
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions of arene ruthenium metallaprisms with human proteins†
Lydia E. H.
Paul
a,
Bruno
Therrien
*b and
Julien
Furrer
*a
aDepartement für Chemie und Biochemie, Universität Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: julien.furrer@dcb.unibe.ch
bInstitut de Chimie, Université de Neuchâtel, Avenue de Bellevaux 51, CH-2000 Neuchâtel, Switzerland. E-mail: bruno.therrien@unine.ch
First published on 12th November 2014
Abstract
Interactions between three hexacationic arene ruthenium metallaprisms, [(p-cymene)6Ru6(tpt)2(dhnq)3]6+, [(p-cymene)6Ru6(tpt)2(dhbq)3]6+ and [(p-cymene)6Ru6(tpt)2(oxa)3]6+, and a series of human proteins including human serum albumin, transferrin, cytochrome c, myoglobin and ubiquitin have been studied using NMR spectroscopy, mass spectrometry and circular dichroism spectroscopy. All data suggest that no covalent adducts are formed between the proteins and the metallaprisms. Indeed, in most cases electrostatic interactions, leading to precipitation of protein-metallaprism aggregates, have been observed. In addition, with the smallest proteins, ubiquitin, myoglobin and cytochrome c, the presence of the hexacationic arene ruthenium metallaprisms induces structural changes of the proteins, as emphasized by circular dichroism. The results suggest that proteins are certainly a biological target for these metalla-assemblies.
Introduction
Since the fortuitous discovery of the chemotherapeutic properties of platinum complexes by Rosenberg in the 1960s,1 a lot of effort has been devoted to identify cellular targets and mechanisms of action of metal-based drugs. Initially, DNA was thought to be the primary and most effective target. Consequently most of the research was focused on improving the DNA binding properties of metal-based drugs. This approach resulted in rather unselective compounds that often exhibit significant side effects.2 It is now well accepted that one should not focus on DNA when developing new metal-based drugs. Indeed, current research aims at developing new compounds with new biological targets. This quest for finding truly selective compounds has resulted in what is today known as targeted therapy.3,4 The increased selectivity represents the most important feature of the newly developed drugs in order to minimize side effects and to prevent the development of resistance mechanisms.5,6As emphasized in the literature, in order to successfully design and develop new and selective metal-based drugs, it is of the highest importance to understand the mode of action of established and also of newly developed anticancer drugs.7,8 Among the new generation of metal-based drugs, ruthenium derivatives appear to be quite promising. These complexes selectively bind to proteins or inhibit enzymes known to be overexpressed in many types of cancer.7,9–13 To date, much work has been done to elucidate the mode of action of various ruthenium-based drug candidates,14,15 but the way they exert their antitumoural or antimetastatic effects is not yet fully understood, even for NAMI-A and NKP-1339, which have successfully completed clinical trials.14,16–18 By analogy with platinum complexes, it was originally assumed that DNA binding was the main reason for the anticancer activity of ruthenium complexes.16,19–21 While the ability of ruthenium complexes to bind to DNA has been demonstrated,22–25 in particular for arene ruthenium ethylenediamine complexes,26,27 it was also observed that DNA binding of ruthenium was weaker and different from that observed for platinum complexes.22 Recent examples of rationally designed metal complexes that inhibit enzymatic activities involved in cancer proliferation were investigated,28,29 including ruthenium half-sandwich complexes that bind to specific enzymes.30,31 Dyson and co-workers have shown that several complexes of the RAPTA family bind rather selectively to Cathespin B,9 a prognostic marker for various types of cancer.32 The interactions between metal-based drugs and enzymes or proteins have to be expected since proteins are ubiquitous in every tissue and in the blood stream of the human body and any potential anticancer drug will be facing various proteins either in the blood or in the cytosol on their way to cancer cells.
Our previous work describing the reactivity of the metallaprisms [(p-cymene)6Ru6(tpt)2(dhnq)3]6+ ([1]6+; tpt = 2,4,6-tris(pyridyl-4-yl)-1,3,5-triazine; dhnq = 5,8-dihydroxy-1,4-naphthoquinonato), [(p-cymene)6Ru6(tpt)2(dhbq)3]6+ ([2]6+; dhbq = 2,5-dihydroxy-1,4-benzoquinonato) and [(p-cymene)6Ru6(tpt)2(oxa)3]6+ ([3]6+; oxa = oxalato) (Fig. 1) towards free amino acids suggests an increasing reactivity from [1]6+ to [3]6+, with in some cases the formation of degradation products, especially in the presence of basic amino acids (Arg, His and Lys).33–35 For this reason, interactions can be expected with proteins, especially with ubiquitin. This small protein possesses a C-terminal tail and seven lysine residues which are potentially accessible for the metallaprisms.
