Cucurbituril chemistry: a tale of supramolecular success
Eric
Masson
*,
Xiaoxi
Ling
,
Roymon
Joseph
,
Lawrence
Kyeremeh-Mensah
and
Xiaoyong
Lu
Department of Chemistry and Biochemistry, Ohio University, Athens Ohio, 45701, USA. E-mail: masson@ohio.edu; Fax: +1-740-593-0148; Tel: +1-740-593-9992
First published on 9th December 2011
Abstract
This review highlights the past six year advances in the blossoming field of cucurbit[n]uril chemistry. Because of their exceptional recognition properties in aqueous medium, these pumpkin-shaped macrocycles have been generating some tremendous interest in the supramolecular community. They have also become key units in various self-organizing and stimulus-controlled assemblies, as well as in advanced materials and drug carriers. The scope of this review is limited to the main family of cucurbit[n]urils (n = 5, 6, 7, 8, 10). The reader will find an overview of their preparation, their physicochemical and biological properties, as well as their recognition abilities towards various organic and inorganic guests. Detailed thermodynamic and kinetic considerations, as well as multiple applications including supramolecular catalysis are also discussed.
 Eric Masson | Eric Masson was born in Lausanne, Switzerland. In 2001, he obtained a Master’s degree in chemistry from the University of Lausanne, and in 2005, a Ph. D. in organic chemistry from the Swiss Federal Institute of Technology Lausanne (EPFL) under the guidance of Prof. Manfred Schlosser. He then spent two years as a post-doctoral associate at Yale University under the supervision of Prof. Andrew D. Hamilton, and joined Ohio University as an Assistant Professor in 2007. His research interests revolve around supramolecular and recognition chemistry, with a particular affinity for the cucurbituril family of macrocycles. |
 Xiaoxi Ling | Xiaoxi Ling was born in Shanghai, China. He obtained a Bachelor of Engineering degree in Applied Chemistry from East China University of Science and Technology in 2007. In September of the same year, he joined Ohio University, and became the first graduate student pursuing a Ph. D. degree under the guidance of Prof. Eric Masson. His current research is focused on the kinetics and thermodynamics of cucurbituril recognition, as well as on the preparation and applications of cucurbituril-containing molecular switches and advanced materials. |
 Roymon Joseph | Roymon Joseph was born in Kerala, India, and received his Bachelor’s and Master’s degrees from St. Berchmans College Changanacherry affiliated to Mahatma Gandhi University, Kerala. He obtained his Ph. D. degree from the Indian Institute of Technology Bombay in 2010 under the supervision of Prof. C. P. Rao, and continued to work there as a research associate for another year. He is currently a postdoctoral researcher with Prof. Eric Masson at Ohio University. His research interests include supramolecular chemistry, ion and molecular recognition, as well as bioorganic and bioinorganic chemistry. |
 Lawrence Kyeremeh-Mensah | Lawrence Kyeremeh-Mensah was born in Dormaa-Koraso, in the Brong Ahafo region of Ghana. He received a Bachelor’s degree in 2005, followed by a Master’s degree in chemistry in 2009 from the University of Ghana, Legon, Accra. He is currently pursuing his Ph. D. under the guidance of Prof. Eric Masson at Ohio University. His research interests focus on the catalytic properties of cucurbiturils on benchmark organic and organometallic reactions. |
 Xiaoyong Lu | Xiaoyong Lu was born in Shanxi, China, studied chemistry in Xinzhou Teachers University from 1999 to 2002, and completed his undergraduate studies after a final evaluation from Shanxi University in 2003. He received a Master’s degree in organic chemistry from Shanghai University in 2007, and worked in the pharmaceutical company Shanghai Chempartner before joining Ohio University as a graduate student. Xiaoyong has been an active member of the Masson group since 2008. His research interests include the recognition and catalytic properties of cucurbiturils, and the synthesis and evaluation of cucurbituril-based rotaxanes and nanomaterials. |
1. Introduction
In 1905, Behrend and coworkers characterized the condensation products of glycoluril (1) and formaldehyde under strongly acidic conditions as “white, amorphous compounds, which are weakly soluble in dilute acid and base, and absorb large quantities of water without losing their dusty powdery character”.1 One of those products was found to contain “at least three molecules of glycoluril”, condensed with twice as many formaldehyde units, thereby corresponding to the formula C18H18N12O6.1 More than a century later, this characterization of what was likely a mixture of cucurbit[n]urils (CB[n]), is still remarkably valid. In 1981, Mock and coworkers revisited Behrend's experiments and, upon complexation with calcium sulfate, successfully crystallized a hydrated macrocycle bearing six glycoluril units linked by twelve methylene bridges, and interacting with the calcium cations via its two carbonylated rims. The authors named the structure “cucurbituril” for its resemblance to “a gourd or [a] pumpkin” (which belong to the Cucurbitaceae family), and to a cucurbit, a vessel connected to an alembic used by alchemists for distillations;2 it is now known as curcurbit[6]uril (commonly abbreviated CB[6], or in some cases CB6, Q[6], Q6 or Cuc6, ‘6’ representing the number of glycoluril units in the macrocycle). In the same study, CB[6] was already found to encapsulate alkylammonium cations.2 Although other CB[n] analogs must have been formed together with CB[6] under the conditions reported by Mock,3 one had to wait until 2000 for the isolation and X-ray characterization of three new members of the CB[n] family by Kim and coworkers (CB[5], CB[7] and CB[8]; see Fig. 1 for the structure of CB[7]).4 Less than two years later, Day et al. eventually identified3 and crystallized5 the interlocked complex CB[5] ⊂ CB[10] (as much as 65 g isolated from 1 kg glycoluril!).3![Preparation of CB[n]s from glycoluril (1) and formaldehyde under acidic conditions. Structure of CB[7] from X-ray diffraction (carbon atoms in grey, hydrogens in white, nitrogens in blue and oxygens in red).](/image/article/2012/RA/c1ra00768h/c1ra00768h-f1.gif) | ||
| Fig. 1 Preparation of CB[n]s from glycoluril (1) and formaldehyde under acidic conditions. Structure of CB[7] from X-ray diffraction (carbon atoms in grey, hydrogens in white, nitrogens in blue and oxygens in red). | ||
Thanks in part to these exciting developments, CB[n] chemistry has been blossoming at a remarkable rate since the beginning of our millennium, with a growth rate that does not pale in comparison to resorcinarenes and calixarenes, approximately seven and twelve years earlier: since 1997, the number of articles, reviews and patents related to CB[n]s has grown from less than 10 per year to 124 in 2010 (an average yearly growth rate of 10 documents, vs. 28 and 5.8 in the case of calixarenes and resorcinarenes, respectively; see Fig. 2). These numerical data support Kim's wish expressed during the last evening of the 1st International Conference on Cucurbiturils (July 10–11, 2009) held at POSTECH in Pohang, Korea, that CB[n]s would be to the next decade what calixarenes have been to the previous one.
![Histograms representing the number of reviews (blue), patents (green) and articles (red) published each year, in the case of (a) calixarenes, (b) resorcinarenes and (c) CB[n]s. (d) Total number of published documents y vs. time t [year] for calixarenes (black), resorcinarenes (blue) and CB[n]s (red); the yearly growth rate k is determined by fitting the data with the discontinuous equation y = k(t–t0) when t > t0, and y = 0 when t ≤ t0.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f2.gif) | ||
| Fig. 2 Histograms representing the number of reviews (blue), patents (green) and articles (red) published each year, in the case of (a) calixarenes, (b) resorcinarenes and (c) CB[n]s. (d) Total number of published documents y vs. time t [year] for calixarenes (black), resorcinarenes (blue) and CB[n]s (red); the yearly growth rate k is determined by fitting the data with the discontinuous equation y = k(t–t0) when t > t0, and y = 0 when t ≤ t0. | ||
2. Scope and limitations of this review
Several reviews describing the amazing recognition properties and applications of CB[n]s have been published in the past few years.6–15 Yet the functionalization of CB[6]13,16–18 and the remarkable synthesis of new CB[n] analogues (inverted iCB[n]s,19,20nor-seco-CB[n]s21–23 and various cyclic24–27 and acyclic28–30 congeners) have inevitably shrunk the review space solely dedicated to the theoretical studies and applications of the main family of CB[n]s (n = 5, 6, 7, 8 and 10). Therefore, the aim of the present review is to provide the reader with an overview of those studies carried out during the past six years, approximately since the publication of Isaacs' landmark article “The Cucurbit[n]uril Family”.12 For more in-depth information about selected aspects of CB[n] chemistry, we also recommend the very recent series of articles published in the Israel Journal of Chemistry on the occasion of the 2nd International Conference on Cucurbiturils held at the University of Cambridge, UK (June 29–July 2, 2011).313. Synthesis and characterization of CB[n]s
3.1 Preparation
Various procedures have been proposed for the preparation of mixtures of CB[n]s, all based on general protocols developed by Day,3 Kim4 and Isaacs.14 Generally, a mixture of glycoluril, aqueous formaldehyde or paraformaldehyde, and hydrochloric or sulfuric acid (concentrated, or diluted to approximately 5 M) is heated to 80–100 °C during 10–100 h. Evaporation and consecutive precipitations in water and methanol afford a mixture of CB[n]s (n = 5–8, CB[6] being the major component of the mixture), as well as traces of CB[5] ⊂ CB[10], iCB[6] and other oligomers. Separation of each component is based on their differential solubility in water, water/methanol and diluted hydrochloric acid solutions, according to Fig. 3a.14 A useful variation was proposed by Day,3 and repeated by Halterman32 and Leventis,33 in which the authors use a hot 20% aqueous solution of glycerol to extract CB[7] from the mixture of CB[n]s with good selectivity. Recently, Scherman reported an alternate environmentally friendly separation of CB[5] and CB[7] (see Fig. 3b): CB[7] could be precipitated selectively upon complexation with 1-alkyl-3-methylimidazolium bromides (Im+Br− in Fig. 3b) and anion exchange with ammonium hexafluorophosphate (NH4+PF6−), and CB[5] was recrystallized from the aqueous phase. The imidazolium/CB[7] complex was subjected to Br−/PF6− anion exchange, and CB[7] was released from the Im+Br−/CB[7] complex upon reverse anion exchange with NH4+PF6− in dichloromethane under heterogeneous conditions.34![Purification of CB[n]s: (a) General procedure;14 (b) alternate method for the separation of CB[5] and CB[7].34 Curved arrows indicate precipitation.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f3.gif) | ||
| Fig. 3 Purification of CB[n]s: (a) General procedure;14 (b) alternate method for the separation of CB[5] and CB[7].34 Curved arrows indicate precipitation. | ||
Free CB[10] was obtained in 2005 by Isaacs upon treatment of CB[5] ⊂ CB[10] with an excess amount of melamine derivative 2a (yielding the ternary complex CB[10]·2a2), followed by the ejection of the first guest with methanol, and of the second one after reaction with acetic anhydride.35 The crystal structure of free CB[10] was reported in 2009 by the same group.15 A more recent procedure indicates that CB[5] can be ejected from CB[10] using commercially available 1,12-dodecanediamine (2b) at low pH; free CB[10] is obtained upon repetitive washing with a hot ethanolic solution of sodium hydroxide, and subsequent recrystallization in concentrated hydrochloric acid.36
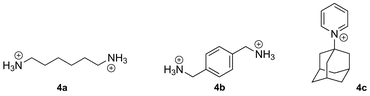
In addition to elementary analysis, the purity of the macrocycles can be assessed by 1H nuclear magnetic resonance spectroscopy (NMR), since the chemical shifts of the methylene hydrogens differ along the CB[n] series.4,14 CB[n]s are hygroscopic, and may still interact with some water molecules even after several drying cycles; they may also be contaminated with hydrochloric acid and various cations and anions. Therefore, we recommend that the molecular weight of the isolated CB[n] (e.g., the molecular weight of a CB[n]·xH2O·yHCl complex) be determined by titration with a high affinity guest. For example, the Kaifer group uses UV-Vis titration with a solution of cobaltocenium hexafluorophosphate (3; 15 μM) and varying concentrations of CB[7] and CB[8],37 while the Masson group usually opts for 1H NMR titration with solutions containing a known concentration of 1,6-hexanediammonium (4a), p-xylylene diammonium (4b) or 1-adamantylpyridinium (4c) cations, in the case of CB[6], CB[7] and CB[8], respectively. The concentration of CB[n]-bound species can be obtained from the signals of free and bound guests, or by comparison and calibration with an inert standard present at a known concentration (N,N-dimethylformamide, dimethyl sulfoxide, sodium methanesulfonate, etc.). One should finally note that CB[n]s can be detected at concentrations as low as 10 ppb using surface enhanced Raman scattering (SERS) on Klarite™ nanostructured surfaces.38
3.2 Physical properties
CB[n]s bear two hydrophilic carbonylated rims and a hydrophobic cavity. Their total depth is 9.1 Å if one includes the van der Waals radii of the oxygen atoms, and the depth of the cavity is 7.4–7.8 Å if one considers the separation between the two planes of local electrostatic potential minima at both portals.15,39–41 The width of the CB[5]–CB[8] cavities vary between 4.4 and 8.8 Å, and the ellipsoidal CB[5] ⊂ CB[10] complex has transverse and conjugate diameters of 10.7 and 12.6 Å, respectively;8,41 free CB[10] is also ellipsoidal, with transverse and conjugate diameters of 11.3 and 12.4 Å.15 The diameter of the CB[n] portal is approximately 2 Å narrower than the cavity of the macrocycle (see Table 1). Depending on their size, the inner cavity of CB[n]s can host between 2 and 22 “high-energy” water molecules (“high-energy” relative to their stability in the aqueous environment), as calculated using a 55% packing coefficient42 (i.e. a 55% ratio between the volume of the combined water guests and the volume of the CB[n] cavity; values are in excellent agreement with those extracted from X-ray crystal structures).43 The water content of CB[5] and CB[8] parallels α- and γ-cyclodextrins (CD), respectively, while β-CD accommodates 6–7 water molecules, vs. 4 and 8 in the case of CB[6] and CB[7].43 It is of course much more difficult to evaluate the number of water units at the carbonylated portals of CB[n]s, because of the complex network of possible dipole–dipole interactions in this region. As the reader will appreciate in the next sections, ejection of water from CB[n]s plays a critical role in the recognition properties of these macrocycles.| a | b | c | S | ng | K a | |
|---|---|---|---|---|---|---|
a Portal diameter [Å].8,12,15
b Cavity diameter calculated from the X-ray crystal structures,8,12,15 and in parentheses, diameter corresponding to the distance between electrostatic potential minima [Å].41
c Cavity depth determined from electrostatic potential minima [Å];41 the total CB[n] depth, which includes the van der Waals radii of oxygen atoms, is 9.1 Å in all cases.8,12,15
d Solubility in water [mM],8,12,35 and in parentheses, in hydrochloric acid (3 M).12
e In a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and formic acid, instead of hydrochloric acid.
f From ref. 35.
g Number of water molecules in the CB[n] inner cavity, calculated using a 55% packing coefficient.43
h Highest reported binding affinity towards organic guests [M−1].
i in LiCl 0.20 M.56
j in pure water.57
k in acetate buffer (pD 4.74, 50 mM).58 :1 mixture of water and formic acid, instead of hydrochloric acid.
f From ref. 35.
g Number of water molecules in the CB[n] inner cavity, calculated using a 55% packing coefficient.43
h Highest reported binding affinity towards organic guests [M−1].
i in LiCl 0.20 M.56
j in pure water.57
k in acetate buffer (pD 4.74, 50 mM).58
|
||||||
| CB[5] | 2.4 | 4.4 (3.9) | 7.4 | 20–30 (60) | 2 | |
| CB[6] | 3.9 | 5.8 (5.5) | 7.5 | 0.018 (60)e | 4 | 5.4 × 1010i |
| CB[7] | 5.4 | 7.3 (7.1) | 7.6 | 20–30 (700) | 8 | 5.0 × 1015j |
| CB[8] | 6.9 | 8.8 (8.6) | 7.7 | < 0.01 (1.5) | 12 | 4.3 × 1011k |
| CB[10] | 9.5–10.6 | 11.3–12.4 | 7.8 | < 0.05f | 22 | |
The cavity of CB[n]s is a remarkably unpolar and unpolarizable environment: based on the bathochromic shift observed when rhodamine 6G is encapsulated within CB[7], Nau determined that the dielectric constant in the cavity is equal or lower than 10.44 CB[7] was also found to have an extremely low polarizability, even lower than perfluorohexane!45,46 This is not totally surprising if one remembers that no bonds or lone pairs are present inside the cavity of the macrocycle. Using the density functional method (DFT) at the B3LYP/6-311G(d,p) level, Nau also reported extremely high negative quadrupole moments for CB[n]s.43
CB[5], CB[6] and CB[8] form stable crystals with well-organized 1D channels. Two different polymorphs of CB[6] could be obtained, both revealing a honeycomb-like structure (the orientation of CB[6] differs in the two cases)47,48 with large channels filled with water. CB[8] units can be located at the center and vertex of a perfectly square parallelepiped,48 or adopt a distorted honeycomb structure with partially self-closed cavities.49 CB[5], among other arrangements, forms a distorted honeycomb structure with water-filled channels, which transforms to a more stable layered phase upon heating.50 This honeycomb organization, which maximizes interactions between the outer methylene and methine hydrogens and the carbonylated portal of the neighboring macrocycle, is a general characteristic of CB[n]s in the solid state. Although such CH/O interactions are very weak, their sheer number is responsible for the remarkable thermal stability of CB[n] crystals.49 Nau also mentions that the orientation of the CB[n] units maximizes quadrupolar interactions between the macrocycles.43 In the case of CB[5], CB/water interactions significantly compete against weak CB/CB interactions, while CB[7]/CB[7] interactions are strong, but outnumbered by the CB[7]/water interactions. To the contrary, CB[6] crystals are stabilized by strong CB/CB interactions, with limited CB[6]/water competition.49 CB[8]/CB[8] contacts are weaker than CB[6]/CB[6] ones, but CB[8]/water interactions are also limited. Both CB[6] and CB[8] remain crystalline upon drying, while CB[5] and CB[7] become amorphous. These structural considerations likely explain why CB[5] and CB[7] are much more water-soluble than CB[6] and CB[8] (20–30 mM vs. less than 0.01 mM in pure water, see Table 1).8 Fortunately, the solubility dramatically improves as the ionic strength of the medium is increased, or upon encapsulation with amphiphilic guests (see Table 1).
CB[n]s are virtually insoluble in all organic solvents, with a few exceptions. Kaifer is the first investigator to have reported a CB[7]-based rotaxane that is soluble in acetonitrile and dimethyl sulfoxide; the central station was p-xylylene dipyridinium, its counteranion hexafluorophosphate, and the unit was decorated with bulky hydrophobic stoppers.51 The same group published several additional examples in a recent past, with millimolar solubilities in dimethyl sulfoxide, acetonitrile and N,N-dimethylformamide; in all cases, hexafluorophosphate was the counteranion to the positively charged guests.52 Our group also recently determined that the solubility of CB[7]-bound p-xylylene diammonium (4b; trifluoromethanesulfonate as the counteranion) reaches 60 mM in dimethyl sulfoxide.53 To the best of our knowledge, there has been only one example of a CB[8] interlocked assembly partially soluble in a non-aqueous solvent (a viologen unit linked to two tris(2,2′-bipyridine)ruthenium substituents in acetonitrile).54
Finally, we note that CB[6] and CB[8] crystals grown from acidic solutions display excellent proton conductivities, with very high anisotropicities (up to 8.6 × 103-fold higher conductivities were measured along the channels, than perpendicular to those).55
3.3 Biological properties
CB[n]s are remarkably inert in vitro and in vivo: Nau and Day determined that the IC50 value of CB[7] towards Chinese hamster ovary CHO-K1 cells after 48 h is 0.53 mM.59 At shorter incubation times (3 h), concentrations as high as 1.0 mM were found viable, and no cytotoxic effects were detected at concentrations of 0.50 mM or less. CB[7] is also inert at 1.0 mM towards human kidney HEK293, human hepatocyte HepG2 and murine macrophage RAW264.7 cells after 48 h of incubation.60 A similar result was obtained with CB[5]. At concentrations greater than 0.10 mM, CB[7] is equally inert towards human A549 non-small lung cells, SKOV-3 ovarian cells, SKMEL-2 melanoma, XF-498 brain cells and HCT-15 human colon cells.61 Intravenous injections into mice of a single dose of a CB[7] solution at 250 mg kg−1 showed little sign of toxicity, with a body weight loss of 5% 4 days after injection and subsequent recovery.59 Single oral doses of 600 mg kg−1 of a 1:1 mixture of CB[7] and CB[8] led to no adverse effect; the even lower toxicity of CB[7] via the oral route is probably due to the low absorption of CB[7] across the gastrointestinal system.59 Monitoring the toxicity of CB[8] is more problematic due to its very low solubility in aqueous medium, yet at 20 μM, no sign of in vitro toxicity was observed. Unfortunately, we haven't found any toxicological information related to CB[6].
