
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and physico-chemical properties of the first water soluble Cu(II)@hemicryptophane complex†
Aline
Schmitt
a,
Solène
Collin
a,
Christophe
Bucher
a,
Vincent
Maurel
b,
Jean-Pierre
Dutasta
*a and
Alexandre
Martinez
*ac
aLaboratoire de Chimie, École Normale Supérieure de Lyon, CNRS, UCBL, 46, Allée d'Italie, F-69364 Lyon, France. E-mail: Alexandre.martinez@centrale-marseille.fr
bLaboratoire de Résonances Magnétiques, CEA-Grenoble/INAC/SCIB/LRM, UMR-E 3 CEA-UJF, 17, rue des Martyrs 38054, Grenoble Cedex 09, France
cAix Marseille Universite, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France
First published on 10th December 2014
Abstract
A hemicryptophane ligand soluble in water at neutral pH was obtained thanks to the derivatization of the cyclotribenzylene unit with three carboxylate groups. The corresponding Cu(II) complex was then synthesized and its spectroscopic and electrochemical properties in water were investigated, showing that water solubilisation retains the geometry of the complex around the metal center but strongly affects its redox properties, compared to previously reported Cu(II)@hemicryptophane complexes soluble in organic solvents.
Introduction
Hemicryptophanes1 have revealed to be attractive molecular receptors2 and they have been used for the design of supramolecular catalysts3 and molecular propellers or gyroscopic systems.4 In particular, the introduction of a vanadium atom in the cavity of hemicryptophanes has for instance been found to yield oxidovanadium-hosts acting as haloperoxidase enzyme mimics catalyzing sulfoxidation reactions.3aAs a general statement, the versatility and modularity of these host compounds have proven particularly suitable to mimic enzymatic systems by combining a lipophilic cavity and a catalytic site.5–9 By this way, original nanoreactors can be synthesized and give rise to artificial metallo-enzymes. Such systems have been already studied in organic solvents.10 Obviously, most biocatalytic processes in nature occur in aqueous media and to obtain a better understanding and control of such biological processes, any synthetic systems should be able to work in water.11 Water solubilisation of biomimetic entities is highly challenging,12 and very few water-soluble metalloenzyme models have been reported in the literature.13
Recently, we have reported several supramolecular systems, based on hemicryptophane ligands, combining a biomimetic copper(II) coordination core and a lipophilic cavity.14 Cu(II) complexes have been found to be efficient catalysts in organic solution for the conversion of cyclohexane to cyclohexanol/cyclohexanone, using H2O2 as the oxygen source. Both the stability and the selectivity of the catalyst have been improved by encapsulating the active site.3d We have also described the first synthesis of a water-soluble hemicryptophane host and its recognition properties toward choline in a basic aqueous medium.15
By combining both water solubility and Cu(II) complexation in a unique cage molecule we expected a water-soluble metallo-enzyme model, presenting a copper site encaged in a closed-shell cavity. Herein, we report the synthesis of the first water-soluble copper@hemicryptophane complex together with its spectroscopic characterization and redox properties.
Results and discussion
Synthesis of the water-soluble complex
Hemicryptophane 1 was synthesized in 6 steps, following a previously published procedure (Scheme S-1†).15 The strongly basic conditions (pH = 12) which are required to solubilize 1 in water are not compatible with its use in biological environments. The substitution of the phenol groups in 1 by carboxylate moieties was performed in order to obtain the hemicryptophane 3, which was found to be soluble in water at physiological pH (pH ≈ 7). The strategy used to synthesize compound 3 is depicted in Scheme 1. The first step involves the reaction of compound 1 with tert-butyl-bromoacetate in DMF in the presence of Cs2CO3 to yield the protected tri-ester derivative 2. Deprotection of both the amine and the ester functions in 2, using trifluoroacetic acid, afforded the water-soluble hemicryptophane 3. Following this synthetic pathway, host 3 has been obtained from commercially available products in 8 steps with an overall yield of 6%. | ||
| Scheme 1 Synthesis of the water soluble ligand 3. | ||
The 1H NMR spectrum of 3 in D2O (pD ≈ 7) indicates that this molecule is on average of C3 symmetry. It displays the expected signals for the cyclotribenzylene unit (Fig. 1): two singlets for the aromatic protons, and the characteristic AB system for the ArCH2 bridges. Two doublets for the aromatic protons of the linkers and the multiplets for the OCH2 groups are also observed in the spectra. The NCH2 protons of the tren unit appear as a complex pattern between 2.0 and 2.25 ppm.
