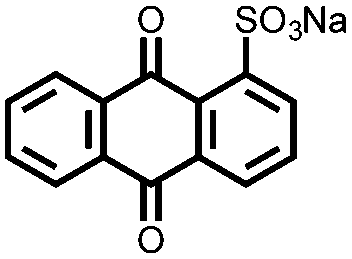
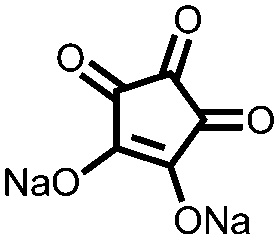
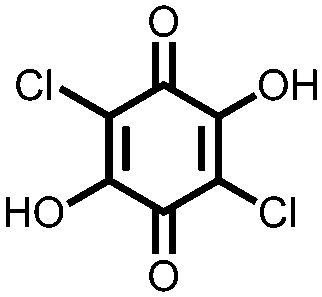
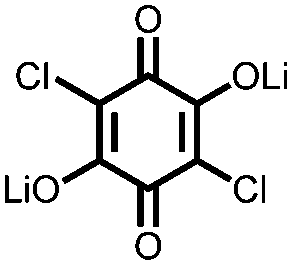
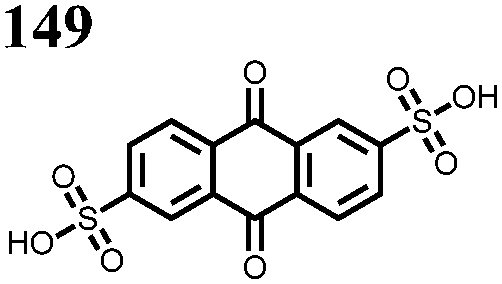
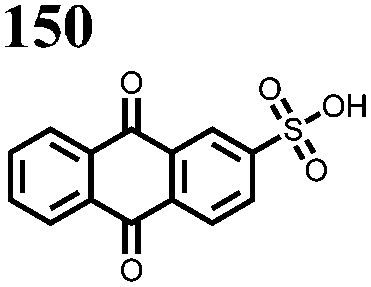
Another strategy to improve the cycling stability and also raise the reduction potential is to functionalize quinones with ionic groups to prevent dissolution in organic electrolytes. When anthraquinone was functionalized to form the mono- and disodium sulfate anthraquinones 2 and 3, both compounds have high Csp's, 130 and ∼150 mA h g−1 at 0.2C for 2 and 3 respectively, when studied in a lithium-ion battery.30 Compared to the mono-substituted compound, further substitution of the anthraquinone ring raises the average reduction potential by 150 mV due to the extra electron-withdrawing effect afforded by the additional sodium sulfate group. The additional ionic group also improves the cycling performance from ∼50% to 92% after 100 cycles at 0.1C. This is an effective way to both increase cycling stability and redox potential, however the heavy sulfate groups have a significant impact on the Ctheor. The trade-off between Ctheor and redox potential, and its effect on energy density, is a general concern when designing electrodes materials using this strategy.
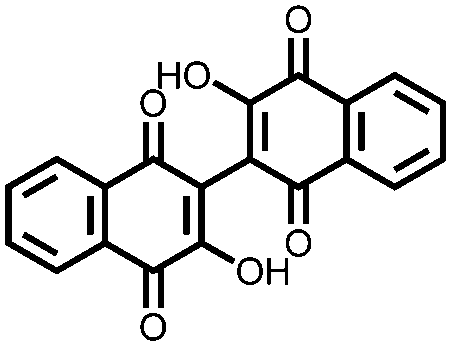
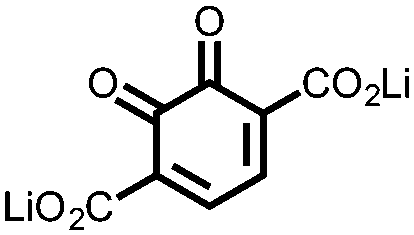
Using porous carbons that can entrap the redox active molecules to prevent dissolution is another strategy to improve cycling stability. Li et al. attempted to improve the cycling stability of bis-naphthoquinone 4 by impregnating it within mesoporous carbon CMK-3.31 They found that it achieves almost 100% of its Ctheor at 0.1C, which is an improvement from electrodes prepared by simply mixing with carbon black. The capacity retention is also improved compared to that of the conventional electrode, retaining 65.7% of the initial capacity after 50 cycles at 0.1C. In a more recent report, this method was extended towards compounds 5, 6, and 7.32 Using a highly concentrated ether based electrolyte, a high capacity (>97% of the Ctheor) is attained for each compound, as well as an impressive 83.9% capacity retention after 100 cycles at 0.2C for 5. The Csp for these compounds are very high, but the cycling stability is inadequate for practical use. An effective way to achieve higher Csp's is impregnating porous carbons with redox active materials. This improves active material usage and cycling stability, but cannot alleviate the problems associated with dissolution; an issue similar to what has been observed in metal–sulfur batteries. A combination of approaches to deal with solubility issues will be required in the future development of small molecule cathodes.
In another effort to improve the cycling stability and working potential of lithium-ion battery cathodes, a carbonyl group on anthraquinone was replaced with a thiocarbonyl group to form 8.33 It was found that the cathode prepared with 8 has a 222 mA h g−1Csp at 0.1C with two discharge plateaus at 2.7 and 2.1 V vs. Li/Li+. This material has improved cycling stability compared to the parent compound, retaining 33.8% of the initial capacity after 40 cycles, however the cycling stability is still relatively poor, attributed to side reactions and dissolution. The use of thiocarbonyls decreases solubility, but also increases charge carrier mobility, as demonstrated by work on thionated arylene diimides.34,35 It is therefore not surprising that thiocarbonyls have improved performance. This work demonstrates that substitution at redox active units modifies redox potential in a favourable manner. However, the cycling stability is still far from ideal, highlighting that the reversibility of the redox unit must be considered along with potential.
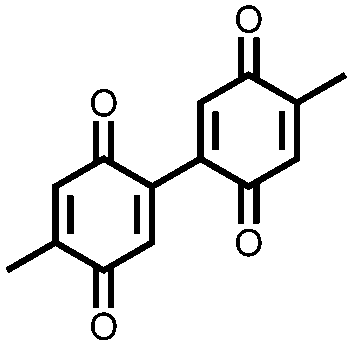
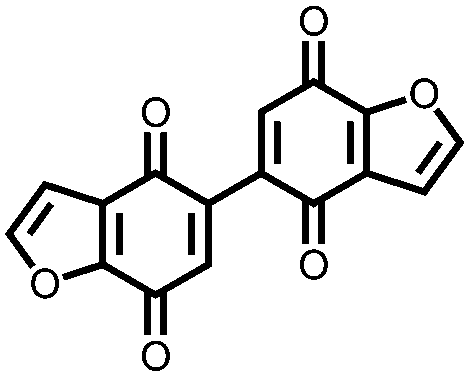
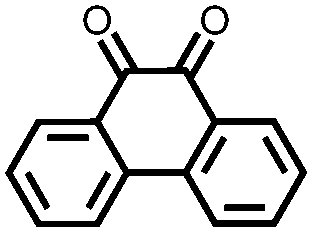
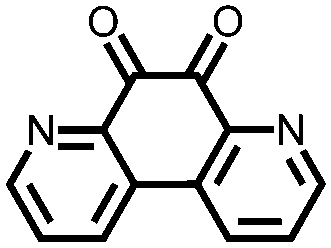
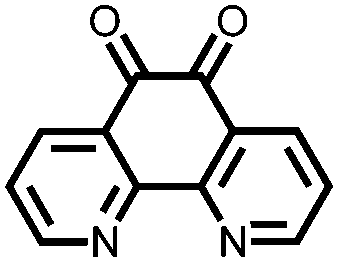
In order to increase the reduction potential of anthraquinone and phenanthrenequinone, 9, nitrogen containing heterocyclic versions were investigated using both computational chemistry and electrochemical analysis in lithium-ion batteries.36 The authors found that replacing the carbon atoms in the 1, 4, 5, and 8 positions with nitrogen to form 10 increases the reduction potential from 2.13 V in anthraquinone to 2.75 V vs. Li/Li+. Similar results were obtained for compounds 11 and 12. In addition to the inherent voltage gain by the electronegative elements, positioning the nitrogens such that they can coordinate with the lithium counter-ions increases the voltage even further. This work demonstrates improving the voltage of organic carbonyl cathodes without having a detrimental effect on Ctheor. Combining this approach with ones that improve cycling stability, such as addition of ionic groups and/or impregnating into porous carbon, might lead to even higher performance electrodes.
Using crystalline nanostructures of small organic molecules is another strategy to enhance cycling stability and performance. Crystalline nanowires of 13 were synthesized in order to accommodate the lithium-ion insertion associated with charging as well as overcome conductivity issues.37 The nanowire electrodes have a 200 mA h g−1Csp at 0.1C and 100 mA h g−1 at 6C. The nanowires retain 100% of their initial capacities after 110 cycles at 0.2C, which is much better than the electrodes fabricated from microwires or the bulk materials. This increased capacity retention is due to the nanowire electrode's ability to accommodate lithium-ion insertion without fracturing (Fig. 3). This is an excellent demonstration of the profound effect that nanostructuring has on the performance of crystalline organic electrode materials. It is important to note that although the polarization of the electrode remains close to 400 mV, it decreases with the size of the nanostructures. Further decreasing the size of the nanostructure should lead to an even lower polarization, allowing for an even higher performing material with an increased rate capability.
 | ||
| Fig. 3 SEM images of 13 (a) micropillars, (b) microwires, and (c) nanowires after 100 cycles. (Reproduced from ref. 37, reprinted with permission, copyright 2014, American Chemical Society). | ||
A clever way to increase the voltage in carbonyl containing molecules without adding EWGs or electronegative atoms was introduced by Gottis et al.38 The authors examined the voltage gain in lithiated enolates with carbonyls in either the ortho- or para-position. They found that when the carbonyl groups were in the ortho-position, compound 14, a voltage gain of ∼300 mV compared to the para regioisomer is observed. This lithium enolate is very stable with a 100% retention of capacity after 30 cycles, however, only 43.6% of the Ctheor is obtained. Although the electrode morphology is not reported, the material usage may be improved by decreasing the domain size of the active material to improve homogeneity. Shimizu and coworkers studied the effect of lithiocarboxy groups on the cycling stability of carbonyl containing compounds 15, 16, and 17.39 In all cases, the cycling stability is improved without significantly affecting the voltage compared to the compounds without lithiocarboxy groups. The improved stability is attributed to strong intermolecular interactions between the lithiooxycarbonyl groups preventing dissolution (Fig. 4). In their best performing compound, 17, the authors observe a 217 mA h g−1Csp at 0.2C with a potential of 2.39 V vs. Li/Li+, which decreases to 34.7 mA h g−1 at 5C and retains 86% capacity after 20 cycles at 0.2C. This shows how to improve cycling stability by taking advantage of the ability of lithium to form an ionically bonded polymeric network. However, the active material usage is low. Although the morphology of the electrode was not reported, the active material usage could be improved by decreasing the domain size of active material in the electrode and improving homogeneity. In an interesting report, Kim and coworkers re-examined the dilithium rhodizonate salt, 18, that has a high Csp (580 mA h g−1 at 50 mA g−1), but only a 20% retention in capacity after 25 cycles.40 By using a combination of first principles and X-ray studies, they concluded that the large capacity fading observed for 18 is due to a crystal structure change causing exfoliation during lithium extraction. This report highlights the importance of designing a crystal structure that does not undergo a phase change after lithium insertion.
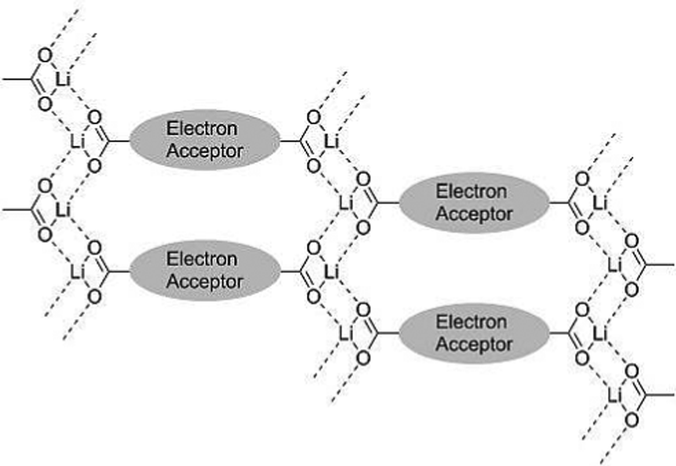 | ||
| Fig. 4 Schematic of the intermolecular interactions provided by lithiooxycarbonyl groups preventing dissolution in 15, 16, and 17. (Reproduced from ref. 39, reprinted with permission, copyright 2014, Elsevier Ltd.). | ||
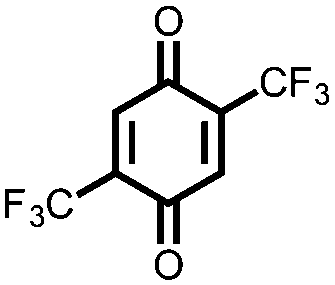
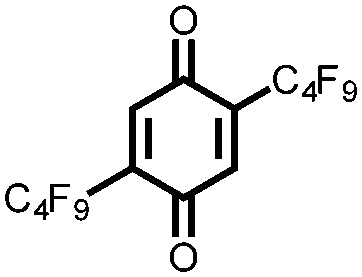
In another effort to increase the voltage of organic cathode materials, Yokoji and coworkers examined the use of electron-deficient benzoquinones 19–22.41 They found that the addition of fluorinated EWGs improves the voltage by up to 600 mV from 2.5 V for 19 to 3.1 V vs. Li/Li+ for 21 and 22, albeit with a decrease in Ctheor due to the addition of redox-inactive mass. However, for compounds 21 and 22, the Csp exceeds the Ctheor, which the authors hypothesize is due to the ability of each molecule to accept extra charges because of the highly EWGs. The extra capacity in the highly fluorinated compounds is interesting, but the extra mass lowers the Ctheor and is not offset by the increased voltage and Csp. The authors also observe an increase in stability for the fluorinated compounds due to a stabilization of the radical and dilithiated intermediate by lithium–fluorine interactions, although capacity fading is still rapid due to dissolution. An alternative route to increase the cycling stability of a battery without significant modification of the chemical structure is to use a solid-state electrolyte. Zhu and coworkers reported the use of pillar[5]quinone, 23, as the cathode material in an all-solid-state battery that exhibits a 418 mA h g−1Csp and a 94.7% capacity retention after 50 cycles at 0.2C.18 Although this is an effective strategy to increase cycling stability, the rate capability of the cathode is relatively low with ∼50% retention of capacity when discharged at a rate of 1C, likely due to the low ionic conductivity of the solid-state electrolyte rather than the inherent properties of the cathode.
To design small organic molecule cathodes with high voltages, Wu and coworkers examined the correlation between the aromaticity and voltage of carbonyl containing polycyclic aromatics with density functional theory (DFT).42 The authors found that molecules that form higher numbers of Clar sextets, or have a positive change in aromaticity, upon reduction have a higher voltage for reduction (Fig. 5). Using this principle, they designed 24, which has a 2.77 V average voltage vs. Li/Li+ and a 243 mA h g−1Csp, 60.3% of its Ctheor. This is a straightforward strategy to design high voltage cathodes that takes into consideration the difference in aromaticity in the neutral and reduced compound. Voltages over 3.0 V vs. Li/Li+ should be attainable by combining this strategy with the introduction of EWGs or heteroatoms to further increase the electron affinity. Wang and coworkers reported an all-organic sodium-ion battery that uses different oxidation states of the quinone compound, 25, as both the cathode and anode material.43 The quinone groups on the molecule afford redox activity at high potentials while the carboxylate groups are redox-active at low potentials (Fig. 6). For the cathode, a 183 mA h g−1Csp at 0.1C is attained, which is ∼98% of its Ctheor. When constructed as a half-cell with sodium as the counter and reference electrode, the cathode has two voltage plateaus at 2.36 and 2.10 V vs. Na/Na+ and retains 84% of its initial capacity after 100 cycles at 0.1C. Improving cycling stability and investigating the capacity fading mechanism are important future topics for this material.
 | ||
| Fig. 5 Correlation between the change in aromaticity and the reduction potential of small molecule quinones. (Reproduced from ref. 42, reprinted with permission from The Royal Society of Chemistry). | ||
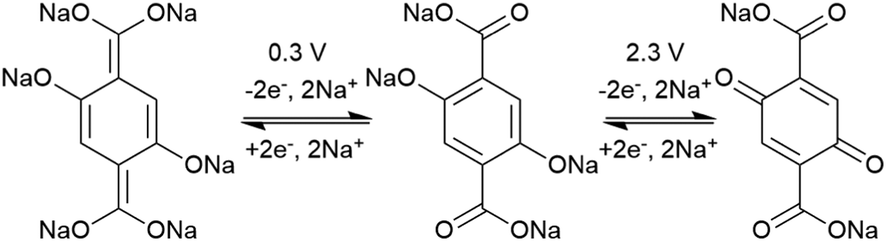 | ||
| Fig. 6 Redox mechanism of compound 25 at low and high potential. Adapted from ref. 43. | ||
To determine the effect of halogen substitution on the reduction potential of quinones for sodium-ion batteries, Kim and coworkers used DFT to design a tetrachloroquinone, 26, for sodium-ion battery cathodes.44 The compound exhibits a 150 mA h g−1Csp at 10 mA g−1 with relatively high voltage plateaus at 2.9 and 2.6 V vs. Na/Na+. Unfortunately, it undergoes rapid capacity fading due to dissolution, retaining only 5% of the capacity after 20 cycles. However, capacity retention is improved to ∼25% when 26 is impregnated into a porous carbon. The positive effect that substitution with chlorine atoms has on voltage is impressive, but capacity fading is still the largest barrier towards a high performance material. Small molecule quinones can also be used for multivalent batteries such as magnesium-ion batteries. Dimethoxybenzoquinone (27) is a promising cathode material for magnesium-ion batteries.45 By cycling in a sulfolane electrolyte, 27 displays a 100 mA h g−1Csp after 10 cycles at 10 mA g−1 and retains 20% of this capacity after 50 cycles. Unfortunately, a high degree of polarization is required for charging and discharging, with a 2.9 V potential needed for charging, while only having a discharge voltage of 0.4 V vs. Mg/Mg2+. In an effort to improve reversibility, 27 was later examined in a magnesium bis(trifluoromethane sulfonyl)imide (Mg(TFSI)2)–MgCl2 dimethoxyethane (DME) electrolyte.46 This electrolyte greatly improved the performance of 27, with the electrode having a 226 mA h g−1Csp at 0.2C on the first cycle and a 2.0 V discharging voltage vs. Mg/Mg2+. However, due to the solubility of 27 in the electrolyte, only 74 mA h g−1 is retained after 30 cycles. Although the performance of quinone materials in magnesium-ion batteries is limited, magnesium-ion battery chemistry is still in its infancy. Magnesium-ion battery electrolytes that promote reversible electrochemistry at both the anode and cathode are needed, and this should allow organic electrode materials to be seriously considered.
Arylene diimides are another popular class of small molecules that have been investigated for metal-ion battery cathodes. Their low cost, ease of functionalization and relatively low solubility make them attractive candidates for energy storage. Due to steric hindrance and electrostatic repulsion, the reduction of the third and fourth carbonyl groups is irreversible, leading to decomposition (Fig. 7). Therefore, the maximum number of electrons that each arylene diimide molecule can reversibly accept is two, limiting the Ctheor.47 Arylene diimides suffer from the same low voltage problem as other organic molecules, having a reduction potential of ∼2.5 V for the unsubstituted naphthalene diimides (NDIs). The most effective strategy for improving the performance of this class of molecules is increasing the reduction potential without adding a large amount of mass.
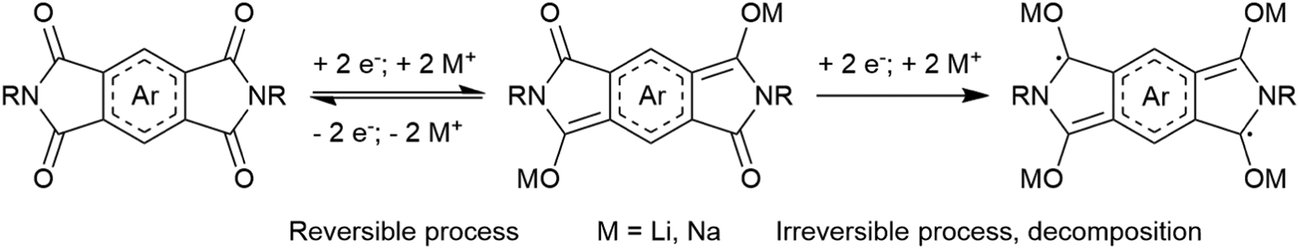 | ||
| Fig. 7 Arylene diimide reduction mechanism showing the decomposition during the third and fourth reductions. Adapted from ref. 103. | ||
In an effort to increase the voltage of NDIs in lithium-ion batteries, Vadhera and coworkers introduced different substituents onto the NDI core in compounds 28–34.48 They observed that attachment of cyano groups to the NDI core in compound 34 increases the voltage from 2.55 V in the parent compound 32 to 2.90 V vs. Li/Li+ for the first reduction. It was also observed that when the diimide nitrogens were functionalized with hexyl groups, the capacity faded rapidly due to dissolution. However, when the unsubstituted compounds are used, a higher cycling stability is observed but with a low capacity due to an unfavourable crystal packing. The use of hexyl side chains will undoubtedly lead to problems with dissolution and lower the Ctheor. Although the unsubstituted NDIs reported in this work performed poorly, better results may be obtained by improving the morphology. Improved morphology can be achieved with processing techniques that improve homogeneity and decrease the domain size of the active material. When a benzoic acid functionalized perylene diimide (PDI) 35 is treated with hydrazine, it performs better compared to an untreated sample.49 Hydrazine increases the conductivity of the electrode and therefore improves material usage as well as the rate capabilities with an 85 mA h g−1Csp at 1C and 68 mA h g−1 at 10C with an 88% capacity retention after 200 cycles at 5C. The low Ctheor of 35 limits performance, but the hydrazine treatment that improves conductivity could also be applied to NDI analogs that have higher Ctheor's, and should be investigated in the future.