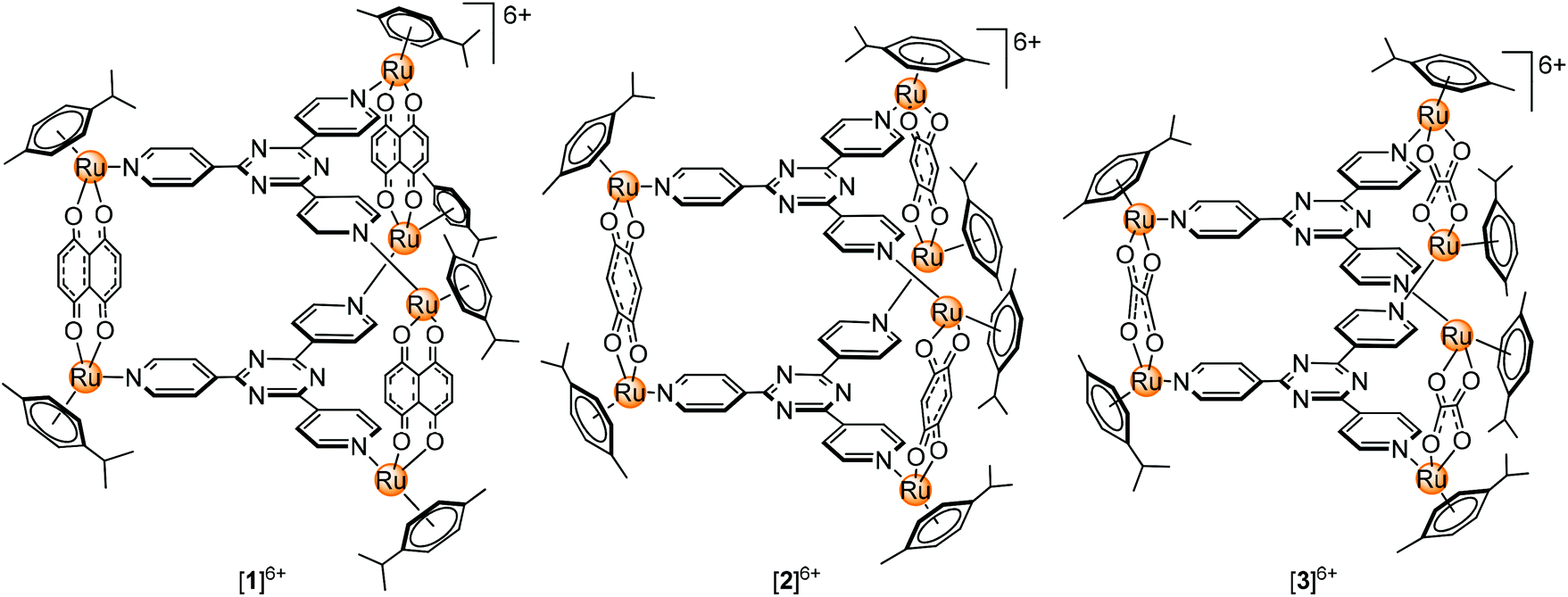 | ||
| Fig. 1 Structures of the hexacationic metallaprisms [1]6+, [2]6+ and [3]6+. | ||
Herein we report the NMR, CD and MS study of the interactions between the hexanuclear metallaprisms [1]6+–[3]6+, with two serum proteins, human serum albumin (HsA) and transferrin (Tf), and three cytosolic proteins, cytochrome c (Cyt c), myoglobin (Mb) and ubiquitin (Ub). All proteins are water-soluble and stable in solution under physiological conditions. Moreover, high-resolution crystal structures or NMR structural data are available, making them ideal candidates for such an investigation.36–40
Albumin, known to transport numerous important biomolecules and drugs in the cytoplasm,41 and transferrin, which controls the level of free iron in biological fluids, may react immediately with the metallaprisms, if they are administrated intravenously. Cytosolic proteins also possess important functions: cytochrome c is involved in apoptotic pathways,42 while ubiquitin is relevant in posttranslational modifications together with the proteasome system,43 and myoglobin is the primary oxygen-carrying pigment of muscle tissues.44
Results and discussion
NMR and MS investigations of the interaction between metallaprisms and proteins
Interactions between the proteins and the metallaprisms were first monitored in aqueous solution at 37 °C by 1H NMR spectroscopy over a time frame of 96 h. As an example, Fig. 2 shows the 1H NMR spectra obtained after mixing HsA and [1]6+. Over time, the intensity of the protein's resonances and of the tpt and dhnq ligands of the metallaprism decreased considerably. The 1H NMR spectra suggest that the metallaprism [1]6+ induces the precipitation of HsA without formation of a covalent bond between the metallaprism and HsA. Accordingly, in the NMR tube a precipitate was visible. Similar to [1]6+, precipitation of the proteins were observed for the mixtures [2]6+/HsA and [3]6+/HsA (Fig. S1 and S2†).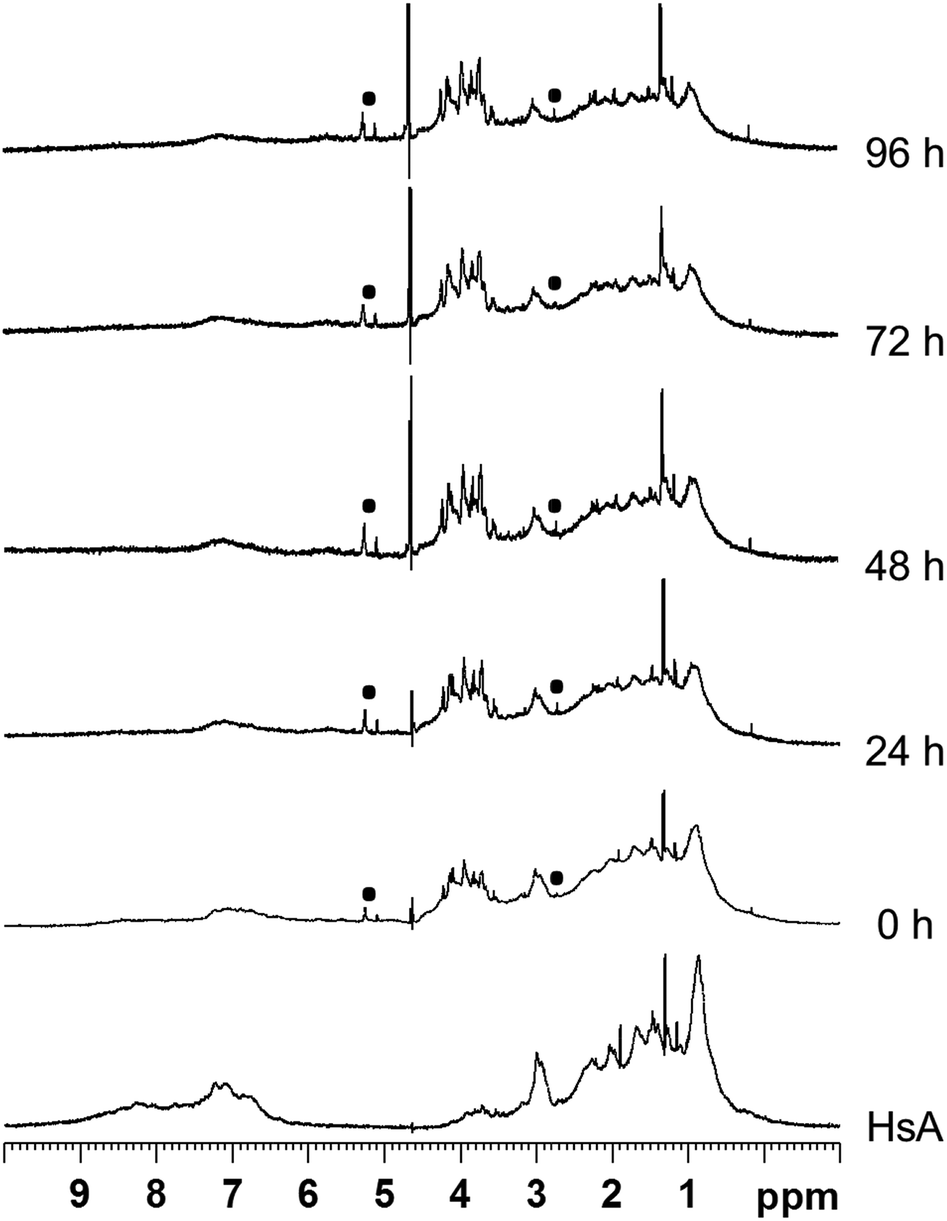 | ||
Fig. 2
1H NMR spectra of the mixture HsA/[1]6+ (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) recorded in D2O at 37 °C between 0 h and 96 h. The 1H NMR spectrum of free HsA is shown at the bottom as reference. The p-cymene resonances of the metallaprism are labelled by (■). :10) recorded in D2O at 37 °C between 0 h and 96 h. The 1H NMR spectrum of free HsA is shown at the bottom as reference. The p-cymene resonances of the metallaprism are labelled by (■). | ||