3.4 Growth mechanism
The mechanism of CB[n] formation, a step growth cyclo-oligomerization, has been studied in detail by Isaacs and coworkers.20,62,63 The first step is the dimerization of glycoluril (1) in the presence of formaldehyde under acidic conditions, which can afford the pair of diastereomers 5a and 5b, the “curved” isomer 5b being more stable than the S-shaped analog 5a by at least 2 kcal mol−1.62 Isaacs then managed to isolate further intermediates of the oligomerization process (glycoluril trimer to hexamer), which all adopt the “curved” conformation. Since (1) CB[n] can only be formed with a fully curved oligomer, and (2) the probability for one or more “S-shaped” mismatches increases during oligomerization, an intramolecular “S-to-curved shape correction” isomerization must be operational. The same authors propose the fragmentation of S-isomer 5a to iminium 5c, which undergoes S-to-curved isomerization via the spiro intermediate 5d (see Fig. 4).20 The mixture of CB[n]s thus obtained results from a subtle interplay between kinetics and thermodynamics. Day3 and Isaacs62 clearly showed that, while CB[8] can undergo reorganization to smaller analogs, CB[5]–CB[7] are more stable, or behave as kinetic traps (i.e. their respective formation is irreversible under all experimental conditions tested so far): cyclization attempts with glycoluril dimers and trimers afford a high ratio of CB[6] (2 × 3 = 3 × 2 = 6), and a high yield of CB[8] is obtained with glycoluril tetramers (2 × 4 = 8); also, a high ratio of CB[5] is obtained with glycoluril pentamer, and CB[6] is the only cyclization product from the glycoluril hexamer. Yet, the formation of small amounts of size-mismatched CB[n]s (such as CB[5] when condensing glycoluril trimers, for example), indicates that oligomers can undergo concomitant fragmentation and recombination before cyclization.62 When discussing the ratios of CB[n] formed during the oligomerization process, one should also consider plausible template effects3 caused by other components of the reaction mixture, such as glycoluril oligomers, CB[n]s, cations and anions, and especially water!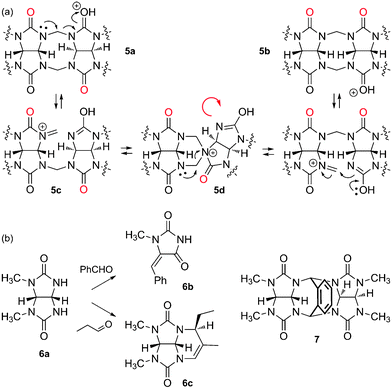 | ||
| Fig. 4 (a) “S-to-curved shape” correction mechanism, operational during the cyclooligomerization of glycoluril (1) and formaldehyde. (b) Undesired reactions between glycoluril derivatives and aldehydes. | ||
Although it would be tempting to prepare CB[n] derivatives bearing functionalized methylene bridges from glycoluril and various aldehydes in a one-pot reaction, Isaacs recently showed that failure is very likely. Indeed, methylated glycoluril 6a did not afford any dimer in the presence of various aldehydes, and small amounts of hydantoin 6b were detected instead. Performing the reaction in anhydrous trifluoroacetic acid (in an attempt to favor condensation) was equally unsuccessful, and in the case of propanal, compound 6c was isolated in a 67% yield (see Fig. 4). While condensation of glycoluril derivative 6a in the presence of phthalaldehyde was successful, the thermodynamically more stable S-shaped conformation of dimer 7 precludes any incorporation into CB[n]s.63 Yet, Isaacs and coworkers have just proposed a very elegant two-step procedure that circumvents those functionalization obstacles: the authors described the gram-scale preparation of the fully curved open glycoluril hexamer (similar to structure 5b, with six glycoluril units instead of two), and showed that it readily undergoes ring closure with o-phthalaldehydes and naphthalene-2,3-dicarbaldehyde in 9 M sulfuric acid or concentrated hydrochloric acid to afford the corresponding monofunctionalized CB[6] hosts in excellent yields (up to 83%).64
4. Recognition properties of CB[n]s
We divide this chapter into three sections, which describe the recognition properties of CB[n]s towards (1) inorganic species, such as metallic cations, their counteranions and various clusters, (2) organic guests in solution, and (3) organic guests in the gas phase.4.1 Inorganic cations, counteranions and clusters
An impressive number of crystal structures depicting interactions between CB[n]s, metallic cations, metal clusters and their corresponding counteranions, have been published during the past decade, in particular by Fedin and coworkers.7 CB[n]s bind to metallic cations via their two carbonylated portals; however, most metals interact with only a fraction of the oxygen atoms at the CB[n] rim (i.e. the metal does not usually sit at the center of the portal, with the notable exception of cesium).65–67 In the case of alkali and alkali-earth metals, several cations may occupy the same portal.9,68–71 Transition and group 13 metal ions do not usually interact directly with the oxygens of the CB[n] rim, and binding takes place between the carbonyl groups and the coordinated water molecules of the metal aqua complexes (CB[n] behaves as an outer-sphere ligand).66,72,73 In the case of lanthanides, both direct metal–portal and metal–water–portal interactions have been observed.67,74 Large metallic clusters also interact with CB[n]s via their coordinated water molecules, with the cluster often sitting right above the center of the portal (see Fig. 5 for an example).9,71,75–83 Contrary to clusters involving CB[5] and CB[6], crystal structures of CB[7] complexes are still rare, with a few uranyl/CB[7] assemblies84–86 and one rubidium/hydroquinone ⊂ CB[7] complex87 representing the only examples published so far. Finally, we want to stress that CB[7] and CB[8] can even encapsulate metallic cations and organometals such as metallocenes (from iron,88,89 cobalt,90 molybdenum91 and titanium),91 as well as several complexes of tin, nickel, cobalt, copper, iron and palladium.92–97![Clusters interacting with CB[n]s via their coordinated water molecules: structure of the CB[6]/{[Mo3(Ni(P(OH)3)S4(H2O)8Cl]3+}2 adduct (only one cluster shown; a second complex interacts with the opposite CB[6] portal). Violet lines represent metal–ligand and metal–metal bonds, and dashed black lines hydrogen bonding interactions between water and the CB[6] rim.78](/image/article/2012/RA/c1ra00768h/c1ra00768h-f5.gif) | ||
| Fig. 5 Clusters interacting with CB[n]s via their coordinated water molecules: structure of the CB[6]/{[Mo3(Ni(P(OH)3)S4(H2O)8Cl]3+}2 adduct (only one cluster shown; a second complex interacts with the opposite CB[6] portal). Violet lines represent metal–ligand and metal–metal bonds, and dashed black lines hydrogen bonding interactions between water and the CB[6] rim.78 | ||
Binding affinities of metallic cations towards CB[n]s are highly dependent on the composition of the solvent (pure water, 50% formic acid, etc.), and the stoichiometry of the interaction is unclear (Kim and Inoue suggest that alkali metal ions probably form 2:1 complexes with CB[6],56 while Buschmann and Schollmeyer indicate the formation of 1:1 complexes;98 also, metal/CB stoichiometries of up to 5:1 have been reported with some transition metals).99 If one applies a 1:1 binding model, affinities of alkali metal ions towards CB[6] range from 3.3 × 102 to 3.1 × 103 M−1 in pure water,100 and binding constants of up to 1.7 × 105 M−1 have been reported in the case of barium.101 We also note that affinities of alkali and alkaline earth metal ions towards CB[5] are much lower (7.9–70 M−1 only), and that data related to CB[7] and CB[8] are almost inexistent; in fact, our group recently had to measure the binding affinity of sodium towards CB[7] and CB[8] (7.7 × 102 and 4.2 × 102 M−1, respectively, in deuterium oxide).102
Anions and anionic clusters can occupy the void between stacks of CB[n]s,103,104 or may be encapsulated inside the cavity of the macrocycle. The anionic unit may even affect the recognition properties of the CB[n] cavity: in a recent example, a large electron-rich polyoxovanadate cluster interacting with the equatorial periphery of CB[8] was found to enable the reduction and encapsulation of two viologen radical cations.105 As mentioned before, several crystal structures show the encapsulation of anions into the cavity of CB[n]s; recent examples include chloride and nitrate inclusion within CB[5],106–110 as well as perrhenate ReO4− within CB[6] and CB[7].111,112 Yet, to the best of our knowledge, evidence of anion encapsulation in solution has been reported on only one occasion: the affinity of chloride and nitrate anions towards CB[5] was monitored by fluorescence spectroscopy (λex = 240 nm, λem = 340 nm), and a 1:1 binding model could be used to fit the interaction with the nitrate anion (binding affinity 1.7 × 102 M−1 in 4.0 M sulfuric acid).108
4.2 Organic guests
CB[n]s can encapsulate numerous organic guests, and in most cases, thermodynamic parameters can be determined by UV-Visible spectroscopy, isothermal titration calorimetry, and 1H NMR spectroscopy. In the latter case, hydrogen atoms sitting near the center of the CB[n] cavity undergo a strong upfield shift (up to 1.6 ppm),113 decentered hydrogens are affected by a more moderate upfield shift,114 and hydrogen atoms located outside the cavity undergo a significant downfield shift (up to 0.7 ppm), that weakens as the distance between the hydrogens and the portal increases.115 Although less frequently evaluated, kinetic parameters can also be measured in some instances, and can be used to assess plausible mechanisms for the formation, threading and dethreading of CB[n] complexes. Therefore, we split this section into the thermodynamic and kinetic components of the recognition mechanisms.
Binding enthalpy. We now realize that the nature of the CB[n]–guest interaction is much more subtle, especially since Kaifer and Kim have shown that the affinity between CB[7] and the neutral hydroxymethylferrocene 8a reaches 3.0 × 109 M−1!88 In a landmark article,116 Kaifer, Isaacs, Gilson, Kim and Inoue reported that the enthalpies of the interaction between ferrocene derivatives 8a–8c and CB[7] are virtually identical (−21.5 kcal mol−1), although their total charges are radically different (see Fig. 6)! Very recently, some of these authors observed a similar behavior with substituted adamantanes 9 and bicyclo[2.2.2]octanes 10 (binding enthalpies −19.0 to −20.1, and −15.6 to −16.3 kcal mol−1, respectively; see Fig. 6).57 A very likely explanation is that the strong coulombic attractions between positively charged substituents and the partially negative CB[7] rim (approximately 60 kcal mol−1 per positive unit) are perfectly counterbalanced by the dramatic losses in solvation enthalpy upon binding; therefore, ion-dipole interactions in water are not the main driving force of the CB[n]–guest interaction per se, and the loss of solvation may or may not surpass the coulombic attraction (in the case of guests 8–10, the sum of the coulombic and solvation energies varies between −7.0 and +7.2 kcal mol−1,57 according to the empirical Mining Minima algorithm M2).122,123 The complementarity between the size and shape of the CB[n] cavity and its guest, possibly leading to favorable van der Waals interactions (−27 to −39 kcal mol−157 in the 8–10 series, still according to M2 calculations), has a much stronger impact on the binding affinity. However, one should remember that CB[n]s have an extremely low polarizability, therefore (1) interactions between hydrophobic guests and the bulk should be slightly more favorable compared to interactions with the CB[n] cavity, and (2) dispersion interactions between the guest and the cavity should be weak. Nau even suggests that the driving force of the cavity–guest interaction is solely caused by the ejection of high-energy water molecules from the cavity, a non-classical enthalpic hydrophobic effect!43 The significantly negative average binding enthalpy of selected guests towards CB[7] compared to CDs (see Fig. 6) seems to corroborate this model. Accordingly, Keinan reported the remarkable thermodynamic differences between the interaction of 1,6-hexanediammonium 4a and a series of diyne dications such as 1,6-hexa-2,4-diynediammonium (11) with CB[6]:124 the binding enthalpy of diammonium 4a was found to be −14 kcal mol−1, but only −0.70 kcal mol−1 in the case of dication 11; the authors suggested that the interaction between the electron-rich diyne rod and the macrocycle walls may even be repulsive!124 This effect can be justified since, due to the low polarizability of the cavity, the cavity-to-bulk enthalpic gain in dispersion interaction between unsaturated systems like dication 11 and water is greater than the one between saturated dication 4a and water. Macartney proposed that quadrupole–dipole interactions play a role in the stability of CB[n]–neutral guest complexes and in their orientation within the cavity of the macrocycle.125 The optimum geometry is reached when the dipole of the guest is perpendicular to the quadrupole moment of CB[n]. For example, acetone, pentan-3-one and 3,3-dimethylbutan-2-one, albeit much less hydrophobic than common CB[n] guests, display unexpectedly significant binding affinities towards CB[7] (5.8 × 102, 2.1 × 103 and 6.7 × 103 M−1, respectively), and according to the force-field MM2 model, their carbonyl units are likely to be located within the equatorial plane of the macrocycle and perpendicular to the CB[7] quadrupole.125 As far as other neutral guests are concerned, alcohols and carboxylic acids bind weakly to CB[n]s (binding constants in the 101–102 M−1 range). The length of the alkyl chain in the aliphatic alcohol and carboxylic acid series does not have a significant effect on binding affinities towards CB[6] (4.1–5.4 × 102 M−1 in the propanol to heptanol series, and 5.0–6.2 × 102 M−1 in the propanoic to nonanoic acid group, both measured in 50% formic acid).126 Binding enthalpies and entropies are also remarkably constant among both series (ΔH = −0.49 ± 0.22 and −0.32 ± 0.08 kcal mol−1; TΔS = + 3.1 ± 0.2 and 3.4 ± 0.1 kcal mol−1, respectively). These values suggest that the encapsulation of these guests triggers the ejection of the same number of water molecules from the host cavity, and that dispersion interactions are insignificant.
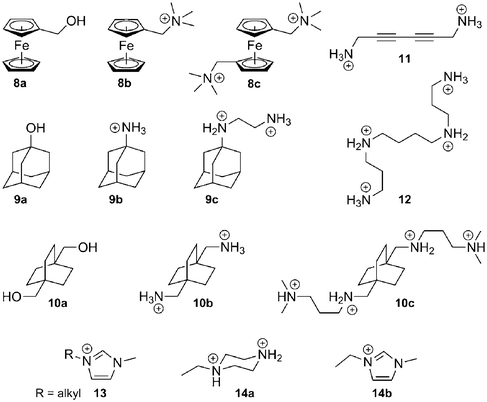
![Enthalpy–entropy compensation plot for the complexation of α-CD (pale pink dots), β-CD (pale orange dots), γ-CD (pale green dots),128,130 CB[6] (red dots)56,65,101,124,132–148 and CB[7] (blue dots)57,141,142,149–156 with various guests. The dashed green line connects the 1-alkyl-3-methylimidazolium 13 data points (length of the alkyl chain: 1, 2, 3, 4, 6, 8, 9, 10, 12, 14 carbons atoms).149](/image/article/2012/RA/c1ra00768h/c1ra00768h-f6.gif) | ||
| Fig. 6 Enthalpy–entropy compensation plot for the complexation of α-CD (pale pink dots), β-CD (pale orange dots), γ-CD (pale green dots),128,130 CB[6] (red dots)56,65,101,124,132–148 and CB[7] (blue dots)57,141,142,149–156 with various guests. The dashed green line connects the 1-alkyl-3-methylimidazolium 13 data points (length of the alkyl chain: 1, 2, 3, 4, 6, 8, 9, 10, 12, 14 carbons atoms).149 | ||
Binding entropy. The changes in configurational entropy upon binding (i.e. the loss of mobility of both CB[n] and its guest after complexation) do not significantly depend on the charge of the guest, and rigid CB[n]s interacting with constrained guests afford high affinities.57,116 For example, the more flexible dication 4a suffers from a 6.1 kcal mol−1 entropic penalty (at 25 °C) upon binding to CB[6], while the encapsulation of diyne 11 is entropically favorable by 7.0 kcal mol−1 (Fig. 6).124 However, the total binding entropy, as measured by isothermal titration calorimetry, becomes less and less unfavorable as positively charged units are added to the guests (for example, TΔS = −8.6→−0.5, −4.9→+ 1.4 and −2.4→+ 4.3 kcal mol−1, in the case of series 8, 9 and 10, respectively; see Fig. 6);57 therefore, since the effect of the configurational entropy can be neglected, the parameter that has the most dramatic effect on the variations in binding affinities is the difference in solvation entropies, caused by the hydration water molecules being ejected from the host and guest upon binding. This observation is in stark contrast to the common enthalpy–entropy compensation model valid for most supramolecular systems, where binding entropies and enthalpies are closely linked.121,127–129 For example, large sets of thermodynamic data corresponding to the interactions between guests and α-, β- or γ-CD indicate that in general, gains in binding enthalpies are approximately compensated by losses in binding entropies (see Fig. 6),128,130 leading to a narrower range of binding affinities (102.1±0.9, 102.6±1.0 and 102.8±1.1 M−1;131 Pearson product–moment correlation coefficients r of the enthalpy–entropy compensation: 0.91, 0.88 and 0.91, respectively). However, in the case of CB[6] and especially CB[7], deviations from the enthalpy–entropy correlation are significant, with broader ranges of binding affinities (103.6±1.5 and 107.1±3.5 M−1, respectively, corresponding to correlation coefficients r equal to 0.83 and 0.56; see Fig. 6). Even if the combined sets of thermodynamic data pertaining to CB[n]s are approximately 15 times smaller than the combined CD sets, and therefore create a bias towards lower correlation coefficients, we think that the latter are not merely due to a statistical effect, but really indicate that variations in solvation entropy upon binding cause the deviations from the common enthalpy–entropy compensation model.
Summary. In short, extreme binding affinities observed with CB[n]s are mainly due to: (1) the ability of the guests and their substituents sitting close to the CB[n] portals, in particular positively charged ones, to return as many hydration water molecules as possible to the bulk upon binding (a process that is both enthalpically and entropically favorable), (2) the rigidity of the macrocycles and some selected guests, (3) a minimally penalizing loss of solvation energy upon encapsulation, and (4) favorable ion-dipole interactions between positively charged substituents and the CB[n] rims, as well as multiple hydrogen bonding (see sections 4.2.1(e) and (f) for a discussion about the impact of hydrogen bonding on the geometry of CB[8] and CB[10] assemblies).
The impact of positively charged substituents can be appreciated when the binding affinity of various ammonium cations is compared to their corresponding neutral amines; several studies carried out by Nau153,157–160 and Macartney161,162 indicate that affinity ratios between both forms towards various CB[n]s range from 16 to 32000;159 the decimal logarithm of these values correspond to the pKa shift of the ammonium cation upon encapsulation by CB[n]s (1.2–4.5 pKa units). The latter value is one of the highest ever reported for both synthetic and natural systems.163
One should finally note that attempts to predict binding affinities in silico, using the M2 algorithm for example,57,116,123 remain unpractical, with errors usually greater than 2 kcal mol−1; considering the subtlety of the CB[n]–guest interaction as far as solvation is concerned, this error is in fact remarkably low. Nau also rightfully notes that a precise evaluation of the possible, albeit very minor dispersion interactions between guests and the inner wall of the CB[n] cavity should be evaluated using the second order Møller–Plesset theory (MP2) with extended basis sets.43
(b) CB[5]. As discussed on numerous occasions, the CB[n] series displays remarkable and selective recognition properties, yet CB[5] is too small to accommodate many organic guests, and the richness of its chemistry is mostly inorganic, as described in section 4.1. We note, however, that CB[5] can encapsulate xenon (approximate binding affinity 1.3 × 103 M−1),164 as well as methane, ethylene and ethane (binding constants in the 103, 102 and 101 M−1 range, respectively).165
(c) CB[6]. The recognition properties of CB[6] have been studied during the past thirty years, and numerous guests have been identified.12 Among those, the positively charged form of spermine 12 displays the strongest interaction (5.4 × 1010 M−1 in 0.20 M LiCl, and 3.3 × 109 M−1 in 50 mM NaCl), followed closely by 1,6-hexane- and 1,5-pentanediammonium (2.9 and 1.5 × 108 M−1, respectively, in 50 mM NaCl).56 We note that the shortest 1,ω-alkanediammonium dication to strongly interact with CB[6] is 1,4-butanediammonium, which binds 6.0 × 104 times stronger than its 1,3-analog (2.0 × 107 M−1vs. 3.3 × 102 M−1 in 50 mM NaCl); according to Kim and Inoue, this per-methylene difference is the highest ever measured in supramolecular chemistry.56 In fact, 1,3-propanediammonium does not form an inclusion complex with CB[6], but interacts with its rim externally.166 As the alkyl chain gets longer (ω ≥ 7), the binding affinity decreases due to a subtle interplay between enthalpic losses and entropic gains. Those incremental entropic gains may suggest that long 1,ω-alkyldiammoniums interact with CB[6] via only one of their portals, and do not adopt a much more constrained S-shape which would have allowed CB[6] contact at both ammonium groups. Enthalpic losses suggest that in this series, coulombic attractions between the CB[6] portal and the ammonium groups compete advantageously against losses in solvation energy. Similar trends are observed with 1-alkyl-3-methylimidazolium 13149 and alkylammonium cations,56 with a marked drop in binding affinity between hexyl- and heptylammonium (1.7 × 105vs. 5.8 × 103 M−1), most probably because longer alkyl chains dislodge coordinated alkali metals from the CB[6] rim opposite to the ammonium group. In those cases, binding affinities follow the classical enthalpy–entropy compensation model (see green dashed line in Fig. 6). Although curling of alkyl chains inside the CB[6] cavity has never been reported, we found four cases where two guests form ternary complexes with CB[6]: the first example is the well-known encapsulation of a terminal alkyne together with an organic azide, which leads to a dramatic rate enhancement of their [3 + 2] cycloaddition, affording the corresponding 1,2,3-triazole.167 This CB[6]-catalyzed reaction has been exploited on several occasions, in particular by Tuncel and coworkers,168–170 for the design of CB[6]-based self-organizing systems and molecular switches (see sections 5.1 and 5.2). The second and third cases of double encapsulation are the formation of 1:2 complexes between CB[6] and N-ethylpiperazines (14a)166 or the ionic liquid 1-ethyl-3-methylimidazolium (14b),171 with the two ethyl groups co-existing within the cavity of the macrocycle; the last example is the capture of two molecules of carbon dioxide by solid CB[6] exposed to the gas172 (as a matter of fact, a similar sorption has been recently reported with CB[7]).173 In addition to the impossibility for alkyl chains to curl inside CB[6], the formation of stable ternary complexes with two CB[n] units thread consecutively along the same 1,ω-alkyldiammonium chain has never observed upon combination of the three individual components; however, in a unique example, Tuncel and coworkers managed to lock two CB[6] units along a polyaminated axle 15, and subsequently force the two macrocycles to shuttle and share the same 1,12-dodecanediamine station upon full deprotonation of the axle and subsequent reprotonation (see Fig. 7a).169 Also, to the best of our knowledge, there has been only one report where a disubstituted ammonium could share two CB[n] units (a transient, unstable complex between a polyaminated axle, CB[6] and CB[8];174 see section 5.1), and 1:1 complexes are always the most stable configurations.175,176 For example, Kim and Inoue showed that the dihexylammonium cation (16) forms a 1:1 assembly in the presence of an excess amount of CB[6], with one hexyl chain encapsulated, and the other one sitting at the periphery of the macrocycle.177 However, the outer hexyl chain can be encapsulated within β-CD, whose rim interacts with CB[6] via multiple hydrogen bonds, and benefits from the weakened positive charge of the ammonium group at the CB[6] portal; the presence of CB[6] actually improves the binding affinity of β-CD towards the hexylammonium moiety by 33 times, a remarkable case of supramolecular positive cooperativity (see Fig. 7b).177
![(a) Two CB[6] units locked along the 1,12-dodecanediammonium station of axle 15.169 (b) Formation of a 1 : 1 complex between dihexylammonium (16) and CB[6], and of a ternary assembly with β-CD.177](/image/article/2012/RA/c1ra00768h/c1ra00768h-f7.gif) | ||
| Fig. 7 (a) Two CB[6] units locked along the 1,12-dodecanediammonium station of axle 15.169 (b) Formation of a 1:1 complex between dihexylammonium (16) and CB[6], and of a ternary assembly with β-CD.177 | ||
As far as neutral guests are concerned, Luhmer and coworkers showed that gases such as sulfur hexafluoride,132 and to a lesser extent xenon,178,179 form adducts with CB[6] (binding affinities 3.1 × 104 and 2.1 × 102 M−1 in a 0.20 M sodium sulfate solution). Nau and coworkers recently identified 15 hydrocarbons capable of interacting with CB[6] with affinities greater than 3 × 103 M−1; binding constants of propane, butane, isobutane and cyclopentane reach 1.5 × 105, 2.8 × 105, 8.5 × 105 and 1.3 × 106 M−1, respectively, in a 1.0 mM hydrochloric acid solution!165 Scherman also reported that a 1:1 complex of CB[6] and diethyl ether, with no cation or anion attached, could be precipitated upon vapor diffusion of the organic solvent into a solution of CB[6] interacting with a positively charged imidazolium ionic liquid.180
Finally, we note the unique case of CB[6]-mediated chiral recognition described by Kim and Inoue,142 a phenomenon nicknamed by the authors as “assembled enantiorecognition”: the binding affinity of CB[6] towards (S)-2-methylbutylammonium ((S)-17a) is 19 times higher when one of the CB[6] portals interacts with an excess of (R)-2-methylpiperazine ((R)-17b) compared to its (S)-enantiomer (1.5 × 104vs. 8.0 × 102 M−1, a 95% enantioselectivity, the highest discrimination ever reported in supramolecular chemistry using an achiral macrocyclic mediator!).142 In other terms, the CB[6]/(S,R)-17a/17b ternary complex is 1.7 kcal mol−1 more stable than its corresponding CB[6]/(S,S)-17a/17b diastereoisomer.