In an effort to decrease solubility and improve cycling stability of NDIs without adding excess mass, a triangular shaped NDI, 36, was synthesized.50 The active material usage is ∼95% with a 146.4 mA h g−1Csp at 0.1C and an excellent rate capability with a 58.1 mA h g−1Csp at an enormous rate of 100C. The low solubility of this compound affords good cycling stability with ∼60% retention after 300 cycles at 10C. 36 is slightly soluble in the neutral and reduced states and this leads to capacity fading. The high rate performance is attributed to lithium-ion diffusion through the triangular channels in the NDI triangle. Additionally, this triangular arrangement electronically couples the redox units allowing for electronic conductivity. Synthesizing rigid materials with inherent porosity is an effective way to make insoluble materials that can have high rate capabilities. The cycling stability could be improved by using the perylene diimide derivatives that are generally less soluble, or by covalently linking triangular shaped units together to form a network-like structure.
Arylene dianhydrides and diimides can also be used for sodium-ion battery cathodes. Luo and coworkers successfully developed a sodium-ion battery cathode with perlyene dianhydride 37.51 The compound exhibits a high rate capability with a 145 mA h g−1Csp at 10 mA g−1 and 91 mA h g−1 at 1000 mA g−1. 37 also has a moderate cycling stability with a 69% retention of the initial capacity after 200 cycles at 1.4C and a high CE of ∼100% after the first few cycles. This study demonstrates that inexpensive organic pigments can be successfully applied as electrode materials for sodium-ion batteries. 37 has also been used as a potassium-ion battery cathode.52 Using potassium as the anode, the Csp is 131 mA h g−1 at 10 mA g−1 and 73 mA h g−1 at 500 mA g−1. Deng and coworkers examined crystalline PDI 38, also as a cathode for sodium-ion batteries.5338 has a relatively low voltage compared to the dianhydride 37, with a 1.7 V reduction plateau vs. Na/Na+ (2.3 V for 37). 38 has a 138.6 mA h g−1Csp at 10 mA g−1, which slightly exceeds its Ctheor, and a 90% retention of its initial capacity after 300 cycles. Organic pigments can be used as inexpensive battery electrodes with respectable performances, however issues such as cycling stability and voltage still need to be addressed.
An increasingly popular strategy for developing sustainable lithium-ion battery electrodes is to use bio-derived electrode materials. Lee and coworkers built upon their previous work using a flavin unit, 39, as a cathode material. By hybridizing the flavin unit with single-walled carbon nanotubes (SWCNTs), the electrode uses 98% of the Ctheor to provide a high Csp of 204 mA h g−1 at 1C compared to 84% of the Ctheor at 10 mA g−1 in the previous report.54,55 Hybridizing with SWCNTs also improved the cycling stability, with a 99.7% retention of capacity after 100 cycles at 0.2 A g−1, due to the strong π–π interactions between the aromatic rings in 39 and the SWCNT surface (Fig. 8). This is a general strategy that can be applied to both anthraquinone and (−)-riboflavin, 40. In an effort to increase the Csp of the bio-derived molecules, the same group reported the use of alloxazinic forms of flavin with more simplified structures 41, 42, and 43.56 They observed that while the Ctheor increases going from 41 to 42 to 43, the Csp only increases for 42, suggesting that there is strong electronic repulsion between molecules of 43 when accepting two electrons. A high Csp (236 mA h g−1 at 1C) and a 92% capacity retention after 200 cycles at 1.0 A g−1 is reported for 42, partly due to the use of the SWCNT hybridization strategy for electrode preparation. The authors also reported that 41, 42, and 43 were useful for sodium-ion battery cathodes. Although a higher Csp for 42 is achieved in the sodium-ion battery (255 mA h g−1 at 50 mA g−1), the cycling stability is poor, with only a 50% retention after 20 cycles. This is an excellent way to incorporate redox units found in biological systems into metal-ion batteries, and addresses issues such as cycling stability, voltage, and capacity.
 | ||
| Fig. 8 (a) Schematic representation of the hybridization between 39 and SWCNTs giving better conductivity and strong π–π interactions. (b) Schematic of the charge storage mechanism of 39. (Reproduced from ref. 55, reprinted with permission, copyright 2014, Wiley-VCH). | ||
Indigo carmine 44 is widely used as a food dye and was investigated as a lithium and sodium-ion battery cathode. 44 has a 110 mA h g−1Csp at 10 mA g−1 and a 2.2 V potential vs. Li/Li+ with a lithium electrolyte and 106 mA h g−1 at 10 mA g−1 at a 1.8 V potential vs. Na/Na+ using a sodium electrolyte.57 After 40 cycles, the compound retains 82.7% and 81.1% of its initial capacity at 10 mA g−1 in lithium and sodium-ion batteries respectively. The sulfate groups on 44 decrease the Ctheor and their electron-withdrawing character does not raise the voltage enough to compensate for the low Csp.
In a recent report, hypervalent sulfur, 45, and selenium, 46, compounds were isolated and used as cathode materials for lithium-ion batteries.58 The compounds have 64.4 mA h g−1 and 76.7 mA h g−1Csp's at 1.2C for 45 and 46 respectively, with a high stability of ∼90% retention after 50 cycles for both materials. Although these compounds are interesting, their low Csp's need to be improved. Decreasing their molecular weight by removing the perfluoroalkyl chains are suggested future areas of inquiry. Fullerene C60, 47, was recently studied as a magnesium-ion battery cathode.59 Using a Grignard reagent/AlCl3 electrolyte, it has a 50 mA h g−1Csp in the first cycle but decreases quickly after 10 cycles to ∼5 mA h g−1 because of dissolution. Due to the lack of electrophilic functionalities in 47, this compound is chemically stable in the Grignard reagent-containing electrolyte required for reversible magnesium stripping and plating. More work is needed in the area of magnesium-ion batteries, but the relatively low voltage hysteresis in 47 is promising.
Arylene diimides can be readily incorporated into non-conjugated polymeric materials. This is because the parent dianhydrides can be condensed with a diamine to form an insoluble material in a one-step reaction. In an effort to form a flexible and free standing cathode film, Wu and coworkers performed an in situ polymerization of pyromellitic dianhydride with ethylene diamine and SWCNTs to form 48.60 The in situ polymerization formed a hierarchical structure with 48 wrapped around the SWCNTs to form a freestanding film, which was used as a binder-free cathode. This material exhibits good rate capabilities with a high Csp of 226 mA h g−1 at 0.1C and 120 mA h g−1 at 20C. The material also retains 85% of its initial capacity after 200 cycles at 0.5C due to its insolubility in the electrolyte. In a continuation of the work, 49 was synthesized by polymerizing pyromellitic dianhydride with a triamine in the presence of SWCNTs using the same in situ polymerization strategy.61 Similarly, the electrode forms a freestanding, flexible film where 49 is wrapped around the SWCNTs (Fig. 9). Although the Csp is slightly lower than the previous work (179 mA h g−1 at 0.1C), the cycling stability is slightly improved (86.6% retention after 200 cycles at 0.5C), and the electrode is very durable, with 80% retention of the initial capacity after 1000 bending cycles. This work demonstrates that the in situ polymerization of arylene diimides is a promising technique to make flexible electrodes. A series of different non-conjugated arylene diimide polymers with pyromellitic, NDI, and PDI cores with different lengths of diamine spacers (compounds 48, 50–53) were studied for sodium-ion battery cathodes by Wang and coworkers.62 By examining different arylene cores, a systematic increase in voltage when increasing the ring size from pyromellitic (48) to PDI (51) from 1.73 V to 1.94 V vs. Na/Na+ is observed because the increasing ring size increases the electron affinity. Additionally, the Csp varies from 124 mA h g−1, 132 mA h g−1, to 107.7 mA h g−1 in 48, 50, and 51 respectively, although the capacity in the last case can be increased to 148.9 mA h g−1 when the carbon content is increased from 30% to 60%. Importantly, as the size of the arylene core increases, the cycling stability also increases with an 83% retention after 150 cycles at 200 mA g−1 for 51. When varying the alkyl spacer length from propyl to butyl with the PDI core, the Csp decreases to 116 mA h g−1 and 100 mA h g−1 at 25 mA g−1 for 52 and 53 respectively, likely due to the insulating effect of the alkyl chains that prevents efficient charge transfer. The use of arylene diimide polymers connected through the nitrogen atoms is a straightforward way to synthesize inexpensive cathode materials that can solve some of the capacity fading issues with small molecules, especially dissolution. However, the cycling stability still needs to be improved, and the low voltages of these materials are still a concern. Future work should focus on increasing the voltage through substitution of the aromatic rings and increasing cycling stability.
 | ||
| Fig. 9 (a and b) Photograph of hybrid 49/SWCNT film, scanning electron microscope image of (c) a cross-section of the hybrid film with a SWCNT current collector, (d) the top of the bare SWCNT film, and (e) the hybrid 49/SWCNT film. (f) Transmission electron microscope image of hybrid 49/SWCNT film. (Reproduced from ref. 61, reprinted with permission, copyright 2015, Wiley-VCH). | ||
To determine the effect of block copolymer structure on the performance of arylene diimide lithium-ion battery cathodes, 54 and 55 were synthesized with varying poly(ethylene oxide) (PEO) block lengths and incorporated into binder-free, low carbon cathodes.63 Although the pyromellitic compound 54 has a higher Ctheor, the NDI 55 performs the best, with a 196 mA h g−1Csp at 0.1C and a 54% capacity retention after 100 cycles at 0.1C. Interestingly, it was found that better performance is achieved with longer PEO blocks due to an increase in ionic conductivity. These polymers may find use in devices such as thin film batteries due to their high ionic conductivity. However, the active material content is still low, and increasing the amount of redox active material while retaining ionic conductivity should be the focus of future work.
A PDI derivative polymerized with hydrazine, 56, was reported as a cathode material in an all organic sodium-ion battery using sodium terephthalate, 57, as the anode.64 The cathode has a 126 mA h g−1Csp at 100 mA g−1 with two voltage plateaus at 2.45 and 1.86 V vs. Na/Na+. When used in the all-organic battery, the full cell has an initial Csp of 73 mA h g−1 with a 1.35 V open circuit voltage. This device has a relatively low open circuit voltage, but this could be further improved by using a different anode material.
Pendant polymers have become very popular in the field of organic radical electrodes, with extensive work performed by the Nishide group.65 This has recently been extended towards the “rocking chair” style of metal-ion batteries. In an interesting example of this class of molecules, a dendronized polymer was synthesized with anthraquinone groups pendant to the dendrons (58).66 Compound 58 exhibits an 84 mA h g−1Csp at 0.1C and a high capacity retention (∼90–95% after 100 cycles at 0.5C). This is the first example of a dendronized polymer for energy storage applications and, due to the large macromolecular nature, the cycling stability is high. The low usage of active material is an issue that may be caused by large polymer domains that are electrically isolated from the conductive pathway of carbon black particles. A dithiophenedione containing polymer, 59, was synthesized by the Schubert group and exhibits a 219 mA h g−1Csp at 1C and has a high rate capability, retaining 190 mA h g−1 at 10C when used as a lithium-ion battery cathode.67 Unfortunately, the capacity fading was significant, only 52% of the initial capacity is retained after 100 cycles at 1C. The polymer was insoluble in the electrolyte solution, ruling out dissolution as the capacity fading mechanism, but spectroelectrochemistry results point to side reactions as the main contributor to the degradation of performance. Although this polymer degrades rapidly in the lithium-containing electrolyte, its high capacity and rate capability may allow it to be used in other battery electrolytes where the redox chemistry is fully reversible. This will depend on the exact mechanism of chemical degradation, and if it can be prevented by using different salts or solvents. Schmidt and coworkers reported the synthesis and use of 60 as a lithium-ion battery cathode with a 258.5 mA h g−1Ctheor.68 The polymer exhibits a two-step reduction process in solution, but in the composite electrode with the conductive carbon and binder the reduction process only has one step. Since the Csp is ∼50% of the Ctheor (137 mA h g−1), this suggests that each repeat unit accepts only one electron. The polymer retains 86% of the initial capacity after 100 cycles at 5C. Interestingly, when the polymer is cross-linked, the Csp decreases but the cycling stability is improved. The performance of 60 changes from solution to the uncross-linked and cross-linked film. This indicates that there is room to improve this system. If conditions are found to improve the capacity and the cycle life of 60, it should become useful in some applications.
Recently, we have reported the first bio-derived pendant polymer cathode, 61, using a norbornene-based backbone with pendant flavin units.69 When built into a device with a lithium anode, 61 has a 125 mA h g−1Csp at 0.1C and 77 mA h g−1 at 1C. The capacity degrades fairly quickly, with only 28.3% of the initial capacity after 200 cycles at 1C, but this is improved to 110% when cycled with a limited potential window. The increase in capacity upon cycling is due to an increased electrolyte penetration into the electrode. Degradation is caused by geometry changes in the repeating unit upon reduction (Fig. 10). This causes a the flavin redox units to become isolated from the conductive carbon as evidenced by the Raman, XPS, and FTIR spectra, and this also causes an increase in charge transfer resistance. Changing the connectivity to reduce this geometry change should increase stability and future work is focussed on addressing this issue, as well as rate capability.
 | ||
| Fig. 10 Geometry changes in the repeating unit of 61 when discharging. These geometry changes are expected to be responsible for capacity fading. Adapted from ref. 69. | ||
Chloroanilic acid and dilithium chloranilate, 62 and 63 respectively, were examined as lithium-ion battery cathodes.7062 has a 119 mA h g−1Csp at 50 mA g−1 with two voltage plateaus at 3.0 V and 2.0 V vs. Li/Li+, while 63 has a 193 mA h g−1Csp with a sloping voltage plateau between 2.3 and 1.8 V. 63 also has a better cycling stability with ∼75% capacity retention after 20 cycles due to the strong ionic interactions helping to prevent dissolution. When polymerized with sulfur, these oligomers exhibit 214 mA h g−1 and 247 mA h g−1Csp's at 50 mA g−1 for 64 and 65 respectively. The lithiated derivative 65 exhibits a very high rate capability and cycling stability with a 124 mA h g−1Csp at a high rate of 10000 mA g−1 and a 90% capacity retention after 1500 cycles at 500 mA g−1. This is in contrast to the non-lithiated derivative 64 that has a 47% capacity retention after 20 cycles at 50 mA g−1. In a follow up study, the same authors polymerized different isomers of anthraquinone with either sulfur or by a condensation polymerization forming compounds 66–68.23 The 1,4-isomer of the anthraquinone formed by the condensation polymerization, 68, performs the best with a 263 mA h g−1Csp at 0.2C and a 98.3% capacity retention after 100 cycles at 0.2C. They attribute the superior performance of 68 to highly reversible redox reactions and the insolubility of both the neutral and the reduced forms of the polymer (Fig. 11). Additionally, 66 can be used as a magnesium-ion battery cathode.71 The polymer displays a 225 mA h g−1Csp in the first cycle at 50 mA g−1 with a 1.5 V to 0.5 V sloping voltage plateau vs. Mg/Mg2+. However, 66 suffers from poor cycling stability, with only ∼22% capacity retention after 100 cycles. Song and coworkers also reported the synthesis of polybenzoquinonyl sulfide, 69, and its use as a lithium-ion battery cathode.72 Due to low amount of inactive mass in 69, it has a high Ctheor of 388 mA h g−1 making it an attractive electrode material. The Csp reaches 275 mA h g−1 at 50 mA g−1 and 198 mA h g−1 at 5000 mA g−1, with an 86% retention of the initial capacity after 1000 cycles at 500 mA g−1 and a high CE of 99.5%. As a sodium-ion battery cathode, 69 has a 268 mA h g−1Csp at 50 mA g−1 and a 68% capacity retention after 100 cycles at 500 mA g−1. This work shows that polymerization with sulfur is an attractive strategy to design high performance organic cathodes. These materials are currently the highest capacity conjugated polymers for metal-ion battery cathodes and are excellent examples of stable, high capacity battery materials. Obtaining polymers with higher molecular weight and high purity should improve performance and commercial applicability. Additionally, in order to gain insight into the development of organic sodium-ion batteries, the reasons for incompatibility with sodium-ion battery electrolytes should be resolved.
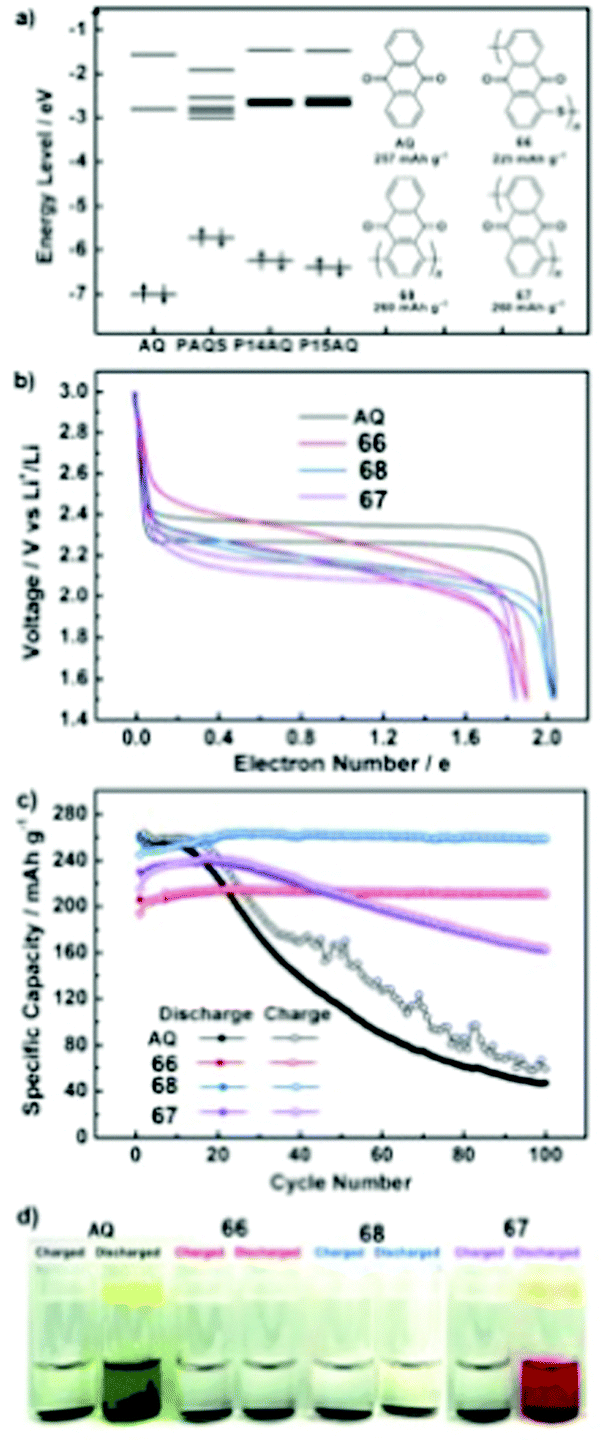 | ||
| Fig. 11 (a) Electronic configuration of anthraquinone, 66–68, calculated by DFT. (b) Charging/discharging and (c) cycling performance of the electrodes fabricated using anthraquinone and compounds 66–68. (d) Solubility of the charged and discharged electrodes in a 1 M LiTFSI DOL:DME 2:1 electrolyte. (Reproduced from ref. 23, reprinted with permission, copyright 2015, Wiley-VCH). | ||
Donor–acceptor copolymers 70 and 71 have also been studied as ultrafast lithium-ion battery cathodes.73 Although the Ctheor of these polymer are low, 54.2 and 52.7 mA h g−1 for 70 and 71 respectively, the very high rate capabilities of 70 allows it to have a 42.8 mA h g−1Csp at 500C. Additionally, 70 has a 96% capacity retention after 3000 cycles at 10C. The authors found that when the polymer conjugation is interrupted with a saturated ethylene linker such as that in 71 (Fig. 12), the rate capability decreases. This work introduces the first use of donor–acceptor copolymers for use in lithium-ion battery electrodes. Although the stability and rate capability is excellent, the Csp is relatively poor and could be improved if the solubilizing alkyl chains were removed. Arylene diimide-co-anthraquinone alternating copolymers were also studied for sodium-ion battery cathodes.74 Surprisingly, the voltages were very similar for the pyromellitic (72) and the NDI (73) polymers. This is in contrast to other reports comparing different arylene cores, which suggests that the anthraquinone unit has more influence on the redox potentials. The NDI polymer 73 did, however, have a higher Csp (179 mA h g−1 at 50 mA g−1) and also a slightly better capacity retention (95% after 150 cycles at 50 mA g−1). The concept of polymerizing arylene diimides with other redox active groups is unique, and provides for materials with high capacities. However, the low redox potential of these materials need to be addressed.
