Enantioselective organocatalytic α-heterofunctionalization of active methines
Alessio
Russo
,
Claudia
De Fusco
and
Alessandra
Lattanzi
*
Dipartimento di Chimica e Biologia, Università di Salerno, Via Ponte don Melillo, 84084, Fisciano, Italy. E-mail: lattanzi@unisa.it; Fax: +39 089 969603; Tel: +39 089 969563
First published on 18th November 2011
Abstract
The enantioselective α-heterofunctionalization of α-substituted 1,3-dicarbonyl compounds is an attractive area of organic chemistry, thanks to the undoubted value of the final compounds as versatile intermediates for the synthesis of natural products and pharmaceutical targets. In this context, the organocatalytic approach is gaining importance, alongside the asymmetric metal-based catalysis, for the production of molecules with highly functionalized chiral quaternary stereocenters containing a carbon-heteroatom bond. This review aims to illustrate progress in the enantioselective α-amination, α-hydroxylation, α-sulfenylation and α-halogenation reactions of α-substituted 1,3-dicarbonyl and related compounds mediated by mono-, bi-, multi-functional organocatalysts, phosphoric acids and phase transfer catalysts appeared from 2002 to mid-2011.
 Alessandra Lattanzi | Alessandra Lattanzi was born in Rome. She graduated and received her PhD degree from University of Rome “La Sapienza”. In 1996 she was appointed as a Lecturer at University of Salerno. As a visiting scientist she joined the groups of Prof. V. K. Aggarwal at University of Sheffield (UK, 1999–2000) and Dr N. E. Leadbeater at King’s College in London (2001). In 2005 she was promoted to Associate Professor. Her research interests are in the fields of asymmetric organocatalysis, stereoselective metal-catalysed oxidations and description of chiral structures through algebraic and geometrical methods. |
 Alessio Russo | Alessio Russo was born in Avellino. He is a postdoctoral fellow working at the University of Salerno in Italy. He received his PhD in Chemistry (2009) from the University of Salerno and a MD in Chemistry (2005) from the same institution. He has wide experience in organic chemistry and his current research activities are focused on organocatalytic asymmetric techniques, especially noncovalent chiral base promoted reactions. |
 Claudia De Fusco | Claudia De Fusco was born in Naples. She completed a MSc in Organic Chemistry at the University of Naples, Federico II, Italy in 2009 with a thesis on synthesis of indole-based systems. She is currently a PhD student under the supervision of Prof. Alessandra Lattanzi at the University of Salerno, Italy. Her research focuses on development of new organocatalytic asymmetric reactions with particular attention on epoxidation and Michael addition reactions. |
Introduction
The formation of tertiary and quaternary carbon stereocenters, bearing a heteroatom at the α-position of a carbonyl compound, is a process of utmost synthetic value.1 Indeed, it enables the access to a wide number of small precious optically active building blocks amenable to further manipulations. Over recent years, exceptionally rapid development of asymmetric organocatalysis,2 as a mild and simple synthetic tool for organic chemists, has met with a lot of successful examples for the direct α-heterofunctionalization of simple carbonyl compounds, such as aldehydes and ketones.3 The highly effective strategy employed relies on the activation of the α-carbon of the carbonyl compound with a chiral secondary or primary amine to generate the nucleophilic enamine, which then attacks the heteroatom containing electrophile (Scheme 1). The most successful organocatalysts for these transformations are L-proline and its derivatives leading to products with excellent enantioselectivity.4 The stereocontrolled formation of the new carbon–heteroatom bond is assured by effective shielding of one of the two prochiral faces of the enamine by sterically demanding moiety or by a hydrogen bonding directing group placed at the α-position of the pyrrolidine core.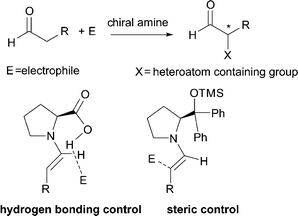 | ||
| Scheme 1 | ||
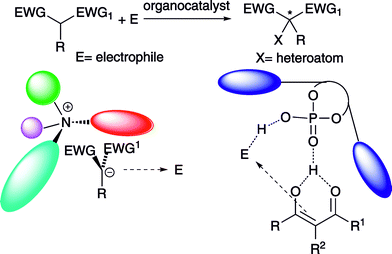
Differently, activation of the reagents by the catalyst through noncovalent interactions5 has been exploited in the α-heterofunctionalization of α-substituted 1,3-dicarbonyl compounds, leading to important products bearing quaternary stereocenters.6 The generation of chiral ion pairs, when using Brønsted bases and under phase transfer catalysis, has been proposed (Scheme 2). Alternatively, hydrogen bonded prochiral enols, have been invoked to justify the stereocontrol observed in the carbon-heteroatom bond formation with chiral Brønsted acids (Scheme 2).