(d) CB[7]. Among all the members of the CB[n] family, CB[7] displays the strongest interactions towards positively charged amphiphilic guests, with affinities greater than 1015 M−1.57,116 Unlike all other synthetic hosts, CB[7] displays common affinities between 107 and 1012 M−1, with adamantyl-, ferrocenyl-, p-xylylenyl- and trimethylsilyl-containing guests forming the tightest complexes.58 The affinity and selectivity of trimethylsilylmethylammonium (18a) and 3-(trimethysilyl)propionic acid (18b) towards CB[7] is unprecedented (8.9 × 108 and 1.8 × 107 M−1, respectively, while no interaction is observed towards CB[6] and CB[8]!).58 The interaction between CB[7] and the neutral silane 18b illustrates again the importance of a good fit between guests and CB[n]s. Another outstanding feature of CB[7] is its ability to encapsulate positively charged units instead of interacting with them via its portals.181 Examples of such guests include a series of tetraalkylammonium, tetraalkylphosphonium and trialkyl-sulfonium cations, with tetraethylammonium, tetramethyl-phosphonium and triethylsulfonium displaying the highest affinities (1.0, 2.2 and 5.2 × 106 M−1, respectively, in pure water).181 The fact that hydrophobic interactions and possibly favorable coulombic interactions between the alkyl substituents (where most of the positive charge is located) and the inner portion of the CB[7] rim can overcompensate the drastic loss in cation solvation energy is remarkable. CB[7] forms complexes with other guests bearing a diffuse positive charges, such as tricylic basic dyes 19 proflavine, pyronine, acridine, oxonine, thionine and some of their derivatives (binding affinities 106–107 M−1), yet it is unclear whether the heteroatom(s) of the tricylic units are located at the portal or inside the cavity of CB[7].182–184
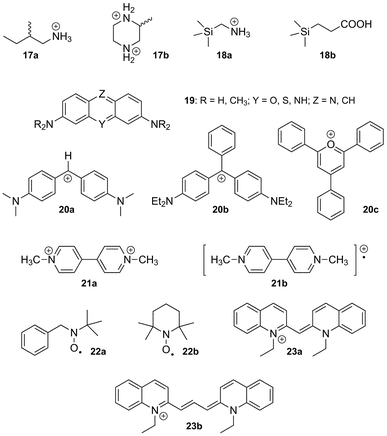
We note that CB[7] has also been found to interact with diphenylmethane (20a), triphenylmethane (20b)185,186 and triphenylpyrylium (20c)187,188 carbocations (binding affinities 2.0 × 104, 1.7 × 104 and 7.5 × 105 M−1, respectively), as well as several radicals. For example, Kaifer showed that methylviologen radical cations (21b; noted MV+ thereafter), obtained upon reduction of dicationic viologen 21a (noted MV2+ in the following sections), form stable inclusion complexes with CB[7] (binding affinity 5.0 × 104 M−1);189 Anderson reported that CB[7] threading along an oligoaniline axle increased the thermodynamic and kinetic stability of its oxidized radical cation form, with a first oxidation potential reduced by 0.57 V!190 Similarly, Liu showed that the conductive doped form of polyaniline (i.e. its radical cation) is stabilized when surrounded by multiple CB[7] units.191 Finally, Lucarini reported the CB[7] encapsulation of nitroxides 22a and 2,2,6,6-tetramethyl piperidine-N-oxyl (TEMPO; 22b)192 as well as some derivatives,193 and monitored the interaction by electron paramagnetic resonance; important changes in the nitrogen hyperfine splitting were observed upon encapsulation. We note that CB[7] has also been used to disrupt aggregates and other non-covalent interactions such as π–π stacking: Kaifer showed that CB[7] could efficiently break the J-aggregate formed upon stacking of the pseudoisocyanine dye 23a, and the H-aggregate formed with the pinacyanol dye 23b. In both cases, the absorption bands of J- and H-aggregates (575 and 473 nm, respectively) vanished upon addition of the macrocycle.194,195
(e) CB[8]. Like CB[7], CB[8] displays some remarkably strong binding affinities towards large amphiphilic positively charged guests, such as adamantane derivatives 24a and 24b (up to 4.3 × 1011 M−1).58 It can also encapsulate macrocycles such as fully protonated cyclen (25a) and cyclam (25b), as well as their Cu(II) and Zn complexes.7
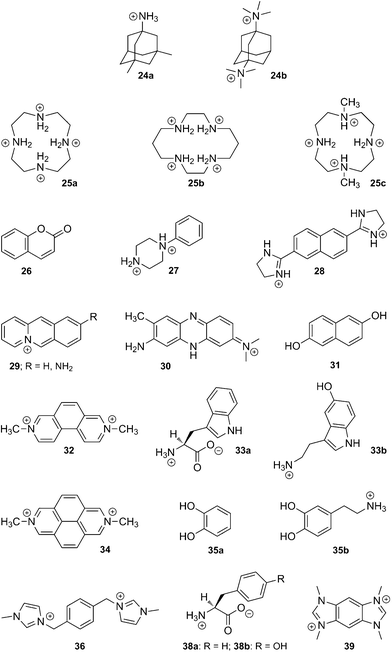
Interestingly, while the protonated cyclam (25b) adopts the more stable trans-III configuration even while encapsulated,196 subsequent complexation with Cu(II) affords a 7:3 mixture of the unusual trans-I and trans-II configurations in the solid state.197 Incorporation of the partially methylated cyclen 25c into the cavity of CB[8] has also been reported.198 When set in the presence of CB[8], long alkylammonium chains (8–16 carbon atoms in the alkyl unit) curl inside the cavity of the macrocycle, and the entropic penalty caused by conformational restriction, which peaks at 12 carbon atoms, is overcompensated by enthalpic gains; affinities are remarkably similar along the C8–C16 series (1.0–4.8 × 106 M−1), and thus follow the classical enthalpy–entropy compensation model.199,200 Van der Waals contact between the curled hydrophobic chain and the cavity has been proposed as the main cause of the enthalpic gain. One could also argue that curling maximizes the number of ejected high-energy water molecules from the cavity. Similarly to alkylammoniums, but also to 1,4-butylidene- and 1,10-decylidenedipyridinium cations,201 this curling behavior is observed in the case of the 1,12-dodecanediammonium cation (binding affinity 1.1 × 106 M−1), which adopts a U-shaped conformation within CB[8], with both ammonium heads located at the same portal and separated by a water molecule.202 We note that the CB[8] encapsulation of nitroxide radicals has also been reported on several occasions,203–205 including by Kaifer and coworkers.205
Despite these findings, the richness of CB[8] chemistry is attributed without any doubt to its ability to encapsulate two guests into its cavity, and to form highly stable ternary complexes. A series of applications, including supramolecular catalysis and the design of new polymeric materials, have exploited this property, and will be described in sections 5.3.2 and 5.5.1. Several guests have been found to undergo double encapsulation into CB[8], such as two equivalents of coumarin (26),206N-phenylpiperazine (27),207 naphthyl derivative 28,4 aminoacridiziniums 29,208 and neutral red 30 under acidic conditions.209 Hetero-ternary complexes are particularly stable when favorable interactions between both guests add to the stability of the assembly. For example, the charge-transfer complex between electron-deficient MV2+ (21a) and electron-rich 2,6-dihydroxynaphthalene (31) is readily encapsulated by CB[8].210 An elegant variation of this interaction is the formation of a ternary complex between CB[8], an MV2+ unit linked to yellow fluorescent proteins, and a dihydroxynaphthalene unit linked to cyan fluorescent proteins, as published by Brunsveld and coworkers very recently.211 Two other examples reported by Kaifer are the CB[8] encapsulation of 2,7-dimethyldiazaphenanthrenium (32) and indole derivatives such as tryptophan (Trp; 33a) and serotonin (33b),212 as well as the formation of ternary CB[8]/dimethyldiazapyrenium (34) complexes with catechol (35a) and dopamine (35b).213 Recently, Scherman reported the encapsulation of bisimidazolium salt 36 and various small guests, such as phenol, acetone, diethyl ether and tetrahydrofuran, which fill the small void left by the large bisimidazolium in the CB[8] cavity.214 Looped structures can be readily obtained when both electron-poor and electron-rich units are linked; several examples based on the viologen/naphthol motif have been reported (see axle 37 in Fig. 8).215–220 Another interesting feature of CB[8] is its ability to encapsulate two MV+ radical cations (21b), while the dicationic MV2+ (21a) forms a binary complex with CB[8]; electrochemical or light-induced reduction of the latter affords the corresponding radical cation.220–226
![Formation of a looped structure triggered by CB[8], and stabilized by charge transfer interactions.218](/image/article/2012/RA/c1ra00768h/c1ra00768h-f8.gif) | ||
| Fig. 8 Formation of a looped structure triggered by CB[8], and stabilized by charge transfer interactions.218 | ||
Finally, we describe the remarkable recognition properties of the MV2+ ⊂ CB[8] [2]pseudorotaxane towards selected amino acids, as unveiled by Urbach and coworkers. MV2+ ⊂ CB[8] forms ternary complexes with Trp (30a), phenylalanine (Phe; 38a) and tyrosine (Tyr; 38b), with a marked preference for Trp (30a; binding affinity 4.3 × 104vs. 5.3 and 2.2 × 103 M−1 in the case of Phe (38a) and Tyr (38b), respectively);227,228 moreover, in a peptide sequence, MV2+ ⊂ CB[8] targets N-terminal Trp with very good selectivity over internal or C-terminal Trp residues (up to a 40-fold specificity). Formation of the ternary complex leads to the emergence of a charge-transfer band (λmax 420–450 nm) and to the quenching of Trp fluorescence.227 CB[8] alone was also found to interact with two short peptides bearing N-terminal Trp or Phe residues with high affinity (3.6 × 109 and 1.5 × 1011 M−2, respectively).229
In a subsequent study, Urbach studied the CB[8]-mediated pairing of peptides containing one or more Trp residues with a series of synthetic analogs bearing one or more MV2+ side-units (see Fig. 9), thereby affording discrete “peptide duplexes”.230 Peptide and small molecule recognition experiments were also carried out using benzobis(imidazolium) 39 instead of MV2+; binding affinities were found to parallel those of MV2+-containing assemblies.150 Brunsveld showed that the yellow fluorescent protein, once linked to a Phe-glycine-glycine peptide (Phe-Gly-Gly) dimerizes in the presence of CB[8], since the latter can encapsulate two Phe units; dimerization caused a decrease of the fluorescence anisotropy by intermolecular energy transfer (homo-FRET). A similar interaction between Phe-Gly-Gly-labeled yellow and green fluorescent proteins was monitored by hetero-FRET.231 Finally, we note that Urbach recently prepared the first example of a CB[8]-containing rotaxane, by “clicking” extremely bulky alkyne-substituted tetraphenylmethane stoppers to a CB[8]-bound MV unit substituted with azide-terminated linkers.232
![Formation of “peptide duplexes” upon CB[8] interaction with Trp-rich (residues in blue) and MV2+-decorated peptides (MV2+ units in red). Reprinted with permission from ref. 230. Copyright 2009 American Chemical Society.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f9.gif) | ||
| Fig. 9 Formation of “peptide duplexes” upon CB[8] interaction with Trp-rich (residues in blue) and MV2+-decorated peptides (MV2+ units in red). Reprinted with permission from ref. 230. Copyright 2009 American Chemical Society. | ||
(f) CB[10]. Not much of the chemistry of CB[10] has been unveiled since its isolation by Isaacs and coworkers in 2005.35 In the same article, the authors described the remarkable formation of an inclusion complex between calix[4]arene derivative 40 and CB[10], as well as ternary assemblies between a selection of adamantyl derivatives and assembly 40 ⊂ CB[10] (see Fig. 10 for a force-field minimized structure of ternary complex 9b·40·CB[10]).35 Besides the isolation of a potassium-coordinated water ⊂ CB[5] ⊂ CB[10] complex,233 there has been only three new reports of CB[10] recognition: Isaacs described the conformational behavior of some triazene-arylene units in the presence of CB[n] hosts (including guest 41), and showed it exclusively adopted the quadruple anti conformation inside the cavity of CB[10] (see guest a,a,a,a-41a in Fig. 10).234 In agreement with Urbach's discussion about the structure of CB[8]–peptide complexes,229 the a,a,a,a conformation maximizes N–H/O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and NH3+/OC interactions, while no intramolecular π–π interaction between aromatic rings is observed (see the red arrows in Fig. 10, which point towards the three phenylene units). The authors judiciously show that the CB[10]–guest interaction modes parallel the three-dimensional folding of proteins: ejection of solvating water molecules to the bulk is a critical driving force of both protein folding and CB[n] encapsulation, and hydrogen bonds and coulombic interactions are responsible for the exact geometry of the folded (or interlocked) structures.234
C and NH3+/OC interactions, while no intramolecular π–π interaction between aromatic rings is observed (see the red arrows in Fig. 10, which point towards the three phenylene units). The authors judiciously show that the CB[10]–guest interaction modes parallel the three-dimensional folding of proteins: ejection of solvating water molecules to the bulk is a critical driving force of both protein folding and CB[n] encapsulation, and hydrogen bonds and coulombic interactions are responsible for the exact geometry of the folded (or interlocked) structures.234
![(a) MMFF-optimized structure of ternary complex 9b·40·CB[10] (1-adamantylammonium (9b) in bright green and calixarene 40 in brown and violet).35 (b) Conformers of triazene-arylene 41 and X-ray structure of complex a,a,a,a-41a ⊂ CB[10].234 Red arrows point towards the three phenylene units. Structures of Pt(ii) and Ru(ii) complexes 42 and 43; both can be encapsulated by CB[10].36](/image/article/2012/RA/c1ra00768h/c1ra00768h-f10.gif) | ||
| Fig. 10 (a) MMFF-optimized structure of ternary complex 9b·40·CB[10] (1-adamantylammonium (9b) in bright green and calixarene 40 in brown and violet).35 (b) Conformers of triazene-arylene 41 and X-ray structure of complex a,a,a,a-41a ⊂ CB[10].234 Red arrows point towards the three phenylene units. Structures of Pt(II) and Ru(II) complexes 42 and 43; both can be encapsulated by CB[10].36 | ||
Wagner, Kaifer and Isaacs also showed that porphyrins (metal free, or coordinated to Zn(II), Fe(III) and Mn(III)), bearing four methylpyridinium substituents, form an inclusion complex with CB[10].235 According to force-field MMFF optimization, the plane of the porphyrin macrocycle is perpendicular to the equatorial plane of CB[10], and two positively charged pyridinium units interact with each CB[10] portal, while CB[10] is significantly distorted into an oval shape. The electrochemical and spectroscopic properties of the porphyrin derivative were barely affected by the surrounding CB[10]. Interestingly, ternary complexes could be obtained upon addition of pyridine derivatives, MV2+ (21a), quinoline and isoquinoline, and affinities were surprisingly high (up to 4.8 × 105 M−1); yet, those guests did not coordinate to the metallic center, but merely interacted with the porphyrin ring via π–π stacking.235
Keene, Day and Collins showed that Pt(II) and Ru(II) complexes 42 and 43 could slip through CB[10], and interact with the macrocycle via their alkyl central station.36 In the case of guest 43, part of the bipyridyl ligands were also located in the CB[10] cavity. A fast exchange on the NMR time scale was observed with platinum guest 42, while the very bulky ruthenium/bipyridyl head groups caused guest 43 to exchange slowly; despite the complexity of the 43 ⊂ CB[10] pseudorotaxane geometry, remarkably clear 1H NMR spectra could be recorded for the free guest and the CB[10]-bound assembly.36
![Plausible ingression mechanism of alkylamines into CB[n]s.236,237](/image/article/2012/RA/c1ra00768h/c1ra00768h-f11.gif) | ||
| Fig. 11 Plausible ingression mechanism of alkylamines into CB[n]s.236,237 | ||
We have recently studied the kinetics of CB[6] slippage along polyaminated axles 44, and have identified two mechanisms for the CB[6] translation from station 1 to station 2 (see Fig. 12).240 Although the threading of the protonated ammonium guest through CB[6] first comes to mind, we favor an alternate process, involving (1) the “intra-rotaxanic” deprotonation of the ammonium cation by the carbonyl portal of CB[6], followed by acid–base equilibrium with the aqueous medium, (2) the slippage of the neutral amine through the CB[6] cavity, and (3) the fast protonation of the opposite CB[6] carbonyl followed by the reprotonation of the amine guest (see Fig. 12). Since we have shown that incorporation of water molecules within the cavity of CB[6] during the slippage process is unlikely,240 CB[6] could have to overcome a penalty as high as 60 kcal mol−1 (which corresponds to the loss of solvation of the cation) when slipping over the ammonium group. When threading takes place over the neutral amine, the loss of solvation is limited to 4 kcal mol−1.241 We also found the slippage rates to be highly dependent on even minor sterical alterations of the N-terminal substituent R, with free Gibbs energies of activation ranging from 24 to 29 kcal mol−1 (which correspond to half-lives of [2]pseudorotaxanes 44 ⊂ CB[6] at 100 °C ranging from 2 s to 3 h). Similar barriers of 24 and 26 kcal mol−1 were determined in two other cases of CB[6] threading over a nitrogen atom.170,242
![CB[6] slippage along a polyaminated axle, between station 1 and station 2 of guest 44. A deprotonation–reprotonation mechanism of the ammonium unit is favored (pathway 2) over direct CB[6] translation along the positively charged group (pathway 1).240](/image/article/2012/RA/c1ra00768h/c1ra00768h-f12.gif) | ||
| Fig. 12 CB[6] slippage along a polyaminated axle, between station 1 and station 2 of guest 44. A deprotonation–reprotonation mechanism of the ammonium unit is favored (pathway 2) over direct CB[6] translation along the positively charged group (pathway 1).240 | ||
4.3 CB[n] recognition in the gas phase
Much of the gas phase chemistry of CB[n]s has been studied by Dearden and coworkers.243 Several features of the host–guest interaction, such as the formation of inclusion or exclusion complexes in the gas phase, can be readily determined: Dearden showed that inclusion complexes of CB[6] undergo guest exchange with tert-butylamine much slower than exclusion complexes (we note that tert-butylamine does not penetrate into the cavity of CB[6]).244 Sustained off-resonance irradiation-collision-induced dissociation experiments (SORI-CID) afford similar information, since exclusion complexes readily dissociate, and inclusion complexes require higher energies that may even trigger the fragmentation of the macrocycle.244,245 1,4-Butanediammonium was found to form 2:1 doubly charged exclusion complexes with CB[5], with the two singly charged 1,4-butanediamines interacting with each CB[5] portal. To the contrary, CB[6] encapsulates the guest, and the doubly charged 1:1 complex is virtually the only product (a trace of a doubly charged 2:1 adduct was detected).245 Exchange and SORI-CID experiments confirmed the structure of both inclusion and exclusion complexes. Interestingly, the optimum length of 1, ω-alkyldiammonium cations for binding to CB[6] is ω = 4, as determined by SORI-CID, and not ω = 6 like in solution. This difference is most likely due to the greater energy penalty for additional loss of cation solvation when 1,4-butanediammonium interacts with CB[6] in solution, since the two ammonium units sit deeper in the macrocycle and are less exposed to water.246 The interaction between lysine (Lys; 45a), CB[5] and CB[6] was also studied: Lys forms a singly charged exclusion complex with CB[5] and a doubly charged pseudorotaxane with CB[6]. The authors also propose that pentalysine 45b interacts with both portals of CB[6] in a “clamp”- or “forceps”-like conformation without slipping through the macrocycle.247Ortho- and meta-phenylenediamine were found to form exclusion complexes with CB[6], while the para-isomer inserted into the macrocycle cavity.244 Dearden showed that CB[7] could encapsulate benzene, fluorobenzene and toluene, while having its portals covered with two guanidinium units. According to DFT calculations at the B3LYP/6-31+G(d) level, those complexes are not thermodynamically stable in the gas phase, and result from their formation in solution.248 Scherman showed that complexes of CB[8], a MV dimer and some naphthol derivatives were stable in the gas phase and could be readily assessed by mass spectrometry.249 Finally, Da Silva recently reported that aggregation of CB[n]s as dimers, trimers or tetramers in the gas phase is a general phenomenon, as long as the portals are available for interactions with neighboring CB[n] units.250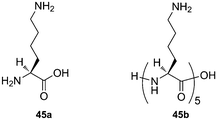