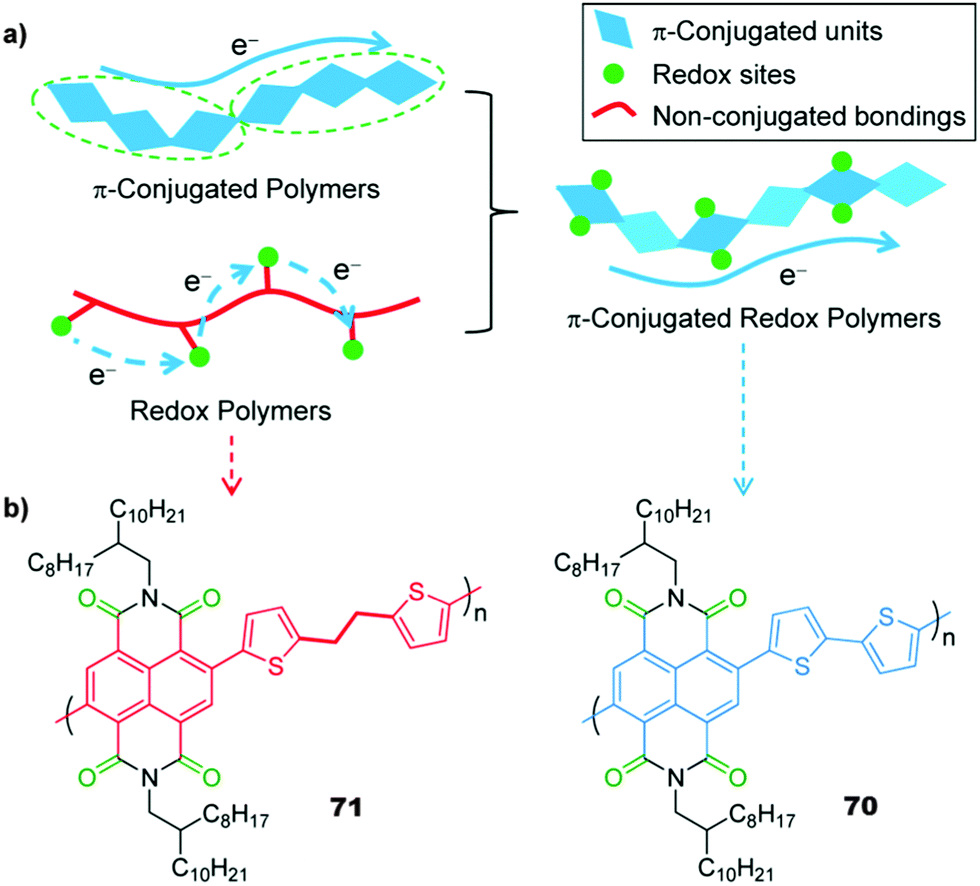 | ||
| Fig. 12 (a) Illustration of the conduction pathways of π-conjugated polymers, redox polymers, and π-conjugated redox polymer. (b) Structure of 70 and 71 in relation to the above illustrations. (Reproduced from ref. 73, reprinted with permission, copyright 2015, American Chemical Society). | ||
Vlad and coworkers reported the polymerization of a polyaniline-type monomer with methoxy groups attached to the benzene ring, followed by deprotection to reveal the hydroquinone polymer 74, which displays quinone-like redox activity.75 The polymer's electron conduction pathway is along the polyaniline-type backbone while the redox activity is due to the quinone groups on the benzene ring (Fig. 13). The polymer exhibits a 270 mA h g−1Csp at 0.1C in the first cycle, however, it has poor cycling stability with only 7% retention after 5 cycles attributed to an irreversible keto–enol tautomerization. The concept of using a polyaniline backbone in a novel redox-active polymer is very interesting and could lead to high performance organic electrodes without conductive carbon additives. The stability of 74 could be improved through chemical modification to drive the equilibrium towards the keto form. The use of self-doped polymers provides a way to integrate positive charge-accepting polymers into the “rocking chair” style of metal-ion batteries. A polyaniline polymer with phenyl sulfonate groups attached to the polyaniline nitrogen, 75, was used as a cation-exchanging organic cathode for sodium-ion batteries.76 The cathode has a very high redox potential, with a sloping voltage plateau between 3.3 and 3.6 V vs. Na/Na+, and a 100 mA h g−1Csp at 50 mA g−1. The capacity retention is 72% after 100 cycles at 50 mA g−1. In an effort to improve upon this performance, sulfonated polyaniline 76 was synthesized.77 Here, the sodium ions are ionically bonded to the sulfonate groups when discharged. Upon charging, the sodium ions migrate out of the electrode and charge on the polyaniline backbone is compensated by the sulfonate groups (Fig. 14). This polymer has an improved Csp of 133 mA h g−1 with a similar voltage as 75. The capacity retention is also greatly improved to 96.7% after 200 cycles at 100 mA g−1. The use of self-doped polymers is an excellent strategy to attain high voltages. However the use of polymers that have highly delocalized polarons and require heavy sulfate groups limits the Csp.
 | ||
| Fig. 13 Molecular structure of 74 showing the redox active groups and the electronic conduction pathway. (Reproduced from ref. 75, reprinted with permission from Royal Society of Chemistry). | ||
 | ||
| Fig. 14 The sodium insertion mechanism of the self-doped polymer 76. Adapted from ref. 77. | ||
Conjugated polymers have also been investigated for multivalent metal-ion battery cathodes. Polypyrrole, 77, and polythiophene, 78, were investigated for aluminium-ion battery cathodes.78 Used in AlCl3 and 1-ethyl-3-methylimidazolium chloride electrolyte, a ‘rocking-chair’ mechanism can occur by reducing chloroaluminate ions at the anode to form aluminium metal and using the chloroaluminate ions to dope the polymers when they are positively charged. At 0.2C, 77 has a 50 mA h g−1Csp and 78 has an 80 mA h g−1Csp with sloping voltage plateaus between 0.6 to 1.8 V and 1.1 to 1.9 V vs. Al/Al3+, respectively. A bio-derived melanin polymer, 79, with a high amount of redox-active carbonyl groups, was investigated as a cathode material for magnesium-ion batteries.79 This polymer displays a ∼60 mA h g−1Csp at 0.1 A g−1 and ∼15 mA h g−1 at 5.0 A g−1, with a high cycling stability (98–99.5% retention after 50 cycles at 0.1 A g−1). While the development of multivalent metal-ion batteries is still in its early stages, and the performance of these materials are poor compared to lithium- and sodium-ion batteries, these studies represent important advances in the field.
A comparative study was carried out on the application of arylene diimide frameworks for lithium-ion battery cathodes as well as for gas storage.81 When varying the arylene core from pyromellitic (81) to NDI (82) and PDI (83), a noticeable trend in Csp is not observed and the voltage remains relatively constant at ∼2.35 V vs. Li/Li+. The capacity retention, however, increases when increasing the size of the aromatic core, with 83 having a 74.1% capacity retention after 65 cycles at 25 mA g−1. The introduction of carbon nanotubes (CNTs) into NDI-based framework, 84, was shown to increase the performance compared to materials synthesized without CNTs.82 The material has a 69 mA h g−1Csp at 2.4C, 81% of Ctheor, and incredible stability with 100% capacity retention after 700 cycles at 2.4C. The low Csp and low voltage means that these materials require further improvement. The introduction of carbon additives in situ increases performance by forming well-dispersed conductive pathways. This strategy could be one solution to increase the active material content in organic electrodes and should be explored with high capacity materials.
DeBlase and coworkers synthesized a β-ketoenamine-linked NDI framework, 85, that operates in a wide variety of electrolytes for electrochemical energy storage.83 This work demonstrates the versatility of porous aromatic frameworks in energy storage with 100, 120, 95, and 110 mA h g−1Csp's for magnesium, lithium, potassium and tetrabutyl ammonium electrolytes respectively. The redox potentials shift depending on the electrolyte, with the highest redox potential for the magnesium salts and the lowest being for the tetrabutyl ammonium salts due to the differences in coordination energy with the reduced framework (Fig. 15). The use of arylene diimide frameworks reduces solubility and creates well-defined pores to enhance ionic conductivity. These materials still require research to increase the Csp and the voltage in order to compete with other classes of materials.
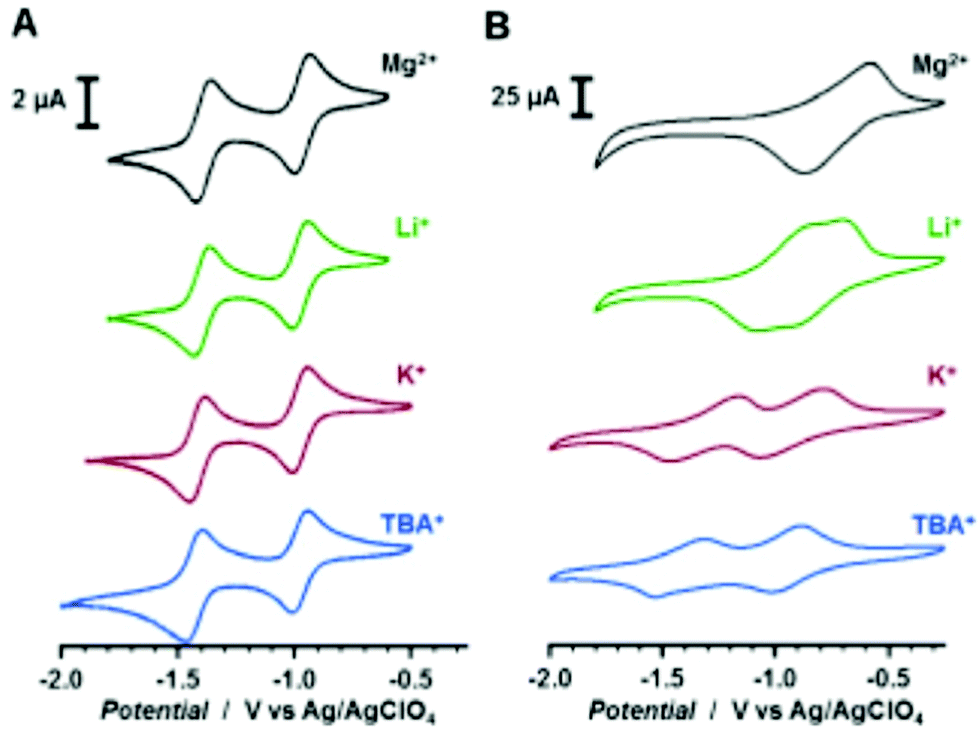 | ||
| Fig. 15 Changes in redox potential for 85 with different electrolytes in (A) solution and (B) solid state. (Reproduced from ref. 83, reprinted with permission, copyright 2015, Wiley-VCH). | ||
There have been a number of studies on terephthalates for use in metal-ion batteries due to their high abundance, simplicity, and the redox activity of the carboxylate groups (∼1.2 V and 0.01 V vs. Li/Li+). Lithium terephthalate (86) has been heavily studied as a lithium-ion battery anode. In an improvement of previous work on 86, Zhang and coworkers synthesized porous microspheres consisting of 86 nanoparticles via a spray drying method, followed by coating them in an N-doped carbon layer to improve the electronic conductivity and diffusion of lithium ions.84 The goal of this study was to improve the cycling stability and rate capability of the resultant electrode. The Csp reaches 259 mA h g−1 at 0.05C and 121 mA h g−1 at 1C. The capacity retention after 50 cycles at 0.5C is 150 mA h g−1. This value is an improvement upon the electrode fabricated by standard electrode processing and formulations. This work shows that changes in processing methods can result in large changes in performance. Although more work needs to be done to improve the rate capabilities, optimizing electrode fabrication and formulation offers an alternative and complementary way to improve performance beyond chemical modification.
Investigating excess capacity in conjugated carboxylates, Lee and coworkers studied compounds 86, 87, and 88 at voltages below 0.7 V vs. Li/Li+.85 This follows that of others who report the excess capacity obtained in some conjugated systems.86 In 86, they found that when they discharged the electrode to 0 V, there is a reduction plateau at 0.81 V and another sloping voltage plateau from 0.8 V to 0.0 V that gives a 522 mA h g−1Csp after 15 cycles at 30 mA g−1. This is much higher than the Ctheor (302 mA h g−1) based on the insertion of one lithium ion per carboxylate group. When they performed the same cycling experiments with the linear compound 87, they did not observe excess capacity. However, with the thiophene derivative 88, the Csp is 850 mA h g−1. Through a combination of X-ray, solid-state 13C NMR, and isotope labelling, they were able to determine that the extra capacity is due to the insertion of lithium ions into the internal alkene of the cyclic compounds. This excess capacity is not observed with the linear compound, likely because it causes a break in the conjugation of the structure (Fig. 16). This ‘superlithiation’ has also recently been demonstrated in dilithium benzenedipropiolate, 89.87 This material exhibits the highest Csp for a lithium carboxylate (1363 mA h g−1), with 11.5 lithium ions inserted per molecule. Using DFT, the authors determined that lithiation occurs at the carbonyls first as expected, followed by the triple bonds, and finally the aromatic rings. ‘Superlithiated’ compounds open up a new strategy to design organic anodes where the carboxylate groups are not the sole contributors to the redox-activity. However, this mechanism of charge storage is rate limited. The long sloping discharge plateau for most of the ‘superlithiated’ compounds could impose limits on practical use. Further research into these compounds could prove to be fruitful.
 | ||
| Fig. 16 Mechanism of extra capacity in conjugated carboxylates exemplified by 86. (Reproduced from ref. 85, reprinted with permission, copyright 2014, American Chemical Society). | ||
Inspired by work suggesting that extension of the electronic conjugation between carboxylates can increase the rate capability of an organic electrode, Fédèle and coworkers studied 2,6-naphthalene dicarboxylate, 90, as an anode material for lithium-ion batteries.88 The morphology and homogeneity of the electrode is much better when it is fabricated by a freeze drying technique than ball milling. The electrode has a 200 mA h g−1Csp at 0.1C and 176 mA h g−1 at 1C, retaining 115 mA h g−1 after 50 cycles at 1C. The extended conjugation of the naphthalene core provides higher rate capabilities compared to that of 86. By preparing highly crystalline 90, the electrode behaves as an insertion metal–organic framework (iMOF).89 This iMOF has a short distance between aromatic rings, allowing for efficient electron conduction. Also, favourable crystal packing allows for high lithium ion conduction throughout the crystal (Fig. 17). The iMOF of 90 has a 213 mA h g−1Csp at 0.1C and 100% capacity retention after 10 cycles at 0.1C. Additionally, due to its reduction potential at 0.8 V vs. Li/Li+, 90 can be cast on an aluminium foil current collector allowing 90 to operate in a bipolar electrode.90 Coupled with a LiNi0.5Mn1.5O4 cathode, 90 exhibits a voltage of 8 V in a stacked cell using a bipolar electrode. This work highlights the ability to use organic electrodes to make high voltage devices using less inactive material for the cell components compared to current lithium-ion battery anodes. Additionally, this shows that compounds with favourable crystal structures can allow high ionic and electronic conductivity. However, more cycling experiments should be performed in order to prove that the cycling stability is high. In an effort to extend the aromatic core of organic carboxylate anodes to achieve even higher rate capabilities, a perylene tetracarboxylate, 91, was synthesized and tested as a lithium-ion battery anode.91 This compound achieves ∼95% of its Ctheor at 1.25C with a 222 mA h g−1Csp. The authors also demonstrate that 91 retains 125 mA h g−1 after 100 cycles at 1.25C, although they note that the cycling stability increases when a much deeper cycling is used. Although the authors succeed at increasing the rate capabilities of the electrode by using a larger aromatic core, they do so at the expense of the cycling stability, and this is an important area for future improvement.
 | ||
| Fig. 17 (a) Rietveld refinement for lithium-intercalated 90; the experimental diffraction pattern, the calculated diffraction pattern and the difference (red line, green solid line and pink solid line respectively). (b and c) The structure of the lithium-intercalated 90 in two proposed models. (d and e) The coordination geometry of the lithium-intercalated state of 90 of the two corresponding models depicted in (b) and (c) respectively. (Reproduced from ref. 90, reprinted with permission, copyright 2014, Wiley-VCH). | ||
The need for anode materials for sodium-ion batteries is even greater than that for lithium-ion batteries because sodium ions typically cannot insert into the commonly used anode, graphite, like lithium ions. However, some recent work has shown that under certain conditions, insertion is possible.92–94 Wang and coworkers demonstrated that 92 can be used as an anode material in sodium-ion batteries.43 At higher potentials and at a different oxidation state (compound 25) this compound can also function as the cathode in order to make symmetric, all-organic sodium-ion batteries. With an oxidation potential of 0.40 V vs. Na/Na+, 92 has a 207 mA h g−1Csp at 0.1C and decreases to 117 mA h g−1 at 5C. Additionally, the compound is very stable, with an 89% capacity retention after 100 cycles. The exceptional performance of this material and its chemical simplicity make 92 one of the most promising anode materials for sodium-ion battery anodes.
Croconic acid, 13, has also been used as a sodium-ion battery anode.95 The best performance is achieved by preparing graphene oxide (GO) covered 13 by spray pyrolysis to prevent particle pulverization. A 293 mA h g−1Csp is observed at 20 mA g−1 with ∼40% capacity retention after 100 cycles. The high Csp of this material is attractive for sodium-ion battery anodes but the rapid capacity fading and the complex electrochemical profile over a wide potential range are concerns. Capacity fading could be improved by optimizing the particle size and/or nanostructuring, varying the GO content, and spray pyrolysis conditions. The complex electrochemical profile, however, is a harder issue to solve. Biphenyl dicarboxylate, 93, can also be used as a sodium-ion battery anode.96 The compound has a 187 mA h g−1Ctheor while the Csp exceeds this at 0.1C when cycled at 30 °C. It has a high rate capability, retaining 100 mA h g−1 at 20C and also a very high stability with ∼100% capacity retention after 150 cycles at 0.1C. The compound needs to be fully deprotonated to be stable when cycling, likely because the protonated form reacts with the electrolyte at low potentials. The well-defined, low-potential voltage plateau, along with high rate capability, high stability, and Csp give this material advantages as a sodium-ion battery anode.
The sodiated salt of benzene diacrylate, 94, was also reported as an anode material for sodium-ion batteries.97 The compound exhibits a 177.7 mA h g−1Csp at 0.025C with a 91% CE, but decreases to ∼40 mA h g−1 after 40 cycles. The authors suggest that the capacity fading is due to dissolution of the active material in the electrolyte or a decomposition reaction. Interestingly, the lithiated analog of 94 does not suffer from capacity fading. This is likely because lithium salts form a polymeric network, which leads to less dissolution. Changing the counter-ion from bis(fluorosulfonyl)imide (FSI) for the sodium salt compared to the bis(trifluoromethane)sulfonamide (TFSI) for the lithium salt may result in a different SEI and/or electrochemical degradation products. It is important to perform comparisons between different compounds using as similar conditions as possible in order to rule out any effects not related to the structure.
The quinone derivative 95 was also reported to operate as a sodium-ion anode.98 The compound has a 265 mA h g−1Csp at 0.1C and also has a good rate capability, retaining 159 mA h g−1 at 5C. 95 has a relatively high potential for anode materials, with anodic peaks at 1.59 V and 1.28 V vs. Na/Na+, and has good stability with 81% capacity retention after 300 cycles. Although the high Csp, good rate capability, and stability make this material attractive, the relatively high reduction potential will result in a very low voltage if paired with sodium-ion battery cathodes. For example, a maximum voltage of ∼2.3 V will result if 95 is paired with the highest voltage sodium-ion battery cathode reported in this review (compound 75).
Biomolecules can also be used for sodium-ion battery anodes. Juglone, 96, is a promising high capacity anode material.99 The Csp of 96 reaches 398 mA h g−1 at 0.05 A g−1, which is higher than the Ctheor (290 mA h g−1), although the authors attribute the excess capacity to the contribution from reduced GO from the electrode formulation. The anodic peak for this compound occurs at 1.5 V vs. Na/Na+ and it retains 69.5% of its initial capacity after 300 cycles at 0.1 A g−1. The large change in voltage over discharge and capacity retention are concerns for this material, but the investigation of biomolecules for battery materials remains important for low cost, sustainable batteries.
In an effort to examine the effect of extended π conjugation on the performance of sodium-ion battery anodes, a comparative study between 97 and 57 was performed.100 The authors found that 97 exhibits a 260 mA h g−1Csp at 50 mA g−1 and 72 mA h g−1 at 10 A g−1 compared to 57 which has a 192 mA h g−1Csp at 50 mA g−1 and 22 mA h g−1 at 10 A g−1. This work highlights the importance of extended π conjugation on charge transport, demonstrating that enhanced intermolecular interactions and packing facilitate sodium-ion diffusion in compound 97. In a separate study to improve the performance of 57, nanosheets were synthesized that provide a 248 mA h g−1Csp at 25 mA g−1 and 59 mA h g−1 at 1250 mA g−1.101 This is greatly improved compared to the electrodes prepared from the bulk material. 57 was also reported as an anode for an all-organic sodium-ion battery using 56 as the cathode.64 The anode provides a 180 mA h g−1Csp at 50 mA g−1 with a reduction potential of ∼0.25 V vs. Na/Na+. Organic dicarboxylates are important sodium-ion battery anodes because they have high electrochemical activity at low potentials, high Csp's, and moderate rate capability and cycling stability. For the most part, the Ctheor can be attained for these compounds. The cycling stability and rate capability could be further improved by nanostructuring, coating with conductive materials, and optimizing electrode formulation and fabrication.
The sodium salt of NDI, 98, can also be used as an aqueous sodium-ion battery anode because its reduction potential occurs in the operating potential of aqueous electrolytes.10298 provides a 62 mA h g−1Csp at 6C and has high rate capability with a 40 mA h g−1Csp at 24C. The device with a Prussian blue analog as the cathode has a 1.1 V operating potential. The reduction potential of 98 is close to the water reduction potential, so this material is uniquely suited for an aqueous sodium-ion battery anode. Improvements in material structure to attain higher Csp's need to be considered in order to make aqueous batteries viable. A sodium salt of pyromellitic diimide, 99, was also used as a sodium-ion battery anode.103 With anodic peaks at 1.6 and 1.2 V vs. Na/Na+, it delivers a 128.9 mA h g−1Csp at 0.025C. However, due to irreversible side reactions, it only retains 70% of its initial capacity after 100 cycles at 0.025C. This material has obstacles that need to be overcome before it can be relevant for a sodium-ion battery, such as its cycling stability, and its relatively high reduction potential. However, 99 may prove to be useful in an aqueous device, much like compound 98.
Perylene dianhydride, 37, can also be used as an anode material for sodium-ion batteries.104 The compound exhibits a 361 mA h g−1Csp when taking into account the contribution from the conductive carbon additive. This is much higher than the Ctheor, 273 mA h g−1, which suggests that another mechanism is contributing to the increased capacity of the compound. The authors conduct additional experiments to prove that the addition of sodium ions across the unsaturated double bonds of the aromatic core, a mechanism suggested for lithium ions in a number of different aromatic anode materials, does not occur.86,105 The authors did not observe an increase in capacity with the size of the aromatic cores. They therefore conclude that the extra capacity is due to the formation of a SEI and the decomposition of the electrolyte when reducing the compound. This is supported by the Csp stabilizing to 250.5 mA h g−1 after 140 cycles at 25 mA g−1. They also study 100, which has a 350.6 mA h g−1Csp in the first cycle, but decreases to 131.1 mA h g−1 after 120 cycles at 25 mA g−1. Although 37 does not undergo ‘supersodiation’, it still has good performance in terms of voltage and Csp. 37 is a prime candidate for high performance sodium-ion battery anodes if the cycling stability issues are alleviated.
Oligomeric-Schiff bases have also been examined as anode materials for sodium-ion batteries.106 By examining the different connectivities and lengths of compounds 101 to 104 in sodium-ion batteries and by DFT, the authors find that the redox activity comes from coplanar Hückel groups, giving high Csp's for 101 and 104 above 250 mA h g−1 at 0.1C. Additionally, the sodium salt compounds have much better performance due to reduced hydrogen bonding between units allowing for higher ionic conductivity. The simplicity and high Csp of these materials make them promising sodium-ion battery anodes. This work provides a solid foundation upon which the further development of Schiff base anodes can be built. However, more detailed electrochemical characterization is required in order to fully judge their applicability in sodium-ion batteries.
Biopolymers are also useful for lithium and sodium-ion battery anodes. Humic acid, 106, can function as both a lithium-ion and sodium-ion battery anode, providing a 484 mA h g−1 and 208.3 mA h g−1Csp respectively in an organic electrolyte.110 The capacity retention is 70% and 80% of the initial capacity after 200 cycles at 40 mA g−1 respectively for lithium and sodium-ion batteries. This is an interesting example of using a relatively undefined biopolymer as an energy storage electrode. The performance metrics are very good, especially considering the 85% active material loading. Further investigation into these types of biopolymers is expected to yield very promising candidates for electrode materials. In general, non-conjugated polymers are used less often as anode materials for metal-ion batteries. This is because the backbone is susceptible to decomposition at low voltages and therefore they are mostly used as aqueous battery anodes making 106 a very interesting compound.