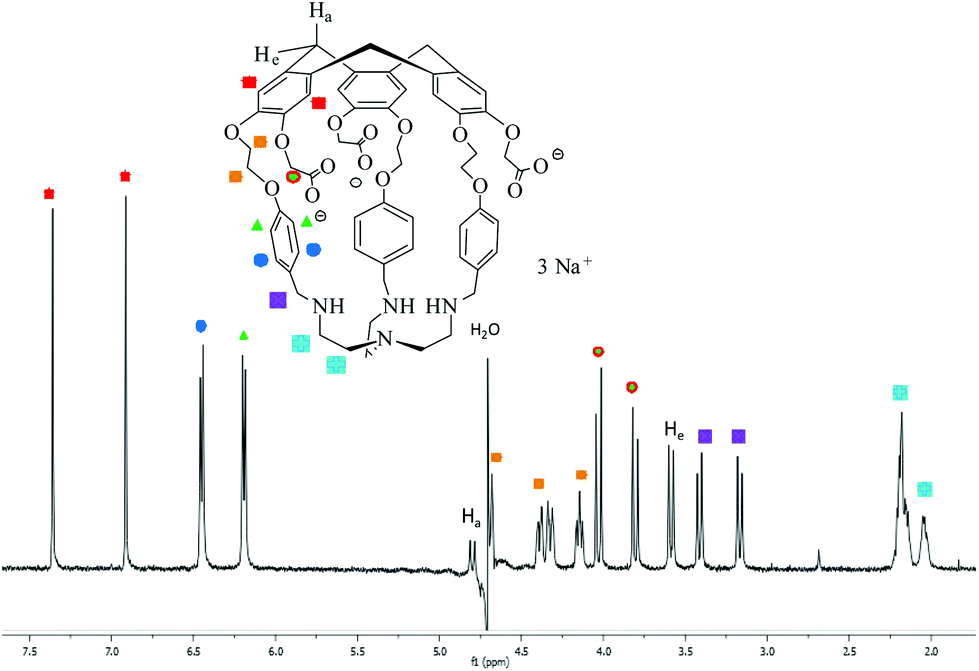 | ||
| Fig. 1 1H NMR spectrum (500.1 MHz, D2O, 298 K, pD ≈ 7) of hemicryptophane 3. | ||
The hemicryptophane copper(II) complex was then synthesized by the reaction of 3 with a stoichiometric amount of copper(II) perchlorate in water to produce Cu(II)@3 isolated in a 50% yield as a pale green solid (Scheme 2).
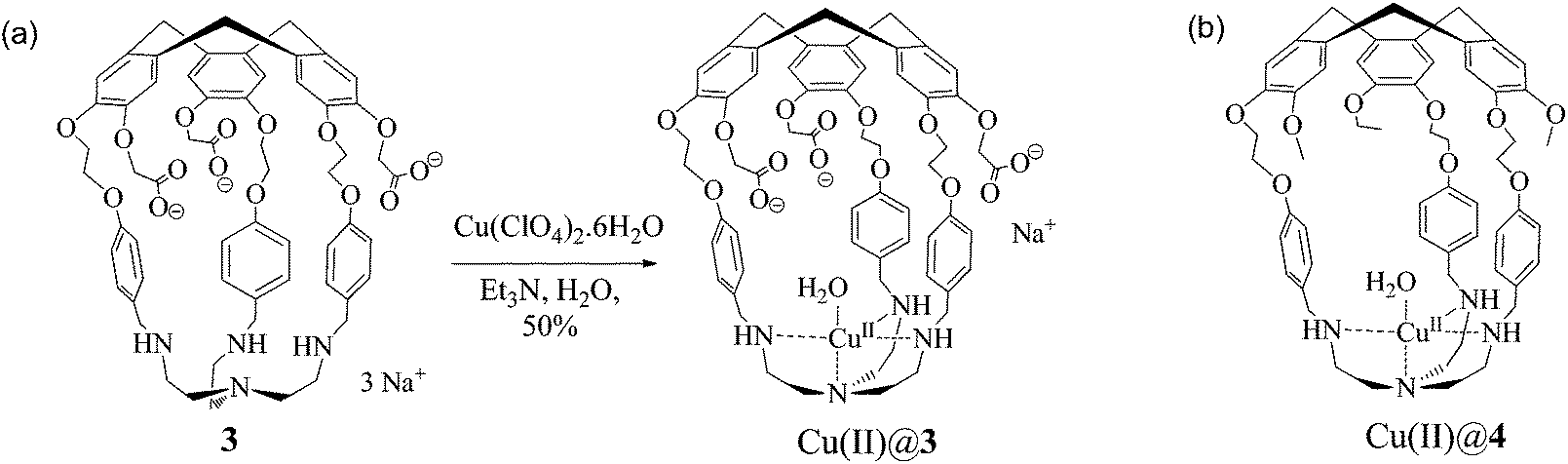 | ||
| Scheme 2 (a) Synthesis of the water soluble Cu(II)@hemicryptophane 3 complex. (b) Structure of its non-water soluble parent Cu(II)@4. | ||
Electronic spectroscopy
The near-IR/vis absorption spectrum of the hemicryptophane complex Cu(II)@3 in CH2Cl2 displays a broad asymmetrical band centred at 850 nm which is a diagnostic signature supporting the trigonal-bipyramidal geometry of the complexed copper(II) ions (Fig. 2). These observations are fully consistent with those previously observed with the parent Cu(II)@4 complex soluble in organic solvents (Scheme 2(b)).14,16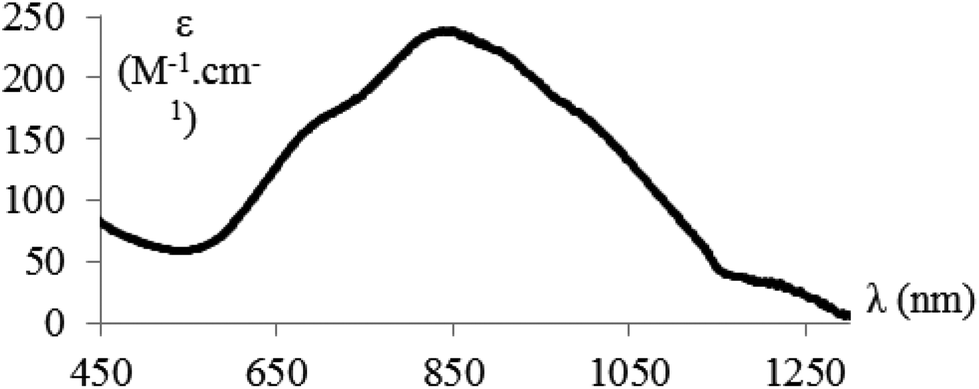 | ||
| Fig. 2 Near-IR/vis spectrum of Cu(II)@3 (H2O–NaOH, pH ≈ 8, 298 K). | ||
EPR measurements
The EPR spectrum of the Cu(II)@3 complex was recorded in frozen water at 70 K (Fig. 3). A pseudo-axial EPR signal (g1 = 2.015, g2 = 2.135 and g3 = 2.220) with a resolved hyperfine structure (A1 = 193 MHz, A2 = 158 MHz and A3 = 363 MHz) was obtained, giving a R value (g2–g1)/(g3–g2) equal to 1.41.17 These values (g1 < 2.04 and R > 1) are typical for a N4X coordination sphere with a distorted geometry close to trigonal-bipyramidal geometry.18 The g-values are very similar to that previously obtained with the Cu(II)@4 complex (g1 = 2.010, g2 = 2.136, g3 = 2.220; R = 1.50),14 indicating that water solubilisation did not induce strong distortion of the initial geometry. Complexes Cu(II)@3 and Cu(II)@4 present a five-coordination sphere with the copper ion bound to the four nitrogen atoms of the tren unit and to one solvent molecule. | ||