5. Applications of CB[n] chemistry: a progress report
The outstanding recognition properties of CB[n]s have prompted the rapid development of exciting applications in the supramolecular, synthetic, medicinal and material science fields. In this chapter, we highlight the recent progress in the design of new self-organizing systems and stimulus-responsive switches (sections 5.1 and 5.2), in the development of CB[n]-promoted and CB[n]-catalyzed organic reactions (section 5.3), and in the exploitation of CB[n]s as key units in novel drug carriers (section 5.4), and advanced materials (section 5.5).5.1 Self-organizing systems
The supramolecular community identifies self-sorting or self-organizing systems, as one or several hosts that can discriminate between a set of guests, and form well-defined assemblies. Nuances about self-sorting have been carefully described by Isaacs,251,252 who pioneered self-sorting with CB[n]s252 and invented the notion of “social self-sorting” (i.e. heteromeric complex formation)251 as the counterpart to Anderson's “narcissistic self-sorting” (i.e. homomeric aggregation).253 Schalley clearly summarized these notions in a recent article.219A series of examples involving CB[n]s have been published during the past six years. Isaacs and coworkers showed that CB[6] and CB[7] respectively target adamantanebutylammonium (46a) at its butyl moiety and cyclohexanediammonium (46b); yet this self-organizing is only kinetically favored, and the macrocycles exchange their respective guest in the course of 56 days to afford the thermodynamically favored combination, with the adamantyl unit encapsulated inside CB[7] and the cyclohexyl unit in CB[6] (see Fig. 13a).254 The driving force of the reaction is the 4.3 × 103-fold gain in binding affinity when CB[7] is displaced from the cyclohexane to the adamantane unit; this largely overcompensates the 14-fold loss of affinity suffered by CB[6] after the exchange. The rate-limiting steps are the egression of guest 46a and the ingression of guest 46b from and into CB[6] (2.2 × 10−3 and 1.2 × 10−3 M−1 s−1, respectively, at room temperature).254 The relatively strong affinity of cyclohexanediammonium (46b) towards CB[6] (1.4 × 106 M−1) and the bulkiness of the guest were found to cause an extremely slow egression of this guest from CB[6], with a rate as low as 8.5 × 10−10 M−1 s−1, two orders of magnitude slower than the dissociation of the benchmark avidin–biotin pair!118
![(a) Kinetic vs. thermodynamic self-sorting of CB[6] and CB[7] towards guests 46a and 46b.254 (b) Interaction between acetylcholinesterase inhibitor 47 and CB[7] (1.0 and 2.0 equiv., respectively).256 (c) Self-assembly between axle 48, CB[6] and CB[8].174 (d) Self-organization between adamantyl/MV derivative 49, 2,6-dihydroxynaphthalene (31), CB[8] and β-CD.260](/image/article/2012/RA/c1ra00768h/c1ra00768h-f13.gif) | ||
| Fig. 13 (a) Kinetic vs. thermodynamic self-sorting of CB[6] and CB[7] towards guests 46a and 46b.254 (b) Interaction between acetylcholinesterase inhibitor 47 and CB[7] (1.0 and 2.0 equiv., respectively).256 (c) Self-assembly between axle 48, CB[6] and CB[8].174 (d) Self-organization between adamantyl/MV derivative 49, 2,6-dihydroxynaphthalene (31), CB[8] and β-CD.260 | ||
Kim and Inoue showed that CB[7] binds to the aromatic residue of dipeptide Phe-Gly, and efficiently discriminates between Phe-Gly and Gly-Phe (binding affinities of 3.0 × 107 and 1.3 × 103 M−1, respectively, a 2.3 × 104-fold difference!); CB[7] can also recognize dipeptides Tyr-Gly and Trp-Gly over Gly-Tyr and Gly-Trp with 1.8 × 104 and 2.0 × 103 fold selectivities, respectively.156 Very recently, Urbach showed that CB[7] targets selectively the N-terminal Phe residue of the human insulin B-chain against all other surface-exposed residues, and forms a 1:1 complex with the protein (binding affinity 1.5 × 106 M−1)!255 For additional recognition mechanisms involving peptides and CB[8], we recommend the other publications from the Urbach group (see section 4.2.1(e)). Macartney showed that some α,ω-bis(trialkylammonium)alkane bolaamphiphilic acetyl-cholinesterase inhibitors 47 (or their phosphonium analogs) interact with 1.0 equivalent CB[7] via their alkyl central station; however, upon addition of more than 2.0 equivalents CB[7], the macrocycles bind to the terminal ammonium stations and leave the central alkyl unit exposed to the solvent (see Fig. 13b).256 A similar scenario was observed with a series of α,ω-bis(pyridinium)alkane dications.257 For these reorganization mechanisms to take place, the strength of CB[7] binding to the central station must be (1) greater than the affinity towards one terminal station, and (2) weaker than the combined CB[7] affinities towards both terminal stations (i.e. lower than the product of the binding constants). Tuncel described the self-sorting properties of polyaminated axle 48 towards CB[6], CB[7] and CB[8]: CB[6] interacts exclusively with the terminal triazole stations, while CB[7] and CB[8] bind to the central 1,12-dodecanediammonium station. Remarkably, upon addition of 2.0 equivalents CB[6] to [2]pseudorotaxane 48 ⊂ CB[8], unstable [4]pseudorotaxane 48·(CB[6])2·CB[8] is initially formed (the only example where two CB units share the same ammonium cation, see section 4.2.1(a)), and CB[8] is then slowly ejected, with the dethreading rate obviously depending on the exchange kinetics of the two CB[6] ‘valves’ sitting at the terminal stations (see Fig. 13c).174 In a recent study,219 Schalley discussed the self-sorting processes taking place between CB[7], CB[8], 2,6-dihydroxynaphthalene (31), MV2+ (21a), and several guests bearing the latter electron-withdrawing and electron-donating units on a single axle, a process called integrative self-sorting.258,259 Also this year, Liu and coworkers described a very elegant self-organizing system involving both CB[8] and β-CD; upon combination of CB[8]-bound adamantyl/MV derivative 49 with β-CD-bound 2,6-dihydroxynaphthalene (31), quaternary complex 31 49 CB[8] β-CD was formed, with β-CD now encapsulating the adamantyl unit, and CB[8] the charge transfer complex between MV and 2,6-dihydroxynaphthalene (see Fig. 13d)!260
Isaacs reported a remarkable hybrid natural/synthetic self-sorting system involving CB[7], an enzyme (bovine carbonic anhydrase (BCA) or acetylcholinesterase (AChE)), and inhibitors bearing both enzyme- and CB[7]-binding units (see compounds 50 and 51 in Fig. 14). CB[7] can efficiently disrupt the interaction between BCA and inhibitor 50via the transient formation of a ternary BCA 50 ⊂ CB[7] assembly (this interaction actually enhances the rate of dissociation of BCA and inhibitor 50); after disruption, the catalytic activity of the enzyme is restored (Fig. 14a). In the case of AChE, the AChE (51 ⊂ CB[7])4 assembly is stable, and CB[7] does not dislodge the inhibitor from the more open sites of the enzyme, which remains inactive (Fig. 14b).117
![(a) Successful CB[7]-mediated control of inhibitor activity towards the BCA enzyme. (b) Unsuccessful control of AChE activity. Reprinted with permission from ref. 117. Copyright 2010 American Chemical Society.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f14.gif) | ||
| Fig. 14 (a) Successful CB[7]-mediated control of inhibitor activity towards the BCA enzyme. (b) Unsuccessful control of AChE activity. Reprinted with permission from ref. 117. Copyright 2010 American Chemical Society. | ||
5.2 Molecular switches
When an external stimulus, such as a pH or a temperature change, light irradiation, etc. triggers self-sorting, allows some re-organization towards a new well-defined system, or induces a change in detectable output, the overall mechanism becomes a switch. A series of examples involving CB[n]s have been reported during the past few years.![Kinetic vs. thermodynamic self-sorting of polyaminated axle 52 in the presence of CB[6] and CB[7].261](/image/article/2012/RA/c1ra00768h/c1ra00768h-f15.gif) | ||
| Fig. 15 Kinetic vs. thermodynamic self-sorting of polyaminated axle 52 in the presence of CB[6] and CB[7].261 | ||
![pH-controlled CB[n] switches.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f16.gif) | ||
| Fig. 16 pH-controlled CB[n] switches. | ||
Although the following example by Nau does not involve the displacement of CB from one station to another, the controlled on/off fluorescence output of this system makes it pertinent to this section: benzimidazole derivative 56 is barely fluorescent at high pH, due to a photoinduced electron transfer between the benzimidazole unit and the excited naphthalimide fluorophore; at lower pH, protonation of benzimidazole prevents the electron transfer, and fluorescence increases. Upon addition of CB[7], which encapsulates the benzimidazole moiety, an additional increase in fluorescence is observed, probably due to constraints in rotational and vibrational freedom, and to the proximity between the naphthalimide fluorophore and the CB[7] portal. Therefore, the system behaves as an AND logic gate, with the two inputs being CB[7] and the concentration of protons: both protons (lower pH) and CB[7] are needed in order to generate a high fluorescence output (see Fig. 16d).267
Finally, we report two pH-driven switches involving both CB[n]s and β-CD. Isaacs showed that disubstituted ammonium 57 interacts with CB[6] via its alkyl substituent, and quaternary ammonium 58 with β-CD via its adamantyl unit, when all four components are combined at pH < 7; however, under basic conditions (pH > 13), disubstituted ammonium 57 gets deprotonated, interacts now preferentially with β-CD (via its adamantyl unit), and leads CB[6] to interact with the alkyl substituent of quaternary ammonium 58 (see Fig. 17a)!268 Thompson, Kim and Yui prepared mixed CB[7]/β-CD polypseudorotaxane 61·CB[7] β-CD by “clicking” alkyne-substituted pseudorotaxane 59 ⊂ CB[7] with azide-substituted adduct 60 ⊂ β-CD. CB[6] could then be shuttled from the xylylene to the triazole/propylene glycol units by varying the pH from 2 to 11 without any concomitant dethreading, thanks to the steady position of the β-CD macrocycle along the polymer (see Fig. 17b).269
![(a) pH-mediated selective host/guest pairing between secondary and quaternary ammonium cations 57 and 58, CB[8] and β-CD.268 (b) CB[7]-shuttling along a β-CD-shielded polymer.269](/image/article/2012/RA/c1ra00768h/c1ra00768h-f17.gif)
![Redox-controlled shuttling of CB[7] along a ferrocene derivative.270](/image/article/2012/RA/c1ra00768h/c1ra00768h-f18.gif)
Most redox-controlled switches involve CB[8], and exploit its ability to encapsulate two MV+ radical cations, obtained upon reduction of the corresponding dications MV2+. For example, Kim showed that axle 63 forms loop 63·CB[8] upon interaction with CB[8] (the two possible donor–acceptor combinations are present in solution), and undergoes a reorganization to loop 64·CB[8] upon electrochemical reduction (see Fig. 19a);216 similarly, Sun and Peng showed that guest 65 adopts a looped conformation inside CB[8], and forms 2:1 complex 662·CB[8] upon reduction (Fig. 19b). The authors also noted the presence of looped radical cation 66·CB[8].220 In another report, Sun described the shuttling of CB[8] from the 3,3′-dimethylviologen station to the neighboring MV station of axle 67 upon reduction, with concomitant formation of dimer 682·CB[8] (Fig. 19c).225 We note that reductions have also been carried out by light irradiation on axles linked to a photo-sensitizer (a Ru(II)-tris(bipyridine) unit for example), in the presence of a sacrificial electron donor such as triethanolamine.218,224
![Redox-controlled switches, exploiting the contrasted recognition properties of CB[8] towards MV2+ cations and MV+ radical cations.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f19.gif) | ||
| Fig. 19 Redox-controlled switches, exploiting the contrasted recognition properties of CB[8] towards MV2+ cations and MV+ radical cations. | ||
Finally, we describe a remarkable redox-controlled self-sorting switch recently reported by Kim and coworkers. CB[8] readily encapsulates MV2+ (21a) and electron-donating tetrathiafulvalene (69); upon reduction with sodium dithionite, CB[8] frees tetrathiafulvalene (69), and forms a ternary complex with the dimer of the MV+ radical cation (21b); the ternary adduct 21a·69·CB[8] is regenerated upon treatment with oxygen. The donor–acceptor complex can also be oxidized with Fe(III); CB[8] then liberates MV2+ (21a) and forms a ternary complex with the dimer of radical cation 69; reduction with sodium metabisulfite regenerates the original ternary assembly 21a·69·CB[8] (see Fig. 20)!273
![A redox-controlled three-position switch, involving CB[8], MV2+ (21a), tetrathiafulvalene (69)and their respective radical cations.273](/image/article/2012/RA/c1ra00768h/c1ra00768h-f20.gif)
![Tandem assay for diamine oxidase monitored by CB[n] and a fluorescent dye.276](/image/article/2012/RA/c1ra00768h/c1ra00768h-f21.gif) | ||
| Fig. 21 Tandem assay for diamine oxidase monitored by CB[n] and a fluorescent dye.276 | ||
Very recently, Urbach and Nau adapted this tandem assay to the continuous monitoring of the enzymatic cleavage of enkephalin-type peptides by metallopeptidase thermolysin, using acridine orange as the fluorescent probe.277 As noted by the authors, thermolysin metallopeptidases play critical roles in reproduction and cardiovascular homeostasis mechanisms, and enkephalin-type peptides are involved in pain perception, emotional behavior, and play a role in dementia caused by the Alzheimer's disease. The affinity of the reacting peptide towards CB[7] is only moderate (approximately 104 M−1) compared to the cleaved peptide (affinity greater than 106 M−1), which competes with acridine orange for CB[7] binding. Therefore, upon enzymatic cleavage, the dye is released from CB[7] and the fluorescence decreases. The kinetics of the cleavage can be readily monitored, and can be used to assess peptide sequence specificity, the effect of terminal charges (neutral amide vs. carboxylate units) on the degradation rates, stereospecificity, as well as endo-vs. exopeptidase activity. The tandem assay was also used to determine the inhibition constant of the protease inhibitor phosphoramidon (17.8 ± 0.4 nM).277
5.3 Impact of CB[n]s on organic reactivity
CB[n]s impact the distribution of reactants and products at equilibrium (a thermodynamic effect, section 5.3.1), as well as reaction rates (see section 5.3.2 for kinetic effects); both inhibition and rate enhancement are discussed below.:2 complex 74b·(CB[7])2 is detected; to the best of our knowledge, this is the only example where one pyridinium unit participates in the stabilization of two CB[7] macrocycles.280 The same authors also reported that CB[7] could trigger the partial tautomerization of lumichrome 75a to the corresponding isoalloxazine 75b.281 Isaacs and coworkers showed that CB[7] could promote the trans→cis isomerization of 4,4'-diaminoazobenzene by overcompensating the higher stability of the free trans isomer, at least between pH 3 and 6.282 They also described the impact of CB[8] on the ratios of N-substituted ureas conformers, such as guest 76, and note for example that in the presence of a stoichiometric amount of CB[8], the (E,E)-76b·CB[8] adduct is formed exclusively.283 In addition, Isaacs and coworkers beautifully showed that triazene-arylene 41, as well as a few analogs, exclusively adopt (1) the fully anti conformation in the cavity of CB[10] (see guest a,a,a,a-41a in Fig. 10, section 4.2.1(f)), (2) the anti-anti-anti-syn conformation inside CB[8] (guest a,a,a,s-41b in Fig. 10), and (3) the anti-syn-syn-anti conformation when interacting with two CB[7] units!234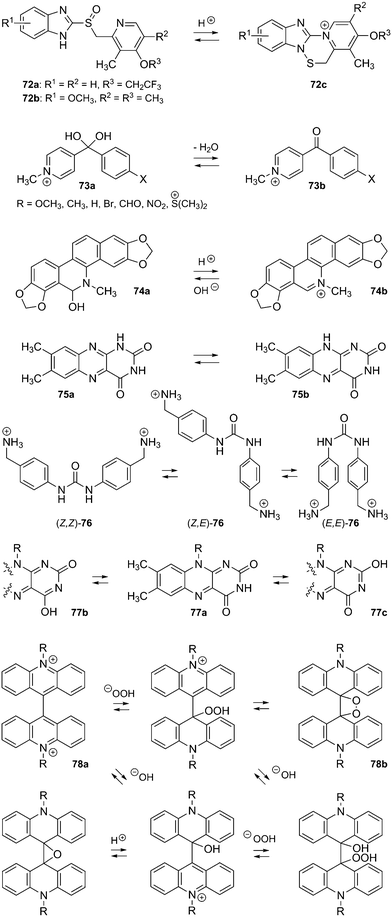
Choudhury and Pal showed that the equilibrium between lactam 77a and lactims 77b and 77c was shifted towards the lactims in the presence of CB[7], despite all three species only forming exclusion complexes with CB[7]. Hydrogen bonding between the hydroxy groups of the lactims and the CB[7] rim may be responsible for this effect.284 Finally, our group has just reported that CB[7] could stabilize positively charged lucigenin derivatives 78a to the expense of the corresponding neutral 1,2-dioxetanes 78b in a complex network of equilibria promoted by the addition of hydrogen peroxide. 1,2-Dioxetanes 78b undergo a chemiluminescent degradation pathway, which can therefore be interrupted or dimmed in the presence of CB[7].285
000-fold rate increase was observed relative to the cycloaddition in the absence of CB[6].167,286 This reaction, which allows the preparation of [2]rotaxanes with no possibility for CB[6] to escape as long as bulky stoppers are connected to the ammonium groups, has been applied on numerous occasions to the design of complex interlocked systems, in particular by Steinke and Tuncel.11,168,170,174,287–291
In 2001, Kim reported that two equivalents of (E)-diaminostilbene dihydrochloride 79 could be encapsulated by CB[8], and that irradiation at 300 nm during 30 min triggered [2 + 2] cycloaddition and the formation of cyclobutane derivative 80 (syn/anti ratio > 95:5);292 the reaction was found to be much slower within γ-CD with a poorer stereoselectivity (72 h, syn/anti ratio 4:1),293 and in the absence of any macrocycle, isomerization to (Z)-diaminostilbene was the main reaction.294 Ramamurthy reported a similar [2 + 2] cycloaddition with trans-1,2-bis(4-pyridyl)ethylenes 81a, and in the presence of CB[8], cyclobutane derivative 82 was obtained in 90% yield. When the reaction was carried out with CB[7], cis-1,2-bis(4-pyridyl)ethylene 81b, 2,9-phenanthroline 83 and hydration product 84 were obtained in a 67:12:21 ratio, while in the absence of macrocycle, the ratio was 17:5:78. 2- and 3-Pyridyl derivatives, as well as trans-n-stilbazoles afforded mostly the syn [2 + 2] cycloaddition products in the presence of CB[8] and the cis-1,2-bis(pyridyl)ethylene or cis-stilbazoles without any macrocycle.295 The product distributions of the [2 + 2] cycloaddition of unsymmetrical azastilbenes were later studied.296 Ramamurthy also reported the dimerization of trans-cinnamic acids 85 in the absence and presence of CB[8], both in solution and in the solid state (i.e. upon grinding of the reaction partners). Irradiation of trans-cinnamic acids 85 in water without macrocycle, or with CB[7], triggered isomerization to their cis analog, while irradiation of their crystals afforded the anti head-to-tail dimer 86b, if a reaction takes place at all. The same reaction carried out in the presence of CB[8] afforded a mixture of cis-cinnamic acid and syn head-to-head dimer 86a. When the reaction was carried out in the solid state, the anti head-to-tail isomer 86b could also be detected in some cases.297,298 Sivaguru showed that coumarins 87 could undergo [2 + 2] cycloaddition when a pair is encapsulated into CB[8], and syn head-to-head and head-to-tail adducts 88a and 88b are usually the major products.299,300 The formation of the ternary complex is likely the rate determining step of the reaction,301 and excellent syn/anti ratios are obtained with only 10 mol% CB[8].302 In pure water or other organic solvents, various syn/anti ratios were determined, and often, the yield was very poor.