Ladder polymers have recently become of interest for lithium-ion battery anodes. Compound 109 was found to have a 1442 mA h g−1Csp at 0.05C between 1.0 V and 0.0 V vs. Li/Li+.105 The authors suggest the mechanism of charge storage is the insertion of one lithium ion per atom in the structure of the compound (i.e. ‘superlithiation’). In the same report, 110 was found to operate by the same insertion mechanism and provide a 1416 mA h g−1Csp at 0.1C. In a similar report by the same authors, 111 was proposed to operate by the same lithium insertion mechanism and was found to provide a 1550 mA h g−1Csp after 100 cycles with a sloping profile between 1.5 V and 0 V vs. Li/Li+.113 These are some of the first reports of ‘superlithiation’ in organic materials and of ladder-type polymers. These reactions have slow kinetics and therefore could find more in use in batteries that operate at elevated temperatures. Additionally, these materials also suffer from a large change in voltage over discharge. However, their high capacities make them useful as lithium-ion battery anodes.
Polymeric Schiff bases were also investigated for use as sodium-ion anode materials.114 A systematic study on the variation of both backbone and substituents was conducted on compounds 112 to 117. Increasing the length in the non-conjugated spacer results in poor electrochemical performance, and inverting the Schiff base in 112 leads to very little electrochemical activity. The best performing polymer, 114, has a 180 mA h g−1Csp at 0.1C with 100% retention after 25 cycles at 0.1C. The use of polymeric Schiff bases for sodium-ion battery anodes is promising. While more work is needed to achieve the Ctheor, the design principles outlined in this work, along with other work from the same group on oligomeric Schiff bases, is expected to aid in the further development of these materials.
3.2 Metal–sulfur batteries
There has been a great deal of interest in metal–sulfur batteries in the research community due to their high theoretical energy density, making them attractive candidates for replacing lithium-ion batteries. The main focus is improving sulfur cathodes, specifically increasing the usage of sulfur to attain the Ctheor and decreasing polysulfide dissolution. Metal–sulfur cathodes have the same requirement as metal-ion batteries: the anode must contain metal ions in order for the system to function. To improve active material usage, organic materials have been used as redox shuttles to direct the growth of lithium sulfide deposits and facilitate charge transport between sulfur and the current collector in both lithium–sulfur batteries and dissolved polysulfide batteries.115,116 The use of pyrite (FeS2) as a cathode additive sequesters lithium polysulfides via S–S covalent bonds, thereby increasing the cycling stability.117 Since these materials are not used to store the charge, this is beyond the scope of this review.Organic materials can be incorporated into the sulfur cathode in an effort to improve conductivity, increase active material usage, and prevent dissolution. This was first studied in the seminal report by Chung and coworkers has increased interest in polymerizing sulfur with unsaturated organic compounds.118 Heating molten sulfur with diisopropenylbenzene leads to the formation of a highly cross-linked polymer network 118 (Fig. 18). At a current of 0.1C, 118 displays a 1100 mA h g−1Csp on the first cycle and, importantly, retains 74.8% capacity after 100 cycles. In a later publication, the same group improved the Csp to 1225 mA h g−1 at 0.1C.119 The capacity fading improves on prior work, and polymerization with unsaturated organic molecules is a promising alternative compared to encapsulation with conductive carbon. The ability to mould the electrode and form free-standing films is an opportunity to conform to different form factors for a range of battery architectures. Following a similar procedure, allyl terminated poly(3-hexylthiophene) (P3HT) was heated with molten sulfur to form 119 in order to improve the conductivity of the electrode.120 The Csp is 1212 mA h g−1 at 0.5C and decreases to 739 mA h g−1 at 1C. The capacity retention was similar to the reports of 118, retaining 799 mA h g−1 after 100 cycles. The advantage of 119 over 118 is that the use of a conjugated polymer allows the material to form a microstructure due to the phase separation of the P3HT and the sulfur domains, and the semiconducting P3HT phase can transport charge into the sulfur domains. In another report, poly(acrylonitrile) was ball milled with sulfur followed by a heating step to form 120 which can be used as a lithium–sulfur cathode.121 The material has an ∼9 mA h cm−2 areal capacity at 4.2 mA cm−2 and retains 73.3% of its capacity after 90 cycles at 0.42 mA cm−2 for charging and 4.2 mA cm−2 for discharging. It should be noted that to prevent polysulfide shuttling, the investigators coated the separator in polydopamine and added 0.05 M CsNO3 to the electrolyte to prevent lithium dendrites. It is expected that the use of organic compounds for metal–sulfur batteries will increase due to the groundwork being laid by polymerizing sulfur with unsaturated organic compounds.118 However, the introduction of a commercially viable metal–sulfur battery is expected to result from a combination of strategies to encapsulate polysulfides, prevent dissolution and shuttling, and increase conductivity of the cathode.
 | ||
| Fig. 18 (a) Synthesis of 118 by a melt polymerization of sulfur and diisopropenylbenzene. (b) Images of the molten sulfur and resultant 118 after polymerization. (Reproduced from ref. 118, reprinted with permission, copyright 2013, Nature Publishing Group). | ||
3.3 Metal–air batteries
Metal–air batteries are a promising new class of batteries that use an oxygen cathode. The oxygen cathode is usually a conductive carbon support with an oxygen reduction/oxidation catalyst. It is likely that only the anode can be an organic material since the generation of superoxides and peroxides at the cathode would likely deteriorate organic compounds. With metal–air batteries, the anode needs to be in the reduced state to contain metal ions in order for the system to function, similar to metal-ion and metal–sulfur batteries.The first example of an organic metal–air battery was a poly(vinylanthraquinone), 121, designed as an aqueous sodium–air battery anode.122121 was prepared as a thin film with a thickness of 30 nm and has a 214 mA h g−1Csp at current rates of 15 to 150C. The device had an operating voltage of 0.63 V, and maintained ∼70% of its capacity after 500 cycles at 15 A g−1. The use of a norbornene-based polymer with pendant anthraquinone units, 122, has also been demonstrated.123 A 50 nm thick film of the polymer exhibited a 210 mA h g−1Csp at current rates of 1 to 600C. The electrode is stable with respect to cycling, retaining 95% of its capacity after 500 cycles. When used in a full device, the operating voltage was 0.68 V. Although the Csp, cycling stability and rate capability is very high in both 121 and 122, the films are very thin. Thicker films will be required for metal–air batteries, and the increased thickness may have a dramatic effect on performance.
3.4 Dual-ion batteries
Dual-ion batteries rely on the diffusion of both anions and cations in the electrolyte to balance charge at the cathode and anode respectively. This requires a relatively large amount of electrolyte compared to metal-ion batteries because the ions are not replenished by a ‘rocking-chair’ type mechanism. Overall, this increases the mass of the device, decreasing the Csp of a full cell. However, the use of a positive charge-accepting cathode does have advantages. Dual-ion batteries do not require one electrode to contain metal ions. The electrodes can both be in the uncharged state and assembled without pre-treatment steps, such as electrochemical doping. They also are able to be paired with a diverse set of electrode materials. Additionally, these cathodes operate at high voltages, achieving device voltages of over 3.5 V when an alkali metal is used as the anode. Much work has been carried out on radical-type polymers, especially the use of 2,2,6,6-tetramethylpiperdinyloxyl (TEMPO) as the redox-active unit.65,124–126 While we acknowledge the importance of this work in the field of organic energy storage devices, we will focus on recent examples of materials for dual-ion batteries. | ||
| Fig. 19 The redox mechanism of compound 124. The bold lines indicate the pathway of conjugation that gives aromaticity or antiaromaticity. Adapted from ref. 128. | ||
 | ||
| Fig. 20 (a) Schematic of 125 (red) and PEDOT:PSS (blue) entangled. Transmission electron microscope images of (b) 125, (c) PEDOT:PSS, and (d) the mixture of 125 and PEDOT:PSS. (Reproduced from ref. 129, reprinted with permission from the PCCP Owner Societies). | ||
A battery using only anions to balance charge was designed using a TEMPO-based polymer 127 as the cathode and magnesium as the anode.131 TFSI ions decompose on the metal anode forming a polymeric network that can store charge by the insertion of anions. This is in contrast to the reversible stripping and plating of magnesium on the anode to create a dual-ion battery. The battery has an 84.2 mA h g−1Csp with a 1.7 V voltage vs. Mg/Mg2+ and 50% capacity retention after 10 cycles. Since reversible magnesium stripping and plating is one of the downfalls of magnesium-ion batteries, this could be a solution, however cycling stability issues still need to be resolved. This will require characterization of the anion insertion network.
Main-chain benzidine polymer, 128, was demonstrated to attain a high rate capability.132 A 165 mA h g−1Csp, 91.2% of the Ctheor, is reached at a rate of 100 to 1000C. Additionally, 128 retains 92% of its capacity after 100 cycles at 1000C. It is curious that increasing the length of alkyl chain spacer between aniline groups facilitates the electropolymerization and results in better electrochemical performance. The high rate capability, capacity, and electrochromic nature open up the possibility of constructing a high performance electrochromic, thin film battery using 128. In order to investigate the effect of block copolymer self-assembly on electrochemical performance, films of a polystyrene–TEMPO block copolymer, 129, were prepared and tested.133 With different annealing conditions, different morphologies can be obtained (Fig. 21). The total Csp of the film is ∼27 mA h g−1 at 23.7 μA cm−2, with ∼95% capacity retention after 50 cycles. The spuncast films were ∼80 nm thick and formed cylindrical nanostructures when the sample was solvent annealed. The polymer films are expected to have superior mechanical properties due to the nanoscale morphology. This work demonstrates the application of block copolymer self-assembly towards organic electrodes for energy storage. It is expected that an even greater performance will be achieved by selecting blocks that can be used to provide ionic/electronic conduction pathways. However, the amount of active material in the electrode needs to be considered in order to maximize the capacity of the device.
 | ||
| Fig. 21 Atomic force microscopy images of 129 (a) after spin coating without annealing, (b) after annealing with diethyl carbonate and water, (c) after annealing with dimethylformamide and diethyl carbonate. (Reproduced from ref. 133, reprinted with permission from Royal Society of Chemistry). | ||
A high voltage thianthrene pendant polymer 130 was synthesized and tested for dual-ion batteries with a lithium metal anode.134 The electrode has a 4.10 V and 4.05 V charging and discharging voltage vs. Li/Li+ respectively, with a 66 mA h g−1Csp after 14 cycles with >96% CE. The fast capacity fading after 100 cycles (only 30% retention) is attributed to irreversible anion intercalation. The geometry change upon cycling may play an important role in this capacity fading. It is also interesting that the polymer with two thianthrene units in the repeat unit did not perform as well. This suggests that charge repulsion may not allow complete charging in this system.
An oligomeric pyrene polymer, 132, was investigated as a conjugated polymer cathode for dual-ion batteries.136132 reaches 90.2% of its Ctheor, having a 120 mA h g−1Csp at 20 mA g−1 at a high discharging voltage of 3.54 V vs. Na/Na+ and ∼70% capacity retention after 50 cycles at 20 mA g−1. Interestingly, the best performance is observed for amorphous 132, rather than crystalline, which is unexpected based on results from small molecule electrodes. The performance of the crystalline pyrene suggests that the ionic conductivity through the electrode is more important that the electrical conductivity, since crystalline materials typically have higher electronic conductivity. The cycling performance needs to be addressed. Investigating a number of phenazine polymers, the authors found that having slightly different substituents has a significant impact on performance.137 Compound 133 has the best performance because it has the smallest calculated geometry change upon charging, and therefore the largest electron transfer rate. Formulating 133 into a cathode gives an 80 mA h g−1Csp at 0.1C with a sloping discharging plateau between 4.0 V and 3.2 V vs. Li/Li+. This work provides important insight into the use of DFT to rationalize the behaviour of different compounds with small variations in structure. Although the capacity of this material limits its applicability, this work emphasizes that geometry changes upon charging can greatly influence electron transfer rates, which is an important consideration when designing organic battery electrodes.
In an interesting report, 134 was used as both the anode and cathode in an all-organic dual-ion battery.138 The device exhibits a 65 mA h g−1Csp at 0.05 A g−1 and 17 mA h g−1 at 2.0 A g−1. Incredibly, it has a high stability with ∼100% retention after 1000 cycles and a voltage of up to 3.5 V. The low capacity is likely due to the minimal redox activity of the material, as indicated by the lack of voltage plateaus in the charge/discharge curve. A polytriphenylamine derivative 135 was reported as a cathode material for dual-ion batteries, using lithium as the anode, that had a better performance than polytriphenyl amine itself.139 Due to the higher radical density and Ctheor than polytriphenyl amine, 135 achieves a much higher Csp (129.1 mA h g−1 at 20 mA g−1 and 92.8 mA h g−1 at 500 mA g−1). This material has two voltage plateaus at 3.8 V and 3.3 V vs. Li/Li+ and an 85.6% capacity retention after 50 cycles at 20 mA g−1. The high radical density allows 135 to transport charge rapidly through the film allowing for high rate capabilities.
In an effort to understand the performance of phenoxy radical polymers as aqueous anode materials, a series of polymer structures were synthesized bearing different backbones and phenoxy radicals (144–146).141 Bulky alkyl groups are required on the aromatic ring to stabilize the radical species. Compound 146 has the best performance, with a 60 mA h g−1Csp at 1C and 100% capacity retention after 100 cycles. The low Csp is in part due to the bulky groups required to stabilize the radicals and the redox unit's ability to accept only one charge. The low content of active material is a concern, which may be due to the hydrophobic polymer backbone and carbon additives. This draws attention to the importance of the wettability of the electrode.
4. Redox flow batteries
Traditional RFBs use vanadium and bromine as the redox couples in an aqueous electrolyte. These systems have the disadvantage of being vulnerable to changes in the price of the metal, they typically use highly corrosive electrolytes, and bromine itself is volatile and hazardous. RFBs have recently come back into the spotlight for research. This is likely due to the push towards renewable energy and the need for inexpensive grid storage that is reliable and can be easily scaled up without the safety concerns such as those associated with large-scale lithium-ion batteries. Consequently, the development of organic materials for RFBs has increased, with most research focused on quinones. There is great interest in replacing both the anolyte and catholyte with organic materials and also increasing the operating potential of RFBs by replacing the aqueous electrolyte with an organic electrolyte. Here we will highlight some of the novel structures and major advances.4.1 Aqueous electrolyte RFBs
Aqueous RFBs are the most popular type of flow battery system. This is because water is highly abundant and inexpensive, relatively non-volatile, has high ionic conductivity, and can dissolve high concentrations of the metal salts usually used as the redox couples in commercial RFBs. The use of organic redox couples has in the past been plagued with the low solubility of organic compounds in aqueous solutions causing low energy density and high cost. However, in a seminal paper by Huskinson and coworkers, the ability to use a 2,6-bisulfonated anthraquinone, 147, in an aqueous electrolyte was demonstrated.142 They showed that not only can 147 be dissolved in high concentrations on the order of 1 M, but by using it as the anolyte and bromine as the catholyte, an operating voltage of 0.92 V at 90% SOC is achieved. This RFB has a high cycling stability of >99% retention per cycle and an energy density of >50 W h kg−1. This research spurred the development of numerous quinone derivatives and highlights that solubility can be imparted by sulfonation of aromatic rings. Additional work was conducted on improving the performance of the device in terms of power density, attaining 1 W cm−2, which is about 75% of the highest value reported in the literature for vanadium RFBs.143 Although the voltage of the battery was moderate, it could be improved with the use of a different catholyte. This work is the first to demonstrate that organic materials can be used in high performing aqueous RFBs. In order to address the issues of using molecular bromine as the redox couple in aqueous RFBs, an organic catholyte was introduced to construct an all-organic RFB.144 The disulfonated benzoquinone, 148, was used as the catholyte in conjunction with either 149 or 150 as the anolyte. This all-organic RFB has a 0.6 V voltage at 100% SOC using 150 as the anolyte, with no noticeable change in capacity after 12 charge/discharge cycles. The voltage of this device is low due to the relatively low redox potential of the catholyte, however this work demonstrates that the development of an aqueous RFB using only organic redox couples is feasible. The development of high voltage, and highly soluble catholytes is an area of research that has been met with significant challenges. This is because the ionic groups typically used to impart high solubility in aqueous electrolytes are electron-withdrawing, which lowers the redox potential.A significant hurdle in RFB research is reducing cost, which has been difficult in the past due to the expensive Nafion membrane typically used in aqueous RFBs. In an effort to reduce this cost, a polymeric catholyte, 151, and anolyte, 152, were developed.145,146 These polymers are soluble in a 2 M NaCl solution and the device has a 1.1 V open circuit voltage at 100% SOC. The RFB made with 151 and 152 has an 8.2 A h L−1Cv, an 8.0 W h L−1 energy density, and a high capacity retention of 80% after 10000 cycles at 20 mA cm−2. Importantly, due to the macromolecular nature of the polymers, the Nafion membrane can be replaced with a dialysis membrane, preventing crossover by size exclusion (Fig. 22). This represents a significant step forward in the design of aqueous RFBs using only organic redox couples for two reasons: (1) the replacement of the Nafion membrane will significantly lower costs; and (2) the operating potential is 1.1 V, which is very close to reaching the limits imposed by aqueous electrolytes. Further improvement in this area could be achieved by using polymers that do not require solubilizing blocks, as these inherently lower the redox active group content in the polymer and decrease the Cv. This could be achieved by using ionic polymer backbones with pendant redox groups or by using redox active backbones with pendant solubilizing groups.
 | ||
| Fig. 22 (a) Working principle of the polymer based on 151 and 152 as the catholyte and anolyte respectively. (b) The redox mechanism occurring in polymers 151 and 152 respectively. (Reproduced from ref. 146, reprinted with permission, copyright 2015, Nature Publishing Group). | ||
An example of using an alkaline electrolyte in RFBs was reported where 153 is used as the anolyte and Fe(CN)64− is used as the catholyte.147 The authors postulated that insignificant crossover would occur with a Nafion membrane since both redox species are negatively charged. The RFB has a 1.2 V open circuit voltage at 50% SOC, and the 153 anolyte has a 27 A h L−1Cv. The capacity retention was 84% after 100 cycles, attributed to hydraulic leakage from the gaskets rather than chemical degradation or crossover. Additionally, the full cell has a 6.8 W h L−1 energy density and a 0.7 W cm−2 power density at 45 °C. This high performance redox flow battery prevents crossover and uses non-toxic chemicals to store energy. Due to the basicity of the electrolyte and the use of carbon electrodes, the potential window can be expanded to a maximum of ∼2.0 V before electrolyte breakdown is observed. This large voltage can be achieved through chemical design and it is expected that alkaline redox flow batteries will be the subject of further investigation because of their high voltage limit. Exploring low cost redox-active materials, a TEMPO based small molecule, 154, was synthesized and used as a catholyte with methyl viologen, 155, as the anolyte in an aqueous RFB.148 The RFB displays a high Cv of 9.58 A h L−1 with 62.1%, 62.5%, and >99% VE, EE, and CE's respectively. The flow battery using 0.5 M of each redox couple has an 89% capacity retention after 100 cycles. Interestingly, using a lower concentration increases the cycling stability, but decreases the other performance parameters. The high diffusion coefficients of these molecules should allow them to attain high power densities, however, a detailed investigation of the capacity degradation mechanism along with the low VE and EE should be undertaken.
4.2 Organic electrolyte RFBs
The inherent problem with any aqueous device is the limitation of the operating voltage due to water oxidation and reduction. By using organic electrolytes, the voltage window can be increased significantly and thereby increase the energy and power density. Much of the research in organic electrolyte RFBs has been focussed on improving the redox potentials of the active materials, increasing the capacity, and, possibly the most challenging, improving solubility of the redox-active species. An interesting strategy to overcome solubility issues is to use polythiophene, 78, microspheres suspended in a propylene carbonate electrolyte.149 By taking advantage of 78's ability to be both n- and p-doped, a symmetric device can be constructed that mitigates problems associated with crossover. The symmetric device attains a large operating potential of 2.5 V and has a 110 mA h g−1Csp at 0.5 mA cm−2 based on the mass of 78, with a 77.5% CE, a 78.6% VE, and a 60.9% EE. The high voltage of this battery is a significant advantage, but the instability of 78 is a concern, and it may explain the low efficiency values. In another effort to construct an all organic RFB, 156 and 157 were used as the catholyte and anolyte respectively to achieve a 2.37 V operating voltage.150 When the anolyte was cycled in acetonitrile, significant decomposition reactions occur, but when a DME electrolyte is used, the stability increases. Using this optimized electrolyte, the Cv reaches 1.04 A h L−1 and a 90% capacity retention is achieved after 50 cycles at 10 mA cm−2. An important part of this work is the investigation of the capacity fading mechanism and suggestions of possible solutions to this problem. The establishments of guidelines that can be used to avoid capacity fade are of paramount importance to the development of highly stable and high performing materials for RFBs. Using a phenothiazine derivative, 158, as a catholyte and 159 as the anolyte, another all organic RFB was developed.151 This RFB has a 0.62 mA h g−1Csp based on the mass of the limiting solution and an average of 1.4 V discharging voltage with a 92% CE. Unfortunately, the cycling stability is poor, losing most of the initial capacity after 100 cycles. However, the authors found that using a higher concentration slightly improves stability. This system has a low voltage and low cycling stability. By coupling each material with a different anolyte/catholyte, an improved device could be constructed.An interesting study on the development of an RFB anolyte was performed by Sevov and coworkers taking into account the redox potential, solubility, stability, and Ctheor.152 After a series of optimization steps, the authors designed pyridinium derivative, 160, which has reduction potentials centred at −1.1 and −1.48 V vs. Ag/Ag+ and can be dissolved up to 1.6 M in acetonitrile. The compound has diffusion coefficients of 1.1 × 10−5 cm2 s−1 and 1.8 × 10−5 cm2 s−1 and electron transfer rate constants of 6 × 10−3 cm s−1 and 4.7 × 10−3 cm s−1 for the first and second reductions respectively. Although the solubility of the neutral species was optimized, the authors note that the solubility of the fully reduced species is low and this needs to be taken into account in the future. The stepwise design process of organic RFB redox couples is described especially well in this work and provides great insight into the consideration of almost all aspects of optimizing performance metrics. This should be a general strategy for the design of future organic RFB redox couples.
A symmetric all organic RFB was investigated using 161 as both the anolyte and the catholyte.153 By using a combination of simulations and experimental studies, the authors outline the definition, properties and advantages of symmetric RFBs. The battery displays relatively low EE's between 43% and 28%, likely due to a number of factors contributing to polarization, including the low solubility of the compound in the electrolyte. The design of symmetric RFBs has clear advantages as outlined in this work.