For the mixtures metallaprism/Cyt c and metallaprism/Tf (Fig. S3–S5 and Fig. S6–S8†), the 1H NMR spectra indicate that metallaprisms [1]6+ and [3]6+ as well as the proteins precipitate, as visible in the respective NMR tubes. However, for metallaprism [2]6+ the intensity of the signals associated to the metallaprism remains relatively strong over the 96 hours period, suggesting a slower interaction between [2]6+ and the two proteins (Fig. S4 and S7†).
In the case of Mb, a different behaviour was observed for the three metallaprisms. In the presence of [1]6+, minimal precipitation of Mb was observed (Fig. S9†), the intensity of the protein signals remaining strong after 96 h. However, the resonance's intensity of the metallaprism [1]6+ dropped rapidly, and the resonances were overlapped with those of the protein after only 24 hours. A similar observation was made with metallaprism [2]6+ but at a much slower rate (Fig. S10†). Additionally, the resonances of the protein became gradually broadened, indicating an ongoing reaction or an interaction with the metallaprism [2]6+. On the other hand, in the presence of [3]6+, Mb precipitated almost completely while the signals of the metallaprism remained sharp and well resolved (Fig. S11†).
For ubiquitin, the 1H NMR spectra (Fig. 3, and Fig. S12 and S13†) revealed that Ub remained unaffected upon addition of the metallaprisms and did not precipitate in any case. This outcome is rather surprising because Ub possesses a C-terminal tail and several lysine residues potentially accessible for the metallaprisms. Ub was therefore anticipated to react with the metallaprisms, since it was previously shown that metallaprisms rapidly react with lysine.33–35 The only changes observed in the NMR spectra can be ascribed to the disappearance of the signals associated to the metallaprism [1]6+, and to a lesser extent to a reduction of the intensity of the signals of metallaprisms [2]6+. However, for metallaprism [3]6+, additional signals in the aromatic region of the p-cymene protons (5–6 ppm) are observed, together with the signals of the p-cymene ligands of the intact metallaprism. These new signals can either be tentatively ascribed to decomposition products or to protein-metallaprism adducts.