 | ||
| Scheme 2 | ||
In both cases, one of the enantiotopic faces of the nucleophile is shielded by the chiral environment of the ammonium ion or 3,3′-substituted BINOL scaffold of the phosphoric acids. Moreover, the electrophile is thought to be steered towards the nucleophile by hydrogen bonding interactions provided by additional groups of the organocatalysts. The enantioselective metal-based α-heterofunctionalization of carbonyl compounds has been the subject of intense investigation and this area has been recently reviewed.7 Comprehensive or general reviews including, among other approaches, the organocatalytic direct α-heterofunctionalization of carbonyl compounds have been reported.8 This article specifically focuses on the organocatalytic α-heterofunctionalization of α-substituted 1,3-dicarbonyl compounds and strictly related compounds in consideration of the interesting new developments recently achieved in this area.
α-Amination
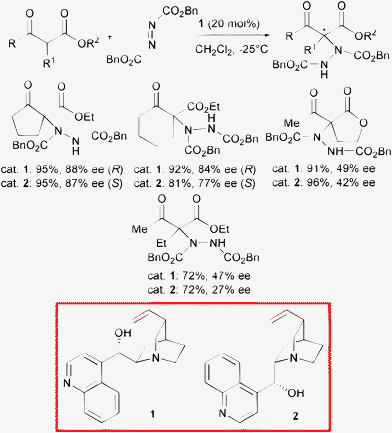
The introduction of a nitrogen containing group at the α-position of a carbonyl compound is a particularly attractive transformation. Indeed, unusual non-natural and more interestingly quaternary α-amino acids are obtained, which are impossible to isolate from natural sources.9 These products found important applications in the synthesis of conformationally restricted peptides and bioactive compounds.10 Moreover, chiral β-amino alcohols can be produced after reduction as another valuable class of derivatives.11 Thus, the electrophilic α-amination of 1,3-dicarbonyl compounds represents a facile and direct method to effect this transformation.Cinchona alkaloids and their derivatives are privileged ligands and catalysts which have found extensive application in asymmetric synthesis.12 In 2004, Pihko and co-authors described the α-hydrazination of cyclic β-ketoesters and lactones with dibenzyl azodicarboxylate in the presence of natural cinchona alkaloids such as cinchonine 1 and cinchonidine 2 (Scheme 3).13
 | ||
| Scheme 3 | ||
Cyclic β-ketoesters readily afforded both enantiomerically enriched products in high yield and moderate to good enantioselectivity. It is interesting to note that the generation of a quaternary center with high enantiocontrol in acyclic systems is a very difficult task to attain.6 Indeed, open chain β-ketoesters and cyclic lactones demonstrated to be less suitable substrates as only a modest level of asymmetric induction was observed. In 2004, Jørgensen and co-authors illustrated the first highly enantioselective α-hydrazination of α-substituted α-cyanoacetates with di-tert-butyl azodicarboxylate employing 5 mol% of β-isocupreidine 3 as a highly effective organocatalyst easily obtained from quinidine (Scheme 4).14
 | ||
| Scheme 4 | ||
The products bearing an α-aryl substituent were isolated in excellent yield and enantioselectivity. Moreover, catalyst loading could be lowered to 0.5 mol% without compromising either yield and enantiocontrol. Compound 3 proved to be far more effective than cinchonine 1 and cinchonidine 2 illustrated in Scheme 3 in the hydrazination of open chain β-ketoesters, although when employing catalyst 3 with common cyclic β-ketoesters the enantioselectivity ranged from 83 to 89% ee.
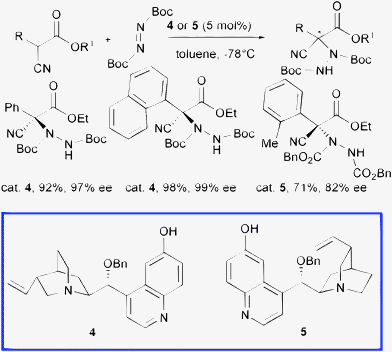
Deng and co-authors disclosed a powerful class of organocatalysts, namely 9-O-protected cupreine 4 and 9-O-protected cupreidine 5, easily accessible in a few steps from cinchona alkaloids, which served as highly enantioselective promoters in various transformations.15 Interestingly, in analogy to β-isocupreidine 3, the presence of the free phenolic 6′-OH group and the protection of the 9-OH residue have been found to be of crucial importance to achieve high asymmetric induction (Scheme 5).