Hybrid devices using a solid-state anode have also been reported, with the intention of developing a specific catholyte and highlighting the ability to construct a hybrid flow-solid electrode battery. Wei and coworkers reported the use of TEMPO, 162, a catholyte that is soluble up to 2.0 M in a carbonate electrolyte mixture.154 With lithiated graphite as the anode, 162 has a high voltage of 3.5 V vs. Li/Li+ and a high energy density of 126 W h L−1 with an 84% CE, 82% VE, and a 69% EE. The high voltage of the device along with the respectable energy density is certainly an advantage. The capacity fading may be caused by charge-shuttling by the soluble catholyte. A series of alkoxybenzenes were investigated for redox flow catholytes using lithium as the anode.155 It was found that 163 functions well as a catholyte because it is a liquid, and can be dissolved in higher amounts than a solid compound of similar structure. It has a 3.9 V vs. Li/Li+ voltage with a ∼75% capacity retention after 30 cycles at 0.4 mA, and an 80.9% EE after the first cycle. The high voltage is very attractive, but an investigation into the capacity fading mechanism is very important in order to design future materials. In an attempt to construct a flexible hybrid battery containing a solid lithium anode and a liquid organic catholyte, compounds 164 and 165 were studied (Fig. 23).156 Out of the two compounds, the most promising is 165 with a 169 mA h g−1Csp at 24 mA g−1 and two voltage plateaus at 2.43 V and 2.24 V vs. Li/Li+. The flexible device has a 93.5% capacity retention after 100 cycles when cycled at an elevated temperature of 60 °C. This work demonstrates that liquid catholyte-containing devices are not limited to stationary grid storage and can also be used in portable electronics. In general, these hybrid devices seem to be fairly unstable compared to most the other RFB systems. The reasons for this are rarely discussed in detail, and in order for these systems to become viable this issue needs to be addressed.
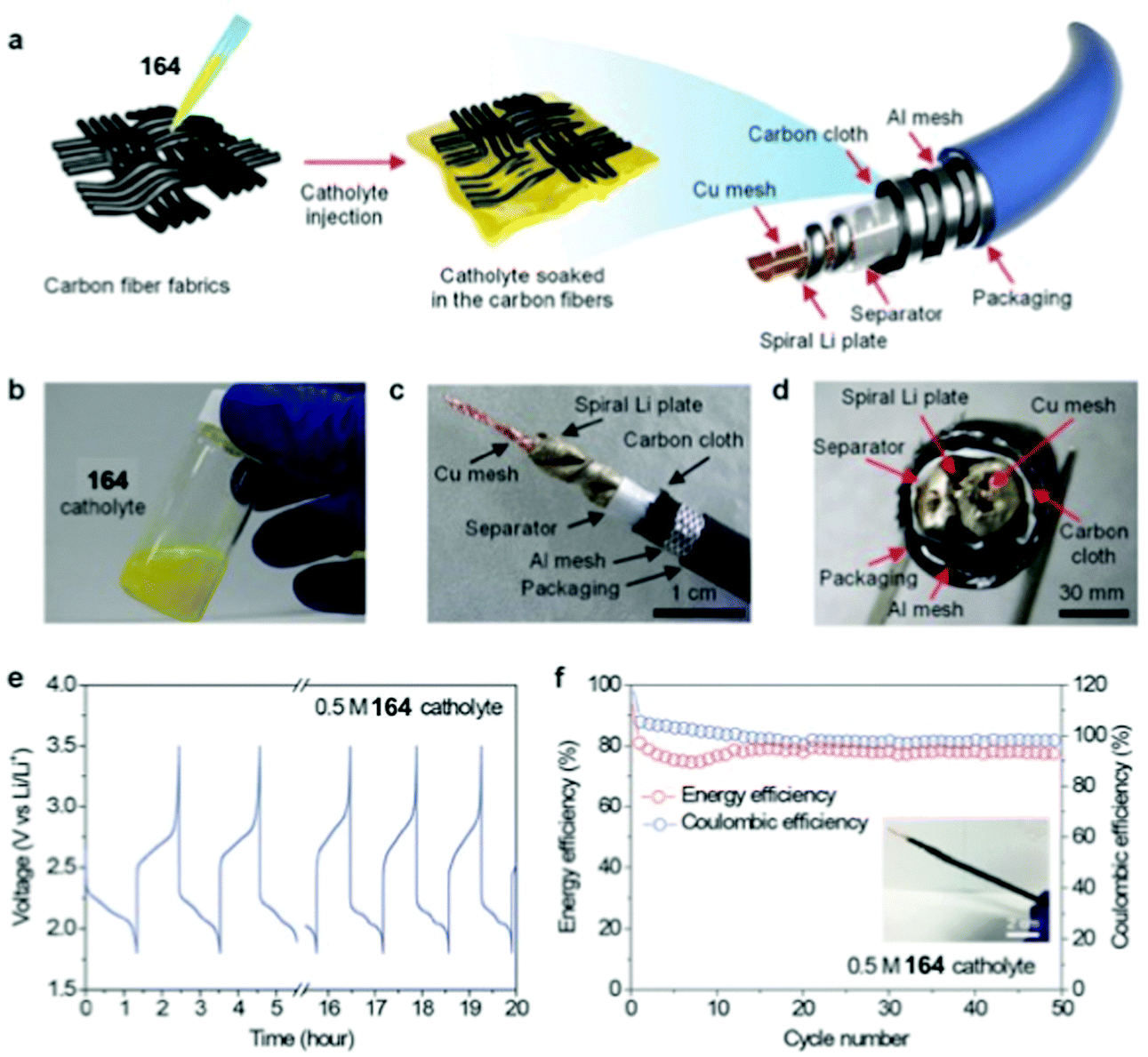 | ||
| Fig. 23 (a) Schematic of battery construction and configuration using 164 impregnated into the carbon fiber fabric as the catholyte and lithium as the anode. (b) Photograph of the 164 catholyte. (c and d) Photograph of the battery components. (e) The charge/discharge cycle behavior and (f) Cycling performance of 164 catholyte for 50 cycles at a current rate of 0.41 mA cm−2 and voltage window from 1.6 to 3.5 V. (Reproduced from ref. 156, reprinted with permission, copyright 2015, Wiley-VCH). | ||
5. Supercapacitors
Due to their redox activity, organic materials used in SCs typically store charge by pseudocapacitance. Important pseudocapacitive materials include metal oxides and conjugated polymers. Metal oxides, such as RuO2, have some of the highest specific capacitances, however they typically have low flexibility and conductivity (leading to limited rate capability), contain rare materials, and can be expensive.157 Conjugated polymers are promising alternatives to metal oxides to store energy in SCs because they can be synthesized from abundant materials, their properties can be tuned through synthetic modification, they are flexible, light weight, and are potentially inexpensive. Conjugated polymers are promising in different roles for SC electrodes other than to store energy, such as transparent current collectors.158Most research on organic SC materials has focused on p-dopable conjugated polymers such as polyaniline, polythiophene, and polypyrrole. However, the key to improvement and widespread implementation of pseudocapacitive SC technologies is innovation at the materials level. This hinges on uncovering the fundamental relationship between polymer structure and device performance. From an organic materials design standpoint, it is difficult to predict pseudocapacitive behavior in organic compounds because, to the best of our knowledge, there are no quantitative guidelines or certain electronic properties of organic materials that can be modeled computationally to distinguish between battery-like or SC-like behaviour. Qualitatively, however, highly conductive organic materials seem to possess pseudocapacitive electrochemical profiles and we suggest this could be a guideline for designing new materials. This is supported by the fact that the majority of organic SC publications have focused on highly conductive conjugated polymers such as PEDOT, polyaniline, polypyrrole and derivatives of these materials. In fact, the majority of new SC organic materials are conjugated polymers, compared to the relatively small amount studied for battery applications. Current research in supercapacitors focuses on increasing cycling stability, energy density, power density, and decreasing the cost of the device. To this end, we will review recent advances in novel materials for applications in SCs.
5.1 Positive charge-accepting materials
Functionalization of various carbons, such as graphene and carbon nanotubes, with pseudocapacitive materials is an important area of research, aimed at harnessing the favourable properties of HSACs (high surface area and pore structure), while increasing their capacitance. In a study by Duan et al., graphene hydrogels were functionalized with hydroquinones 166.159 The high Cpc of a symmetric SC constructed using 166 (441 F g−1 at 1 A g−1) compared to unfunctionalized graphene hydrogels (211 F g−1) is attributed to a large surface area for hydroquinone π–π interactions, leading to a large pseudocapacitive contribution. The device exhibits exceptional stability, with only a 14% decrease in Cpc over 10000 cycles. In a solid-state device using a gel electrolyte, 166 demonstrates similar Cpc (412 F g−1 at 1 A g−1) and stability (87% retention over 10000 cycles) (Fig. 24). Interestingly, the device displays excellent mechanical flexibility, performing exceptionally at a 150° bending angle. Non-covalently functionalizing conductive carbons with pseudocapacitive materials is an effective way to combine high surface area and redox activity, without affecting the conductivity.
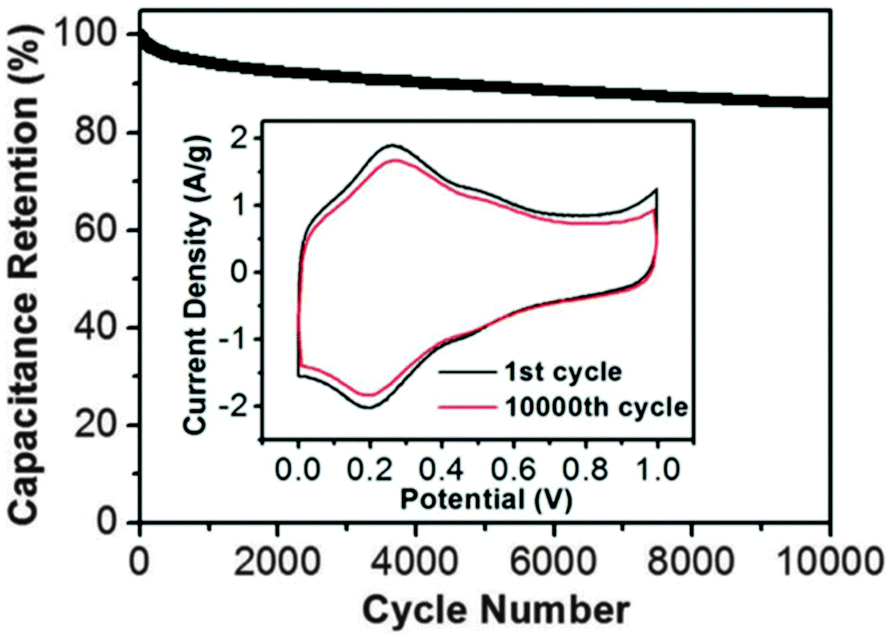 | ||
| Fig. 24 Cycling stability of FGH-based SC at a current density of 10 A g−1. Inset shows a cyclic voltammogram of the device at 5 mV s−1 after the 1st and 10000th galvanostatic charge/discharge cycle. (Reproduced from ref. 159, reprinted with permission, copyright 2013, Wiley-VCH). | ||
Functionalized graphene nanoplatelets with the thiophene-based polymer 167 has an enhanced Cpc of 206 F g−1 at 1 A g−1 compared to unfunctionalized graphene nanoplatelets.160 The composite electrode has a 78% capacitance retention over 1000 cycles. This is similarly attributed to the strong π–π interaction between the nanoplatelets and 167, increasing the effective conjugation length of 167, as evidenced by Raman spectroscopy. Furthermore, the electron donating functionality of this polymer helps stabilize the doped state. Graphene nanoplatelets increase the conjugation length of the polymer, decrease the resistance of the electrode, and make the morphology of the electrode more favourable for ion diffusion. Thiophene moieties could lead to stability issues due to coupling and cross-linking reactions at the unsubstituted positions upon oxidation.
Similar electrostatic interactions between carboxylate functional groups on few-walled nanotubes and ammonium groups on an amino-functionalized pyrene polymer 168b leads to strong electrostatic interactions and uniform coating.161 A hybrid device using a lithium negative electrode attains a 210 F g−1Cpc with 100% CE at 0.05 A g−1. This composite electrode demonstrates superior cycling stability, with minimal capacitance loss after 1000 cycles, and only a 15% loss after 11000 cycles. Using functional groups that have strong electrostatic interactions with carbon composites can provide better interfacial interactions, facilitate charge transfer, and increase Cpc and stability.
Well-defined, high surface area materials with tunable pore sizes such as covalent organic frameworks are attractive candidates for capacitive energy storage. The effect of synthesis temperature on the capacitance of a triazine based framework, 169, was investigated and tested in a symmetric SC.162 Increasing the synthesis temperature from 550 to 700 °C was found to increase crosslinking, conductivity, and surface area, and also tune pore surfaces (Fig. 25). However, only a slight increase in Cpc from 147.1 F g−1 to 151.3 F g−1 at 0.1 A g−1 was observed. This is attributed to the decrease in nitrogen content with increasing temperature, causing a decreased interaction between the electrode and electrolyte, therefore decreasing capacitance and counteracting the increase in surface area. A symmetric SC made with 169 synthesized at 700 °C had high energy and power densities of 47.4 W h kg−1 and 7.5 kW kg−1 at 3 V, and 62.7 W h kg−1 and 8.75 kW kg−1 at 3.5 V, respectively. At 10 A g−1 and 3 V the 700 °C SC maintains 85% of the capacitance after 10000 cycles, indicating the impressive stability of this material. This work provides insight into the relationship between heat treatment temperature, heteroatom content, and surface area for synthesizing triazine frameworks. Increasing microporosity while preserving a high nitrogen content, would be a very important contribution to the field.
 | ||
| Fig. 25 The effect of reaction temperature of 169 on (a) pore size, determined by DFT (with inset specific capacitance vs. effective specific surface area at 0.1 A g−1) and (b) nitrogen content, determined using elemental analysis and X-ray photoelectron spectroscopy. (Reproduced from ref. 162, reprinted with permission, copyright 2014, American Chemical Society). | ||
In 2015, the Jiang group developed the first radical-functionalized porous material for energy storage.163 By first synthesizing a porphyrin-based framework and then functionalizing it with TEMPO groups, compound 170 was synthesized. The framework 50% functionalized with TEMPO has a 124 F g−1Cpc at 0.1 A g−1, with 81% capacity retention when increasing the current density to 2 A g−1. The framework 100% functionalized with TEMPO groups has a 167 F g−1Cpc, however only 68% capacity retention upon increasing the current density to 2 A g−1 due to a decrease in porosity, leading to slower ion transport. Importantly, the Cpc of 170 functionalized 50% with TEMPO groups is completely retained over 100 cycles at 500 mA g−1 due to the insolubility of the redox active groups. Increasing the pore size should improve the ionic conductivity and would increase the rate performance of the 100% functionalized frameworks. Future work should focus on decreasing the mass of the redox inactive components while retaining conductivity and insolubility. Our group recently developed a novel 3-D framework containing thiophene, 171, and heavier group 16 heterocycles selenophene and tellurophene.164171 was found to be a promising positive electrode for asymmetric supercapacitors. An asymmetric device using 171 as the positive electrode and carbon black as the negative electrode has a 4.01 ± 0.05 mF cm−2 areal capacitance at 0.1 A g−1, with 80% capacity retention after 500 cycles. This loss occurs within the first few cycles, and remains constant thereafter, highlighting the stability of this novel material (Fig. 26). Furthermore, areal capacitance is highly dependent on the thickness of the electrode, and could be improved by increasing the film thickness. Improving conductivity, device and film optimization, and pore size optimization are expected to lead to improved capacity and rate capability. This novel material demonstrates that 3-D porous materials can be used successfully as SC materials.
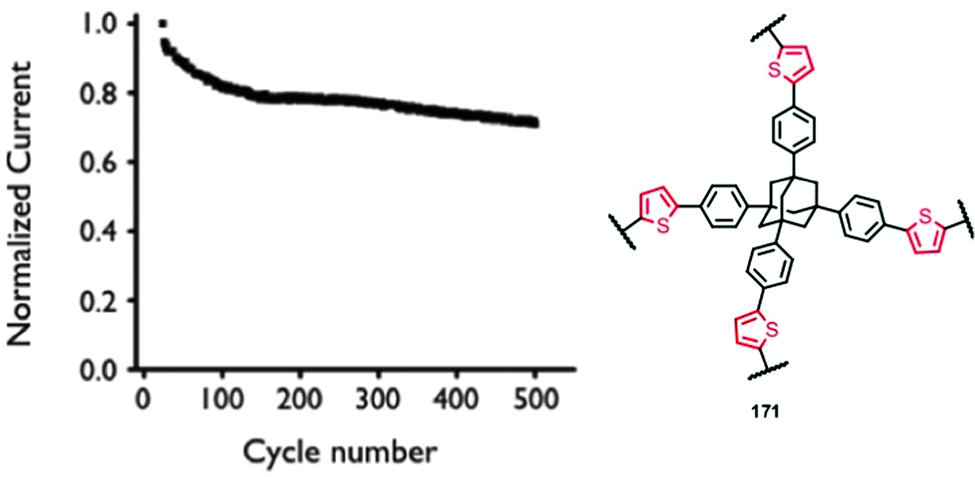 | ||
| Fig. 26 Cycling stability of SC using 171 as the positive electrode and carbon black as the negative electrode at 0.1 A g−1. Adapted from ref. 164. | ||
Zhang and coworkers demonstrated the ability to synthesize a high surface area, cross-linked structure of pyrroles, 172, by thermal cyclodebromination of polybromopyrroles.165 The electrode has a 423 F g−1Cpc and a 143 F cm−3 volumetric capacitance at 0.1 A g−1, retaining 88.2% of its initial Cpc after 2100 cycles at 1 A g−1. A symmetric device of 172 has a 216 F g−1Cpc and a 101 F cm−3 volumetric capacitance at 0.5 A g−1. Stability tests reveal a 78.3% and 77.4% capacitance retention after 2000 and 4000 cycles respectively at 0.5 A g−1. The impressive performance of this material in terms of Cpc and cycling stability makes it very attractive. The device voltage should be improved by using a different counter electrode material or by switching to an organic electrolyte. A high surface area, 3-D structured electrode consisting of polypyrrole functionalized with a catechol derivative through non-covalent interactions, 173, attains a similarly high Cpc of 385 F g−1 at 0.4 A cm−3.166 Functionalization facilitates a synergetic charge storage mechanism of polypyrrole and quinone–hydroxyquinone redox reactions, while the 3-D structure enables fast ion transport and charge transfer. Impressively, 173 retains 75% of its initial Cpc after 10000 cycles at 2 A cm−3, with 97% CE. The authors predict that this stability is due to passivation of polypyrrole by the catechol. The strong non-covalent interactions between polypyrrole and the catechol derivatives make this material very attractive for SC electrodes and limits the possibility for charge shuttling. A conjugated microporous polymer film containing zinc porphyrins and carbazole moieties, 174, reaches a 142 F g−1Cpc at 5 A g−1, with 70% capacity retention of the initial Cpc upon increasing from 10 to 50 A g−1.167 The highly cross-linked, porous network can be peeled off of the current collector to form free-standing, flexible films and can be used directly as the electrode, making it promising for flexible SC applications. This material uses the redox activity from both the zinc porphyrin rings and the carbazole moieties, which gives it a series of overlapping redox peaks and ideal-like SC behavior. The material should be tested in an electrolyte that is more feasible for commercial applications, rather than DCM.
A poly(aminoanthraquinone) polymer, 175, demonstrates excellent charge accepting ability, with a high Cpc of 406.3 F g−1 at 0.2 A g−1.168 The optimized synthesis of this polymer with chemical oxidant Ce(SO4)2 at 20 °C led to homogenous submicron particles and higher conductivity than previously reported. The material exhibits good rate capability with a 63% capacity retention at an increased current density of 50 A g−1. A symmetric SC also demonstrates only a 9.3% loss in capacity over 20000 cycles at 1 A g−1. This superior cycling stability is attributed to the strong π–π stacking of anthracene rings and hydrogen bonding between N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, forming a supramolecular structure. This polymer is a promising material for SCs because of its low cost, no required additives, industrially scalable synthesis and outstanding stability. The negative charge-accepting capabilities of the anthraquinone groups should be investigated. Xu and coworkers demonstrated that a furan–thiophene copolymer, 176, could be electrochemically synthesized and the resultant polymer is promising for supercapacitor electrodes.169 Electrodes tested in acetonitrile lithium perchlorate electrolytes display moderate Cpc's, however, only 25.5% of the initial Cpc is retained after 500 cycles, thought to be due to side reactions with residual water. When cycled with an electrolyte containing boron trifluoride diethyl etherate, the Cpc reaches 392 F g−1 at 5 A g−1. Additionally, the cycling stability increases to 67% retention after 500 cycles at 10 A g−1. This improved performance is attributed to the high conductivity of the electrolyte, its ability to increase ion diffusion and the reduced oxidation potential of 176, thereby increasing its stability. The high oxidation potential of unsubstituted polythiophene may be the reason for the instability during electrochemical cycling. Increasing stability, and improving other performance metrics by lowering the oxidation potential with a Lewis acid is very interesting. However, boron trifluoride diethyl etherate may generate hydrofluoric acid.
O groups, forming a supramolecular structure. This polymer is a promising material for SCs because of its low cost, no required additives, industrially scalable synthesis and outstanding stability. The negative charge-accepting capabilities of the anthraquinone groups should be investigated. Xu and coworkers demonstrated that a furan–thiophene copolymer, 176, could be electrochemically synthesized and the resultant polymer is promising for supercapacitor electrodes.169 Electrodes tested in acetonitrile lithium perchlorate electrolytes display moderate Cpc's, however, only 25.5% of the initial Cpc is retained after 500 cycles, thought to be due to side reactions with residual water. When cycled with an electrolyte containing boron trifluoride diethyl etherate, the Cpc reaches 392 F g−1 at 5 A g−1. Additionally, the cycling stability increases to 67% retention after 500 cycles at 10 A g−1. This improved performance is attributed to the high conductivity of the electrolyte, its ability to increase ion diffusion and the reduced oxidation potential of 176, thereby increasing its stability. The high oxidation potential of unsubstituted polythiophene may be the reason for the instability during electrochemical cycling. Increasing stability, and improving other performance metrics by lowering the oxidation potential with a Lewis acid is very interesting. However, boron trifluoride diethyl etherate may generate hydrofluoric acid.
Three similar conjugated polymers based on PEDOT, containing different alkyl side chains, 177–179, all display similar Cpc's of 132.5, 135.4, and 129.3 F g−1 at 1 A g−1 respectively, and a decrease to 123.6, 112.4, and 108.9 F g−1 at 10 A g−1.170 Symmetric SCs were fabricated with low Cpc's of 31.2, 31.8, and 30.2 F g−1 at 0.5 A g−1 for 177, 178, and 179 respectively, with maximum specific energies of 11.1, 11.3 and 10.7 W h kg−1. These energies exceed those of most commercial SCs. Specific power values of 11.6, 13.8 and 13.9 kW kg−1 are obtained with 76.3%, 84.5% and 86.4% capacity retentions upon increasing the current density to 17.5 A g−1. Although the performance is acceptable, the use of polymers with solubilizing side chains does not seem to offer many advantages over PEDOT itself.