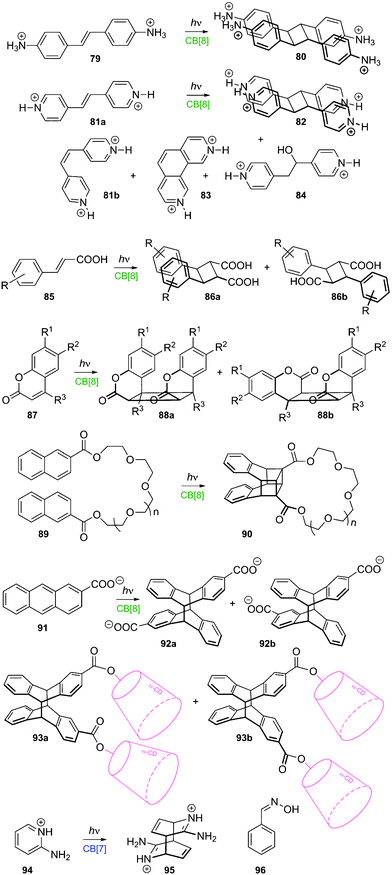
Wu showed that 2-naphthoate 89 could form a 1:1 complex with CB[8] by adopting a looped shape, with both naphthyl units encapsulated. 500 W irradiation at λ > 280 nm for 12 min triggered a [4 + 4] cyclodimerization, which afforded cubane-like product 90 in a 96% yield, while no adduct was formed in a host-free environment.303 Methyl and ethyl 2-naphthoate,304 as well as 2-cyanonaphthalene,305 could also be photodimerized upon encapsulation with CB[8], much faster than in the absence of the macrocycle (no reaction was observed with 2-cyanonaphthalene in pure water). Kim and Inoue reported the CB[8]-mediated [4 + 4] photocyclodimerization of 2-anthracene carboxylate 91, linked (or not) to α-CD. While CB[8] encapsulation does not significantly effect the distribution of syn/anti, head-to-head/head-to-tail adducts with 2-anthracene carboxylate (head-to-tail isomers 92a and 92b are the major products), dimerization of the α-CD-linked derivative in the presence of CB[8] affords almost exclusively the head-to-head isomers 93a and 93b, while the corresponding head-to-tail products are obtained when the anthracene units are bound to γ-CD!306 This example illustrates how interactions remote from the reaction sites can dramatically affect the stereochemistry of photoreactions in confined spaces. A recent extension by Inoue shows that when the anthracene carboxylate units are connected to the same chiral anchor (α and β-CD), and the cycloaddition is promoted by double encapsulation of the aromatic units into CB[8], syn- and anti head-to-head dimers can be respectively obtained with excellent yields and enantioselectivity.307 Finally, Macartney reported a unique example of a CB[7]-mediated [4 + 4] photodimerization of the 2-aminopyridinium cation (94), which affords exclusively the anti-trans adduct 95 in 90% after irradiation at 365 nm during 21 h. In a host-free medium, a 4:1 mixture of anti-trans and syn-trans isomers is obtained, and the conversion is twice slower.308
Compared to the number of reported CB[n]-promoted photoreactions, rationalized examples of CB[n]-catalyzed reactions that are not triggered by irradiation are still scarce, despite an early take-off with the [3 + 2] azide-alkyne cycloaddition described above. Nau recently showed that CB[n]s can catalyze the hydrolysis of amides, carbamates and oximes with acceleration factors ranging from 4 to 285, as long as substituents are chosen judiciously, and the reactive units are positioned close to the CB[n] rim.309 The macrocycle then facilitates the protonation of the reacting unit due to favorable interactions between the positively charged guest and the carbonylated rim of CB[n]s (another case of a CB[n]-mediated pKa shift). In the case of benzaldoxime (96), a 10-fold acceleration was observed with only 10 mol% CB[7], at least at low conversion (< 30%); however, the hydrolysis product, benzaldehyde, displays a stronger binding affinity towards CB[7] than the oxime, and acts as a catalyst poison.309 The same author also showed that the formation of sulfenamide 72c (see previous section) from benzimidazoles 72a and 72b can be catalyzed by CB[7], which enhances the basicity of the encapsulated benzimidazole units, increases the concentration of their protonated forms, and thereby accelerates the intramolecular nucleophilic attack by the neighboring pyridine ring.158 García-Río reported the CB[7]-catalyzed hydrolysis of benzoyl chlorides, as long as those are substituted with electron-donating groups (acceleration factors up to 5.5-fold in the case of p-methoxybenzoyl chloride). Using Hammett plots, the authors determined that the mechanism was dissociative (i.e. SN1-like; ρ+ = −3.1), and that the favorable interaction between the partially negative CB[7] portal and the acylium cation developing at the transition state decreased the activation barrier of the reaction.310 Finally, we note that rate enhancements of the oxidation of aryl and allyl alcohols to the corresponding aldehydes with hypervalent o-iodoxybenzoic acid (IBX) have also been reported in the presence of CB[8].311
A few organometallic reactions promoted or catalyzed by CB[n]s have been published recently. Demets showed that pentane, unlike cyclohexane, cyclooctene or styrene, could be oxidized to a mixture of 2-pentanol, 2-pentanone and 3-pentanone upon treatment with hydrogen peroxide or iodosylbenzene in the presence of an oxovanadium/CB[6] complex and various solvents; the author proposed that CB[6] was responsible for the selectivity, since only pentane could readily enter the cavity of the macrocycle.312 The Ru(II)-catalyzed reduction of aldehydes to the corresponding alcohols was also reported to be facilitated in the presence of CB[6], but no mechanism was suggested.313 Our group has recently published the first mechanistically rationalized case of an organometallic reaction catalyzed by CB[n]s.314 We found that CB[6], CB[7] and CB[8] could catalyze the Ag(I)-promoted desilylation of trimethylsilylalkynyl derivative 97, and proposed the following mechanism (see Fig. 22): (1) a fraction of guest 97 interacts with CB[n] (depending on the size of the macrocycle, different binding sites are targeted; in the case of CB[7], the trimethylsilyl unit is encapsulated, but the mechanism is valid for all CB[n]s); (2) Ag cations form π-complex Ag·97 ⊂ CB[7], since favorable interactions between silver and the oxygen lone pairs of the CB[n] portal can stabilize the complex; (3) assembly Ag·97 ⊂ CB[7] undergoes a nucleophilic substitution when water displaces the trimethylsilyl unit (water probably crosses the CB[7] portal and reaches the interior of the cavity before displacing the trimethylsilyl group); products of the substitution are CB[7]-bound trimethylsilanol-d1, deuterium cations and presumably alkynylsilver 98;315 (4) alkynylsilver 98 is hydrolyzed in the presence of D+, and phenylacetylene derivative 99 is obtained quantitatively, while CB[7] liberates trimethylsilanol-d1, and can interact with guest 97 as a new cycle begins.314
![Plausible cycle for CB[n]-catalyzed desilylations in the presence of Ag(i) salts.314](/image/article/2012/RA/c1ra00768h/c1ra00768h-f22.gif) | ||
| Fig. 22 Plausible cycle for CB[n]-catalyzed desilylations in the presence of Ag(I) salts.314 | ||
While not an organometallic reaction per se, the photolysis of azoalkanes 100a and 101a encapsulated within CB[7] was found to be significantly affected by metallic cations (the latter interact with the CB[7] portal and the nitrogen atoms of the azoalkanes).316 The reactions, reported by Nau and coworkers, were carried out in a biphasic mixture of water and pentane, from which the products were collected and analyzed by gas chromatography. Remarkably, some metals had a significant effect on the product distribution: in the presence of Tl(I), Fe(III), Co(II), Ni(II), Cu(II) and Ag(I), photolysis of CB[7]-bound azoalkane 100a afforded a mixture of bicyclo[2.2.0]hexane (100b) and 1,5-hexadiene (100c) in an averaged ratio of 13:87, while 30:70 ratios were obtained with free azoalkane 100a (the products are formed after ring closure and opening of the 1,4-cyclohexadiyl biradical). CB[7] encapsulation thus tends to favor reactions from the triplet excited state by selective metal-induced intersystem crossing. In the case of azoalkane 101a, all conditions afforded exclusively housane (101b), with one amazing exception: in the presence of Ag(I), a 59:41 mixture of housane (101b) and cyclopentene (101c) was obtained.316 The authors proposed that CB[7]-bound Ag(I) triggers a one-electron oxidation of singlet azoalkane 101a; the resulting radical cation would afford a 1,3-cyclopentanediyl radical cation upon nitrogen elimination, and cyclopentene would be formed after subsequent rearrangement.317
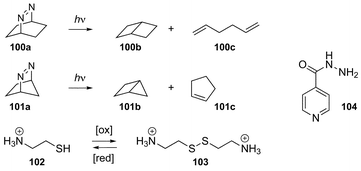
While catalysis with CB[n]s shows signs of a very promising future, the opposite effect, namely reaction inhibition or retardation by CB[n]s, should not be overlooked. Such reactions illustrate that CB[n]s may be used as “protecting groups” in organic synthesis. A remarkable example by Macartney is the inhibition of hydrogen/deuterium exchange at the C(2) positions of bis-imidazolium 36 upon interaction with CB[7]. Rate retardation reaches 1.3 × 103, which translates into a 3.1 pKa shift at the C(2) position, from 22.3 to 25.4! The author attributes the extra basicity to C(2)–H/OC hydrogen bonding interactions between the guest and the CB[7] portal.318 García-Río reported that the solvolysis of 1-bromoadamantane and electron-poor benzoyl chlorides were slowed down by 103- and 102-fold, respectively, when bound to CB[7].310 Kaifer showed that CB[6] encapsulation of cysteamine (102) completely inhibits its oxidation to cystamine (103) with Fe(III) chloride; only trace amounts of cystamine (103) were detected when oxidation was carried out with oxygen or chloropicrin (in a host-free medium, reactions are complete after 7 h, 3 h and 40 min, respectively). CB[6] also totally inhibits the reduction of cystamine (103) to cysteamine (102) when treated with dithiothreitol, while the same reaction without CB[6] is completed in 1 h.319 Tao and coworkers have recently reported that acylation of the anti-tuberculosis drug isoniazid (104), a hydrazide, could be slowed down by 5.5–77 times in the presence of CB[6] or CB[7] and various acetylating agents. Interestingly, the mode of interaction (exclusion complex in the case of CB[6] and encapsulation with CB[7]) does not have a significant effect on the inhibition.320 Finally, Biczók showed that the alkanolamine form of sanguinarine 74a could be protected against photooxidation with oxygen if surrounded by CB[7].280
5.4 CB[n]s as key units for drug delivery
The lack of target specificity and solubility of hydrophobic drugs in biological medium are serious impediments to the treatment of various pathologies, including cancer. These problems can be circumvented in part by using drug carriers such as nanoparticles, micelles, liposomes, carbon nanotubes, quantum dots, as well as amphiphilic macrocycles like cyclodextrins.321–324 It is thus not surprising that CB[n]s have been considered as potent drug delivery vehicles during the past few years.Although CB[n]s are virtually non-toxic, they readily cross cell membranes, as shown by Scaiano and García in the case of mouse embryonic 3T3 cells; cell penetration was monitored by fluorescence using CB[7]- and CB[8]-bound acridine orange and pyronine Y dyes.183 Similarly, CB[7] is also internalized by murine macrophage RAW264.7 cells.60 The effect of CB[n]s on a series of antitumoral platinum complexes has been studied on several occasions, in particular by Wheate, Day and Collins.61,325–330 CB[n] encapsulation usually inhibit the degradation of the bioactive agents, and has a weak positive or negative effect on their cytotoxicity, depending on the size of the macrocycle and the nature of the platinum complex. Day and Collins also showed that CB[7] had little impact on the biological properties of albendazole, an anti-cancer agent plagued by its very low solubility in aqueous medium. However, CB[6]- or CB[7]-encapsulation enhanced its solubility by 2000-fold!331 Similar conclusions were reached with an albendazole derivative,332 as well as the anti-cancer drug camptothecin.333 Nakamura showed that CB[6] had a significant effect on selected enzymatic reactions of DNA. For example, the topoisomerization of a supercoiled plasmid catalyzed by calf thymus topoisomerase I is markedly accelerated in the presence of CB[6]-bound spermidine, compared to the free polyamine; to the contrary, the hydrolysis of the plasmid by the endonuclease BanII is slower in the presence of CB[6]-bound spermine compared to the free axle.334 Finally, in a recent article, Isaacs and Rotello showed for the first time that cytotoxicity of a bioactive agent could be regulated by CB[7] encapsulation.335 Functionalized gold nanoparticles, which were decorated with a series of hexanediammonium units (AuNP-NH2 in Fig. 23), readily interacted with CB[7] (approximately 40 macrocycles around each nanoparticle, overall diameter 12 nm; see assemblies AuNP-NH2-CB[7] in Fig. 23), and the large CB[n] units efficiently shielded the gold cores. After 3 h of incubation in the presence of human breast cancer cells MCF-7, the assemblies were internalized, and remained trapped within endosomes even after 24 h, with no toxicity observed at concentrations lower than 50 μM. To the contrary, CB[7]-free particles were released into the cytosol, causing apoptosis at a 1.3 μM IC50 value (34% cell survival at 2 μM after 24 h). Subsequent incubation of the cells containing the CB[7]-protected assemblies with adamantylammonium (9b; 0.40 mM) led to the capture of CB[7] by the competitive guest, to the disruption of the endosome membrane by the now CB[7]-free gold nanoparticles, and to cell death (40% cell survival at 2 μM, comparable to the 34% survival obtained in the control experiment)!335
![CB[7]-controlled cytotoxicity of functionalized gold nanoparticles. Reprinted with permission from ref. 335. Copyright 2010 Nature Publishing Group.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f23.gif) | ||
| Fig. 23 CB[7]-controlled cytotoxicity of functionalized gold nanoparticles. Reprinted with permission from ref. 335. Copyright 2010 Nature Publishing Group. | ||
Du and coworkers,336,337 as well as Zink and Stoddart,338–344 showed a series of examples where mesoporous silica nanoparticles (MSNPs) act as drug containers, and CB[n]s as switchable lids that control the release of the bioactive agent. MSNPs MCM-41 (approximately 0.5 μm, containing hexagonally arranged pores with an average diameter of 2 nm) are readily prepared upon hydrolysis of tetraethyl orthosilicate in the presence of cetyltrimethylammonium bromide; they can then be functionalized with side-chains bearing a CB[n] binding site, soaked into a solution of a drug mimic (in these studies, it was replaced with fluorescent dyes such as rhodamine B or calcein for easy monitoring), capped with CB[n]s, and rinsed. The large macrocycles decorating the surface of the MSNPs efficiently prevent the release of the dye (see Fig. 24). Upon pH increase,337,339–342 addition of a competitive guest,336,337 reductive cleavage of a stopper,338 or magnetically induced heating,344 the CB[n] lids are ejected or shuttled away from the nano-reservoirs at a tunable rate, and the dye is released in solution. In two particularly remarkable examples, Zink and Stoddart described (1) MSNPs, which remain closed at pH 6.5–9, but can be opened at high (> 10) or low pH (< 5),341 and (2) MSNPs interlaced with light-switchable cis/trans azobenzene units in their pores, and decorated with CB[6]-binding substituents at their periphery; the adsorbed dye can only be released upon light irradiation, which triggers the trans→cis isomerization of azobenzenes and opens the pores, coupled to a pH increase, which releases CB[6]; the system thus behaves as an AND logic gate.343 Finally, Cheon and Zink prepared MSNPs containing zinc-doped iron oxide nanocrystals, and functionalized them with 1,6-hexanediammonium chains; the assemblies were then loaded with doxorubicine, capped with CB[6] and internalized into breast cancer cells MDA-MB-231.344 Upon application of an oscillating magnetic field, the nanoparticles generated some local heat, which facilitated the ejection of CB[6] and the release of doxorubicine (see Fig. 24). A 37% cell death was observed after 5 min of magnetic field exposure when the particles were loaded with the cytotoxic agent, vs. 16% with unloaded assemblies, thereby indicating that both hyperthermia and drug release induced cell death.344
![Magnetic MSNPs filled with doxorubicine, linked to 1,6-hexanediammonium chains, and capped with CB[6]. Local heating by an oscillating magnetic field triggers the ejection of CB[6] and the subsequent release of the drug. Reprinted with permission from ref. 344. Copyright 2010 American Chemical Society.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f24.gif) | ||
| Fig. 24 Magnetic MSNPs filled with doxorubicine, linked to 1,6-hexanediammonium chains, and capped with CB[6]. Local heating by an oscillating magnetic field triggers the ejection of CB[6] and the subsequent release of the drug. Reprinted with permission from ref. 344. Copyright 2010 American Chemical Society. | ||
We finally note that the interactions between biologically relevant molecules and CB[n]s have been evaluated on various occasions. Examples include the common fluorescent stain 4',6-diamidino-2-phenylindole,345 vitamin B12,346 a series of fungicidal and anthelmintic benzimidazoles,347 alkaloids palmatine,348 dehydrocorydaline348 and berberine,349,350 antituberculosis drugs pyrazinamide and isoniazid,351β-blocker atenolol, antidiabetic glibenclamide, mydriatic tropicamide,352 the Alzheimer's NMDA glutamate receptor drug memantine, the well-known analgesic paracetamol,353 as well as the anesthetics procaine, prilocaine, tetracaine, procainamide and dibucaine.162
5.5 CB[n]s in nano and advanced materials
In addition to the medicinal applications described above, CB[n]s have been incorporated into promising novel materials. In the following section, we describe the preparation and properties of CB[n]-containing polymers (section 5.5.1), dendrimers (section 5.5.2), metallic nanoparticles (section 5.5.3), fullerenes (section 5.5.4), nanosheets, vesicles, films and surfaces (sections 5.5.5 and 5.5.6) and hydrogels (section 5.5.7).CB[n]s can also be used to connect chains together either covalently or non-covalently, by exploiting two remarkable properties of the macrocycles: (1) CB[6] can catalyze the 1,3-dipolar addition between an alkyne and an azide, and (2) CB[8] can encapsulate two guests in its cavity. Steinke showed that polymer 107 could be obtained upon reaction of an equimolar mixture of dialkyne 105 and diazide 106 in the presence of an excess amount of CB[6].287 Scherman reported that a 5000 g mol−1 MV-terminated poly(ethylene glycol) monomethyl ether chain could be “connected” to a 5000 g mol−1 2-naphthoxy-terminated analog (or to 10500 g mol−1 2-naphthoxy-terminated cis-1,4-poly(isoprene)) by using CB[8], since the macrocycle encapsulates both the electron-deficient MV and the electron-rich naphthol. In the case of the poly(isoprene) derivative, micelles (approximately 250 nm) were likely formed upon connection to the MV unit, as assessed by dynamic light scattering experiments.358 Using the same strategy, these authors could prepare an “ABA” triblock copolymer from two equivalents of a polymer linked to an electron-rich unit (fragment A), one equivalent of an MV dimer (the two MV units being linked via a short triethylene glycol spacer; fragment B), and two equivalents of CB[8]. The elongation process and the gain in molecular weight were monitored using diffusion ordered spectroscopy experiments, and by measuring solution viscosities.359 Scherman also showed that the critical solution temperature of poly(N-isopropylacrylamide) linked to an electron-rich dibenzofuran end group could be increased by 5.7 °C (from 24.5 to 30.2 °C) upon addition of CB[8]-bound MV; the effect is due to an increase in hydrophilicity at the polymer terminus upon encapsulation with CB[8]. The process is fully reversible after addition of a competitive guest such as the adamantylammonium cation (9b).360 Such thermoresponsive materials may be particularly appealing to the design of devices for stimulus-controlled drug delivery. The same author also reported the preparation of 3D viscoelastic polymeric networks from styrene/acrylamide copolymer 108a decorated with MV units, 2-naphthoxy-substituted acrylamide copolymer 108b and CB[8] (see Fig. 25). A spectacular increase in viscosity (up to 103-fold) was observed at concentrations as low as 5% in water with a substoichiometric amount of CB[8] (0.50 equivalent), accompanied by the usual absorbance in the visible range due to charge transfer interactions between the electron-rich and electron-poor CB[8] guests (see Fig. 25, vial d and figures e and f).361
![CB[n]-assisted formation of polymers and 3D polymeric networks. Solutions (5 wt%) of (a) copolymer 108a, (b) copolymer 108b, (c) a 1 : 1 mixture of copolymers 108a and 108b; (d) hydrogel formed upon addition of 0.50 equivalent CB[8] (5% cross-linking) to solution (c). (e) Scanning electron microscopy (SEM) image of the cryo-dried hydrogel. (f) Cartoon illustrating the supramolecular structure of the 3D polymeric network. (a)–(f): Reprinted with permission from ref. 361. Copyright 2010 American Chemical Society.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f25.gif) | ||
| Fig. 25 CB[n]-assisted formation of polymers and 3D polymeric networks. Solutions (5 wt%) of (a) copolymer 108a, (b) copolymer 108b, (c) a 1:1 mixture of copolymers 108a and 108b; (d) hydrogel formed upon addition of 0.50 equivalent CB[8] (5% cross-linking) to solution (c). (e) Scanning electron microscopy (SEM) image of the cryo-dried hydrogel. (f) Cartoon illustrating the supramolecular structure of the 3D polymeric network. (a)–(f): Reprinted with permission from ref. 361. Copyright 2010 American Chemical Society. | ||
Zhang and coworkers described the formation of a polymer from CB[8] and axle 109. Connections between monomers are reinforced with a double charge transfer interaction between anthracene and MV units surrounded by two CB[8] macrocycles (see Fig. 26)!362 The obtained polymer (hydrodynamic radius 30 nm) is not as flexible as traditional polymer chains; also the small segment elasticity shows that the chains are easily lengthened as springs. When the concentration of monomer 109 is increased to 4.0 mM, a deep purple gel is obtained upon CB[8]-assisted polymerization; addition of potassium cations disrupts the interactions between the polymer chains and the gel collapses.362
![Formation of a polymer from monomer 109, stabilized by double charge transfer interaction and double CB[8] encapsulation.362](/image/article/2012/RA/c1ra00768h/c1ra00768h-f26.gif) | ||
| Fig. 26 Formation of a polymer from monomer 109, stabilized by double charge transfer interaction and double CB[8] encapsulation.362 | ||
Finally, several polymeric systems bearing CB[n] units bound to side chains have been reported. For example, Kim described the preparation of a polyacrylamide derivative decorated with spermidine side chains that can interact with CB[6]. Complexation with the macrocycle caused a dramatic increase in the polymer thermal stability (330 °C vs. 150 °C).363 Polyethylene derivatives decorated with CB[6]-bound 1,6-bis(pyridyl)hexane units,364 or CB[7]-bound MV groups,365 as well as copolymers of acrylamide and CB[6]-bound butylammonium methacrylate366 were also prepared. Scherman reported the preparation of a dynamic adduct between a methacrylate copolymer decorated with 2-naphthoxy groups, CB[8] and an α-mannoside viologen; the assembly could self-organize to target Concanavalin A, a tetrameric lectin that interacts specifically with mannose.367 Kaifer showed that p-xylylene sulfonium salt 110 binds extremely strongly to CB[7] (binding affinity 4.0 × 1010 M−1), and since it is a precursor to poly(phenylenevinylene) conducting polymers (PPV) via the Wessling route and intermediate 111 (see Fig. 27),368 the authors were hoping to incorporate CB[7] rings along the polymeric chain. Attempts were fruitless, probably because CB[7] overstabilizes precursor 110 and prevents polymerization to intermediate 111.369 However, addition of CB[7] to axle 111 triggered the encapsulation of the diethylsulfonium substituents, and allowed the formation of PPV (112) upon heating as well as the release of CB[7]-bound diethyl sulfide, which remains close to the PPV main chain. CB[7] was also found to considerably enhance the rate of diethyl sulfide elimination.369
![Preparation of CB[7]-coated poly(phenylenevinylene) (PPV) via the Wessling route.369](/image/article/2012/RA/c1ra00768h/c1ra00768h-f27.gif) | ||
| Fig. 27 Preparation of CB[7]-coated poly(phenylenevinylene) (PPV) via the Wessling route.369 | ||
Kaifer also reported the formation of a binary complex between an MV2+-linked dendrimer and CB[8], and of a ternary complex between two of these dendrimers and CB[8] upon reduction.373 The same group later showed that a ternary complex could be formed between CB[8] and two dendrimers linked to MV2+ and p-dialkoxybenzene units, respectively. Reduction again induced a reorganization of the assembly, with both MV+ radical cations encapsulated into CB[8], and the two identical dendrons dangling at its periphery.217 Finally, Li and coworkers showed that electron transfer from the outer naphthyl units of a dendrimer to a molecule of anthracenecarboxylic acid buried within the dendrimer is hampered by naphthyl-naphthyl interactions, which cause excimer formation and self-quenching; however, upon encapsulation (and isolation) of the naphthyl units by CB[7], the fluorescence quantum yield could be enhanced by up to 100%.377
![Transition electron micrographs (TEM) of Au/CB[5] NPs with CB[5]/Au ratios of (a) 0.0, (b) 0.10, (c) 0.20, (d) 0.50, (e) 1.0 and (f) 1.0 with CB[5] being added after the reduction of tetrachloroauric acid (scale bar: 20 nm).381](/image/article/2012/RA/c1ra00768h/c1ra00768h-f28.gif) | ||
| Fig. 28 Transition electron micrographs (TEM) of Au/CB[5] NPs with CB[5]/Au ratios of (a) 0.0, (b) 0.10, (c) 0.20, (d) 0.50, (e) 1.0 and (f) 1.0 with CB[5] being added after the reduction of tetrachloroauric acid (scale bar: 20 nm).381 | ||
Silver nanoparticles (Ag NPs) can also be prepared and stabilized in the presence of CB[n]s, as our group has recently reported. We showed that 5.3 and 3.7 nm monocrystalline Ag NPs could be prepared upon reduction of silver nitrate with sodium borohydride in the presence of CB[7] and CB[8], respectively (see micrographs in Fig. 29c, e, f and i); solutions of these particles, which display a characteristic SPR band at approximately 415 nm (Fig. 29j), have remained stable during at least 10 months.102 To the contrary, CB[5] and CB[6] induced rapid aggregation and sedimentation. Based on calculations, we proposed that CB[n]s interact with Ag NPs via their carbonylated portal, and that the entropically favorable macrocyclic effect (i.e. the greater affinity of a guest towards a cyclic host bearing n identical binding sites, compared to its affinity towards the n separate fragments)385 enhances the Ag-carbonyl interactions. However, in the case of CB[5] and CB[6], the 5 (or 6) oxygen atoms may not be ideally positioned for Ag binding (an enthalpic impediment). Also, the proximity of the 5 (or 6) partially negative oxygen atoms may enhance partially positive mirror charges on several adjacent silver atoms at the metal surface; such a possible ligand-induced repulsive Ag–Ag interaction may in fact prevent the ligand from properly interacting with the NPs. When the larger and more flexible CB[7] and CB[8] are used, their carbonyl oxygens may better adapt to the structural and electronic requirements of the Ag surface, and the entropic gain attributed to the macrocyclic effect would overcompensate the enthalpic penalty for a slight deformation of the CB[n] unit.