 | ||
| Scheme 5 | ||
These catalysts are able to promote the α-hydrazination of α-aryl-α-cyanoacetates at 5 mol% loading with di-tert-butyl azodicarboxylate in short reaction times (1 min to 12 h) leading to the products in both absolute configurations in excellent yield and high enantioselectivity.16
Attempts to obtain the corresponding enantioenriched products starting from α-alkyl-substituted-α-cyanoacetates failed. The same group tested catalysts 4 and 5, even at 0.1 mol% loading, in the amination of cyclic β-ketoesters achieving the products in excellent yield and enantioselectivity. When using less active α-substituted acyclic β-ketoesters, the reaction proceeded sluggishly and promoter 5 was consumed. Indeed, compound 5 underwent a Friedel–Crafts amination with the azodicarboxylate ester, a reaction previously reported to occur on dihydrocupreidine, which consumed the effective catalyst.14
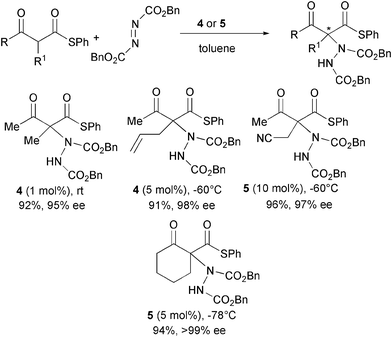
More acidic α-substituted acyclic β-ketothioesters were also reacted with dibenzyl azodicarboxylate and catalysts 4 and 5 (Scheme 6).17
 | ||
| Scheme 6 | ||
In this case, the expected products were rapidly formed (5 min to 27 h) using very low catalyst loading (from 0.05 to up to 10 mol%) in excellent yield and enantioselectivity.
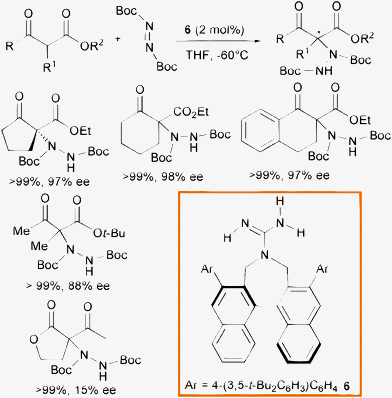
Monofunctional organic bases such as chiral guanidines have demonstrated great synthetic potential as organocatalysts for various transformations.18 Terada and co-authors developed C2-symmetric guanidines bearing axially chiral binaphthyl backbone with a seven-membered ring structure 6.19 They envisioned a plausible discrimination of the enolate faces in the enolate/ammonium generated ion pair provided by the sterically encumbered C2-symmetric axial scaffold of the catalyst, coupled with the multiple hydrogen bonding interactions established among the guanidinium moiety and the carbonyl groups of the enolate. Indeed, the α-hydrazination of cyclic and linear β-ketosters, with the sterically encumbered di-tert-butyl azodicarboxylate, furnished the products in excellent yield and enantioselectivity when using only 2 mol% of the base (Scheme 7). Unfortunately, the system had severe limitations with β-ketolactones, whose products were isolated in poor enantioselectivity.
 | ||
| Scheme 7 | ||
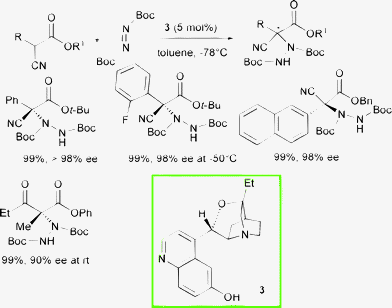
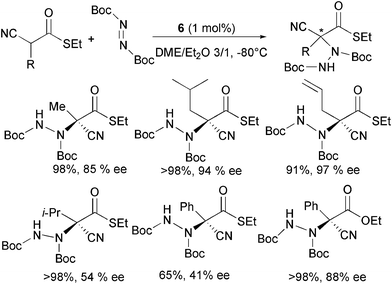
With a view to develop methods for the amination of alkyl-substituted α-cyanoacetate derivatives, Terada and co-authors further investigated the catalytic ability of guanidine 6 to promote the electrophilic amination of α-alkyl-substituted α-cyanothioacetates (Scheme 8).20
 | ||
| Scheme 8 | ||
When reacting α-cyanothioacetates bearing a variety of primary α-alkyl substituents with di-tert-butyl azodicarboxylate and 1 mol% of the organocatalyst in ethereal mixtures, the products were isolated in high yield and good to high enantioselectivity.
It was observed that the selectivity can be improved by employing different solvent mixtures or guanidine catalysts. A secondary alkyl or phenyl α-substituent present in the starting material was found to have a detrimental effect in the enantiocontrol. Quite surprisingly, the product derived from the corresponding α-phenyl cyanoacetate showed high ee, indicating that steric and electronic effects would seem to affect the stereochemical outcome of the reaction.