5.2 Negative charge-accepting materials
The development of negative charge-accepting materials is essential to increasing the operating voltage of SCs and therefore energy and power densities. In 2013, the Dichtel group demonstrated the use of a high surface area, amorphous 2-D covalent organic framework with redox-active moieties, 180, as a negative charge-accepting electrode material.171 Importantly, this was the first example of a 2-D covalent organic framework with reversible redox behaviour of the two electron, two proton reduction of the quinone groups. Low Cpc values (48 ± 10 F g−1 at 0.1 A g−1) result from poor electrical contact between the compound and current collector that leads to the accessibility of only 2.5% of the redox active groups. Cycling experiments reveal an initial decrease in Cpc (40 ± 9 F g−1) in the first 10 cycles, but no further decrease over the next 5000 cycles. Ditchel and co-workers later improved upon this work by forming oriented thin films of the same material, 180.172 They observed a remarkable 400% increase in Cpc compared to the randomly oriented film, attributed to the oriented crystalline morphology facilitating conduction between the redox layers (Fig. 27). Again, excellent cycling stability of films is demonstrated, with only 7% loss over 5000 cycles. The conductivity of the material in the oriented crystal allows for relatively high rate capabilities. However, the thicknesses of the films are fairly low. If the thickness of the films could be increased while retaining high crystallinity, this material would be even more attractive.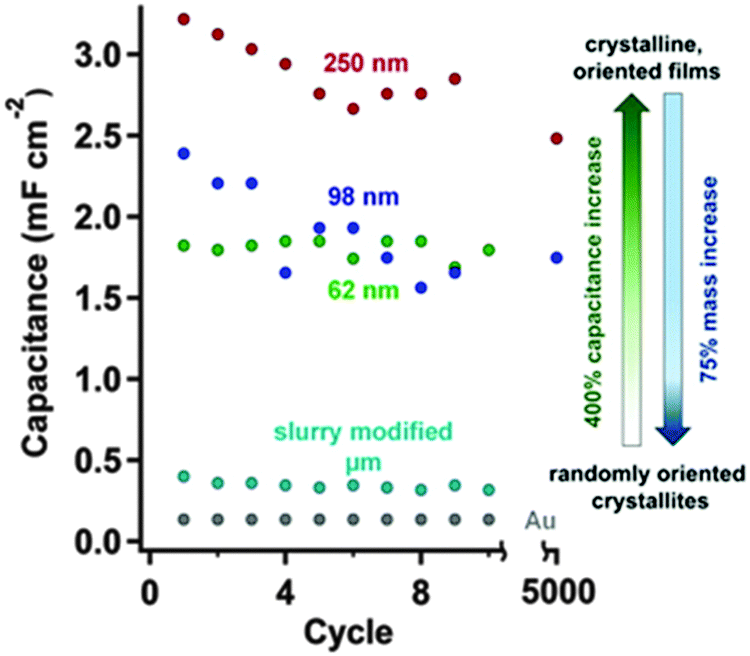 | ||
| Fig. 27 Capacitance retention over 5000 cycles for 180 slurry, blank Au electrode, and oriented thin films as a function of thickness: 250 nm (red), 98 nm (blue), 62 nm (green). (Reproduced from ref. 172, reprinted with permission, copyright 2015, American Chemical Society). | ||
In an effort to reduce the cost of electrode materials, bio-derived lignin, the second most abundant biopolymer and waste product of paper production, can be used as a pseudocapacitive electrode.173 Lignins contain quinone groups that can contribute to the pseudocapacitance afforded by the polypyrrole electrodes and thereby increase Cpc. Using polypyrrole and four lignin samples, 181, with varying concentrations of monolignols syringyl (S), guaiacyl (G), and hydroxyphenyl (H), the electrodes displayed 206, 220, 239, and 282 F g−1Cpc's at 1 A g−1 with increasing S/G ratio from 0.3–2.6. The use of biopolymers in conjunction with conductive polymers is an effective way to increase performance of electrodes without having the charge-shuttling problem associated with small molecule additives. Full characterization of the electrode, including the cycling stability, is required in order to make further conclusions about the practical applicability of this approach.
In 2014, our group reported an electrochemically polymerized fullerene, 182, used as a negative electrode in SCs.174 This highly cross-linked material has high Cpc values ranging from 110–220 F cm−3 at 100–10 A cm−3, due to its ability to accept multiple electrons per monomer unit. The cycling stability of the electrode was poor, with a complete loss in Cpc after 250 cycles, which was attributed to ion influx when charging. An asymmetric type IV device using PEDOT as the positive electrode has a 7.9 ± 1.1 F cm−3Cpc at 0.11 mA. The high device voltage of 2.2 V led to a 5.3 ± 0.7 W h L−1 energy density, and a 4270 kW L−1Pmax. Importantly, the device has a power density two to three times higher than a symmetric PEDOT/PEDOT SC (Fig. 28). Despite the high Cpc of the electrode, the cycling stability and the low film thickness present a barrier. Future work on this system should be focused on improving the film thickness and stability by optimizing synthesis and electrolyte.
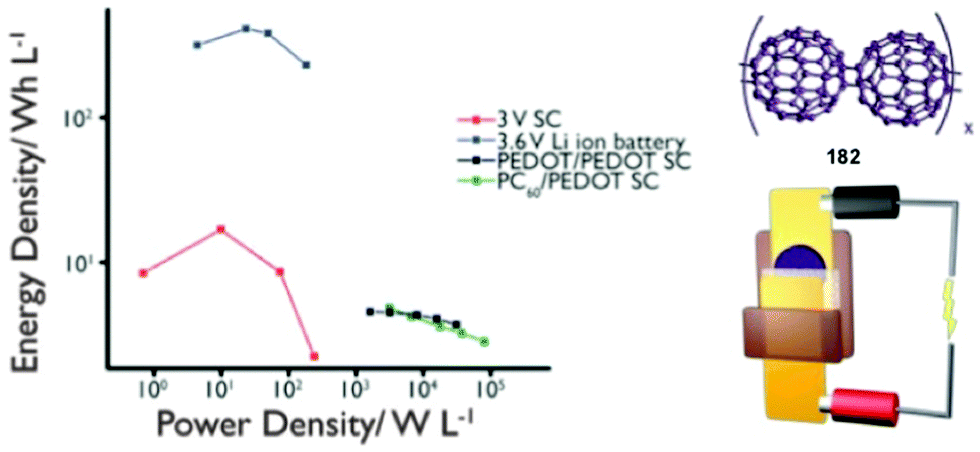 | ||
| Fig. 28 Ragone plot of symmetric PEDOT SC and asymmetric 182 SC plotted with commercially available 3 V 0.2 F activated carbon SC and a 3.6 V 110 mA h lithium-ion battery. Adapted from ref. 174. | ||
Three hyper-branched polymers, 183–185, with increasing numbers of thiophene linkers and terminal NDI groups were fabricated into asymmetric and symmetric SCs.175 Asymmetric devices of 183–185, using activated carbon as the positive electrode yields 22.0, 4.92, and 4.94 F g−1Cpc's at 0.1 mA. Increasing thiophene spacers led to an increase in porosity, but also an increase in charge transfer resistance. While the Cpc values are low, 183 demonstrated high cycling stability, with approximately 90% retention after 500 cycles. This material was fabricated into a symmetric device that has a low Cpc of 0.5 F g−1, but maintains the same high cycling stability. The low Cpc of these materials may exclude them from practical use, however the trends between the polymers with different pore sizes is fundamentally important.
5.3 Donor–acceptor materials
Donor–acceptor polymers, commonly used in organic photovoltaics, accept both positive and negative charge, making them ideally suited for symmetric SCs with high operating voltages. Reynolds and coworkers demonstrated the use of a novel donor–acceptor polymer, 186, in SCs.176 The EDOT functionality allows the material to accept positive charge and the isoindigo functionality allows the material to accept negative charge. As such, 186 was fabricated into a symmetric device with a 2.25 V operating potential due to the negative and positive charge-accepting properties of the polymer. The lack of n-type stability for 186 limits its use in a symmetric device with a large operating voltage, but this work provides fundamental insight into the development of donor–acceptor polymers for symmetric SCs. Our group demonstrated an example of donor–acceptor polymers 187 and 188 in SCs with large potential windows of 2.5 V and impressive Cpc's of 201 F g−1 at 100 mV s−1 and 91 F g−1 at 50 mV s−1 for 187 and 188 respectively.177 A second acceptor unit is found to increase charge stabilization, despite diminished cycle stability of 75% after 100 cycles and 30% after 1000 cycles at 500 mV s−1 (Fig. 29). While the cycling stability is not ideal, this is a significant step forward and provides insight into performance improvement for donor–acceptor polymers by demonstrating that increasing the amount of acceptors in the polymer can lead to greater charge stabilization. Further increasing the acceptor content in donor–acceptor polymers is expected to lead to higher conductivity and stability.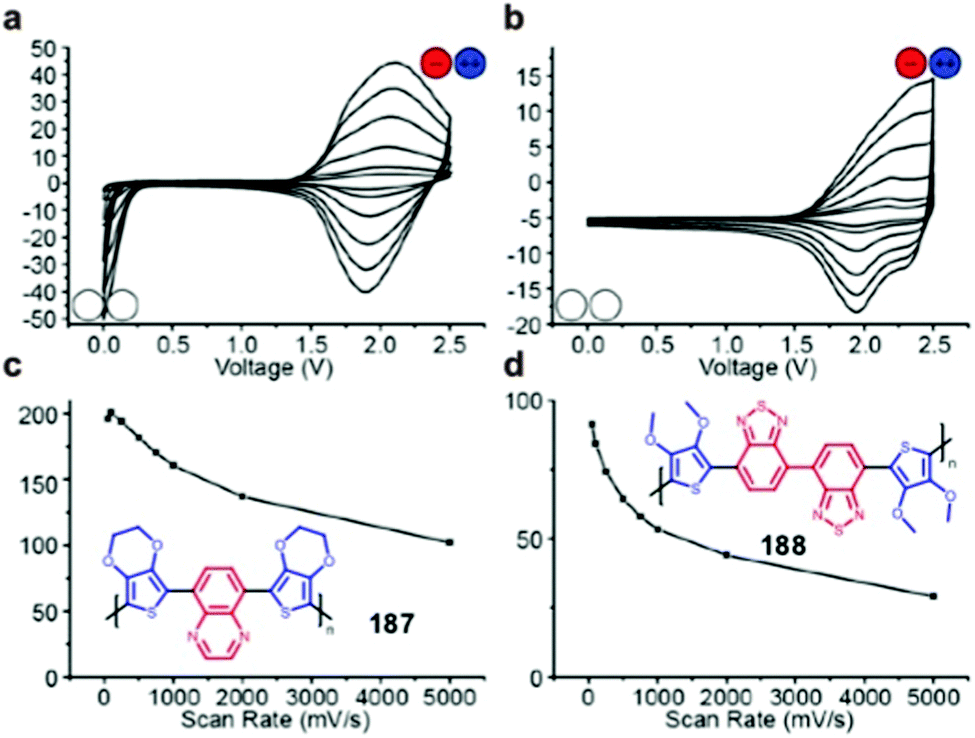 | ||
| Fig. 29 Electrochemical characterization of symmetric 187 and 188 SCs (structure inset in each column). (a and b) Cyclic voltammogram at 50, 100, 200, 500, 750 and 1000 mV s−1. Blue indicates an electrode where the electron donor material stores charge, red indicates where the electron acceptor stores charge, and white indicates a neutral electrode. The SOC of the electrodes are indicated by “+” and “−”. (c and d) Peak capacitance vs. scan rate. Adapted from ref. 158. | ||
The donor–acceptor polymer 189 was studied for its electrochemical and electrochromic properties. With distinct colours in each charged state, 189 is ideal for smart electronics as an energy storage device capable of indicating its immediate capacity.178 While 189 has a modest Cpc (112.4 F g−1 at 1.0 A g−1, and 59.8 F g−1 at 16.0 A g−1), it has excellent cycling stability, with 82% capacity retention after 12500 cycles at 1.0 A g−1, and a CE of 100%. The electrodes reach a 16.6 kW kg−1 maximum power density, and a high maximum specific energy of 49.8 W h kg−1. The exceptional stability of this polymer is only observed in the positive charge-accepting regime. It would be interesting to see the effect of a long alkyl chain on the negative charge-accepting stability of the polymer.
Using the redox-active small molecule alizarin 190, graphene hydrogels were non-covalently functionalized and used as SC electrodes.179 The hydroxyl and carbonyl groups allow 190 to accept both positive and negative charge, enlarging the potential window to 1.4 V in a symmetric device. The resulting macroporous framework facilitates excellent ion diffusion and electron transfer. The electrode exhibits a 350 F g−1Cpc at 1 A g−1, much higher than its individual components, with exceptional rate capability (61% capacity retention at 200 A g−1). The symmetric SC has a 285.6 F g−1Cpc at 1 A g−1, and an impressive energy density of 18.2 W h kg−1 (at a power of 0.7 kW kg−1). The device also shows excellent rate capability (64% retention at 50 A g−1) and cycle stability (88% retention after 1000 cycles at 5 A g−1). The high capacity, relatively large operating potential, and high cycling stability makes this an excellent candidate for SCs.
6. Computational design of electrode materials
Organic compounds are infinitely diverse due to the large number of atom combinations, connectivity, stereochemistry, size, etc. The number of possible organic compounds that can be explored as useful materials for energy storage is immense, even when we only consider the compounds that are synthetically and practically relevant. Fortunately, due to the incredible developments in computational chemistry, the ability to rationally design molecules in silico can greatly reduce the number of candidates for devices, saving valuable time and money. Computational chemistry, particularly DFT, has become a staple for the design and study of molecules for almost all organic electronic devices. DFT and other computational methods can be used as a screening tool to choose the most promising candidate for synthesis or as a tool to rationalize unexpected behaviour from systems already examined experimentally. Below, we highlight some recent work on the use of computational chemistry for the design of all of the above energy storage devices, and how this tool is being used for the design of high performance materials.6.1 Solid electrode batteries
The majority of the work performed on organic materials for energy storage using computational chemistry has focussed on solid electrode batteries. This is not surprising because, overwhelmingly, solid electrode batteries are the most studied in the literature. The recent theoretical work in this field has focussed on a few key points: (1) developing structure–property relationships for classes of organic compounds in order to improve battery performance; (2) assessing the mechanism of charge transport in organic films; (3) screening molecular crystals using new DFT methods in order to determine the effect of crystal structure on performance and; (4) assessing the strength of van der Waals interactions between redox-active molecules and different substrates.Quinones have been studied extensively for use in batteries, however, they typically suffer from low voltages compared to inorganic cathodes. In order to provide structure–property relationships for the development of high performance quinoide-type molecules, DFT was used to analyze different derivatives of quinones that are connected by a two-atom bridge.180 By substituting carbons in the quinone ring with heteroatoms, changing the carbonyl redox group to more electron-withdrawing groups, or by adding substituents to the quinone ring, a wide voltage range between ∼1 to 3 V vs. Li/Li+ can be theoretically attained (Fig. 30). The authors note that placing substituents on the two-atom bridge have little effect on the redox potential, and changes that modify the stabilization energies of the aromatic ring have the largest effect. The same authors later reported the effect of isomerization and nitrogen substitution on the reduction potential of an unexplored class of compounds for energy storage, pentalenediones.181 The highest voltage occurs in isomers where the spin density is mostly located on the oxygen atoms in the reduced form, or in other words, when the five-membered ring is stabilized. In general, nitrogen substitution in the ring raises the reduction potential of the derivatives, except when they appear next to each other in the ring.
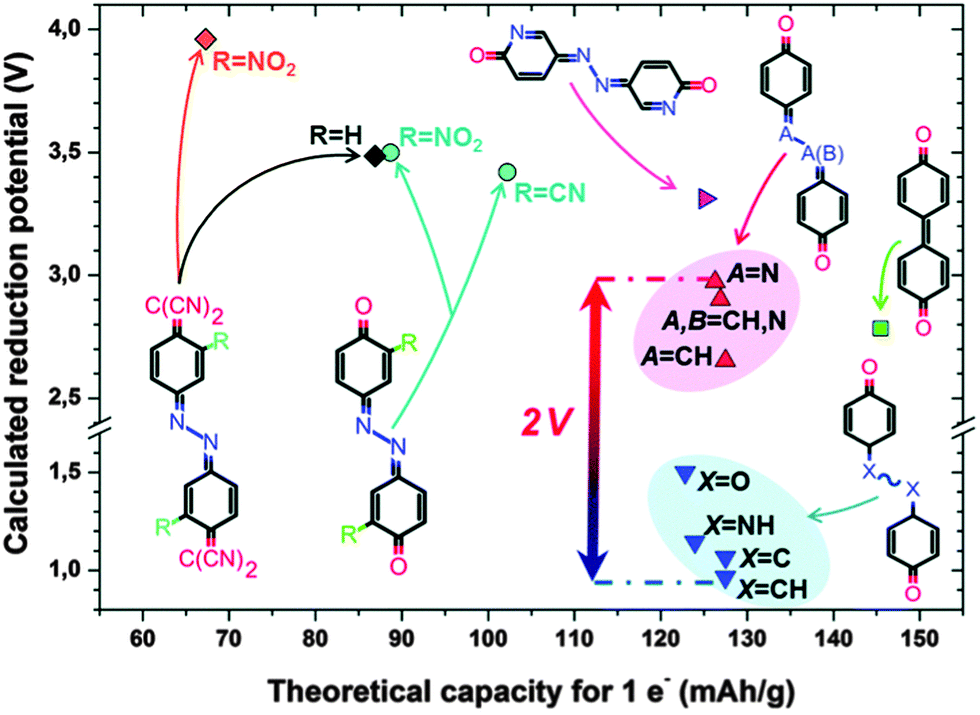 | ||
| Fig. 30 Calculated reduction potential (in V vs. Li+/Li) as a function of the capacity for a one-electron process. (Reproduced from ref. 133, reprinted with permission from the PCCP Owner Societies). | ||
In another effort to elucidate structure–property relationships for designing high-voltage cathodes, a qualitative methodology based on Clar's sextet was developed and validated using DFT.42 The authors hypothesized that an increased number of Clar sextets would result in a higher reduction potential based on the increased aromaticity, or in other words a higher stabilization energy. When tested using DFT, a positive correlation was found between the average change in Clar sextet numbers when two lithium atoms are inserted into the material, denoted as ΔC2Li, and the voltage. The authors also investigated compound 24, which has a positive ΔC2Li, in order to further support their hypothesis. A novel set of carbonyl containing heteroaromatic compounds with a six-membered quinone ring fused with a five-membered heterocycle were also examined by DFT, giving rise to a set of structure–property relationships.182 In general, increasing the aromaticity of the anion leads to greater stability and a higher reduction potential, similar to what others have found. The authors warn against adding heteroatoms and other groups to molecules without considering the effect of the excess mass because it can result in a lower energy density although a higher voltage can be attained.
Radical polymers are an important class of materials for dual-ion batteries. The ability to quickly screen radical compounds for their redox potentials is very useful for the development of radical-based electrodes. Dardenne and coworkers tested a Δ self-consistent field procedure based on DFT with a polarizable continuum model to account for solvent interactions.183 The authors show that this method can be used to accurately predict the redox potentials of organic radicals with little computational cost. This method is expected to be important for the design of high voltage organic radicals for use in batteries.
It is also important to determine the mechanism of charge transport in radical polymers because the morphology of the polymer films and the orientation of redox-active groups relative to each other can impact the rate of charge transport. This knowledge can lead to the development of radical polymers that have high charge carrier mobilities and therefore high rate capabilities. Using a combination of molecular dynamics simulations and DFT, the intermolecular packing and its effect on charge transport were determined in a TEMPO-based pendant polymer.184 Charge transport between pendant TEMPO groups occurs predominantly through intermolecular interactions. Favourable molecular orbital overlap occurs when the TEMPO oxygen–nitrogen bonds stack parallel to each other, and this facilitates charge transport. This work suggests that the stabilizing methyl groups should be replaced with something that allows for closer contact between TEMPO groups to allow these interactions to contribute significantly to the overall charge transport mechanism.
Dispersion-corrected DFT (DFT-D) has been developed previously to study organic crystal systems. Recently, it has been applied to a previously reported lithium 2,6-bis(ethoxycarbonyl)-3,7-dioxo-3,7-dihydro-s-indacene-1,5-bis(olate) battery cathode to determine the method's utility in predicting the properties that are important to function in a lithium-ion battery.185 The authors show that DFT-D is an effective method to predict both the geometry and the discharge potential of the electrode. Additionally, the authors find that modeling the periodic crystal structure is a better means to determine properties than modeling a single molecule. This work was recently extended to nine other crystalline organic materials that were previously studied for lithium-ion batteries and thirty-one other randomly selected organic crystallites.186 The authors also highlight the need for molecules with small band gaps in order to attain sufficient conductivities to facilitate charge transport.
DFT-D was also used to study the diffusion of lithium-ions in tetracyanoquinodimethane.187 Crystals of tetracyanoquinodimethane are highly porous, with an electronic band gap of ∼1.0 eV, suggesting that the compound should have good electrical conductivity, however the dynamic performance is poor. The authors show that there is a high coulombic barrier for lithium ion diffusion through the crystal due to the interaction with the negative charge on the tetracyanoquinodimethane molecules. The authors suggest making nanoparticles of the molecule to shorten diffusion pathways and blending with an ionic conductor in order to improve performance in this promising material.
In order to enhance the cycling stability of organic small molecules for batteries, one strategy is to adsorb them onto a highly conjugated sp2 carbon (or heteroaromatic) network where the π–π stacking will be strong enough to prevent dissolution. In order to quantify this interaction, DFT-D was used to measure the binding energy between different organic molecules on graphene or hexagonal boron nitride.188 The calculations show that there is a strong binding interaction between these molecules and each substrate. The work function is shifted relative to isolated graphene or boron nitride nanosheet, indicating that a strong interaction is in fact occurring. The authors suggest that adding graphene or boron nitride can prevent dissolution and increase the cycling stability of the organic electrode.