 | ||
| Fig. 3
1H NMR spectra of the mixture Ub/[1]6+ (molar ratio 1:10, D2O, 37 °C) recorded between 0 h and 96 h. The 1H NMR spectrum of free ubiquitin is shown as reference at the bottom. The p-cymene resonances of the metallaprism are labelled by (■). | ||
In order to determine the nature of the interaction between the proteins and the metallaprisms, MALDI-TOF mass spectra were recorded in the negative mode. The MALDI-TOF spectra of the free proteins are given in Fig. S14–S18.† We were however unable to get exploitable spectra from the mixtures metallaprism/protein owing to the rapid precipitation of the proteins. In a second attempt, the precipitates were collected, dried and dissolved in DMSO, and MALDI-TOF MS measurements were repeated, but remained silent. The ESI mass spectra of the precipitates were also recorded in the positive mode, giving unusable spectra as well (data not shown). Although not conclusive, the ESI mass spectra support the results obtained by NMR spectroscopy, which suggest that no covalent bonds between the metallaprisms and the proteins are formed. Indeed, upon breakage of the metallaprisms, the ESI mass spectra would have exhibited peaks corresponding to complexes similar to those previously reported in the literature, such as [(p-cymene)Ru-Cyt c + 5H+]8+,45 [(p-cymene)Ru-Ub]+ and [2{Ru(p-cymene)}-Ub]+.46 Interestingly, the results found for these three metallaprisms are in contrast to those observed with other ruthenium complexes. For instance, KP1019 was shown to bind to apoTf as evidenced by ESI-mass and CD spectra,47 and RAPTA-C was shown to bind to Ub and metallothionein-2.48
All NMR and MS experiments strongly suggest that no covalent binding takes place between the metallaprisms and the five selected proteins. It is more plausible that electrostatic interactions between the proteins and the metallaprisms cause the observed precipitation: A phenomenon commonly observed for proteins interacting with cations.49,50
CD investigation of the interaction between the metallaprisms and proteins
Circular dichroism (CD) is an important tool for studying the structural changes of proteins in solution.51,52Fig. 4 shows the CD spectra of the proteins alone in comparison with the spectra measured after addition of the metallaprisms (1:10 ratio). In the case of HsA, upon addition of the metallaprisms the spectra exhibited a strong decrease in the intensity of the negative bands. This reduced intensity can be easily understood, knowing that HsA precipitates in solution in the presence of the metallaprisms, as observed by NMR. According to the CD spectra shown in Fig. 4, it can be assumed that the original α-helical structure of HsA is retained upon addition of metallaprisms [2]6+ and [3]6+. With metallaprism [1]6+, on the other hand, an additional increase of the positive band was observed, suggesting a small alteration of the protein folding. Although the overall α-helical structure of HsA remains, the positive band could suggest the refolding at least in part of the protein into a β-sheet like structure, as previously noted.51,53 The data suggests that electrostatic interactions take place between the metallaprisms and HsA which lead to protein precipitation but not to the formation of covalent bonds. It seems also that HsA undergoes minimal conformational changes upon addition of metallaprism [1]6+.
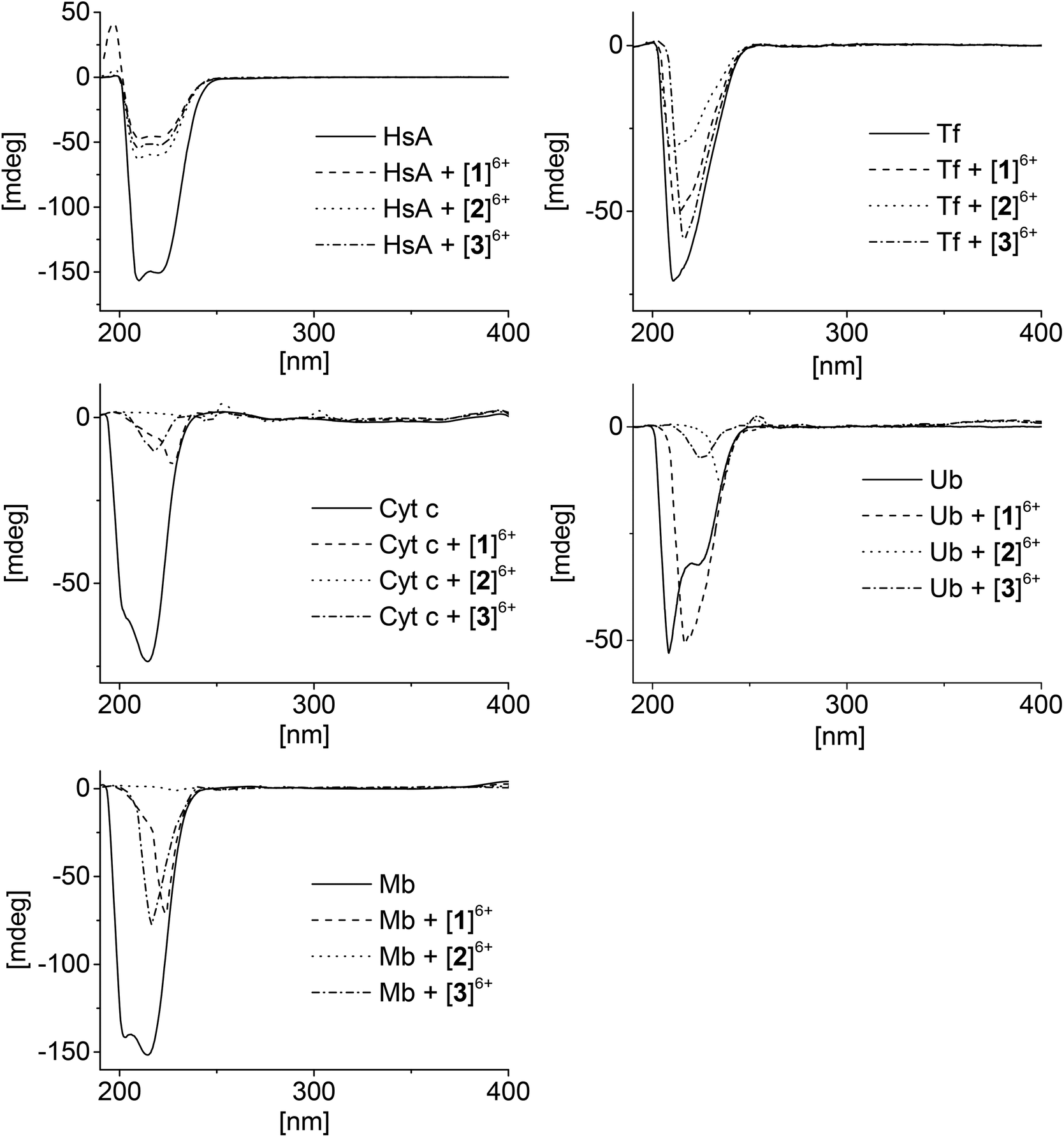 | ||
| Fig. 4 CD spectra of the mixtures containing the proteins and the metallaprisms (molar ratio 1:10). Left column: top = HsA, middle = Cyt c, bottom = Mb; right column: top = Tf, bottom = Ub. | ||
Similar to HsA, the intensity of the CD band associated to Tf decreased upon addition of the metallaprisms. Unlike HsA, the intensity reduction was less pronounced, but a significant decrease could be observed with metallaprism [2]6+ compared to metallaprisms [1]6+ and [3]6+. These observations are consistent with the NMR data, in which the precipitation of Tf was more noticeable with metallaprism [2]6+. In solution, the Tf protein exhibits a random coil-like structure51 which does not change upon addition of the metallaprisms.