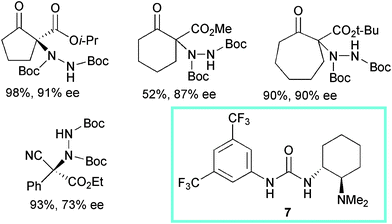
Bifunctional urea 7, bearing a tertiary amine moiety, was reported by Takemoto et al. to be an effective catalyst when used at 10 mol% loading in toluene at −78 °C in the α-hydrazination of cyclic β-ketoesters with di-tert-butyl azodicarboxylate (Scheme 9).21
 | ||
| Scheme 9 | ||
Five-, six- and seven-membered ring β-ketoesters and bicyclic derivatives with bulky ester moieties underwent successful conversion to the products with high enantiocontrol. Again, acyclic β-ketoesters proved to be less reactive compounds and scarce enantiocontrol was observed. Nevertheless, in the case of α-phenyl-α-cyanoacetate derivatives, the product was isolated in good yield and 73% ee. Surprisingly, the corresponding more popular thiourea catalyst showed to be poorly effective in the process.
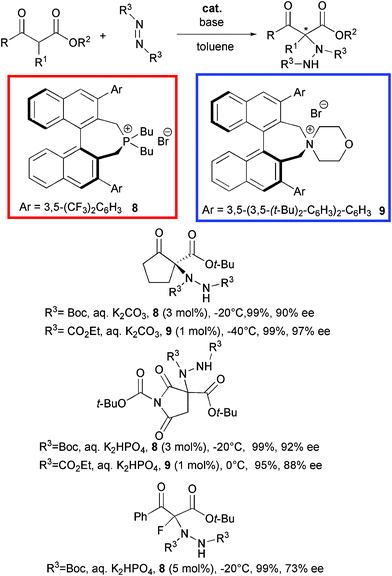
Chiral quaternary tetraalkylphosphonium salts have been rarely studied as phase-transfer catalysts in comparison to the ammonium counterparts. This can be likely ascribed to their easy transformation to ylides under basic conditions.22 Maruoka et al. disclosed the first successful application of binaphthyl modified quaternary phosphonium salts as phase transfer catalysts in the asymmetric α-amination of β-ketoesters (Scheme 10).23
 | ||
| Scheme 10 | ||
Structurally different β-ketoesters smoothly reacted with di-tert-butyl azodicarboxylate using sterically demanding (S)-3,3′-aryl substituted BINOL-based catalyst 8 at 3 mol% loading under basic conditions. The amination products were obtained in excellent yield and good to high ee also in the case of a α-fluorinated acyclic derivative. The imide based product is a useful key intermediate for aldose reductase inhibitor AS-3201.24 Later, the same group synthesized an ammonium bromide 9, structurally similar to catalyst 8.25 Chiral quaternary ammonium salts have been widely recognised as efficient organocatalysts in a variety of synthetic transformations thanks to several advantages: i) operational simplicity and mild reaction conditions with aqueous media, ii) suitable for application to large scale production, iii) convenient chiral source, as most of them are cinchona alkaloids based derivatives.26 Promoter 9 efficiently catalyzed the same process with a comparable level of enantiocontrol, when used at 1 mol% loading with diethyl azodicarboxylate.
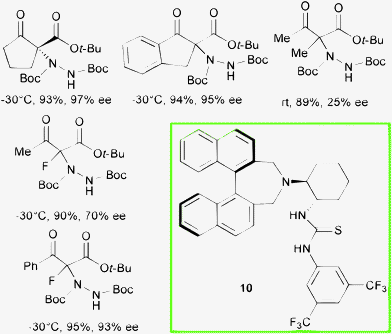
Bifunctional tertiary amine thiourea 10, featuring axial and central chirality backbones, was successfully employed at 10 mol% loading in toluene, in the asymmetric α-hydrazination with di-tert-butyl azodicarboxylate of cyclic β-ketoesters and open chain α-fluorinated β-ketoesters by Kim et al. (Scheme 11).27
 | ||
| Scheme 11 | ||
After screening of different organocatalysts, including the diastereoisomer of compound 10, matching of the chirality of the (R)-binaphthyl moiety and the (1S,2S)-diaminocyclohexane unit in catalyst 10 led to the best level of enantiocontrol in the reactions. The amination product of the corresponding ethyl α-methyl acetoacetate was obtained, after prolonged reaction time and working at room temperature, in 89% yield and 25% ee. In contrast, synthetically important linear derivatives, containing fluorine at the quaternary stereocenter, were obtained in good to high ee when employing this system.28 Indeed, fluorine-containing molecules are very important in the pharmaceutical industry.29 Accordingly, the asymmetric synthesis of chiral fluorinated products is steadily increasing.30 The approach to functionalized products containing a C–F quaternary center is particularly challenging. The direct α-hydrazination of α-fluorinated 1,3-dicarbonyl compounds leads to α-fluorinated amino acids derivatives. In this context, a complementary route to form a chiral quaternary C–F bond is the direct α-fluorination of α-substituted 1,3-dicarbonyl compounds (vide infra).