6.2 Redox flow batteries
The use of DFT to predict the properties of organic molecules for RFBs is becoming increasingly popular. The calculation is somewhat simplified compared to organic crystals, because a solvent model can be used to accurately predict the performance with solvation effects accounted for. Below are some recent examples that exclusively use computational chemistry to give structure–property relationships in order to guide the design of high performance materials for RFBs.Anthraquinones are the mostly widely studied class of compounds for RFBs. Due to their high abundance, facile functionalization, and wide range of synthetically accessible derivatives, they are prime candidates to study via computational chemistry in order to determine the most promising materials. Bachman and coworkers investigated the redox properties of a number of anthraquinone derivatives by DFT.189 The goals of their study were to determine the influence of lithium salts and the decomposition of these salts under normal battery operating conditions on the reduction potential, the influence of various functional groups on the reduction potential, the influence of functional groups on the solvation energy (closely related to solubility), and the identification of stable anthraquinone derivatives. In general, the authors found that lithium salts increase the reduction potential of anthraquinones by ∼0.4 V due to a complexation effect, which is favourable for catholytes. Furthermore, they predicted that substitution of the aryl ring with methyl groups lowers the reduction potential, which is favourable for anolytes. They also identify oxy-methyl dioxolane substituents as the best choice for increasing the solvation energy and suggest that anthraquinone anions are likely to be stable with respect to reduction in solution. Thiophenoquinones were also examined using DFT for aqueous RFBs.190 The authors identified 1056 derivatives that have predicted solubilities of >2 M and out of these, 36 that have reduction potentials lower than 0.25 V, and 15 with reduction potential higher than 0.95 V vs. the standard hydrogen electrode. While substitution on the quinone ring with EDG and EWG groups has the expected trends of lowering and raising the reduction potentials respectively, when substitution occurs on the thiophene ring, sometimes the opposite effect is observed.
In another study, Er and coworkers computationally design an all-quinone RFB by examining the solubility and redox potentials of 1710 quinone and hydroquinone redox couples.191 The author's report a quantitative structure–property relationship using high throughput DFT calculations on different ring sizes, isomers, and substitutions on the quinone rings. It was discovered that substitution near the carbonyl groups on the quinones has the largest effect on the reduction potential of the molecule, with the expected trends for EDGs and EWGs. They also discover that substitution away from the carbonyl groups on the quinone leads to the greatest improvement in solubility.
6.3 Supercapacitors
The majority of the computational predictions on SCs have focused on modelling the charge distribution in the pores of HSACs, the interaction of different electrolytes on charged surfaces, and ion dynamics when under the influence of an external electric field.192–197 Much less work has been performed on the design of organic materials for supercapacitors. This might be because the electronic structure required for pseudocapacitive behaviour is hard to predict and, quite frankly, seems to be poorly understood. As a qualitative observation, it seems as though only highly conducting materials, or materials with broad, overlapping redox peaks, exhibit a pseudocapacitive profile. While this phenomenon has yet to be calculated in detail, to the best of our knowledge, there has been a few recent works that look at various aspects of organic SC materials.Burkhardt and coworkers investigated a method to predict the redox potentials of small violene molecules using DFT.198 The authors examine the applicability of each compound for energy storage based on the predicted redox potential and the molecular weight of the redox unit to give an idea of the expected energy density. The authors found that the heteroatom in the ring has a predictable shift in redox potential following the trend of sulfur > oxygen > nitrogen and that the oxidation potential can be shifted higher when the aromatic system decreases in size, or if additional nitrogen heteroatoms are added to the ring. They also found predictable shifts in redox potential for EDGs. The authors note that the usefulness of these compounds will rely on the ability to attach them to a conjugated polymer backbone to afford additional capacitance and high conductivity.
Our group has also provided some insight into the design of conjugated donor–acceptor polymers for SCs.177 The delocalization of charges on a conjugated polymer backbone is important to determine the conductivity and electrochemical characteristics of the material (i.e. battery or SC-like behaviour). We have quantified the extent of delocalization of charges on a donor–acceptor polymer by the bond length change calculated by DFT when a positive or negative charge is placed on the backbone. We have found that in the case of donor–acceptor polymers, the extent of delocalization is not as great as that of a homopolymer such as poly(3,4-diethoxythiophene) (Fig. 31). This suggests that the conductivity and electrochemical characteristics, such as oxidation/reduction profile and stability, are not as favourable as for a homopolymer. This work was also corroborated with experimental work on donor–acceptor conjugated polymers. We have also performed similar calculations on block-copolymers of donor and acceptor moieties and found that these polymers should have a great extent of charge delocalization. These polymers are prime candidates for future work in the area of conjugated polymer SCs.
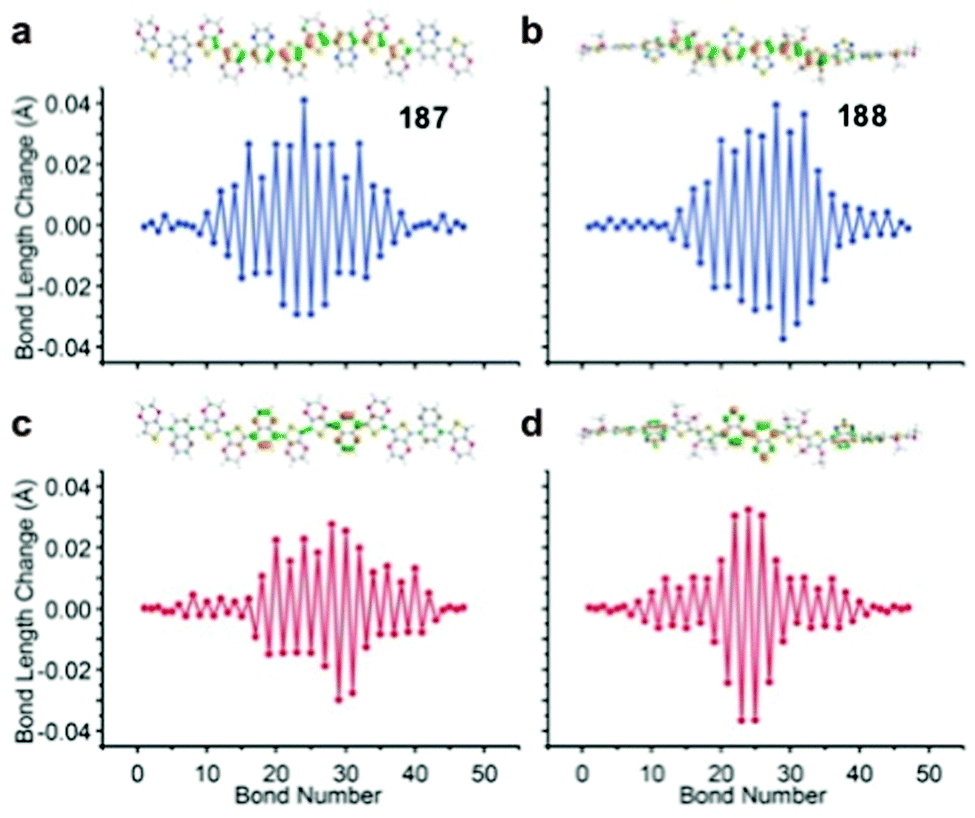 | ||
| Fig. 31 Change in bond length along the conjugated backbone from neutral to +1 (blue) and neutral to −1 (red) doublet states for 187, and 188. The orbital diagram of the highest occupied molecular orbital and lowest unoccupied molecular orbital of the neutral polymer is above the plot neutral minus +1 and neutral minus −1 plot respectively. (Reproduced from ref. 158, reprinted with permission, copyright 2014, American Chemical Society). | ||
7. Summary and outlook
In this review, we have shown that organic materials are promising candidates for low cost and high performance electrodes for all EESSs (Fig. 32). The use of organic electrodes for solid electrode batteries is, by far, the most studied. This is due to the large market share that solid electrode batteries have, in particular lithium-ion batteries. Organic electrodes are important for solid electrode batteries because they can decrease the cost of the device, allow flexibility, and can also open up opportunities for the use of multivalent ions without the problems typically associated with inorganic compounds. Several issues still exist that need to be addressed before organic electrodes can become commercially viable, such as a low cycling stability, low voltage, and low capacity. Strategies have been proposed to address these problems with varying success. The functionalization of small molecules with ionic groups, the incorporation of the redox functionalities into polymers and organic frameworks, and the adsorption of molecules onto highly conjugated, graphitic-like surfaces are all promising ways to improve the cycling stability of redox-active organic molecules and have even shown to improve other performance metrics such as rate capability. Structural modification of organic molecules with EWGs or EDGs, or by the substitution of carbon with more electronegative atoms, can provide for an efficient way to tune the redox potential. Additionally, the use of biologically-derived and industrially abundant materials lends itself to the design of low cost materials for solid electrode batteries and RFBs. This is expected to be an increasingly popular strategy for both academia and industry in order to fabricate sustainable and inexpensive energy storage devices. Investigating structures that possess a high charge:mass ratio is a strategy to overcome capacity issues, however, the discovery of unexpected charge-accepting mechanisms is important for the design of high capacity materials. With a combination of these strategies, voltages comparable to traditional lithium-ion batteries (∼3.6 V), and even higher for dual-ion batteries, stabilities exceeding thousands of cycles, and capacities above 300 mA h g−1 can be realistically achieved. These metrics will require further investigation into both anode and cathode materials and also into electrolytes in order to achieve high voltages, capacities, and stabilities.
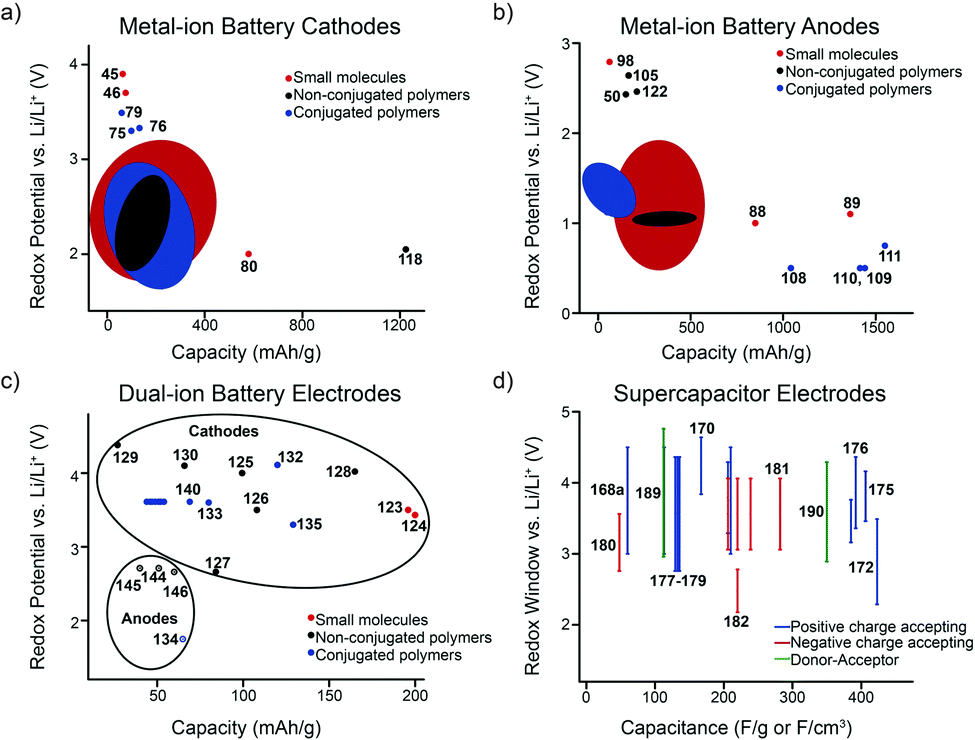 | ||
| Fig. 32 Voltage versus amount of charge stored plots of the various materials reviewed for metal-ion battery (a) cathodes and (b) anodes, (c) dual-ion battery electrodes, and (d) supercapacitor electrodes. The numbers in the plots represent the compound numbers for the data. (a and b) The compound numbers for the plots are displayed for compounds that do not lie in the range typically found for materials of the same class. For (c), anode and cathode materials are enclosed within an area defined by an oval. For (d), the lines represent the redox potential window where the electrode material is electrochemically active. | ||
In the realm of RFBs, organic materials have shown great potential. These materials promise low cost, stable operation, high capacity, and high voltages due to their abundance, reversible electrochemistry, and versatility in structure. Here, the biggest issues are the device voltages, and the low solubility which leads to low capacities and cycling stabilities. Many of the same strategies used in solid electrode batteries can be used to address these issues. Functionalization with ionic groups or organic motifs can afford a greater solubility in the desired electrolyte and can also adjust the redox potentials. The design of high performance materials for RFBs can be greatly simplified by targeting the most promising candidates to synthesize and test by examining the predicted solubility of the compounds at all oxidation states by computational chemistry. The design of symmetric RFBs is a promising strategy to overcome crossover issues, however, this can pose challenges with both solubility and stability for all redox states involved. Additionally, one of the challenges lies in replacing the expensive Nafion separator. An effective strategy is to design soluble redox-active polymers that can be excluded from crossover by size, allowing Nafion to be replaced by an inexpensive dialysis membrane. There have also been developments in new low cost separators to replace Nafion. It is expected that RFBs using organic materials will play a large role in grid energy storage in the future.
Organic materials are also promising for SCs because they can be flexible, and have high capacitances due to their pseudocapacitive properties. The design of new SC materials should focus on improving capacitance through the incorporation of redox motifs into traditional EDLC and known pseudocapacitive polymers. There has been a significant amount of work on novel pseudocapacitive conjugated polymers. However, the design of these novel materials is difficult due to the lack of predictive methods for pseudocapacitive behaviour (compared to battery-like behaviour). For now, the main strategy is focused on using known pseudocapacitive polymers that can be derivatized, although a few novel polymers have been discovered that surprisingly exhibit the necessary electrochemical behaviour for SCs. These discoveries have led to advances in designing pseudocapacitive materials that have larger operating voltages and higher capacities. We hope that going forward, predictive methods for pseudocapacitive behaviour will be developed to propel the design of new pseudocapacitive materials with high capacitance, stability, and voltages.
With the development of more accurate computational methods and the use of existing techniques, we expect that DFT will become a standard technique for designing materials and rationalizing behaviour. This is already becoming apparent in the literature, with a significant proportion of researchers using DFT to investigate energy storage materials. Innovation at the structural level is essential for the commercialization organic energy storage materials. With an interdisciplinary and collaborative effort, we believe that high performance and low cost organic EESSs are attainable in the foreseeable future.
Abbreviations
| BC | Butylene carbonate |
| BMIMPF6 | 1-Butyl-3-methylimidazolium hexafluorophoshate |
| CB | Carbon black |
| CE | Coulombic efficiency |
| CMC | Carboxyl methyl cellulose |
| CNTs | Carbon nanotubes |
| C pc | Specific capacitance |
| CPE | Composite polymer electrolyte |
| C sp | Specific capacity |
| C theor | Theoretical capacity |
| C v | Volumeric capacity |
| DCM | Dichloromethane |
| DEC | Diethyl carbonate |
| DEGDME | Diethylene glycol dimethyl ether |
| DFT | Density functional theory |
| DFT-D | Dispersion-corrected DFT |
| DMC | Dimethylcarbonate |
| DME | Dimethoxyethane |
| DOL | Dioxolane |
| EC | Ethylene carbonate |
| EDGs | Electron donating groups |
| EDLC | Electric double layer capacitors |
| EE | Energy efficiency |
| EESS | Electrochemical energy storage system |
| EiPS | Ethyl isopropyl sulfone |
| EMC | Ethyl methyl carbonate |
| EMIC | 1-Ethyl-3-methylimidazolium chloride |
| EMIMBF4 | 1-Ethyl-3-methylimidazolium tetrafluoroborate |
| EWGs | Electron withdrawing groups |
| F | Faradys's constant |
| Fc | Ferrocene |
| FEC | Fluoroethylene carbonate |
| FWNT | Few-walled carbon nanotubes |
| G | Guaiacyl |
| GNP | Graphene nanoplatelet |
| GO | Graphene oxide |
| H | Hydroxyphenyl |
| HSAC | High surface area carbon |
| iMOF | Insertion metal–organic framework |
| LiTFSI | Lithium bis(trifluoromethane)sulfonimide |
| M | Molecular weight of the compound in g mol−1 |
| MWCNT | Multiwall carbon nanotubes |
| NaFSI | Sodium trifluoromethanesulfonimide |
| NaTFSI | Sodium bis(trifluoromethane)sulfonimide |
| NDIs | Naphthalene diimides |
| P3HT | Poly(3-hexylthiophene) |
| PC | Propylene carbonate |
| PDI | Perylene diimide |
| PEDOT:PSS | Poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) |
| PEO | Poly(ethylene oxide) |
| PMA | Poly(methacrylate) |
| P max | Maximum power |
| PMMA | Poly(methyl methacrylate) |
| PP13 TFSA | N-Methyl-N-propylpiperidinium bis(trifluoromethanesulfonyl)amide |
| PTFE | Poly(tetrafluoroethylene) |
| PVA | Poly(vinyl alcohol) |
| PVdF | Poly(vinylidenefluoride) |
| PVdF-co-HFP | Poly(vinylidene fluoride-co-hexafluoropropylene) |
| RFBs | Redox flow batteries |
| R s | Equivalent series resistance in ohms |
| S | Syringyl |
| SBR | Styrene-butadiene rubber |
| SCE | Saturated calomel electrode |
| SCs | Supercapacitors |
| SEI | Solid electrolyte interface |
| SHE | Standard hydrogen electrode |
| SOC | State-of-charge |
| SWCNTs | Single-walled carbon nanotubes |
| TBAClO4 | Tetrabutyl ammonium perchlorate |
| TBAOH | Tetrabutyl ammonium hydroxide |
| TBASbF6 | Tetrabutyl ammonium hexafluoroantimonate |
| TBAPF6 | Tetrabutyl ammonium hexafluorophosphate |
| TEATFSI | Tetraethyl ammonium bis(trifluoromethylsulfonyl)imide |
| TEABF4 | Tetraethyl ammonium tetrafluoroborate |
| TEGDME | Tetraethylene glycol dimethyl ether |
| TEMPO | 2,2,6,6-Tetramethylpiperdinyloxyl |
| TFSI | Trifluoromethane sulfonyl-amide |
| V | Voltage of the SC in volts |
| VC | Vinylene carbonate |
| VE | Voltage efficiency |
| VGCF | Vapor-grown carbon fibers |
| V i | Initial voltage of the device in volts |
Acknowledgements
We acknowledge the support of the NSERC of Canada (for a Discovery Accelerator Supplement), the Canadian Foundation for Innovation, and the Connaught Foundation. T. B. S. is grateful for an NSERC PGS-D scholarship. B. T. M is grateful for an Ontario Graduate Scholarship.References
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2014, 7, 19–29 CrossRef PubMed.
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116, 2–4 CrossRef CAS.
- H. Alt, H. Binder, A. Köhling and G. Sandstede, Electrochim. Acta, 1972, 17, 873–887 CrossRef CAS.
- T. Ohzuku, H. Wakamatsu, Z. Takehara and S. Yoshizawa, Electrochim. Acta, 1979, 24, 723–726 CrossRef CAS.
- J.-I. Yamaki and A. Yamaji, J. Electrochem. Soc., 1982, 129, 5–9 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- P. J. Nigrey, D. J. MacInnnes, D. P. Nairns and A. G. MacDiarmid, J. Electrochem. Soc., 1981, 128, 1651–1654 CrossRef CAS.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 201–281 CrossRef.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- S. Srivastava, J. L. Schaefer, Z. Yang, Z. Tu and L. A. Archer, Adv. Mater., 2013, 26, 201–234 CrossRef PubMed.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- Z. Guo and E. Takeuchi, ACS Appl. Mater. Interfaces, 2015, 7, 16131–16132 CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 CAS.
- C. Friebe and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1500858 Search PubMed.
- Z. Zhu, M. Hong, D. Guo, J. Shi, Z. Tao and J. Chen, J. Am. Chem. Soc., 2014, 136, 16461–16464 CrossRef CAS PubMed.
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef CAS.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- D. G. Kwabi, N. Ortiz-Vitoriano, S. A. Freunberger, Y. Chen, N. Imanishi, P. G. Bruce and Y. Shao-Horn, MRS Bull., 2014, 39, 443–452 CrossRef CAS.
- K. Oyaizu, T. Suga, K. Yoshimura and H. Nishide, Macromolecules, 2008, 41, 6646–6652 CrossRef CAS.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef CAS PubMed.
- P. Leung, X. Li, C. Ponce de León, L. Berlouis, C. T. J. Low and F. C. Walsh, RSC Adv., 2012, 2, 10125–10156 RSC.
- W. Kangro, German Pat. DE, DE914264C, 1949 Search PubMed.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- R. Ramya, R. Sivasubramanian and M. V. Sangaranarayanan, Electrochim. Acta, 2013, 101, 109–129 CrossRef CAS.
- B. E. Conway, Electrochemical supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publishers, New York, NY, 1999 Search PubMed.
- Q. Zou, W. Wang, A. Wang, Z. Yu and K. Yuan, Mater. Lett., 2014, 117, 290–293 CrossRef CAS.
- W. Wan, H. Lee, X. Yu, C. Wang, K.-W. Nam, X.-Q. Yang and H. Zhou, RSC Adv., 2014, 4, 19878–19882 RSC.
- H. Li, W. Duan, Q. Zhao, F. Cheng, J. Liang and J. Chen, Inorg. Chem. Front., 2014, 1, 193–199 RSC.
- K. Zhang, C. Guo, Q. Zhao, Z. Niu and J. Chen, Adv. Sci., 2015, 2, 1500018 Search PubMed.
- A. Iordache, V. Maurel, J.-M. Mouesca, J. Pécaut, L. Dubois and T. Gutel, J. Power Sources, 2014, 267, 553–559 CrossRef CAS.
- L. M. Kozycz, C. Guo, J. G. Manion, A. J. Tilley, A. J. Lough, Y. Li and D. S. Seferos, J. Mater. Chem. C, 2015, 3, 11505–11515 RSC.
- A. J. Tilley, C. Guo, M. B. Miltenburg, T. B. Schon, H. Yan, Y. Li and D. S. Seferos, Adv. Funct. Mater., 2015, 25, 3321–3329 CrossRef CAS.
- A. Shimizu, Y. Tsujii, H. Kuramoto, T. Nokami, Y. Inatomi, N. Hojo and J.-I. Yoshida, Energy Technol., 2014, 2, 155–158 CrossRef CAS.
- C. Luo, R. Huang, R. Kevorkyants, M. Pavanello, H. He and C. Wang, Nano Lett., 2014, 14, 1596–1602 CrossRef CAS PubMed.
- S. Gottis, A.-L. Barrès, F. Dolhem and P. Poizot, ACS Appl. Mater. Interfaces, 2014, 6, 10870–10876 CAS.
- A. Shimizu, H. Kuramoto, Y. Tsujii, T. Nokami, Y. Inatomi, N. Hojo, H. Suzuki and J.-I. Yoshida, J. Power Sources, 2014, 260, 211–217 CrossRef CAS.