| Fig. 3 EPR spectra of Cu(II)@3 in frozen water solution (H2O–NaOH, pH ≈ 8, T = 70 K) (black line: experimental spectra; red line: simulated spectra). Experimental conditions: ν = 9.655 GHz, power = 2 mW and modulation amplitude = 2.0 G. Best fit parameters: g1 = 2.015, g2 = 2.135 and g3 = 2.220, A1 = 193 MHz, A2 = 158 MHz and A3 = 363 MHz for a lwpp = 70 G Gaussian lineshape. | ||
Electrochemical studies
The CV curve recorded for Cu(II)@3 in an electrolytic aqueous medium exhibits a fully irreversible reduction wave Epc = −0.59 V attributed to the formation of Cu(I)@3 at the electrode interface (Fig. 4). This irreversible feature, observed at all the investigated scan rates, i.e. from 0.02 to 10 V s−1, is due to the existence of an isomerisation process triggered by the electrochemical reduction of the Cu(II) center. The associated EC process19 can indeed be modelled as a classic square scheme involving changes in the coordination sphere around the Cu(I) center in the pentacoordinated Cu(I)@3 species to produce a more stable tetrahedral Cu(I) complex noted Cu(I)@3′.20 These hypotheses are supported by the observation of an irreversible oxidation wave at Epa = 0.14 V on the reverse scan, the large amplitude of the potential shift seen between Epc and Epa being in agreement with a net decrease of the number of coordinated atoms on the copper center, from 5 in Cu(II)@3 to 4 in Cu(I)@3′. It should be mentioned that a similar EC process was postulated for Cu(II)@4 in dichloromethane; the reduction wave and the associated re-oxidation being observed under these conditions at Epc = −0.91 V/ECS and Epa = 0.45 V/ECS, respectively.14 | ||
| Fig. 4 Cyclic voltammogram of Cu(II)@3 recorded under argon in water (NaNO3 (0.1 M), NaOH (1.5 mM)) (carb., ∅ = 3 mm; ν = 0.05 V s−1). | ||
These differences observed between organic and aqueous media highlight the key role of water which is most probably involved both in the solvation and coordination shells of the cupric and cuprous complexes.
Conclusion
In conclusion, the synthesis of the first water soluble copper@hemicryptophane complex has been described. This represents a rare example of water-soluble cage compound trapping a copper ion in the confined space of its cavity. The physico-chemical studies indicated that the solubilisation in water affects only weakly the geometry of the complex, but strongly modifies its redox behavior. This novel water-soluble metallo-enzyme model featuring a copper(II) site encaged in a closed-shell cavity opens up the way to new bio-inspired catalytic systems.Experimental section