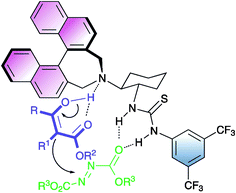
The activation of the reagents provided by bifunctional organocatalyst 10 has been postulated to occur according to the stereochemical model presented in Fig. 1.
 | ||
| Fig. 1 | ||
A carbonyl group of the azodicarboxylate is activated by the thiourea moiety through hydrogen bonding, whereas the β-ketoester by the tertiary amine.
The same group developed the first organocatalytic enantioselective α-amination of α-substituted-α-cyanoketones with di-tert-butyl azodicarboxylate using catalyst 10 (Scheme 12).31
 | ||
| Scheme 12 | ||
Under reaction conditions previously established for β-ketoesters, cyclic and linear α-substituted-α-cyanoketones were converted into the products in generally high yield and enantiomeric excess.
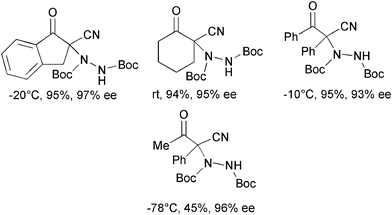
In 2008 Rawal and co-authors developed a new class of bifunctional chiral hydrogen bond (H-bond) donors based on the squaramide scaffold.32 These compounds proved to be generally superior in the activation of nitroalkenes in C–C and C–P bond formation, when compared to double H-bond donors such as thioureas.33 Recently, the same authors showed the effectiveness of catalyst 11, used at 1 mol% loading in toluene, in the α-hydrazination of a variety of cyclic β-ketoesters, lactones and β-cyanoketones with di-tert-butyl azodicarboxylate (Scheme 13).34
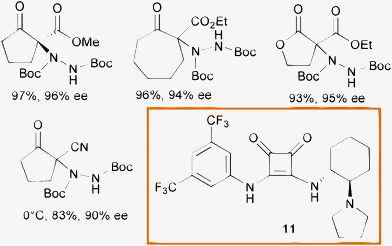 | ||
| Scheme 13 | ||
A good substrate scope, excellent conversion and enantioselectivity have been displayed by this system. The notable feature of catalyst 11 is the high performance under simple reaction conditions. Indeed, it could be used at 1 and as low as 0.1 mol% loading at room temperature without depletion of the enantioselectivity.
Very recently, another class of bifunctional promoters, structurally derived from cinchona alkaloids, have been designed by Lu et al. introducing a guanidine group (Scheme 14).35 The investigation focused on the α-hydrazination of α-fluorinated β-ketoesters using catalyst 12 with di-tert-butyl azodicarboxylate.
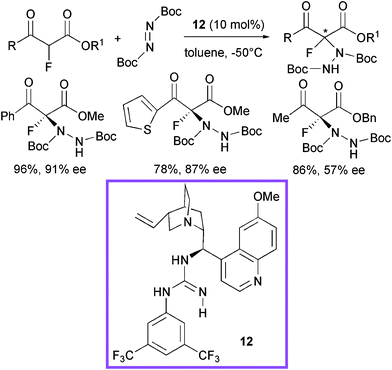 | ||
| Scheme 14 | ||
Aromatic, heteroaromatic β-ketoesters afforded the product in good to high yield and enantiomeric excess. Aliphatic β-ketoesters reacted more sluggishly and modest enantiocontrol was observed. A guanidinium/enolate ion pair was supposed to be formed, after deprotonation of the ketoester by the most basic site of the catalyst and the enolate would be properly complexed by a network of hydrogen bonds (Fig. 2).
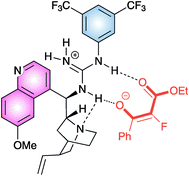 | ||
| Fig. 2 | ||
Wang and co-authors reported the employment of multifunctional organocatalysts, combining structurally different chiral scaffolds and H-bond donors, in the α-amination of cyclic β-ketoesters with dialkyl azodicarboxylates.36 After short reaction times (<10 min) complete conversion of the reactants was observed at room temperature, which confirmed previously observed high catalytic activity of catalysts of type 13 in asymmetric Michael addition and nitro-Mannich reactions. Satisfactory results in terms of stereocontrol have been achieved when performing the reactions at −78 °C in ethyl acetate as solvent with diethyl azodicarboxylate and promoter 13 used at 10 mol% loading (Scheme 15).
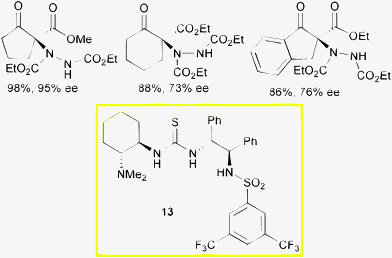 | ||
| Scheme 15 | ||
α-Hydroxylation
The α-hydroxydicarbonyl moiety is a common structural motif in a variety of natural products and pharmaceutical antibiotics.37The most convenient synthetic route to α-hydroxylated products is the direct oxidation of 1,3-dicarbonyl compounds.