For Cyt c and Mb, the CD spectra also exhibited a strong decrease of the band intensity upon addition of the metallaprisms. With metallaprism [2]6+, the CD signal disappeared completely in both cases while in the presence of the other two metallaprisms, an additional change in the structure of the CD band was observed. In the case of [2]6+, the complete vanishing of the CD band cannot be exclusively attributed to the precipitation of the proteins. It is more plausible that soluble protein aggregates are formed in solution due to an interaction with the cationic metallaprisms. The aggregates would no longer be chiral and therefore show no bands in the CD spectrum.54 Both Cyt c and Mb possess a slight α-helical structure51,55 in solution, but after 48 h in the presence of [1]6+ and [3]6+ the CD signals represent a protein folded in a random coil structure.51 This change in the protein folding can be tentatively attributed to a change in the secondary structure of the proteins, caused by electrostatic interactions between Cyt c or Mb and the metallaprisms [1]6+ and [3]6+.
Ubiquitin was assumed to be the most likely protein to react with the metallaprisms, due to the presence of an unstructured C-terminal tail and accessible Lys residues. Surprisingly, neither the mass spectra nor the NMR spectra have indicated covalent interactions between Ub and the metallaprisms. On the other hand, the CD spectra suggest that strong electrostatic interactions take place with the metallaprisms [2]6+ and [3]6+, the CD band being significantly quenched. Moreover, in the presence of [1]6+, the negative band of Ub was shifted, while the intensity of the band remained strong (Fig. 4). As shown by NMR spectroscopy, these significant changes in the CD spectrum of Ub cannot be attributed to the precipitation of the protein. A more plausible explanation for the disappearance could be the formation of soluble Ub aggregates in solution, due to the interaction with the cationic metallaprisms [2]6+ and [3]6+ as described for Cyt c and Mb. Unlike [2]6+ and [3]6+, the intensity of the CD band is almost completely retained but the nature of the band is altered in the presence of metallaprism [1]6+. From the original two negative bands only one remains visible in the spectrum and suggests that the protein adopts a random coil-like structure,51,54 due to electrostatic interactions with [1]6+.
The results collected from the NMR, MS and CD experiments have revealed the occurrence of electrostatic interactions between the proteins and the metallaprisms: the outcomes ranging from precipitations to structural changes of the proteins. In order to get further insight into these protein-metallaprism electrostatic interactions, we modelled the electrostatic surface potentials for the selected proteins (Fig. 5 and Fig. S19–S21†). The ratios of negative and positive surface potentials determined by optical inspection are summarized in Table 1, together with other parametrical data. HsA possesses a high amount of negatively charged surface areas (Fig. S19†) and can thereby strongly interact with the positively charged metallaprisms ([1]6+–[3]6+). These electrostatic interactions most probably lead to the observed precipitation of the protein and of the metallaprisms. Similar arguments can explain the behaviour of Tf (Fig. 5) and Mb (Fig. S20†). Both proteins possess negatively charged areas on the surface which are readily available for interactions with the metallaprisms. The strong effect observed in the CD spectra of Mb could be a result of the spatial distribution of the charged areas. The positive and the negative charges are mainly found on opposite hemispheres of the protein and upon interaction with the highly positively charged metallaprisms a refolding of the protein seems likely, thus minimizing electrostatic repulsion between the positive charges (Fig. S20†). The same explanation could also be applied to the strong structural changes observed for Ub (Fig. 5) and Cyt c (Fig. S21†). The charges are again localized mostly on opposite sides of the protein surface and an interaction between the negative charged area and the metallaprisms should lead to refolding of the protein and even aggregation which can result in the disappearance of the CD signals (Fig. 4). In addition, negatively and positively charged surface fractions are observed in close proximity for Cyt c. This could lead to both electrostatic attraction and repulsion upon contact with the positively charged metallaprisms. The disappearance of the CD and NMR signals could therefore also be attributed to the precipitation of the protein due to electrostatic interactions.