General methods
All reactions were carried out under argon by means of an inert gas/vacuum double manifold and standard Schlenk techniques. Dichloromethane was first dried over molecular sieves and then passed through an activated alumina column followed by an argon flush on a solvent station. 1H and 13C NMR spectra were recorded at 500.10 MHz and 125.76 MHz, respectively. Chemical shifts are reported relative to the residual protonated solvent signal (CDCl3), or relative to residual ethanol for the spectra in D2O. Mass spectra were recorded by the Centre de Spectrométrie de Masse, Institute of Chemistry, Lyon. Compound 1 was prepared according to the published procedure.15 EPR spectra were obtained from a Bruker EMX spectrometer and fitted with Easyspin21 software.Cyclic voltammetry (CV) data were recorded using an ESP-300 Biologic potentiostat equipped with a 1 A/48 V booster and an analog linear scan generator. All the experiments were conducted under an argon atmosphere in a standard one-compartment, three-electrode electrochemical cell placed in a faraday cage. An automatic ohmic drop compensation procedure was systematically implemented prior to recording CV data. All the electrodes were purchased from ALS Co. Ltd. Vitreous carbon (Φ = 3 mm) working electrodes were polished with 1 mm diamond paste before each recording. A saturated ECS electrode was used as a reference. HPLC grade water + sodium nitrate (0.1 M) was used as the electrolyte.
Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O), 157.68 (CArO), 145.85 (CArO), 145.47 (CArO), 133.41 (CAr), 132.79 (CAr), 130.00 (CArH), 129.27 (CAr), 116.38 (CArH), 115.82 (CArH), 114.19 (CArH), 69.12 (O(CH2)2O), 66.71 (OCH2C(O)O), 66.32 (O(CH2)2O), 52.92 (N(CH2)2N), 50.51 (ArCH2N), 44.79 (N(CH2)2N), 35.26 (ArCH2Ar). Mp: 219.3 °C. IR ν = 3400, 2923, 2856, 1681, 1605, 1509, 1425, 1335, 1259, 1204, 1176, 1127 cm−1.