In 2004, Jørgensen and co-authors reported the use of cinchona alkaloids as organocatalysts for the enantioselective hydroxylation of β-ketoesters.38 Optically active products in up to 80% ee were isolated using commercially available dihydroquinine 14 as the catalyst and cumyl hydroperoxide (CHP) as the oxygen donor (Scheme 16).
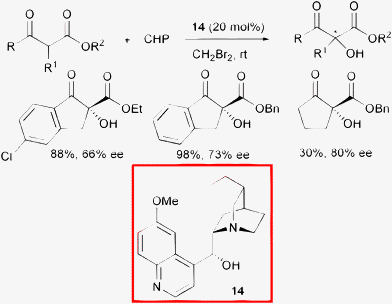 | ||
| Scheme 16 | ||
A variety of different peroxides were used as an oxygen source in the hydroxylation reaction and cumyl hydroperoxide gave the best results. Among the cinchona alkaloid derivatives tested, quinine with the hydroxyl moiety protected afforded a nearly racemic product, whereas dihydroquinine 14 led to the highest enantiomeric excess at room temperature. Of the solvents tested, unusual CH2Br2 gave slightly better results compared to toluene. Furthermore, they demonstrated the synthetic utility of these compounds via highly diastereoselective reduction with BH3-4-ethylmorpholine to produce anti-α,β-diols.
In 2009, Zhong and co-authors developed a highly enantioselective Brønsted acid-catalyzed hydroxylation of β-dicarbonyl compounds using nitroso compounds as the oxygen source.39 By protonation of the basic nitrogen atom of nitroso compound, tandem aminoxylation of β-ketoesters followed by N–O bond heterolysis allowed direct and selective access to enantioenriched cyclic α-hydroxy-β-ketoesters (Scheme 17).
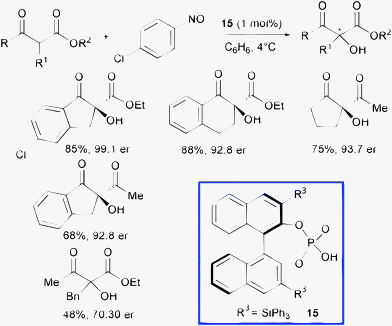 | ||
| Scheme 17 | ||
Catalyst 15 can be used at only 1 mol% loading to furnish the products in excellent enantioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 enantiomeric ratio). Unfortunately, the transformation is significantly less efficient when starting from linear β-ketoesters.
:1 enantiomeric ratio). Unfortunately, the transformation is significantly less efficient when starting from linear β-ketoesters.
Central to the utility of this new catalysis is the mechanistic requirement of selective protonation of the nitrogen atom instead of the oxygen atom in the transition state (TS-1 versus TS-2 in Fig. 3).
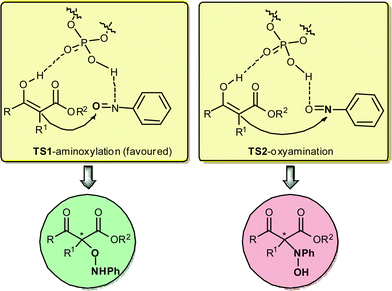 | ||
| Fig. 3 | ||
Notably, catalyst loading as low as 0.5 mol% could be used without particularly affecting the enantioselectivity.
In 2009, Gao et al. showed the first application of the diterpenoid alkaloid lappaconitine 16 as a new densely functionalized chiral base (Scheme 18).40
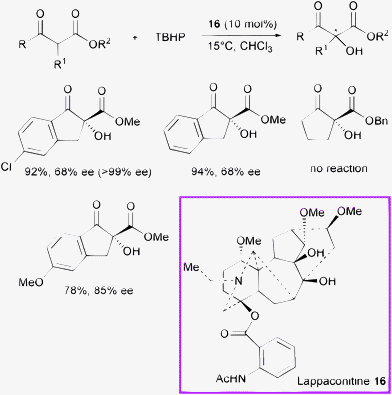 | ||
| Scheme 18 | ||
The α-hydroxylation of β-ketoesters proceeded in high yield, and moderate enantioselectivity (up to 85% ee) when using lappaconitine 16 (a natural product isolated from Aconitum sinomontanum Nakai) as the organocatalyst and tert-butyl hydroperoxide (TBHP) as the oxidant in chloroform. The ee values could be eventually improved to >99% after a single recrystallization.