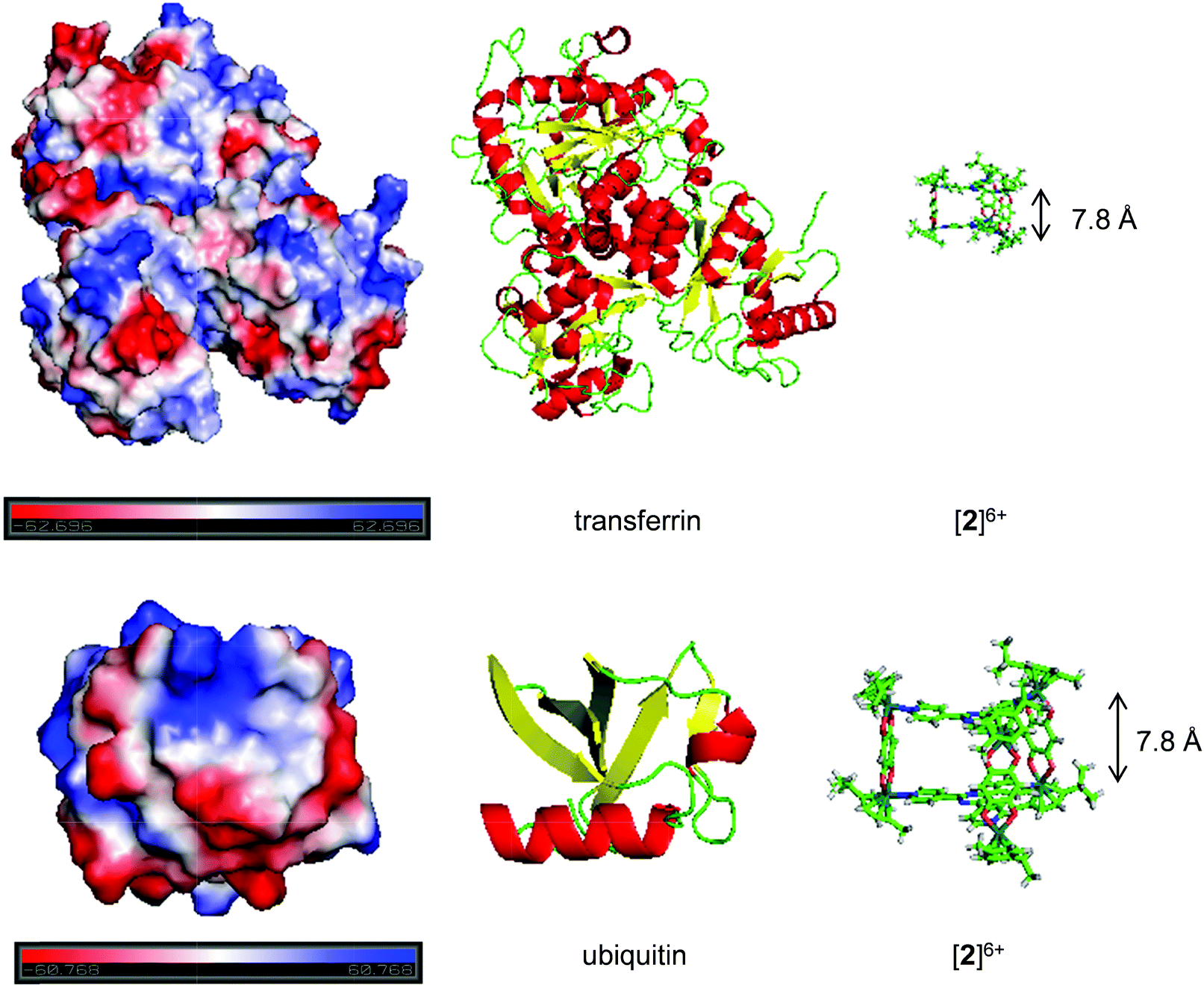 | ||
| Fig. 5 Electrostatic potentials on the protein surface (top: transferrin, bottom: ubiquitin) modelled with PyMOL (v 1.7.2.1) (left). The sequence was modelled from the RSCB pdb files (3QYT for Tf38 and 1UBQ for Ub40). Areas of negative potentials are shown in red, areas of positive potentials in blue and areas of neutral potentials in white. The structure of the protein created from the same pdb files is shown in the middle. The structure of metallaprism [2]6+ (right) was added to highlight the size difference between the two entities. | ||
| Protein | Molecular weighta |
R
minb |
Overall charge1 | Domainsc | Relative positively/negatively charged surfaced |
|---|---|---|---|---|---|
| a Calculated with http://protcalc.sourceforge.net, sequences taken from pdb files (HsA = 1UOR, Cyt c = 1HRC, Tf = 3QYT, Mb = 4DC8, Ub = 2ZCC). b Calculation based on ref. 56, Rmin = minimal radius of sphere that could contain a given mass of protein. c Taken from http://www.uniprot.org, (UniProtKB: HsA = P02768, Cyt c = P00004, Tf = P02787, Mb = P6808, Ub = P0CH28). d Estimated from model structures shown in Fig. 5, S19–S21. | |||||
| Tf | 75.1 kDa | 2.78 nm | 0.4 | 2 | +/− |
| HsA | 66.4 kDa | 2.67 nm | −12.2 | 3 | − |
| Mb | 16.9 kDa | 1.69 nm | 2.6 | 1 | − |
| Cyt c | 11.7 kDa | 1.50 nm | 9.6 | 1 | + |
| Ub | 8.6 kDa | 1.35 nm | 0.2 | 1 | − |
Considering our results, it is unlikely that the metallaprisms [1]6+–[3]6+ bind covalently to proteins. They rather strongly reduce their solubility and lead to the precipitation of the proteins. Proteins often show this behaviour in solution with cations present.49,50,57 Also, the electrostatic interactions cause structural alterations in the proteins’ secondary structure as the CD spectra of Mb, Cyt c and Ub indicate severe spectral changes in solution upon addition of the metallaprisms.