![TEM of (a) Ag/CB[5], (b) Ag/CB[6], (c) Ag/CB[8], (d) Ag/CB[7] (0.10 equiv. CB[7]), (e) Ag/CB[7] (0.50 equiv. CB[7]), (f) Ag/CB[7] (1.0 equiv. CB[7]), (g) Ag/CB[7] (2.0 equiv. CB[7]) and (h) Ag/CB[7] (5.0 equiv. CB[7]). (i) High-resolution TEM of Ag/CB[7] NPs (1.0 equiv. CB[7]). (j) UV-Vis spectra and photographs of Ag/CB[7] assemblies, as represented in TEM (d)–(h). Solutions and suspensions were diluted 10 times immediately before UV-Vis analysis, and suspension (d) was stirred. 1.0 equiv. CB[n] was used in samples (a)–(c). Reprinted with permission from ref. 102. Copyright 2011 American Chemical Society.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f29.gif) | ||
| Fig. 29 TEM of (a) Ag/CB[5], (b) Ag/CB[6], (c) Ag/CB[8], (d) Ag/CB[7] (0.10 equiv. CB[7]), (e) Ag/CB[7] (0.50 equiv. CB[7]), (f) Ag/CB[7] (1.0 equiv. CB[7]), (g) Ag/CB[7] (2.0 equiv. CB[7]) and (h) Ag/CB[7] (5.0 equiv. CB[7]). (i) High-resolution TEM of Ag/CB[7] NPs (1.0 equiv. CB[7]). (j) UV-Vis spectra and photographs of Ag/CB[7] assemblies, as represented in TEM (d)–(h). Solutions and suspensions were diluted 10 times immediately before UV-Vis analysis, and suspension (d) was stirred. 1.0 equiv. CB[n] was used in samples (a)–(c). Reprinted with permission from ref. 102. Copyright 2011 American Chemical Society. | ||
We also showed that the optimal silver nitrate/CB[7] ratio for the formation of stable Ag NPs (Ag concentration 1.0 mM) is 1:1 – 2:1, while large excesses or low substoichiometric amounts of CB[7] trigger aggregation (see Fig. 29). Surprisingly, masking the portals of CB[7] by encapsulating a guest substituted with bulky groups had only a minor effect on the stability of the Ag NPs; in this case, CB[7] probably interacts with Ag via a fraction of its carbonyl oxygen atoms.102 Similarly to Au NPs, Geckeler showed that narrowly-dispersed Ag NPs (approximately 6 nm) could be prepared upon reaction of silver nitrate with sodium hydroxide in the presence of CB[7]; again the mechanism is unclear. These Ag NPs were cytotoxic towards two cancer cell lines (MCF-7 and NCI-H358) at approximately 10 μg mL−1 after a 24 h incubation period.386
De la Rica and Velders recently reported the formation of nanopore assemblies upon combination of silver nitrate (0.10 mM) with CB[7] (1.0 mM). Addition of thioacetamide as a sulfide source triggered the formation of silver sulfide nanoclusters (approximately 20 nm), whose size matches well the dimensions of the nanopore assemblies (i.e. the silver sulfide nanoclusters grow in the nanopores). Using high-resolution TEM, the authors showed that the nanoclusters were composed of perfectly aligned nanocrystals (< 1 nm)!387 Recently, Demets prepared lead iodide nanodisks (5–34 nm wide, 0.7 nm thick) upon reaction of potassium iodide (2.0 mM) with a 1:1 mixture of CB[7] and lead nitrate (1.0 mM) and sedimentation over two days.388 Cao prepared 3 nm palladium NPs upon treatment of palladium chloride with sodium borohydride in the presence of CB[6]; depending on the Pd/CB[6] ratio, contamination with triangular and cubic assemblies took place. These Pd NPs were found to be efficient catalysts in the Suzuki–Miyaura coupling of aryl halides (including less reactive aryl chlorides) with arylboronic acids (0.50 mol% Pd/CB[6] in a 1:1 ethanol-water mixture); the catalyst could be recovered easily, afforded high yields even after 5 cycles, and could be stored under aerobic conditions.389
:1 complex with CB[7] upon addition of C60 to an alkaline solution of CB[7] and stirring during 24 h; unlike C60 which is soluble in toluene, the dark brown complex was insoluble in all common organic solvents and acidic solutions. Alternatively, C60 and CB[7] can be grinded using a mixer mill during 4 h, and the resulting powder washed with water (pH 12) and toluene to eliminate the unreacted partners. Although the structure of the assembly remains unclear, CB[7] may interact with the two fullerenes via its carbonylated portals.390–392 A similar 2:1 complex could also be prepared using CB[8] instead of CB[7].393 Finally, Ogoshi reported that single-walled carbon nanotubes (SWCNTs) could interact with CB[7] upon addition of the macrocycle (3.5 mM) to a suspension of SWCNTs in water (0.20 mg mL−1) and sonication during 3 h. Approximately 80% of the SWCNTs were then removed by centrifugation, leaving a homogeneous black supernatant that remained stable for over a month (while raw SWCNTs are insoluble in water)! Addition of 1-adamantylamine (9b), which binds strongly to CB[7], triggered aggregation, thereby confirming the interaction between the macrocycle and SWCNTs. Even more surprisingly, and contrary to CB[7], CB[5] did not improve the solubility of the SWCNTs.394
![TEM images of (a) CB[8]/quinoline and (b) CB[8]/naphthalene nanosheets. (c) STM image, and (d) proposed molecular packing of the nanosheets.395](/image/article/2012/RA/c1ra00768h/c1ra00768h-f30.gif) | ||
| Fig. 30 TEM images of (a) CB[8]/quinoline and (b) CB[8]/naphthalene nanosheets. (c) STM image, and (d) proposed molecular packing of the nanosheets.395 | ||
Zhang and Zhou recently showed that the critical aggregation concentration of axle 114 is greatly reduced once surrounded by CB[6] (1.8 × 10−5 and 3.2 × 10−7 M−1, respectively), and that the interlocked assembly could form vesicles (diameter 50–200 nm) that remained stable over a week in aqueous medium. Aggregates of the free guest were much smaller (1–4 nm).396
CB[n]s are also promising tools in the separation and purification of high-value compounds, since they can interact with both the surface of stationary phases and the target molecule. As a proof of concept, Feng and Wu showed that the separation of the ortho, meta and para-isomers of nitrotoluene, nitrophenol, nitrophenolate and nitroaniline by capillary electrophoresis could be greatly improved if CB[7] (5.0 mM, 15% methanol in a phosphate buffer) were adsorbed onto the inner wall of the capillary.407 Using the same method, the very similar aristolochic acids 115a and 115b could be separated with a CB[7]-enriched phosphate buffer (3.0 mM CB[7], 10% aqueous acetonitrile).408 CB[n]s can also be applied to the separation of mixtures of peptides, as recently shown by Scherman and coworkers. In this case, a gold surface was decorated with CB[8]-bound viologen units linked to gold-anchored alkanethiol chains; the modified surface was found to selectively recognize a peptide bearing a Trp residue upon formation of the ternary MV Trp CB[8] complex. The peptide could then be released by electrochemical reduction of MV and isolated.400
![X-Ray structure of CB[7]: organization on (a) the ab plane, and (b) the bc plane. Channels between the macrocycles are filled with water, hydronium cations and sulfate anions. (c) and (d) AFM images of the CB[7] gel on a mica substrate. (e) Structure of the CB[7] hydrogel: from macrocycles to bundles of fibrils. Reprinted with permission from ref. 412. Copyright 2007 John Wiley & Sons.](/image/article/2012/RA/c1ra00768h/c1ra00768h-f31.gif) | ||
| Fig. 31 X-Ray structure of CB[7]: organization on (a) the ab plane, and (b) the bc plane. Channels between the macrocycles are filled with water, hydronium cations and sulfate anions. (c) and (d) AFM images of the CB[7] gel on a mica substrate. (e) Structure of the CB[7] hydrogel: from macrocycles to bundles of fibrils. Reprinted with permission from ref. 412. Copyright 2007 John Wiley & Sons. | ||
Slow cooling of a mixture of CB[7], 1.0 M sulfuric acid and 0.10 equivalent trans-4,4′-diaminostilbene dihydrochloride also affords a white gel; yet upon irradiation, trans→cis isomerization triggers gel-to-sol transition. A subsequent heating/cooling cycle regenerates the gel.412 Tan and coworkers also showed that hydrogels could be obtained upon cooling a 50 °C solution of butylammonium tosylate (1.8–2.5 M) and CB[6] (35–70 mM); gel-to-sol transitions ranged from 16–26 °C, and bundles of fibers were observed by scanning electron microscopy. Those fibers were also composed of fibrils, which were formed by stacking interactions between the tosylate units.413
6. Conclusion and outlook
Thirty years ago, Freeman, Mock and Shih unveiled the structure of CB[6], and noticed that this aesthetically appealing macrocycle could encapsulate alkylammonium cations with high affinity. Only two years later, they reported the remarkable 55000-fold rate enhancement of the [3 + 2] cycloaddition between an alkyne and an azide inside the cavity of CB[6], paving the way for the design of various interlocked systems and molecular switches. However, the poor solubility of the macrocycle and its unwillingness to undergo chemical modification kept the field rather dormant until the beginning of our millennium. The successful preparation of CB[7] and CB[8], as well as the functionalization of CB[6] then triggered a dramatic increase in the number of articles, reviews and patents published every year.
The popularity of CB[n]s is largely due to their outstanding recognition properties, and to the exceptional strength of their interaction with various guests. In fact, CB[n]s should appear at a prominent position in any supramolecular or bioorganic chemistry textbook, since (1) they display the strongest non-covalent interaction ever measured between a host and a single stable guest (up to 5 × 1015 M−1!), and (2) the affinity reaches or even slightly surpasses the strength of the landmark biotin–avidin interaction (1015 M−1). These extreme values are due to a subtle combination of factors, such as (1) the ability of the guests and their substituents sitting close to the CB[n] portals to return as many water molecules as possible to the aqueous environment upon binding (this process is both enthalpically and entropically favorable), (2) the rigidity of the macrocycles coupled to restricted conformational mobility of the free guests, (3) a minimally penalizing loss of solvation energy upon binding, and (4) favorable coulombic interactions between positively charged substituents and the CB[n] rims, as well as multiple hydrogen bonding; we should however point out that the coulombic interaction, which is extreme in the gas phase, is negatively affected in solution by a dramatic loss of solvation energy of the positive guest upon encapsulation. Also, as noted by Nau, it is still unclear to what extent dispersion forces impact the interaction between the very weakly polarizable cavity of CB[n]s and their guests. This contribution is anticipated to be either insignificant or very minor at best.
These recognition properties have allowed the design of new self-organizing and stimulus-controlled supramolecular systems, the use of CB[n]s as removable shields for the controlled release of bioactive agents, and the incorporation of these macrocycles into a series of advanced materials, polymers, nanoparticles, films and hydrogels. The increasing focus on CB[n] applications has been welcomed by Scherman and Nau in a joint address during the 2nd International Conference on Cucurbiturils. Both organizers actually stressed that despite a very successful decade, long-term practical applications of CB[n] chemistry would have to be pursued in order to preserve its momentum; we firmly adhere to that statement.
Still, even thirty years after the elucidation of the CB[6] structure, we like to say that wherever CB[n]s are involved, unexpected and sometimes very unusual results will emerge. This has been true on several occasions during our past three years of excursion into CB[n] chemistry, when for instance, CB[n]s were found to catalyze a silver-promoted desilylation reaction, while we were expecting severe rate retardation (see section 5.3.2). As far as recent, yet to be published results are concerned, there is no end in sight for exciting surprises!
Acknowledgements
This work, as well as the unpublished studies summarized herein, were supported by the Department of Chemistry and Biochemistry, the College of Arts and Sciences and the Vice President for Research at Ohio University. It was also supported in part by an allocation of computing time from the Ohio Supercomputer Center in Columbus. We are particularly grateful to Prof. Yoshihisa Inoue, who shared invaluable thermodynamic data with us, and to Prof. Angel Kaifer, who provided us with a draft of one of his manuscripts. We also thank Prof. Lyle Isaacs, Kimoon Kim, Oren Scherman, Adam Urbach, He Tian, Jinwoo Cheon and Guangtao Li for allowing us to use some of their illustrations, and for providing us with high-resolution images. Finally, we thank Prof. Kimoon Kim, Werner Nau and Oren Scherman for reviewing and endorsing the transcripts of their statements in the introduction and conclusion.References
- R. Behrend, E. Meyer and F. Rusche, Justus Liebigs Ann. Chem., 1905, 339, 1 CrossRef.
- W. A. Freeman, W. L. Mock and N.-Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367 CrossRef CAS.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094 CrossRef CAS.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS.
- A. I. Day, R. J. Blanch, A. P. Arnold, S. Lorenzo, G. R. Lewis and I. Dance, Angew. Chem., Int. Ed., 2002, 41, 275 CrossRef CAS.
- K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
- O. A. Gerasko, D. G. Samsonenko and V. P. Fedin, Russ. Chem. Rev., 2002, 71, 741 RSC.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS.
- O. A. Gerasko, M. N. Sokolov and V. P. Fedin, Pure Appl. Chem., 2004, 76, 1633 CrossRef CAS.
- K. Kim, N. Selvapalam and D. H. Oh, J. Incl. Phenom. Macrocycl. Chem., 2004, 50, 31 CAS.
- T. C. Krasia, S. Khodabakhsh, D. Tuncel and J. H. G. Steinke, Cucurbituril: A Versatile “Bead” for Polyrotaxane Synthesis, in: Macromolecular Nanostructured Materials, ed. N. Ueyama and A. Harada, Springer-Verlag: Berlin Heidelberg, 2004, 78, 41 Search PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC.
- W.-H. Huang, S. Liu and L. Isaacs, Cucurbit[n]urils, in: Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH Verlag GmbH & Co., Weinheim, 2008, 113 Search PubMed.
- L. Isaacs, Chem. Commun., 2009, 619 RSC.
- S. Y. Jon, N. Selvapalam, D. H. Oh, J.-K. Kang, S.-Y. Kim, Y. J. Jeon, J. W. Lee and K. Kim, J. Am. Chem. Soc., 2003, 125, 10186 CrossRef CAS.
- H.-K. Lee, K. M. Park, Y. J. Jeon, D. Kim, D. H. Oh, H. S. Kim, C. K. Park and K. Kim, J. Am. Chem. Soc., 2005, 127, 5006 CrossRef CAS.
- Y. J. Jeon, H. Kim, S. Jon, N. Selvapalam, D. H. Oh, I. Seo, C.-S. Park, S. R. Jung, D.-S. Koh and K. Kim, J. Am. Chem. Soc., 2004, 126, 15944 CrossRef CAS.
- L. Isaacs, S.-K. Park, S. Liu, Y. H. Ko, N. Selvapalam, Y. Kim, H. Kim, P. Y. Zavalij, G.-H. Kim, H.-S. Lee and K. Kim, J. Am. Chem. Soc., 2005, 127, 18000 CrossRef CAS.
- S. Liu, K. Kim and L. Isaacs, J. Org. Chem., 2007, 72, 6840 CrossRef CAS.
- W.-H. Huang, S. Liu, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14744 CrossRef CAS.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2007, 46, 7425 CrossRef CAS.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, Org. Lett., 2008, 10, 2577 CrossRef CAS.
- J. Lagona, J. C. Fettinger and L. Isaacs, J. Org. Chem., 2005, 70, 10381 CrossRef CAS.
- J. Zhao, H.-J. Kim, J. Oh, S.-Y. Kim, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 4233 CrossRef CAS.
- H. Isobe, S. Sato and E. Nakamura, Org. Lett., 2002, 4, 1287 CrossRef CAS.
- A. I. Day, A. P. Arnold and R. J. Blanch, Molecules, 2003, 8, 74 Search PubMed.
- C. A. Burnett, D. Witt, J. C. Fettinger and L. Isaacs, J. Org. Chem., 2003, 68, 6184 CrossRef CAS.
- D. Ma, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2010, 75, 4786 Search PubMed.
- D. Lucas and L. Isaacs, Org. Lett., 2011, 13, 4112 Search PubMed.
- 16 consecutive reviews–Isr. J. Chem., 2011, 51, 487–678.
- R. L. Halterman, J. L. Moore and L. M. Mannel, J. Org. Chem., 2008, 73, 3266 CrossRef CAS.
- A. Thangavel, A. M. M. Rawashdeh, C. Sotiriou-Leventis and N. Leventis, Org. Lett., 2009, 11, 1595 Search PubMed.
- D. Jiao, N. Zhao and O. A. Scherman, Chem. Commun., 2010, 46, 2007 RSC.
- S. Liu, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 16798 CrossRef CAS.
- M. J. Pisani, Y. Zhao, L. Wallace, C. E. Woodward, F. R. Keene, A. I. Day and J. G. Collins, Dalton Trans., 2010, 39, 2078 RSC.
- S. Yi and A. E. Kaifer, J. Org. Chem., 2011 DOI:10.1021/jo2018312.
- S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429 RSC.
- R. V. Pinjari and S. P. Gejji, J. Phys. Chem. A, 2008, 112, 12679 CrossRef CAS.
- R. V. Pinjari, J. K. Khedkar and S. P. Gejji, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 371 Search PubMed.
- V. V. Gobre, R. V. Pinjari and S. P. Gejji, J. Phys. Chem. A, 2010, 114, 4464 CrossRef CAS.
- S. Mecozzi and J. Rebek Jr., Chem.–Eur. J., 1998, 4, 1016 CrossRef CAS.
- W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559 Search PubMed.
- J. Mohanty and W. M. Nau, Angew. Chem., Int. Ed., 2005, 44, 3750 CrossRef CAS.
- C. Marquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 4387 CrossRef.
- A. L. Koner and W. M. Nau, Supramol. Chem., 2007, 19, 55 CrossRef CAS.
- S. Lim, H. Kim, N. Selvapalam, K.-J. Kim, S. J. Cho, G. Seo and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 3352 CrossRef CAS.
- D. Bardelang, K. A. Udachin, D. M. Leek and J. A. Ripmeester, CrystEngComm, 2007, 9, 973 RSC.
- D. Bardelang, K. Udachin, D. M. Leek, J. C. Margeson, G. Chan, C. I. Ratcliffe and J. A. Ripmeester, Cryst. Growth Des., 2011 DOI:10.1021/cg201173j ASAP..
- D. Bardelang, K. A. Udachin, R. Anedda, I. Moudrakovski, D. M. Leek, J. A. Ripmeester and C. I. Ratcliffe, Chem. Commun., 2008, 4927 RSC.
- V. Sindelar, K. Moon and A. E. Kaifer, Org. Lett., 2004, 6, 2665 CrossRef CAS.
- W. Wang and A. E. Kaifer, Supramol. Chem., 2010, 22, 710 Search PubMed.
- E. Masson, unpublished results. Search PubMed.
- T. K. Monhaphol, S. Andersson and L. Sun, Chem.–Eur. J., 2011, 17, 11604 Search PubMed.
- M. Yoon, K. Suh, H. Kim, Y. Kim, N. Selvapalam and K. Kim, Angew. Chem., Int. Ed., 2011, 50, 7870 Search PubMed.
- M. V. Rekharsky, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Supramol. Chem., 2007, 19, 39 CrossRef CAS.
- S. Moghaddam, C. Yang, M. Rekharsky, Y. H. Ko, K. Kim, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2011, 133, 3570 CrossRef CAS.
- S. Liu, C. Ruspic, P. Mukhopadhyay, S. Chakrabarti, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2005, 127, 15959 CrossRef CAS.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037 RSC.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PLoS One, 2010, 5, e10514 CrossRef.
- Y. J. Jeon, S.-Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122 RSC.
- W.-H. Huang, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2008, 130, 8446 CrossRef CAS.
- D. Ma, Z. Gargulakova, P. Y. Zavalij, V. Sindelar and L. Isaacs, J. Org. Chem., 2010, 75, 2934 CrossRef CAS.
- D. Lucas, T. Minami, G. Iannuzzi, L. Cao, J. B. Wittenberg, P. Anzenbacher and L. Isaacs, J. Am. Chem. Soc., 2011, 133, 17966 Search PubMed .
- D. Whang, J. Heo, J. H. Park and K. Kim, Angew. Chem., Int. Ed., 1998, 37, 78 CrossRef CAS.
- X. Feng, X.-J. Lu, S.-F. Xue, Y.-Q. Zhang, Z. Tao and Q.-J. Zhu, Inorg. Chem. Commun., 2009, 12, 849 Search PubMed.
- E. A. Mainicheva, A. A. Tripolskaya, O. A. Gerasko, D. Y. Naumov and V. P. Fedin, Russ. Chem. Bull., 2006, 55, 1566 Search PubMed.
- Y.-M. Jeon, J. Kim, D. Whang and K. Kim, J. Am. Chem. Soc., 1996, 118, 9790 CrossRef CAS.
- J. Heo, J. Kim, D. Whang and K. Kim, Inorg. Chim. Acta, 2000, 297, 307 CrossRef CAS.
- J. Heo, S.-Y. Kim, D. Whang and K. Kim, Angew. Chem., Int. Ed., 1999, 38, 641 CrossRef CAS.
- P. A. Abramov, S. A. Adonin, E. V. Peresypkina, M. N. Sokolov and V. P. Fedin, J. Struct. Chem., 2010, 51, 731 Search PubMed.
- D. G. Samsonenko, M. N. Sokolov, A. V. Virovets, N. V. Pervukhina and V. P. Fedin, Eur. J. Inorg. Chem., 2001, 167 CrossRef CAS.
- E. A. Mainicheva, O. A. Gerasko, L. A. Sheludyakova, D. Y. Naumov, M. I. Naumova and V. P. Fedin, Russ. Chem. Bull., 2006, 55, 267 Search PubMed.
- P. Thuéry, Inorg. Chem., 2010, 49, 9078 CrossRef CAS.
- E. V. Chubarova, M. N. Sokolov, D. G. Samsonenko, C. Vicent and V. P. Fedin, J. Struct. Chem., 2006, 47, 939 CrossRef CAS.
- R. Hernandez-Molina, M. Sokolov, P. Esparza, C. Vicent and R. Llusar, Dalton Trans., 2004, 847 RSC.
- M. N. Sokolov, O. A. Gerasko, D. N. Dybtsev, E. V. Chubarova, A. V. Virovets, C. Vicent, R. Llusar, D. Fenske and V. P. Fedin, Eur. J. Inorg. Chem., 2004, 63 Search PubMed.