Recently, the Gao group reported the first enantioselective phase transfer catalyzed direct oxidation of 1-adamantyl (1-Ad) β-oxo esters with CHP (Scheme 19).41
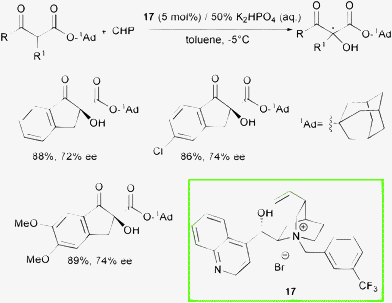 | ||
| Scheme 19 | ||
A range of selectivities was obtained for substituted indanone derivatives using cinchonine-based ammonium salt 17 in combination with K2HPO4 (50% aq) under mild conditions. A bulky ester group in the β-dicarbonyl compound was found to be essential to achieve good enantioselectivity and that was explained with a hypothetical transition state which involves a tight enolate-cinchoninium complex (Fig. 4).
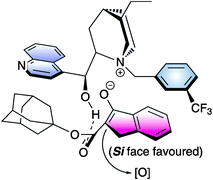 | ||
| Fig. 4 | ||
Although this methodology could be scaled up to a gram-quantity scale, it is restricted to indanone based β-ketoesters.
α-Sulfenylation
Optically active sulfur-containing compounds constitute an important class of chiral ligands, auxiliaries, and synthetic intermediates in organic chemistry.42 Many chiral sulfur-containing molecules exhibit pharmaceutical activities, such as β-lactam antibiotics.In 2005, the Jørgensen group introduced the first enantioselective catalytic asymmetric α-sulfenylation of lactones, lactams, and also β-dicarbonyl compounds (Scheme 20).43 The reactions, carried out at −30 or −40 °C, led to the products in moderate to high yield and enantioselectivity (up to 91% ee) using 1-benzylsulfanyl[1,2,4]triazole as the electrophilic sulfur reagent and the commercially available dimeric cinchona alkaloid (DHQD)2PYR (dihydroquinidine-2,5-diphenyl-4,6-pyrimidinediyl diether) 18 as promoter.
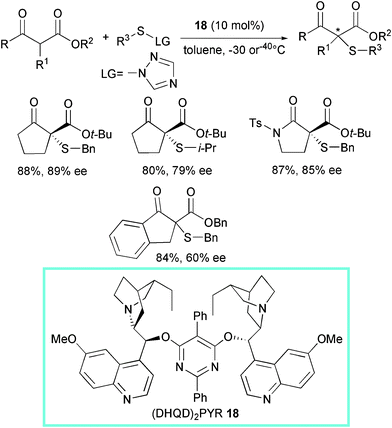 | ||
| Scheme 20 | ||
It was demonstrated that other electrophilic sulfur reagents could be employed such as 1-(2,4-dinitrophenylsulfanyl)-[1,2,4]triazole and isopropylsulfanyl[1,2,4]triazole with comparable efficiency. The diastereoselective reduction of the α-sulfenylated β-keto esters using BH3·DMS gave optically active α-sulfenylated β-hydroxy esters in variable levels of diastereocontrol (dr ranged from 70:30 up to 98:2).
In 2009, Zhu and co-workers reported the first example of α-functionalization of β-ketoesters mediated by a chiral secondary amine. Specifically, the enantioselective α-sulfenylation was catalyzed by easily available α,α-diaryl-L-prolinols,44 which furnished the products in up to 97% ee (Scheme 21).45
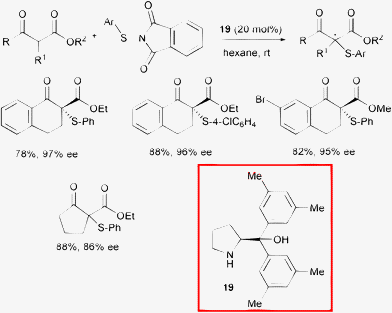 | ||
| Scheme 21 | ||
The optimal protocol required the addition of 20 mol% of commercially available catalyst 19 in hexane at room temperature. The electronic properties of the aryl group of the nucleophiles and their steric features associated with the nature of the ester moiety slightly influenced the level of stereoselectivity. Investigations into the diaryl prolinols stereo- and electronic substitution pattern of the phenyl ring conversely revealed that it has an impact on the performance, with the best results achieved in the case of methyl substituents at the meta positions.
In order to better understand the reaction mechanism, the NMR analysis of an equivalent of compound 19 and a model β-ketoester mixture was performed. A weak intermolecular NOE between the methyl-H group of substrate and the aromatic methyl-H group of the catalyst was observed.
This finding, associated with the absence of signals corresponding to the enamine formation, led them to suggest a plausible noncovalent activation of the β-ketoester by the organocatalyst toward the electrophilic attack by the sulfur reagent (Fig. 5).