Conclusions
The interaction of three hexacationic arene ruthenium metallaprisms with several proteins has been monitored by MS spectrometry, NMR and CD spectroscopies. Electrostatic interactions between the metallaprisms and the proteins, human serum albumin (HsA), transferrin (Tf), myoglobin (Mb), cytochrome c (Cyt c) and ubiquitin (Ub), were observed. In general, with the exception of ubiquitin, the NMR spectra show precipitations of the proteins in D2O. Electrostatic interactions that induce the precipitation of the proteins seem to be the foremost mode of interaction. The CD spectra support these findings, with additional information regarding the experiments for which no precipitation of the proteins was observed. In these particular cases, the metallaprisms induce quite severe changes to the secondary structure of the proteins. For HsA and Tf the observed reduction of the band intensity was attributed to protein precipitation, while for the other three proteins Cyt c, Mb and Ub, the spectral changes were ascribed to structural alterations. These changes seem to be induced by the electrostatic interactions between negatively charged areas on the protein surface and the metallaprisms.Overall, these results suggest that proteins are certainly biological targets for these arene ruthenium metallaprisms. The supramolecular interaction between the hexacationic metalla-assembly and proteins appears to be electrostatic in nature, and this strong interaction in water induces precipitation or unfolding of the proteins, which might in part explain the biological activity of these metallaprisms.
Experimental
General remarks
Chemicals obtained from commercial suppliers were of analytical grade and used as received. The proteins human serum albumin, transferrin, myoglobin, ubiquitin and cytochrome c were obtained from Sigma Aldrich. The arene ruthenium metallaprisms [1](CF3SO3)6, [2](CF3SO3)6 and [3](CF3SO3)6 were synthesised according to published methods.58–60NMR spectroscopy
If not otherwise stated, all experiments were carried out by mixing the metallaprisms [1](CF3SO3)6–[3](CF3SO3)6 in 0.05 mL D2O with 0.1 equivalent of the proteins. All reactions were monitored by 1D 1H NMR spectroscopy over a time period of up to 96 h. All experiments were undertaken at pD = 7, i.e. close to the pH value of the bloodstream. The pD values of D2O solutions were measured by using a glass electrode and addition of 0.4 to the pH meter reading.61,62 To mimic physiological conditions as closely as possible during the experiments, oxygen was not excluded from solutions.NMR data were acquired at a temperature of 37 °C using a Bruker AvanceII 500 MHz NMR spectrometer, equipped with an inverse 1.7 mm triple channel (1H, 31P, 13C) z-gradient microprobehead. All 1D 1H NMR data were measured with 1 k transients into 64 k data points over a width of 25 ppm using a classical presaturation to eliminate the water resonance. A relaxation delay of 2 s was applied between transients.
Mass spectrometry studies
MALDI-TOF mass spectrometry analyses were performed on a Bruker Daltonics Autoflex III MALDI-TOF mass spectrometer equipped with a smartbeam Nd:YAG-laser (355 nm) providing a repetition rate of 200 Hz.The three metallaprisms, using each time 1 mg of the corresponding triflate salts ([1](CF3SO3)6–[3](CF3SO3)6) in 1 mL H2O, were incubated with the selected protein at a 10:1 molar ratio for 48 h at 37 °C. Then, all samples were analysed using various matrices depending on the protein. For HsA, Tf, Cyt c and Ub, CHCA (α-cyano-4-hydroxycinnamic acid) was applied and for Mb a SA (sinapic acid) matrix was used. The Xcalibur software package (Xcalibur 2.0.7, Thermo Fisher Scientific) was used for instrument control and data processing.
Circular dichroism studies
The circular dichroism (CD) spectra of the proteins in the presence or absence of the metallaprisms were accumulated on a J-715 Jasco Spectropolarimeter (with Xe lamp, purged with nitrogen). All experiments were measured using a Hellma Suprasil R 100-QS 0.1 cm cuvette. The metallaprisms [1](CF3SO3)6–[3](CF3SO3)6 and the proteins were dissolved in H2O and incubated for 48 h before data collection. For each sample, the mixture was scanned three times using a rate of 50 nm min−1 at 20 °C. The range of measurement was 190–700 nm, pitch was 0.5 nm, response 16 s and band 1.0 nm. The nitrogen flow was kept around 5 L min−1. All spectra were corrected for solvent signal contribution and measured under identical conditions.Acknowledgements
This work was financially supported by the University of Berne, the Berne University Research Foundation, the University of Neuchatel, and by the Swiss National Science Foundation (project no 200020–143254).Notes and references
- B. Rosenberg, L. V. Camp, E. B. Grimley and A. J. Thomson, J. Biol. Chem., 1967, 242, 1347–1352 CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- W. N. Hait, Cancer Res., 2009, 69, 1263–1267 CrossRef CAS PubMed.