- M. N. Sokolov, E. V. Chubarova, K. A. Kovalenko, I. V. Mironov, A. V. Virovets, E. V. Peresypkina and V. P. Fedin, Russ. Chem. Bull., 2005, 54, 615 Search PubMed.
- E. A. Mainicheva, O. A. Gerasko, L. A. Sheludyakova, D. Y. Naumov, I. I. Karsanova, R. R. Amirov and V. P. Fedin, Russ. Chem. Bull., 2006, 55, 1956 Search PubMed.
- O. A. Gerasko, E. A. Mainicheva, M. I. Naumova, O. P. Yurjeva, A. Alberola, C. Vicent, R. Llusar and V. P. Fedin, Eur. J. Inorg. Chem., 2008, 416 CrossRef CAS.
- A. L. Gushchin, B.-L. Ooi, P. Harris, C. Vicent and M. N. Sokolov, Inorg. Chem., 2009, 48, 3832 CrossRef CAS.
- A. G. Algarra, M. N. Sokolov, J. González-Platas, M. J. Fernández-Trujillo, M. G. Basallote and R. Hernández-Molina, Inorg. Chem., 2009, 48, 3639 CrossRef CAS.
- I. Bernal, U. Mukhopadhyay, A. V. Virovets, V. P. Fedin and W. Clegg, Chem. Commun., 2005, 3791 RSC.
- P. Thuéry, Cryst. Growth Des., 2008, 8, 4132 CrossRef CAS.
- P. Thuéry, CrystEngComm, 2009, 11, 1150 RSC.
- P. Thuéry and B. Masci, Cryst. Growth Des., 2010, 10, 716 CrossRef CAS.
- K. Chen, H. Cong, X. Xiao, Y.-Q. Zhang, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, CrystEngComm, 2011, 13, 5105 RSC.
- W. S. Jeon, K. Moon, S. H. Park, H. Chun, Y. H. Ko, J. Y. Lee, E. S. Lee, S. Samal, N. Selvapalam, M. V. Rekharsky, V. Sindelar, D. Sobransingh, Y. Inoue, A. E. Kaifer and K. Kim, J. Am. Chem. Soc., 2005, 127, 12984 CrossRef CAS.
- L. Cui, S. Gadde, W. Li and A. E. Kaifer, Langmuir, 2009, 25, 13763 CrossRef CAS.
- S. Yi, B. Captain, M. F. Ottaviani and A. E. Kaifer, Langmuir, 2011, 27, 5624 Search PubMed.
- D. P. Buck, P. M. Abeysinghe, C. Cullinane, A. I. Day, J. G. Collins and M. M. Harding, Dalton Trans., 2008, 2328 RSC.
- S. Lorenzo, A. Day, D. Craig, R. Blanch, A. Arnold and I. Dance, CrystEngComm, 2001, 3, 230 RSC.
- T. V. Mitkina, D. Y. Naumov, O. A. Gerasko, F. M. Dolgushin, C. Vicent, R. Llusar, M. N. Sokolov and V. P. Fedin, Russ. Chem. Bull., 2004, 53, 2519 Search PubMed.
- T. V. Mitkina, D. Y. Naumov, N. V. Kurat'eva, O. A. Gerasko and V. P. Fedin, Russ. Chem. Bull., 2006, 55, 26 Search PubMed.
- T. V. Mitkina, N. F. Zakharchuk, D. Y. Naumov, O. A. Gerasko, D. Fenske and V. P. Fedin, Inorg. Chem., 2008, 47, 6748 CrossRef CAS.
- E. A. Kovalenko, T. V. Mitkina, O. A. Geras'ko, D. G. Samsonenko, D. Y. Naumov and V. P. Fedin, Russ. J. Coord. Chem., 2011, 37, 161 Search PubMed.
- T. V. Mitkina, D. Y. Naumov, O. A. Gerasko and V. P. Fedin, Inorg. Chim. Acta, 2010, 363, 4387 Search PubMed.
- H.-J. Buschmann, L. Mutihac and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 343 CrossRef CAS.
- E. Blanco, C. Quintana, P. Hernández and L. Hernández, Electroanalysis, 2010, 22, 2123 CrossRef CAS.
- H.-J. Buschmann, E. Cleve, K. Jansen, A. Wego and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 117 CrossRef CAS.
- H.-J. Buschmann, K. Jansen, C. Meschke and E. Schollmeyer, J. Solution Chem., 1998, 27, 135 CrossRef CAS.
- X. Lu and E. Masson, Langmuir, 2011, 27, 3051 CrossRef CAS.
- E. V. Chubarova, D. G. Samsonenko, H. G. Platas, F. M. Dolgushin, A. V. Gerasimenko, M. N. Sokolov, Z. A. Starikova, M. Y. Antipin and V. P. Fedin, J. Struct. Chem., 2004, 45, 1004 CrossRef.
- D. G. Samsonenko, Y. V. Mironov, O. A. Efremova, D. Y. Naumov, O. A. Gerasko, V. P. Fedin, V. E. Fedorov and W. S. Sheldrick, J. Struct. Chem., 2005, 46, S121 Search PubMed.
- X. Fang, P. Kögerler, L. Isaacs, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2009, 131, 432 CrossRef.
- D. G. Samsonenko, O. A. Gerasko, A. V. Virovets and V. P. Fedin, Russ. Chem. Bull., 2005, 54, 1557 Search PubMed.
- Y.-Q. Zhang, Q.-J. Zhu, S.-F. Xue and Z. Tao, Molecules, 2007, 12, 1325 Search PubMed.
- J.-X. Liu, L.-S. Long, R.-B. Huang and L.-S. Zheng, Inorg. Chem., 2007, 46, 10168 CrossRef CAS.
- Y.-Q. Zhang, J.-P. Zeng, Q.-J. Zhu, S.-F. Xue and Z. Tao, J. Mol. Struct., 2009, 929, 167 CrossRef CAS.
- J. Liu, Y. Gu, R. Lin, W. Yao, X. Liu and J. Zhu, Supramol. Chem., 2010, 22, 130 CrossRef CAS.
- P. Thuéry, Inorg. Chem., 2009, 48, 825 CrossRef CAS.
- P. Thuéry, Inorg. Chem., 2009, 48, 4497 CrossRef CAS.
- K. Moon and A. E. Kaifer, Org. Lett., 2004, 6, 185 CrossRef CAS.
- W. L. Mock and N.-Y. Shih, J. Org. Chem., 1986, 51, 4440 CrossRef CAS.
- H.-J. Buschmann, A. Wego, A. Zielesny and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 241 Search PubMed.
- M. V. Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20737 CrossRef CAS.
- S. Ghosh and L. Isaacs, J. Am. Chem. Soc., 2010, 132, 4445 CrossRef CAS.
- N. M. Green, Methods Enzymol., 1990, 184, 51 CAS.
- Y. Pazy, T. Kulik, E. A. Bayer, M. Wilchek and O. Livnah, J. Biol. Chem., 2002, 277, 30892 CrossRef CAS.
- J. Rao, J. Lahiri, R. M. Weis and G. M. Whitesides, J. Am. Chem. Soc., 2000, 122, 2698 CrossRef CAS.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872 CrossRef CAS.
- C.-E. Chang and M. K. Gilson, J. Am. Chem. Soc., 2004, 126, 13156 CrossRef CAS.
- S. Moghaddam, Y. Inoue and M. K. Gilson, J. Am. Chem. Soc., 2009, 131, 4012 CrossRef CAS.
- I. B. Shir, S. Sasmal, T. Mejuch, M. K. Sinha, M. Kapon and E. Keinan, J. Org. Chem., 2008, 73, 8772 CrossRef CAS.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1796 RSC.
- H.-J. Buschmann, K. Jansen and E. Schollmeyer, Thermochim. Acta, 2000, 346, 33 CrossRef CAS.
- Y. Inoue and T. Wada, Adv. Supramol. Chem., 1997, 4, 55 Search PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Microcalorimetry, in: Cyclodextrins and Their Complexes: Chemistry, Analytical Methods, Applications, ed. H. Dodziuk, Wiley-VCH, Weinheim, 2006, 199 Search PubMed.
- Y. Inoue, personal communication. Search PubMed.
- 0.9, 1.0 and 1.1 are standard deviations..
- L. Fusaro, E. Locci, A. Lai and M. Luhmer, J. Phys. Chem. B, 2008, 112, 15014 CrossRef CAS.
- K. Jansen, H.-J. Buschmann, E. Zilobaite and E. Schollmeyer, Thermochim. Acta, 2002, 385, 177 Search PubMed.
- K. Jansen, H.-J. Buschmann, A. Wego, D. Döpp, C. Mayer, H.-J. Drexler, H.-J. Holdt and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 357 Search PubMed.
- X. X. Zhang, K. E. Krakowiak, G. Xue, J. S. Bradshaw and R. M. Izatt, Ind. Eng. Chem. Res., 2000, 39, 3516 CrossRef CAS.
- H.-J. Buschmann, K. Jansen and E. Schollmeyer, Acta Chim. Solv., 1999, 46, 405 Search PubMed.
- H.-J. Buschmann, K. Jansen and E. Schollmeyer, Thermochim. Acta, 1998, 317, 95 CrossRef CAS.
- M. K. Sinha, O. Reany, G. Parvari, A. Karmakar and E. Keinan, Chem.–Eur. J., 2010, 16, 9056 CrossRef CAS.
- H.-J. Buschmann, L. Mutihac and E. Schollmeyer, Thermochim. Acta, 2009, 495, 28 Search PubMed.
- Y. Kim, H. Kim, Y. H. Ko, N. Selvapalam, M. V. Rekharsky, Y. Inoue and K. Kim, Chem.–Eur. J., 2009, 15, 6143 CrossRef CAS.
- H.-J. Buschmann, L. Mutihac and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2006, 56, 363 Search PubMed.
- M. V. Rekharsky, H. Yamamura, C. Inoue, M. Kawai, I. Osaka, R. Arakawa, K. Shiba, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2006, 128, 14871 CrossRef CAS.
- H.-J. Buschmann, L. Mutihac, K. Jansen and E. Schollmeyer, J. Inclusion Phenom. Macrocyclic Chem., 2005, 53, 281 Search PubMed.
- H.-J. Buschmann, L. Mutihac, R.-C. Mutihac and E. Schollmeyer, Thermochim. Acta, 2005, 430, 79 CrossRef CAS.
- H.-J. Buschmann, K. Jansen and E. Schollmeyer, Inorg. Chem. Commun., 2003, 6, 531 Search PubMed.
- H.-J. Buschmann, E. Schollmeyer and L. Mutihac, Thermochim. Acta, 2003, 399, 203 CrossRef CAS.
- X. He, G. Li and H. Chen, Inorg. Chem. Commun., 2002, 5, 633 CrossRef CAS.
- A. G. Grechin, H.-J. Buschmann and E. Schollmeyer, Angew. Chem., Int. Ed., 2007, 46, 6499 Search PubMed.
- V. Wintgens, L. Biczók and Z. Miskolczy, Supramol. Chem., 2010, 22, 612 Search PubMed.
- F. Biedermann, U. Rauwald, M. Cziferszky, K. A. Williams, L. D. Gann, B. Y. Guo, A. R. Urbach, C. W. Bielawski and O. A. Scherman, Chem.–Eur. J., 2010, 16, 13716 CrossRef CAS.
- J.-S. Yu, F.-G. Wu, L.-F. Tao, J.-J. Luo and Z.-W. Yu, Phys. Chem. Chem. Phys., 2011, 13, 3638 RSC.
- H.-J. Kim, W. S. Jeon, Y. H. Ko and K. Kim, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5007 CrossRef.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132 CrossRef CAS.
- D. M. Bailey, A. Hennig, V. D. Uzunova and W. M. Nau, Chem.–Eur. J., 2008, 14, 6069 CrossRef CAS.
- Y. Huang, S.-F. Xue, Z. Tao, Q.-J. Zhu, H. Zhang, J.-X. Lin and D.-H. Yu, J. Inclusion Phenom. Macrocyclic Chem., 2008, 61, 171 Search PubMed.
- M. V. Rekharsky, H. Yamamura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236 RSC.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, Chem.–Eur. J., 2009, 15, 12362 CrossRef CAS.
- N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398 CrossRef CAS.
- A. Praetorius, D. M. Bailey, T. Schwarzlose and W. M. Nau, Org. Lett., 2008, 10, 4089 CrossRef CAS.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan, V. D. Uzunova, W. M. Nau and H. Pal, Chem. Commun., 2008, 3681 RSC.
- R. Wang and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1955 RSC.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2010, 8, 247 RSC.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 11459 CrossRef CAS.
- G. Huber, F.-X. Legrand, V. Lewin, D. Baumann, M.-P. Heck and P. Berthault, ChemPhysChem, 2011, 12, 1053 Search PubMed.
- M. Florea and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 9338 Search PubMed.
- M. V. Rekharsky, H. Yamamura, T. Mori, A. Sato, M. Shiro, S. V. Lindeman, R. Rathore, K. Shiba, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem.–Eur. J., 2009, 15, 1957 CrossRef CAS.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and T. L. Manimaran, J. Org. Chem., 1983, 48, 3619 CrossRef CAS.
- D. Tuncel, N. Cindir and Ű. Koldemir, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 373 CrossRef CAS.
- D. Tuncel, Ö. Özsar, H. B. Tiftik and B. Salih, Chem. Commun., 2007, 1369 RSC.
- D. Tuncel and M. Katterle, Chem.–Eur. J., 2008, 14, 4110 CrossRef CAS.
- L. Liu, N. Zhao and O. A. Scherman, Chem. Commun., 2008, 1070 RSC.
- H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2010, 132, 12200 CrossRef CAS.
- J. Tian, S. Ma, P. K. Thallapally, D. Fowler, B. P. McGrail and J. L. Atwood, Chem. Commun., 2011, 47, 7626 RSC.
- G. Celtek, M. Artar, O. A. Scherman and D. Tuncel, Chem.–Eur. J., 2009, 15, 10360 CrossRef CAS.
- L. Yuan, R. Wang and D. H. Macartney, J. Org. Chem., 2007, 72, 4539 CrossRef CAS.
- J. Yin, C. Chi and J. Wu, Chem.–Eur. J., 2009, 15, 6050 CrossRef CAS.
- M. V. Rekharsky, H. Yamamura, M. Kawai, I. Osaka, R. Arakawa, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Org. Lett., 2006, 8, 815 CrossRef CAS.
- M. E. Haouaj, M. Luhmer, Y. H. Ko, K. Kim and K. Bartik, J. Chem. Soc., Perkin Trans. 2, 2001, 804 RSC.
- M. E. Haouaj, Y. H. Ko, M. Luhmer, K. Kim and K. Bartik, J. Chem. Soc., Perkin Trans. 2, 2001, 2104 RSC.
- L. Liu, N. Nouvel and O. A. Scherman, Chem. Commun., 2009, 3243 RSC.
- A. D. St-Jacques, I. W. Wyman and D. H. Macartney, Chem. Commun., 2008, 4936 RSC.
- P. Montes-Navajas, A. Corma and H. García, ChemPhysChem, 2008, 9, 713 CrossRef CAS.
- P. Montes-Navajas, M. González-Béjar, J. C. Scaiano and H. García, Photochem. Photobiol. Sci., 2009, 8, 1743 RSC.
- R. L. Halterman, J. L. Moore, K. A. Yakshe, J. A. I. Halterman and K. A. Woodson, J. Inclusion Phenom. Macrocyclic Chem., 2010, 66, 231 Search PubMed.
- R. Wang and D. H. Macartney, Tetrahedron Lett., 2008, 49, 311 CrossRef CAS.
- A. C. Bhasikuttan, J. Mohanty, W. M. Nau and H. Pal, Angew. Chem., Int. Ed., 2007, 46, 4120 CrossRef CAS.
- P. Montes-Navajas, L. Teruel, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 1762 CrossRef CAS.
- P. Montes-Navajas and H. García, J. Phys. Chem. C, 2010, 114, 2034 CrossRef CAS.
- W. Ong, M. Gómez-Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791 CrossRef CAS.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384 CrossRef CAS.
- Y. Liu, J. Shi, Y. Chen and C.-F. Ke, Angew. Chem., Int. Ed., 2008, 47, 7293 CrossRef.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem.–Eur. J., 2007, 13, 7223 CrossRef CAS.
- E. Mileo, C. Casati, P. Franchi, E. Mezzina and M. Lucarini, Org. Biomol. Chem., 2011, 9, 2920 RSC.
- S. Gadde, E. K. Batchelor, J. P. Weiss, Y. Ling and A. E. Kaifer, J. Am. Chem. Soc., 2008, 130, 17114 CrossRef CAS.
- S. Gadde, E. K. Batchelor and A. E. Kaifer, Chem.–Eur. J., 2009, 15, 6025 CrossRef CAS.
- B. Bosnich, C. K. Poon and M. L. Tobe, Inorg. Chem., 1965, 4, 1102 CrossRef CAS.
- S. L. Hart, R. I. Haines, A. Decken and B. D. Wagner, Inorg. Chim. Acta, 2009, 362, 4145 CrossRef CAS.
- X. Wu, K. Hu, X. Meng and G. Cheng, New J. Chem., 2010, 34, 17 RSC.
- Y. H. Ko, H. Kim, Y. Kim and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 4106 CrossRef CAS.
- Y. H. Ko, Y. Kim, H. Kim and K. Kim, Chem.–Asian J., 2011, 6, 652 Search PubMed.
- X. Xiao, Q. Wang, Y.-H. Yu, Z.-Y. Xiao, Z. Tao, S.-F. Xue, Q.-J. Zhu, J.-X. Liu and X.-H. Liu, Eur. J. Org. Chem., 2011, 2366 Search PubMed.
- K. Baek, Y. Kim, H. Kim, M. Yoon, I. Hwang, Y. H. Ko and K. Kim, Chem. Commun., 2010, 46, 4091 RSC.
- E. Mileo, E. Mezzina, F. Grepioni, G. F. Pedulli and M. Lucarini, Chem.–Eur. J., 2009, 15, 7859 CrossRef CAS.
- E. V. Peresypkina, V. P. Fedin, V. Maurel, A. Grand, P. Rey and K. E. Vostrikova, Chem.–Eur. J., 2010, 16, 12481 CrossRef CAS.
- S. Yi, B. Captain and A. E. Kaifer, Chem. Commun., 2011, 47, 5500 RSC.
- R. Wang, D. Bardelang, M. Waite, K. A. Udachin, D. M. Leek, K. Yu, C. I. Ratcliffe and J. A. Ripmeester, Org. Biomol. Chem., 2009, 7, 2435 RSC.
- X. Wu, X. Meng and G. Cheng, J. Inclusion Phenom. Macrocyclic Chem., 2009, 64, 325 CrossRef CAS.
- R. Wang, L. Yuan, H. Ihmels and D. H. Macartney, Chem.–Eur. J., 2007, 13, 6468 CrossRef CAS.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, Phys. Chem. Chem. Phys., 2010, 12, 7050 RSC.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526 CrossRef CAS.
- D. A. Uhlenheuer, J. F. Young, H. D. Nguyen, M. Scheepstra and L. Brunsveld, Chem. Commun., 2011, 47, 6798 RSC.
- Y. Ling, W. Wang and A. E. Kaifer, Chem. Commun., 2007, 610 RSC.
- V. Sindelar, M. A. Cejas, F. M. Raymo, W. Chen, S. E. Parker and A. E. Kaifer, Chem.–Eur. J., 2005, 11, 7054 CrossRef CAS.
- D. Jiao, F. Biedermann and O. A. Scherman, Org. Lett., 2011, 13, 3044 Search PubMed.
- Y. H. Ko, K. Kim, E. Kim and K. Kim, Supramol. Chem., 2007, 19, 287 CrossRef CAS.
- J. W. Lee, I. Hwang, W. S. Jeon, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Chem.–Asian J., 2008, 3, 1277 CrossRef CAS.
- W. Wang and A. E. Kaifer, Angew. Chem., Int. Ed., 2006, 45, 7042 CrossRef CAS.
- D. Zou, S. Andersson, R. Zhang, S. Sun, B. Åkermark and L. Sun, J. Org. Chem., 2008, 73, 3775 CrossRef CAS.
- W. Jiang, Q. Wang, I. Linder, F. Klautzsch and C. A. Schalley, Chem. Eur. J., 2011, 17, 2344 Search PubMed.
- T. Zhang, S. Sun, F. Liu, J. Fan, Y. Pang, L. Sun and X. Peng, Phys. Chem. Chem. Phys., 2009, 11, 11134 RSC.
- W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828 RSC.
- W. S. Jeon, A. Y. Ziganshina, J. W. Lee, Y. H. Ko, J.-K. Kang, C. Lee and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 4097 CrossRef CAS.
- Y. Ling, J. T. Mague and A. E. Kaifer, Chem.–Eur. J., 2007, 13, 7908 CrossRef CAS.
- S. Andersson, D. Zou, R. Zhang, S. Sun and L. Sun, Org. Biomol. Chem., 2009, 7, 3605 RSC.
- S. Andersson, D. Zou, R. Zhang, S. Sun, B. Åkermark and L. Sun, Eur. J. Org. Chem., 2009, 1163 CrossRef CAS.
- S. Sun, W. Gao, F. Liu, J. Fan and X. Peng, J. Mater. Chem., 2010, 20, 5888 RSC.
- M. E. Bush, N. D. Bouley and A. R. Urbach, J. Am. Chem. Soc., 2005, 127, 14511 CrossRef CAS.
- P. Rajgariah and A. R. Urbach, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 251 CrossRef CAS.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574 CrossRef CAS.
- J. J. Reczek, A. A. Kennedy, B. T. Halbert and A. R. Urbach, J. Am. Chem. Soc., 2009, 131, 2408 CrossRef CAS.
- H. D. Nguyen, D. T. Dang, J. L. J. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed., 2010, 49, 895 CAS.
- V. Ramalingam and A. R. Urbach, Org. Lett., 2011, 13, 4898 Search PubMed.
- J.-X. Liu, R.-L. Lin, L.-S. Long, R.-B. Huang and L.-S. Zheng, Inorg. Chem. Commun., 2008, 11, 1085 Search PubMed.
- S. Liu, P. Y. Zavalij, Y.-F. Lam and L. Isaacs, J. Am. Chem. Soc., 2007, 129, 11232 CrossRef CAS.
- S. Liu, A. D. Shukla, S. Gadde, B. D. Wagner, A. E. Kaifer and L. Isaacs, Angew. Chem., Int. Ed., 2008, 47, 2657 CrossRef CAS.
- C. Márquez, R. R. Hudgins and W. M. Nau, J. Am. Chem. Soc., 2004, 126, 5806 CrossRef CAS.
- C. Márquez and W. M. Nau, Angew. Chem., Int. Ed., 2001, 40, 3155 CrossRef CAS.