- H. Kim, D.-H. Seo, G. Yoon, W. A. Goddard III, Y. S. Lee, W.-S. Yoon and K. Kang, J. Phys. Chem. Lett., 2014, 5, 3086–3092 CrossRef CAS PubMed.
- T. Yokoji, H. Matsubara and M. Satoh, J. Mater. Chem. A, 2014, 2, 19347–19354 CAS.
- D. Wu, Z. Xie, Z. Zhou, P. Shen and Z. Chen, J. Mater. Chem. A, 2015, 3, 19137–19143 CAS.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 126, 6002–6006 CrossRef.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- H. Senoh, H. Sakaebe, H. Sano, M. Yao, K. Kuratani, N. Takeichi and T. Kiyobayashi, J. Electrochem. Soc., 2014, 161, A1315–A1320 CrossRef CAS.
- B. Pan, D. Zhou, J. Huang, L. Zhang, A. K. Burrell, J. T. Vaughey, Z. Zhang and C. Liao, J. Electrochem. Soc., 2016, 163, A580–A583 CrossRef CAS.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS PubMed.
- G. S. Vadehra, R. P. Maloney, M. A. Garcia-Garibay and B. Dunn, Chem. Mater., 2014, 26, 7151–7157 CrossRef CAS.
- M. E. Bhosale and K. Krishnamoorthy, Chem. Mater., 2015, 27, 2121–2126 CrossRef CAS.
- D. Chen, A.-J. Avestro, Z. Chen, J. Sun, S. Wang, M. Xiao, Z. Erno, M. M. Algaradah, M. S. Nassar, K. Amine, Y. Meng and J. F. Stoddart, Adv. Mater., 2015, 27, 2907–2912 CrossRef CAS PubMed.
- W. Luo, M. Allen, V. Raju and X. Ji, Adv. Energy Mater., 2014, 4, 1400554 Search PubMed.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS.
- W. Deng, Y. Shen, J. Qian, Y. Cao and H. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 21095–21099 CAS.
- M. Lee, J. Hong, D.-H. Seo, D. H. Nam, K. T. Nam, K. Kang and C. B. Park, Angew. Chem., Int. Ed., 2013, 52, 8322–8328 CrossRef CAS PubMed.
- M. Lee, J. Hong, H. Kim, H.-D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26, 2558–2565 CrossRef CAS PubMed.
- J. Hong, M. Lee, B. Lee, D.-H. Seo, C. B. Park and K. Kang, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- M. Yao, K. Kuratani, T. Kojima, N. Takeichi, H. Senoh and T. Kiyobayashi, Sci. Rep., 2014, 4, 3650 EP Search PubMed.
- Y. Imada, H. Nakano, K. Furukawa, R. Kishi, M. Nakano, H. Maruyama, M. Nakamoto, A. Sekiguchi, M. Ogawa, T. Ohta and Y. Yamamoto, J. Am. Chem. Soc., 2016, 138, 479–482 CrossRef CAS PubMed.
- R. Zhang, F. Mizuno and C. Ling, Chem. Commun., 2014, 51, 1108–1111 RSC.
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed.
- H. Wu, Q. Meng, Q. Yang, M. Zhang, K. Lu and Z. Wei, Adv. Mater., 2015, 27, 6504–6510 CrossRef CAS PubMed.
- H. Wang, S. Yuan, D. Ma, X. Huang, F.-L. Meng and X.-B. Zhang, Adv. Energy Mater., 2014, 4, 1301651 Search PubMed.
- G. Hernández, N. Casado, R. Coste, D. Shanmukaraj, L. Rubatat, M. Armand and D. Mecerreyes, RSC Adv., 2015, 5, 17096–17103 RSC.
- H. Banda, D. Damien, K. Nagarajan, M. Hariharan and M. M. Shaijumon, J. Mater. Chem. A, 2015, 3, 10453–10458 CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- J. Kim, J. Kim, J. Lee, H.-K. Song and C. Yang, ChemElectroChem, 2014, 1, 1618–1622 CrossRef CAS.
- B. Häupler, T. Hagemann, C. Friebe, A. Wild and U. S. Schubert, ACS Appl. Mater. Interfaces, 2015, 7, 3473–3479 Search PubMed.
- D. Schmidt, B. Häupler, C. Stolze, M. D. Hager and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2517–2523 CrossRef CAS.
- T. B. Schon, A. J. Tilley, C. R. Bridges, M. B. Miltenburg and D. S. Seferos, Adv. Funct. Mater., 2016 Search PubMed , submitted.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 CAS.
- J. Bitenc, K. Pirnat, T. Bančič, M. Gaberšček, B. Genorio, A. Randon-Vitanova and R. Dominko, ChemSusChem, 2015, 8, 4128–4132 CrossRef CAS PubMed.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124 Search PubMed.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed.
- F. Xu, J. Xia and W. Shi, Electrochem. Commun., 2015, 60, 117–120 CrossRef CAS.
- A. Vlad, K. Arnould, B. Ernould, L. Sieuw, J. Rolland and J.-F. Gohy, J. Mater. Chem. A, 2015, 3, 11189–11193 CAS.
- Y. F. Shen, D. D. Yuan, X. P. Ai, H. X. Yang and M. Zhou, Electrochem. Commun., 2014, 49, 5–8 CrossRef CAS.
- M. Zhou, W. Li, T. Gu, K. Wang, S. Cheng and K. Jiang, Chem. Commun., 2015, 51, 14354–14356 RSC.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 CAS.
- Y. J. Kim, W. Wu, S.-E. Chun, J. F. Whitacre and C. J. Bettinger, Adv. Mater., 2014, 26, 6572–6579 CrossRef CAS PubMed.
- F. Xu, X. Chen, Z. Tang, D. Wu, R. Fu and D. Jiang, Chem. Commun., 2014, 50, 4788–4790 RSC.
- D. Tian, H.-Z. Zhang, D.-S. Zhang, Z. Chang, J. Han, X.-P. Gao and X.-H. Bu, RSC Adv., 2014, 4, 7506–7510 RSC.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 EP CrossRef PubMed.
- C. R. DeBlase, K. Hernández-Burgos, J. M. Rotter, D. J. Fortman, D. dos S Abreu, R. A. Timm, I. C. N. Diógenes, L. T. Kubota, H. D. Abruña and W. R. Dichtel, Angew. Chem., Int. Ed., 2015, 127, 13423–13427 CrossRef.
- H. Zhang, Q. Deng, A. Zhou, X. Liu and J. Li, J. Mater. Chem. A, 2014, 2, 5696–5702 CAS.
- H. H. Lee, Y. Park, K.-H. Shin, K. T. Lee and S. Y. Hong, ACS Appl. Mater. Interfaces, 2014, 6, 19118–19126 CAS.
- X. Han, G. Qing, J. Sun and T. Sun, Angew. Chem., Int. Ed., 2012, 51, 5147–5151 CrossRef CAS PubMed.
- S. Renault, V. A. Oltean, C. M. Araujo, A. Grigoriev, K. Edström and D. Brandell, Chem. Mater., 2016, 28, 1920–1926 CrossRef CAS.
- L. Fédèle, F. Sauvage, J. Bois and J. M. Tarascon, J. Electrochem. Soc., 2014, 161, A46–A52 CrossRef.
- T. Yasuda and N. Ogihara, Chem. Commun., 2014, 50, 11565–11567 RSC.
- N. Ogihara, T. Yasuda, Y. Kishida, T. Ohsuna, K. Miyamoto and N. Ohba, Angew. Chem., Int. Ed., 2014, 53, 11467–11472 CrossRef CAS PubMed.
- L. Fédèle, F. Sauvage and M. Bécuwe, J. Mater. Chem. A, 2014, 2, 18225–18228 Search PubMed.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- A. P. Cohn, K. Share, R. Carter, L. Oakes and C. L. Pint, Nano Lett., 2016, 16, 543–548 CrossRef CAS PubMed.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963–2969 CAS.
- C. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- A. Choi, Y. K. Kim, T. K. Kim, M.-S. Kwon, K. T. Lee and H. R. Moon, J. Mater. Chem. A, 2014, 2, 14986–14993 CAS.
- V. A. Mihali, S. Renault, L. Nyholm and D. Brandell, RSC Adv., 2014, 4, 38004–38011 RSC.
- Z. Zhu, H. Li, J. Liang, Z. Tao and J. Chen, Chem. Commun., 2014, 51, 1446–1448 RSC.
- H. Wang, P. Hu, J. Yang, G. Gong, L. Guo and X. Chen, Adv. Mater., 2015, 27, 2348–2354 CrossRef CAS PubMed.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130 CrossRef CAS PubMed.
- F. Wan, X.-L. Wu, J.-Z. Guo, J.-Y. Li, J.-P. Zhang, L. Niu and R.-S. Wang, Nano Energy, 2015, 13, 450–457 CrossRef CAS.
- D. J. Kim, Y. H. Jung, K. K. Bharathi, S. H. Je, D. K. Kim, A. Coskun and J. W. Choi, Adv. Energy Mater., 2014, 4, 1400133 Search PubMed.
- S. Renault, V. A. Mihali, K. Edström and D. Brandell, Electrochem. Commun., 2014, 45, 52–55 CrossRef CAS.
- H.-G. Wang, S. Yuan, Z. Si and X.-B. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 CAS.
- J. Wu, X. Rui, C. Wang, W.-B. Pei, R. Lau, Q. Yan and Q. Zhang, Adv. Energy Mater., 2015, 5, 1402189 Search PubMed.
- M. López-Herraiz, E. Castillo-Martínez, J. Carretero-González, J. Carrasco, T. Rojo and M. Armand, Energy Environ. Sci., 2015, 8, 3233–3241 Search PubMed.
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS.
- L. Chen, W. Li, Z. Guo, Y. Wang, C. Wang, Y. Che and Y. Xia, J. Electrochem. Soc., 2015, 162, A1972–A1977 CrossRef CAS.
- L. Chen, W. Li, Y. Wang, C. Wang and Y. Xia, RSC Adv., 2014, 4, 25369–25373 RSC.
- H. Zhu, J. Yin, X. Zhao, C. Wang and X. Yang, Chem. Commun., 2015, 51, 14708–14711 RSC.
- L. Yang, V. A. Mihali, D. Brandell, M. Strømme and M. Sjödin, Macromolecules, 2014, 118, 25956–25963 CAS.
- S. Zhang, W. Huang, P. Hu, C. Huang, C. Shang, C. Zhang, R. Yang and G. Cui, J. Mater. Chem. A, 2015, 3, 1896–1901 CAS.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 127, 7462–7466 CrossRef.
- E. Castillo-Martínez, J. Carretero-González and M. Armand, Angew. Chem., Int. Ed., 2014, 126, 5445–5449 CrossRef.
- P. D. Frischmann, L. C. H. Gerber, S. E. Doris, E. Y. Tsai, F. Y. Fan, X. Qu, A. Jain, K. A. Persson, Y.-M. Chiang and B. A. Helms, Chem. Mater., 2015, 27, 6765–6770 CrossRef CAS.
- L. C. H. Gerber, P. D. Frischmann, F. Y. Fan, S. E. Doris, X. Qu, A. M. Scheuermann, K. Persson, Y.-M. Chiang and B. A. Helms, Nano Lett., 2016, 16, 549–554 CrossRef CAS PubMed.
- S. S. Zhang and D. T. Tran, J. Mater. Chem. A, 2016, 4, 4371–4374 CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- B. Oschmann, J. Park, C. Kim, K. Char and Y. E. Sung, Chem. Mater., 2015, 27, 7011–7017 CrossRef CAS.
- J.-S. Kim, T. H. Hwang, B. G. Kim, J. Min and J. W. Choi, Adv. Funct. Mater., 2014, 24, 5359–5367 CrossRef CAS.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS PubMed.
- T. Kawai, K. Oyaizu and H. Nishide, Macromolecules, 2015, 48, 2429–2434 CrossRef CAS.
- T. Katsumata, M. Satoh, J. Wada, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Rapid Commun., 2006, 27, 1206–1211 CrossRef CAS.
- S. Bahceci and B. Esat, J. Power Sources, 2013, 242, 33–40 CrossRef CAS.
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007, 1730–1732 RSC.
- M. Kato, K.-I. Senoo, M. Yao and Y. Misaki, J. Mater. Chem. A, 2014, 2, 6747–6754 CAS.
- J.-Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 3096–3101 CrossRef CAS PubMed.
- J. Kim, H.-S. Park, T.-H. Kim, S. Yeol Kim and H.-K. Song, Phys. Chem. Chem. Phys., 2014, 16, 5295–5300 RSC.
- B. Häupler, R. Burges, C. Friebe, T. Janoschka, D. Schmidt, A. Wild and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 1367–1371 CrossRef PubMed.
- T. Shiga, Y. Kato, M. Inoue, N. Takahashi and Y. Hase, J. Phys. Chem. C, 2015, 119, 3488–3494 CAS.
- T.-T. Truong, G. W. Coates and H. D. Abruña, Chem. Commun., 2015, 51, 14674–14677 RSC.
- G. Hauffman, A. Vlad, T. Janoschka, U. S. Schubert and J. F. Gohy, J. Mater. Chem. A, 2015, 3, 19575–19581 CAS.
- M. E. Speer, M. Kolek, J. J. Jassoy, J. Heine, M. Winter, P. M. Bieker and B. Esser, Chem. Commun., 2015, 51, 15261–15264 RSC.
- Y. Xuan, M. Sandberg, M. Berggren and X. Crispin, Org. Electron., 2012, 13, 632–637 CrossRef CAS.
- S. C. Han, E. G. Bae, H. Lim and M. Pyo, J. Power Sources, 2014, 254, 73–79 CrossRef CAS.
- T. Godet-Bar, J. C. L. X. tre, O. Le Bacq, J. Y. Sanchez, A. Deronzier and A. Pasturel, Phys. Chem. Chem. Phys., 2015, 17, 25283–25296 RSC.
- K. Sakaushi, E. Hosono, G. Nickerl, H. Zhou, S. Kaskel and J. Eckert, J. Power Sources, 2014, 245, 553–556 CrossRef CAS.
- C. Su, F. Yang, L. Ji, L. Xu and C. Zhang, J. Mater. Chem. A, 2014, 2, 20083–20088 CAS.
- T. Y. Nilsson, M. Wagner and O. Inganäs, ChemSusChem, 2015, 8, 4081–4085 CrossRef CAS PubMed.
- T. Jähnert, B. Häupler, T. Janoschka, M. D. Hager and U. S. Schubert, Macromol. Rapid Commun., 2014, 35, 882–887 CrossRef PubMed.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2013, 505, 195–198 CrossRef PubMed.
- Q. Chen, M. R. Gerhardt, L. Hartle and M. J. Aziz, J. Electrochem. Soc., 2015, 163, A5010–A5013 CrossRef.
- B. Yang, L. Hoober-Burkhardt, F. Wang, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2014, 161, A1371–A1380 CrossRef CAS.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdörfer, B. Häupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- T. Liu, X. Wei, Z. Nie, V. Sprenkle and W. Wang, Adv. Energy Mater., 2015, 6, 1501449 Search PubMed.
- S. H. Oh, C. W. Lee, D. H. Chun, J. D. Jeon, J. Shim, K. H. Shin and J. H. Yang, J. Mater. Chem. A, 2014, 2, 19994–19998 CAS.
- X. Wei, W. Xu, J. Huang, L. Zhang, E. Walter, C. Lawrence, M. Vijayakumar, W. A. Henderson, T. Liu, L. Cosimbescu, B. Li, V. Sprenkle and W. Wang, Angew. Chem., Int. Ed., 2015, 127, 8808–8811 CrossRef.
- A. P. Kaur, N. E. Holubowitch, S. Ergun, C. F. Elliott and S. A. Odom, Energy Technol., 2015, 3, 476–480 CrossRef CAS.
- C. S. Sevov, R. E. M. Brooner, E. Chénard, R. S. Assary, J. S. Moore, J. Rodríguez-López and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 14465–14472 CrossRef CAS PubMed.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, J. Electrochem. Soc., 2015, 163, A338–A344 CrossRef.
- X. Wei, W. Xu, M. Vijayakumar, L. Cosimbescu, T. Liu, V. Sprenkle and W. Wang, Adv. Mater., 2014, 26, 7649–7653 CrossRef CAS PubMed.
- J. Huang, L. Cheng, R. S. Assary, P. Wang, Z. Xue, A. K. Burrell, L. A. Curtiss and L. Zhang, Adv. Energy Mater., 2014, 5, 1401782 Search PubMed.
- M. Park, D.-S. Shin, J. Ryu, M. Choi, N. Park, S. Y. Hong and J. Cho, Adv. Mater., 2015, 27, 5141–5146 CrossRef CAS PubMed.
- D. P. Dubal, O. Ayyad and P. Gómez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC.
- G. Cai, P. Darmawan, M. Cui, J. Wang, J. Chen, S. Magdassi and P. S. Lee, Adv. Energy Mater., 2015, 6, 1501882 Search PubMed.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- Y. Zhang, R. Jamal, W. Shao and T. Abdiryim, Electrochim. Acta, 2013, 113, 382–389 CrossRef CAS.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- L. Hao, J. Ning, B. Luo, B. Wang, Y. Zhang, Z. Tang, J. Yang, A. Thomas and L. Zhi, J. Am. Chem. Soc., 2015, 137, 219–225 CrossRef CAS PubMed.
- F. Xu, H. Xu, X. Chen, D. Wu, Y. Wu, H. Liu, C. Gu, R. Fu and D. Jiang, Angew. Chem., Int. Ed., 2015, 127, 6918–6922 CrossRef.
- P.-F. Li, T. B. Schon and D. S. Seferos, Angew. Chem., Int. Ed., 2015, 54, 9361–9366 CrossRef CAS PubMed.
- S. Wang, L. Gai, J. Zhou, H. Jiang, Y. Sun and H. Zhang, J. Phys. Chem. C, 2015, 119, 3881–3891 CAS.
- S.-K. Kim, J. Cho, J. S. Moore, H. S. Park and P. V. Braun, Adv. Funct. Mater., 2015, 26, 903–910 CrossRef.
- H. Zhang, Y. Zhang, C. Gu and Y. Ma, Adv. Energy Mater., 2015, 5, 1402175 Search PubMed.
- M. Sun, Q. Tang, T. Zhang and G. Wang, RSC Adv., 2014, 4, 7774–7779 RSC.
- D. Mo, W. Zhou, X. Ma and J. Xu, Electrochim. Acta, 2015, 155, 29–37 CrossRef CAS.
- D. Mo, W. Zhou, X. Ma, J. Xu, F. Jiang and D. Zhu, Electrochim. Acta, 2015, 151, 477–488 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed.
- S. Admassie, T. Y. Nilsson and O. Inganäs, Phys. Chem. Chem. Phys., 2014, 16, 24681–24684 RSC.
- T. B. Schon, P. M. DiCarmine and D. S. Seferos, Adv. Energy Mater., 2014, 4, 1301509 Search PubMed.
- D. F. Zeigler, S. L. Candelaria, K. A. Mazzio, T. R. Martin, E. Uchaker, S.-L. Suraru, L. J. Kang, G. Cao and C. K. Luscombe, Macromolecules, 2015, 48, 5196–5203 CrossRef CAS.
- L. A. Estrada, D. Y. Liu, D. H. Salazar, A. L. Dyer and J. R. Reynolds, Macromolecules, 2012, 45, 8211–8220 CrossRef CAS.
- P. M. DiCarmine, T. B. Schon, T. M. McCormick, P. P. Klein and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 8295–8307 CAS.
- R. Yuksel, S. C. Cevher, A. Cirpan, L. Toppare and H. E. Unalan, J. Electrochem. Soc., 2015, 162, A2805–A2810 CrossRef CAS.
- N. An, Y. An, Z. Hu, B. Guo, Y. Yang and Z. Lei, J. Mater. Chem. A, 2015, 3, 22239–22246 CAS.
- D. Tomerini, C. Gatti and C. Frayret, Phys. Chem. Chem. Phys., 2015, 17, 8604–8608 RSC.
- D. Tomerini, C. Gatti and C. Frayret, Phys. Chem. Chem. Phys., 2016, 18, 2442–2448 RSC.
- K. Hernández-Burgos, S. E. Burkhardt, G. G. Rodríguez-Calero, R. G. Hennig and H. D. Abruña, J. Phys. Chem. C, 2014, 118, 6046–6051 Search PubMed.
- N. Dardenne, X. Blase and G. Hautier, Macromolecules, 2015, 119, 23373–23378 CAS.
- T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. C, 2014, 118, 17213–17220 CAS.
- Y. Chen, Z. Wu and S. Sun, J. Phys. Chem. C, 2014, 118, 21813–21818 CAS.
- S. Sun, Y. Chen and J. Yu, J. Phys. Chem. C, 2015, 119, 25770–25777 CAS.
- Y. Chen, S. Sun, X. Wang and Q. Shi, J. Phys. Chem. C, 2015, 119, 25719–25725 CAS.
- Y.-X. Yu, J. Mater. Chem. A, 2014, 2, 8910–8917 CAS.
- J. E. Bachman, L. A. Curtiss and R. S. Assary, J. Phys. Chem. A, 2014, 118, 8852–8860 CrossRef CAS PubMed.
- S. D. Pineda Flores, G. C. Martin-Noble, R. L. Phillips and J. Schrier, J. Phys. Chem. C, 2015, 119, 21800–21809 CAS.
- S. Er, C. Suh, M. P. Marshak and A. Aspuru-Guzik, Chem. Sci., 2015, 6, 885–893 RSC.
- J. Vatamanu, Z. Hu, D. Bedrov, C. Perez and Y. Gogotsi, J. Phys. Chem. Lett., 2013, 4, 2829–2837 CrossRef CAS.
- S. Kerisit, B. Schwenzer and M. Vijayakumar, J. Phys. Chem. Lett., 2014, 5, 2330–2334 CrossRef CAS PubMed.
- D.-E. Jiang and J. Wu, J. Phys. Chem. Lett., 2013, 4, 1260–1267 CrossRef CAS PubMed.
- D.-E. Jiang, Z. Jin and J. Wu, Nano Lett., 2011, 11, 5373–5377 CrossRef CAS PubMed.
- C. Merlet, C. P. E. an, B. Rotenberg, P. A. Madden, B. Daffos, P. L. Taberna, P. Simon and M. Salanne, Nat. Commun., 2013, 4, 1–6 Search PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- S. E. Burkhardt, M. A. Lowe, S. Conte, W. Zhou, H. Qian, G. G. Rodríguez-Calero, J. Gao, R. G. Hennig and H. D. Abruña, Energy Environ. Sci., 2012, 5, 7176–7187 CAS.
| This journal is © The Royal Society of Chemistry 2016 |