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- D. A. Wolters, M. Stefanopoulou, P. J. Dyson and M. Groessl, Metallomics, 2012, 4, 1185–1196 RSC.
- M. Groessl, C. G. Hartinger, K. Poleć-Pawlak, M. Jarosz, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 1609–1614 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- L. Messori, P. Orioli, D. Vullo, E. Alessio and E. Iengo, Eur. J. Biochem., 2000, 267, 1206–1213 CrossRef CAS.
- L. Messori, F. G. Vilchez, R. Vilaplana, F. Piccioli, E. Alessio and B. Keppler, Met.–Based Drugs, 2000, 7, 335–342 CrossRef CAS PubMed.
- G. Mestroni, E. Alessio, G. Sava, S. Pacor, M. Coluccia and A. Boccarelli, Met.–Based Drugs, 1994, 1, 41–63 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- G. Sava, S. Pacor, M. Coluccia, M. Mariggio, M. Cocchietto, E. Alessio and G. Mestroni, Drug Invest., 1994, 8, 150–161 CrossRef CAS.
- O. Nováková, J. Kašpárková, O. Vrána, P. M. van Vliet, J. Reedijk and V. Brabec, Biochemistry, 1995, 34, 12369–12378 CrossRef.
- A. Barca, B. Pani, M. Tamaro and E. Russo, Mutat. Res., 1999, 423, 171–181 CrossRef CAS.
- O. Nováková, H. Chen, O. Vrána, A. Rodger, P. J. Sadler and V. Brabec, Biochemistry, 2003, 42, 11544–11554 CrossRef PubMed.
- P. Nagababu, J. N. L. Latha and S. Satyanarayana, Chem. Biodiversity, 2006, 3, 1219–1229 CAS.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC.
- D. Can, B. Spingler, P. Schmutz, F. Mendes, P. Raposinho, C. Fernandes, F. Carta, A. Innocenti, I. Santos, C. T. Supuran and R. Alberto, Angew. Chem., Int. Ed., 2012, 51, 3354–3357 CrossRef CAS PubMed.
- H. Bregman, D. S. Williams, G. E. Atilla, P. J. Carroll and E. Meggers, J. Am. Chem. Soc., 2004, 126, 13594–13595 CrossRef CAS PubMed.
- J. É. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS PubMed.
- J. E. Koblinski, M. Ahram and B. F. Sloane, Clin. Chim. Acta, 2000, 291, 113–135 CrossRef CAS.
- L. E. H. Paul, J. Furrer and B. Therrien, J. Organomet. Chem., 2013, 734, 45–52 CrossRef CAS PubMed.
- L. E. H. Paul, B. Therrien and J. Furrer, Inorg. Chem., 2012, 51, 1057–1067 CrossRef CAS PubMed.
- L. E. H. Paul, B. Therrien and J. Furrer, J. Biol. Inorg. Chem., 2012, 17, 1053–1062 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CrossRef CAS.
- N. Yang, H. Zhang, M. Wang, Q. Hao and H. Sun, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- D. J. Kissick, C. M. Dettmar, M. Becker, A. M. Mulichak, V. Cherezov, S. L. Ginell, K. P. Battaile, L. J. Keefe, R. F. Fischetti and G. J. Simpson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 843–851 CrossRef CAS PubMed.
- S. Vijay-Kumar, C. E. Bugg and W. J. Cook, J. Mol. Biol., 1987, 194, 531–544 CrossRef CAS.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- X. Liu, C. N. Kim, J. Yang, R. Jemmerson and X. Wang, Cell, 1996, 86, 147–157 CrossRef CAS.
- N. Shigi, J. Biol. Chem., 2012, 287, 17568–17577 CrossRef CAS PubMed.
- J. N. Patton and A. F. Palmer, Biotechnol. Prog., 2006, 22, 1025–1049 CrossRef CAS PubMed.
- A. Casini, A. Guerri, C. Gabbiani and L. Messori, J. Inorg. Biochem., 2008, 102, 995–1006 CrossRef CAS PubMed.
- A. Casini, C. Gabbiani, E. Michelucci, G. Pieraccini, G. Moneti, P. Dyson and L. Messori, J. Biol. Inorg. Chem., 2009, 14, 761–770 CrossRef CAS PubMed.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- A. Casini, A. Karotki, C. Gabbiani, F. Rugi, M. Vasak, L. Messori and P. J. Dyson, Metallomics, 2009, 1, 434–441 RSC.
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CrossRef CAS.
- S. M. Kelly and N. C. Price, Curr. Protein Pept. Sci., 2000, 1, 349–384 CrossRef CAS.
- W. C. Johnson, Proteins: Struct., Funct., Genet., 1990, 7, 205–214 CrossRef CAS PubMed.
- D. H. A. Corrêa and C. H. I. Ramos, Afr. J. Biochem. Res., 2009, 3, 164–173 Search PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- S. L. Thrash, J. C. Otto Jr. and T. L. Deits, Protein Expression Purif., 1991, 2, 83–89 CrossRef CAS.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 4695–4700 CrossRef CAS.
- P. Govindaswamy, D. Linder, J. Lacour, G. Süss-Fink and B. Therrien, Chem. Commun., 2006, 4691–4693 RSC.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
- K. Mikkelsen and S. O. Nielsen, J. Phys. Chem., 1960, 64, 632–637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob02194k |
| This journal is © The Royal Society of Chemistry 2015 |