- O. A. Fedorova, E. Y. Chernikova, Y. V. Fedorov, E. N. Gulakova, A. S. Peregudov, K. A. Lyssenko, G. Jonusauskas and L. Isaacs, J. Phys. Chem. B, 2009, 113, 10149 Search PubMed.
- X. Ling and E. Masson, manuscript in preparation. Search PubMed.
- X. Ling, E. L. Samuel, D. L. Patchell and E. Masson, Org. Lett., 2010, 12, 2730 CrossRef CAS.
- V. S. Bryantsev, M. S. Diallo and W. A. Goddard, J. Phys. Chem. A, 2007, 111, 4422 CrossRef CAS.
- J. W. Lee, K. Kim and K. Kim, Chem. Commun., 2001, 1042 RSC.
- F. Yang and D. V. Dearden, Isr. J. Chem., 2011, 51, 551 Search PubMed.
- D. V. Dearden, T. A. Ferrell, M. C. Asplund, L. W. Zilch, R. R. Julian and M. F. Jarrold, J. Phys. Chem. A, 2009, 113, 989 Search PubMed.
- H. Zhang, E. S. Paulsen, K. A. Walker, K. E. Krakowiak and D. V. Dearden, J. Am. Chem. Soc., 2003, 125, 9284 CrossRef CAS.
- H. Zhang, T. A. Ferrell, M. C. Asplund and D. V. Dearden, Int. J. Mass Spectrom., 2007, 265, 187 Search PubMed.
- H. Zhang, M. Grabenauer, M. T. Bowers and D. V. Dearden, J. Phys. Chem. A, 2009, 113, 1508 CrossRef CAS.
- F. Yang and D. V. Dearden, Supramol. Chem., 2011, 23, 53 Search PubMed.
- S. Deroo, U. Rauwald, C. V. Robinson and O. A. Scherman, Chem. Commun., 2009, 644 RSC.
- J. P. Da Silva, N. Jayaraj, S. Jockusch, N. J. Turro and V. Ramamurthy, Org. Lett., 2011, 13, 2410 Search PubMed.
- A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831 CrossRef CAS.
- P. Mukhopadhyay, A. Wu and L. Isaacs, J. Org. Chem., 2004, 69, 6157 CrossRef CAS.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef CAS.
- P. Mukhopadhyay, P. Y. Zavalij and L. Isaacs, J. Am. Chem. Soc., 2006, 128, 14093 CrossRef CAS.
- J. M. Chinai, A. B. Taylor, L. M. Ryno, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810 CrossRef CAS.
- I. W. Wyman and D. H. Macartney, J. Org. Chem., 2009, 74, 8031 CrossRef CAS.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2009, 7, 4045 RSC.
- W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852 CrossRef CAS.
- W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309 CrossRef CAS.
- Z.-J. Ding, H.-Y. Zhang, L.-H. Wang, F. Ding and Y. Liu, Org. Lett., 2011, 13, 856 Search PubMed.
- E. Masson, X. Lu, X. Ling and D. L. Patchell, Org. Lett., 2009, 11, 3798 CrossRef CAS.
- V. Sindelar, S. Silvi and A. E. Kaifer, Chem. Commun., 2006, 2185 RSC.
- V. Sindelar, S. Silvi, S. E. Parker, D. Sobransingh and A. E. Kaifer, Adv. Funct. Mater., 2007, 17, 694 CrossRef CAS.
- V. Kolman, P. Kulhanek and V. Sindelar, Chem.–Asian J., 2010, 5, 2386 Search PubMed.
- H. Zhang, Q. Wang, M. Liu, X. Ma and H. Tian, Org. Lett., 2009, 11, 3234 CrossRef CAS.
- Z.-J. Zhang, Y.-M. Zhang and Y. Liu, J. Org. Chem., 2011, 76, 4682 Search PubMed.
- U. Pischel, V. D. Uzunova, P. Remón and W. M. Nau, Chem. Commun., 2010, 46, 2635 RSC.
- S. Chakrabarti, P. Mukhopadhyay, S. Lin and L. Isaacs, Org. Lett., 2007, 9, 2349 CrossRef CAS.
- T. Ooya, D. Inoue, H. S. Choi, Y. Kobayashi, S. Loethen, D. H. Thompson, Y. H. Ko, K. Kim and N. Yui, Org. Lett., 2006, 8, 3159 CrossRef CAS.
- D. Sobransingh and A. E. Kaifer, Org. Lett., 2006, 8, 3247 CrossRef CAS.
- S. Gadde, E. K. Batchelor and A. E. Kaifer, Aust. J. Chem., 2010, 63, 184 Search PubMed.
- S. Gadde and A. E. Kaifer, Curr. Org. Chem., 2011, 15, 27 Search PubMed.
- I. Hwang, A. Y. Ziganshina, Y. H. Ko, G. Yun and K. Kim, Chem. Commun., 2009, 416 RSC.
- A. Hennig, H. Bakirci and W. M. Nau, Nat. Methods, 2007, 4, 629 CrossRef CAS.
- N. Barooah, J. Mohanty, H. Pal and A. C. Bhasikuttan, Phys. Chem. Chem. Phys., 2011, 13, 13117 RSC.
- W. M. Nau, G. Ghale, A. Hennig, H. Bakirci and D. M. Bailey, J. Am. Chem. Soc., 2009, 131, 11558 CrossRef CAS.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528 CrossRef CAS.
- P. Lindberg, A. Brändström, B. Wallmark, H. Mattsson, L. Rikner and K.-J. Hoffmann, Med. Res. Rev., 1990, 10, 1 Search PubMed.
- A. M. M. Rawashdeh, A. Thangavel, C. Sotiriou-Leventis and N. Leventis, Org. Lett., 2008, 10, 1131 CrossRef CAS.
- Z. Miskolczy, M. Megyesi, G. Tárkányi, R. Mizsei and L. Biczók, Org. Biomol. Chem., 2011, 9, 1061 RSC.
- Z. Miskolczy, L. Biczók and H. Görner, J. Photochem. Photobiol., A, 2009, 207, 47 CrossRef CAS.
- J. Wu and L. Isaacs, Chem.–Eur. J., 2009, 15, 11675 CrossRef CAS.
- S. Chakrabarti and L. Isaacs, Supramol. Chem., 2008, 20, 191 Search PubMed.
- S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2010, 114, 10717 Search PubMed.
- E. Masson, Y. M. Shaker, J.-P. Masson, M. E. Kordesch and C. Yuwono, Org. Lett., 2011, 13, 3872 Search PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302 CrossRef CAS.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 1999, 1509 RSC.
- D. Tuncel and J. H. G. Steinke, Polymer Preprints, 1999, 40, 585 Search PubMed.
- T. C. Krasia and J. H. G. Steinke, Chem. Commun., 2002, 22 RSC.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 2002, 496 RSC.
- D. Tuncel and J. H. G. Steinke, Macromolecules, 2004, 37, 288 CrossRef.
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, Chem. Commun., 2001, 1938 RSC.
- W. Herrmann, S. Wehrle and G. Wenz, Chem. Commun., 1997, 1709 RSC.
- H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399 CrossRef.
- M. Pattabiraman, A. Natarajan, R. Kaliappan, J. T. Mague and V. Ramamurthy, Chem. Commun., 2005, 4542 RSC.
- M. V. S. N. Maddipatla, L. S. Kaanumalle, A. Natarajan, M. Pattabiraman and V. Ramamurthy, Langmuir, 2007, 23, 7545 CrossRef CAS.
- M. Pattabiraman, A. Natarajan, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2005, 7, 529 CrossRef CAS.
- M. Pattabiraman, L. S. Kaanumalle, A. Natarajan and V. Ramamurthy, Langmuir, 2006, 22, 7605 CrossRef CAS.
- N. Barooah, B. C. Pemberton and J. Sivaguru, Org. Lett., 2008, 10, 3339 CrossRef CAS.
- N. Barooah, B. C. Pemberton, A. C. Johnson and J. Sivaguru, Photochem. Photobiol. Sci., 2008, 7, 1473 RSC.
- B. C. Pemberton, R. K. Singh, A. C. Johnson, S. Jockusch, J. P. Da Silva, A. Ugrinov, N. J. Turro, D. K. Srivastava and J. Sivaguru, Chem. Commun., 2011, 47, 6323 RSC.
- B. C. Pemberton, N. Barooah, D. K. Srivatsava and J. Sivaguru, Chem. Commun., 2010, 46, 225 RSC.
- X.-L. Wu, L. Luo, L. Lei, G.-H. Liao, L.-Z. Wu and C.-H. Tung, J. Org. Chem., 2008, 73, 491 CrossRef CAS.
- L. Lei, L. Luo, X.-L. Wu, G.-H. Liao, L.-Z. Wu and C.-H. Tung, Tetrahedron Lett., 2008, 49, 1502 CrossRef CAS.
- B. Chen, S.-F. Cheng, G.-H. Liao, X.-W. Li, L.-P. Zhang, C.-H. Tung and L.-Z. Wu, Photochem. Photobiol. Sci., 2011, 10, 1441 RSC.
- C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574 CrossRef CAS.
- C. Yang, C. Ke, W. Liang, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 13786 Search PubMed.
- R. Wang, L. Yuan and D. H. Macartney, J. Org. Chem., 2006, 71, 1237 CrossRef CAS.
- C. Klöck, R. N. Dsouza and W. M. Nau, Org. Lett., 2009, 11, 2595 CrossRef.
- N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, J. Org. Chem., 2010, 75, 848 CrossRef CAS.
- Y.-H. Wang, H. Cong, F.-F. Zhao, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, Catal. Commun., 2011, 12, 1127 Search PubMed.
- S. M. de Lima, J. A. Gómez, V. P. Barros, G. de S. Vertuan, M. das D. Assis, C. F. de O. Graeff and G. J.-F. Demets, Polyhedron, 2010, 29, 3008 Search PubMed.
- E. A. Karakhanov, L. M. Karapetyan, Y. S. Kardasheva, A. L. Maksimov, E. A. Runova, M. V. Terenina and T. Y. Filippova, Macromol. Symp., 2008, 270, 106 Search PubMed.
- X. Lu and E. Masson, Org. Lett., 2010, 12, 2310 Search PubMed.
- U. Halbes-Letinois, J.-M. Weibel and P. Pale, Chem. Soc. Rev., 2007, 36, 759 RSC.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545 CrossRef CAS.
- W. Adam and T. Heidenfelder, J. Am. Chem. Soc., 1998, 120, 11858 CrossRef CAS.
- R. Wang, L. Yuan and D. H. Macartney, Chem. Commun., 2006, 2908 RSC.
- L. S. Berbeci, W. Wang and A. E. Kaifer, Org. Lett., 2008, 10, 3721 Search PubMed.
- H. Cong, C.-R. Li, S.-F. Xue, Z. Tao, Q.-J. Zhu and G. Wei, Org. Biomol. Chem., 2011, 9, 1041 RSC.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638 RSC.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223 CrossRef CAS.
- C. O. Mellet, J. M. García Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586 RSC.
- L. Y. T. Chou, K. Ming and W. C. W. Chan, Chem. Soc. Rev., 2011, 40, 233 RSC.
- N. J. Wheate, A. I. Day, R. J. Blanch, A. P. Arnold, C. Cullinane and J. G. Collins, Chem. Commun., 2004, 1424 RSC.
- N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451 RSC.
- N. J. Wheate, R. I. Taleb, A. M. Krause-Heuer, R. L. Cook, S. Wang, V. J. Higggins and J. R. Aldrich-Wright, Dalton Trans., 2007, 5055 RSC.
- N. J. Wheate, J. Inorg. Biochem., 2008, 102, 2060 CrossRef CAS.
- S. Kemp, N. J. Wheate, M. J. Pisani and J. R. Aldrich-Wright, J. Med. Chem., 2008, 51, 2787 CrossRef CAS.
- Y. Zhao, M. S. Bali, C. Cullinane, A. I. Day and J. G. Collins, Dalton Trans., 2009, 5190 RSC.
- Y. Zhao, D. P. Buck, D. L. Morris, M. H. Pourgholami, A. I. Day and J. G. Collins, Org. Biomol. Chem., 2008, 6, 4509 RSC.
- Y. Zhao, M. H. Pourgholami, D. L. Morris, J. G. Collins and A. I. Day, Org. Biomol. Chem., 2010, 8, 3328 RSC.
- N. Dong, S.-F. Xue, Q.-J. Zhu, Z. Tao, Y. Zhao and L.-X. Yang, Supramol. Chem., 2008, 20, 659 CAS.
- H. Isobe, S. Sato, J. W. Lee, H.-J. Kim, K. Kim and E. Nakamura, Chem. Commun., 2005, 1549 RSC.
- C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962 CrossRef CAS.
- J. Liu, X. Du and X. Zhang, Chem.–Eur. J., 2011, 17, 810 CrossRef CAS.
- J. Liu and X. Du, J. Mater. Chem., 2010, 20, 3642 RSC.
- M. W. Ambrogio, T. A. Pecorelli, K. Patel, N. M. Khashab, A. Trabolsi, H. A. Khatib, Y. Y. Botros, J. I. Zink and J. F. Stoddart, Org. Lett., 2010, 12, 3304 CrossRef CAS.
- N. M. Khashab, A. Trabolsi, Y. A. Lau, M. W. Ambrogio, D. C. Friedman, H. A. Khatib, J. I. Zink and J. F. Stoddart, Eur. J. Org. Chem., 2009, 1669 CrossRef CAS.
- N. M. Khashab, M. E. Belowich, A. Trabolsi, D. C. Friedman, C. Valente, Y. Lau, H. A. Khatib, J. I. Zink and J. F. Stoddart, Chem. Commun., 2009, 5371 RSC.
- S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 12912 CrossRef CAS.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222 CrossRef CAS.
- S. Angelos, Y.-W. Yang, N. M. Khashab, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11344 CrossRef CAS.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623 CrossRef CAS.
- Z. Miskolczy, L. Biczók, M. Megyesi and I. Jablonkai, J. Phys. Chem. B, 2009, 113, 1645 CrossRef CAS.
- R. Wang, B. C. MacGillivray and D. H. Macartney, Dalton Trans., 2009, 3584 RSC.
- A. L. Koner, I. Ghosh, N. Saleh and W. M. Nau, Can. J. Chem., 2011, 89, 139 CrossRef CAS.
- C. Li, J. Li and X. Jia, Org. Biomol. Chem., 2009, 7, 2699 RSC.
- M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410 CrossRef CAS.
- N. Dong, L. Cheng, X. Wang, Q. Li, C. Dai and Z. Tao, Talanta, 2011, 84, 684 Search PubMed.
- N. J. Wheate, V. Vora, N. G. Anthony and F. J. McInnes, J. Inclusion Phenom. Macrocyclic Chem., 2010, 68, 359 CrossRef CAS.
- N. Saleh, M. A. Meetani, L. Al-Kaabi, I. Ghosh and W. M. Nau, Supramol. Chem., 2011, 23, 654 Search PubMed.
- F. J. McInnes, N. G. Anthony, A. R. Kennedy and N. J. Wheate, Org. Biomol. Chem., 2010, 8, 765 RSC.
- S. Choi, J. W. Lee, Y. H. Ko and K. Kim, Macromolecules, 2002, 35, 3526 CrossRef CAS.
- D. Tuncel and J. H. G. Steinke, Chem. Commun., 2001, 253 RSC.
- C. Meschke, H.-J. Buschmann and E. Schollmeyer, Macromol. Rapid Commun., 1998, 19, 59 CrossRef CAS.
- C. Meschke, H.-J. Buschmann and E. Schollmeyer, Polymer, 1999, 40, 945 CrossRef.
- U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950 CrossRef CAS.
- J. M. Zayed, F. Biedermann, U. Rauwald and O. A. Scherman, Polym. Chem., 2010, 1, 1434 RSC.
- U. Rauwald, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Commun., 2011, 47, 6000 RSC.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251 CrossRef CAS.
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6576 CrossRef CAS.
- Y. Tan, S. Choi, J. W. Lee, Y. H. Ko and K. Kim, Macromolecules, 2002, 35, 7161 CrossRef CAS.
- Z. Hou, Y. Tan and Q. Zhou, Polymer, 2006, 47, 5267 Search PubMed.
- H. Yang, Y. Tan, J. Hao, H. Yang and F. Liu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2135 Search PubMed.
- X. Huang, Y. Tan, Y. Wang, H. Yang, J. Cao and Y. Che, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5999 CrossRef CAS.
- J. Geng, F. Biedermann, J. M. Zayed, F. Tian and O. A. Scherman, Macromolecules, 2011, 44, 4276 Search PubMed.
- R. A. Wessling, J. Polym. Sci., Polym. Symp., 1985, 72, 55 CAS.
- Y. Ling and A. E. Kaifer, Chem. Mater., 2006, 18, 5944 CrossRef CAS.
- J. W. Lee, Y. H. Ko, S.-H. Park, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 746 CrossRef CAS.
- Y.-B. Lim, T. Kim, J. W. Lee, S.-M. Kim, H.-J. Kim, K. Kim and J.-S. Park, Bioconjugate Chem., 2002, 13, 1181 CrossRef CAS.
- W. Ong and A. E. Kaifer, Angew. Chem., Int. Ed., 2003, 42, 2164 CrossRef CAS.
- K. Moon, J. Grindstaff, D. Sobransingh and A. E. Kaifer, Angew. Chem., Int. Ed., 2004, 43, 5496 CrossRef CAS.
- D. Sobransingh and A. E. Kaifer, Chem. Commun., 2005, 5071 RSC.
- D. Sobransingh and A. E. Kaifer, Langmuir, 2006, 22, 10540 CrossRef CAS.
- W. Wang and A. E. Kaifer, Adv. Polym. Sci., 2009, 222, 205 CAS.
- Y. Zeng, Y. Li, M. Li, G. Yang and Y. Li, J. Am. Chem. Soc., 2009, 131, 9100 CrossRef CAS.
- A. Corma, H. García, P. Montes-Navajas, A. Primo, J. J. Calvino and S. Trasobares, Chem.–Eur. J., 2007, 13, 6359 CrossRef CAS.
- P. Montes-Navajas, L. C. Damonte and H. García, ChemPhysChem, 2009, 10, 812 Search PubMed.
- P. Montes-Navajas and H. García, J. Phys. Chem. C, 2010, 114, 18847 Search PubMed.
- T.-C. Lee and O. A. Scherman, Chem. Commun., 2010, 46, 2438 RSC.
- R. W. Taylor, T. C. Lee, O. A. Scherman, R. Esteban, J. Aizpupurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878 CrossRef CAS.
- R. J. Coulston, S. T. Jones, T.-C. Lee, E. A. Appel and O. A. Scherman, Chem. Commun., 2011, 47, 164 RSC.
- T. Premkumar and K. E. Geckeler, Chem.–Asian J., 2010, 5, 2468 CrossRef CAS.
- J. Liu, W. Ong and A. E. Kaifer, Langmuir, 2002, 18, 5981 CrossRef CAS.
- T. Premkumar, Y. Lee and K. E. Geckeler, Chem.–Eur. J., 2010, 16, 11563 CrossRef CAS.
- R. de la Rica and A. H. Velders, J. Am. Chem. Soc., 2011, 133, 2875 CrossRef.
- E. M. S. dos Santos, L. S. Pereira and G. J.-F. Demets, J. Braz. Chem. Soc., 2011, 22, 1595 Search PubMed.
- M. Cao, J. Lin, H. Yang and R. Cao, Chem. Commun., 2010, 46, 5088 RSC.
- F. Constabel and K. E. Geckeler, Tetrahedron Lett., 2004, 45, 2071 CrossRef CAS.
- F. Constabel and K. E. Geckeler, Fullerenes, Nanotubes, Carbon Nanostruct., 2004, 12, 811 Search PubMed.
- F. Constabel and K. E. Geckeler, Nanoencapsulation of Fullerene with the Hosts Cyclodextrin and Cucurbituril, in: Functional Nanomaterials, ed. K. E. Geckeler and E. Rosenberg, American Scientific Publishers, Valencia, USA, 2006, 377 Search PubMed.
- G. Jiang and G. Li, J. Photochem. Photobiol., B, 2006, 85, 223 Search PubMed.
- T. Ogoshi, A. Inagaki, T. Yamagishi and Y. Nakamoto, Chem. Commun., 2008, 2245 RSC.
- Q. An, Q. Chen, W. Zhu, Y. Li, C. Tao, H. Yang, Z. Li, L. Wan, H. Tian and G. Li, Chem. Commun., 2010, 46, 725 RSC.
- Q. Zhou, H. Wang, T. Gao, Y. Yu, B. Ling, L. Mao, H. Zhang, X. Meng and X. Zhou, Chem. Commun., 2011, 47, 11315 RSC.
- Q. An, G. Li, C. Tao, Y. Li, Y. Wu and W. Zhang, Chem. Commun., 2008, 1989 RSC.
- K. Kim, W. S. Jeon, J.-K. Kang, J. W. Lee, S. Y. Jon, T. Kim and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 2293 CrossRef CAS.
- K. Kim, D. Kim, J. W. Lee, Y. H. Ko and K. Kim, Chem. Commun., 2004, 848 RSC.
- F. Tian, M. Cziferszky, D. Jiao, K. Wahlström, J. Geng and O. A. Scherman, Langmuir, 2011, 27, 1387 Search PubMed.
- J. F. Young, H. D. Nguyen, L. Yang, J. Huskens, P. Jonkheijm and L. Brunsveld, ChemBioChem, 2010, 11, 180 CrossRef CAS.
- M. Freitag and E. Galoppini, Langmuir, 2010, 26, 8262 CrossRef CAS.
- G. J.-F. Demets, B. V. S. Schneider, H. D. Correia, R. R. Gonçalves, T. M. Nobre and M. E. Darbello Zaniquelli, J. Nanosci. Nanotechnol., 2008, 8, 432 Search PubMed.
- M. del Pozo, P. Hernández, L. Hernández and C. Quintana, J. Mater. Chem., 2011, 21, 13657 RSC.
- L. F. S. da Silva, G. J.-F. Demets, C. Taviot-Guého, F. Leroux and J. B. Valim, Chem. Mater., 2011, 23, 1350 Search PubMed.
- H. D. Correia and G. J.-F. Demets, Electrochem. Commun., 2009, 11, 1928 CrossRef CAS.
- L. Xu, S.-M. Liu, C.-T. Wu and Y.-Q. Feng, Electrophoresis, 2004, 25, 3300 Search PubMed.
- F. Wei and Y.-Q. Feng, Talanta, 2008, 74, 619 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473 RSC.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908 RSC.
- I. Hwang, W. S. Jeon, H.-J. Kim, D. Kim, H. Kim, N. Selvapalam, N. Fujita, S. Shinkai and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 210 CrossRef CAS.
- H. Yang, Y. Tan and Y. Wang, Soft Matter, 2009, 5, 3511 RSC.
| This journal is © The Royal Society of Chemistry 2012 |