O)O), 157.68 (CArO), 145.85 (CArO), 145.47 (CArO), 133.41 (CAr), 132.79 (CAr), 130.00 (CArH), 129.27 (CAr), 116.38 (CArH), 115.82 (CArH), 114.19 (CArH), 69.12 (O(CH2)2O), 66.71 (OCH2C(O)O), 66.32 (O(CH2)2O), 52.92 (N(CH2)2N), 50.51 (ArCH2N), 44.79 (N(CH2)2N), 35.26 (ArCH2Ar). Mp: 219.3 °C. IR ν = 3400, 2923, 2856, 1681, 1605, 1509, 1425, 1335, 1259, 1204, 1176, 1127 cm−1.
Notes and references
- J. Canceill, A. Collet, J. Gabard, F. Kotzyba-Hibert and J.-M. Lehn, Helv. Chim. Acta, 1982, 65, 1894–1897 CrossRef CAS
.
-
(a) S. Le Gac and I. Jabin, Chem. – Eur. J., 2008, 14, 548–557 CrossRef CAS PubMed
-
(a) A. Martinez and J.-P. Dutasta, J. Catal., 2009, 267, 188–192 CrossRef CAS PubMed
-
(a) A. Martinez, V. Robert, H. Gornitzka and J.-P. Dutasta, Chem. – Eur. J., 2010, 16, 520–527 CrossRef CAS PubMed
- M. Raynal, P. Ballester, A. Visal-Ferran and P. W. N. M. Van Leeuwen, Chem. Rev., 2014, 43, 1660–1733 RSC
- M. Raynal, P. Ballester, A. Visal-Ferran and P. W. N. M. Van Leeuwen, Chem. Rev., 2014, 43, 1734–1787 RSC
- A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707–724 CrossRef CAS
- J. K. M. Sanders, Chem. – Eur. J., 1998, 4, 1378–1383 CrossRef CAS
- J.-M. Lehn, Rep. Prog. Phys., 2004, 67, 245–249 CrossRef
-
J. W. Steed and J. L. Atwood, in Supramolecular Chemistry, Wiley-VCH, Weinheim, 2nd edn, 2009 Search PubMed
- G. V. Oshovky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef PubMed
- E. Klein, Y. Ferrand, N. P. Barwell and A. P. Davis, Angew. Chem., Int. Ed., 2008, 47, 2693–2696 CrossRef CAS PubMed
-
(a) R. Breslow, Acc. Chem. Res., 1995, 28, 146 CrossRef CAS
- O. Perraud, J.-B. Tommassino, V. Robert, L. Khrouz, B. Albela, L. Bonneviot, J.-P. Dutasta and A. Martinez, Dalton Trans., 2013, 42, 1530–1535 RSC
- A. Schmitt, V. Robert, J.-P. Dutasta and A. Martinez, Org. Lett., 2014, 16, 2374–2377 CrossRef CAS PubMed
-
(a) M. Duggan, N. Ray, B. Hathaway, G. Tomlinson, P. Brint and K. J. Pelin, J. Chem. Soc., Dalton Trans., 1980, 8, 1342–1348 RSC
- D. E. Billing, R. J. Dudley, B. J. Hathaway and A. Tomlinso, J. Chem. Soc. A, 1971, 691–696 RSC
- R. J. Dudley, B. J. Hathaway, P. G. Hodgson, P. C. Power and D. J. Loose, J. Chem. Soc., Dalton Trans., 1974, 1005–1009 RSC
-
(a) C. Bucher, E. Duval, J.-M. Barbe, J.-N. Verpeaux, C. Amatore and R. Guilard, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 211–222 CrossRef CAS
- The driving force of most reorganization reactions reported so far in the literature for electrogenerated Cu(I) complexes is the stabilization of the Cu(I) ion in a tetrahedral environment: see for instance ref. 18 or J.-P. Sauvage, J.-P. Collin, S. Durot, J. Frey, V. Heitz, A. Sour and C. Tock, C. R. Chim., 2010, 13, 315–328 CrossRef CAS PubMed
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1, 1H and 13C NMR spectra of compounds 2 and 3. See DOI: 10.1039/c4ob02085e |
| This journal is © The Royal Society of Chemistry 2015 |