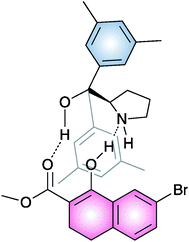 | ||
| Fig. 5 | ||
Increasing use of chiral phosphonic acids and derivatives in peptide, medicinal chemistry and as intermediates for the synthesis of natural products requires the development of new enantioselective methods for their production.46 Recently, Zhu, Cheng and co-authors enlarged the applicability of α,α-diaryl-L-prolinol 19 as an organocatalyst in the α-sulfenylation of β-keto phosphonates (Scheme 22).47
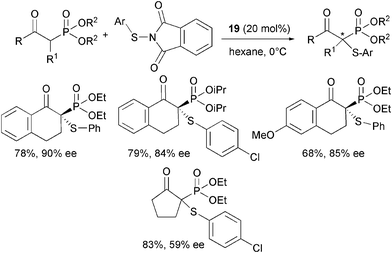 | ||
| Scheme 22 | ||
Highly functionalized products were isolated in good yield and moderate to high enantioselectivity. Different N-(arylthio)phthalimides could be used as a sulfur source with a variety of cyclic β-keto phosphonates. The enantiocontrol proved to be affected by the nature of the ester group and also the ring size of the β-keto phosphonate was important as the aliphatic five-membered ring derivative led to the product in modest enantiomeric excess. Finally, acyclic β-keto phosphonates did not react under usual conditions.
α-Halogenation
The important role of halogen containing compounds in drug synthesis, metabolism studies and material science has led to the development of strategies for halogen–carbon bond formation.29,30,48 Moreover, compounds bearing halogens attached to a stereogenic center are amenable to further synthetic manipulations.A variety of procedures for enantioselective fluorination, chlorination and bromination of organic compounds were reported in the last decade.49 The incentive to the development of this area was given by the discovery of easily accessible and highly efficient electrophilic halogen sources such as N-fluorobenzene sulfonimide (NFSI), N-chlorosuccinimide (NCS) and N-bromosuccinimide (NBS).
In 2002, Kim et al. reported the phase transfer catalyzed α-fluorination of β-ketoesters, using quaternary ammonium salts derived from cinchona alkaloids 20a and 20b using NFSI as the fluorine source and an inorganic base, in the first economic and clean organocatalytic methodology for this process (Scheme 23).50
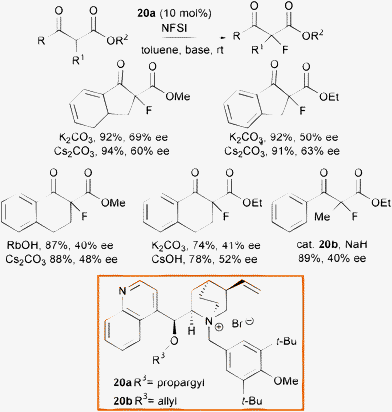 | ||
| Scheme 23 | ||
The nature of the inorganic base slightly influenced the outcome of the reaction and cyclic α-fluoro β-ketoesters were obtained in high yield and moderate enantioselectivity. One example of fluorination of acyclic β-keto esters was reported to proceed with only modest enantiocontrol. The drawback of the process is the long time, up to 10 h, to generate the enolate although, after the slow addition of NFSI it was claimed the product formated within 10 min.
Recently, Maruoka et al. used the same fluorinating agent with a new phase transfer catalyst obtained by combining the thiomorpholine moiety with a C2-symmetric scaffold bearing the axially chiral binaphthyl backbone 21 (Scheme 24).51
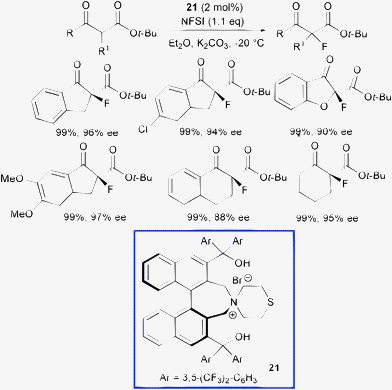 | ||
| Scheme 24 | ||
Notably, all cyclic α-fluoro β-ketoesters were obtained in quantitative yield and high enantioselectivity employing 2 mol% loading of compound (S)-21, although for the acyclic β-ketoesters low enantiocontrol was attained (up to 20% ee). The bis(diarylhydroxymethyl) substituents, placed at the 3,3′-positions of the chiral binaphthyl core, play a crucial role to obtain high stereocontrol. Indeed, catalysts simply bearing aryl groups at 3,3′-positions, which are important to regulate the stereochemical outcome of reactions organocatalyzed by chiral phosphoric acids,52 provided nearly racemic fluorinated product.
In the postulated transition state model, the Z-enolate is stabilized by both the ionic interaction with the ammonium ion and hydrogen bonding of one of the hydroxy groups of the catalyst with the enolate oxygen. Then, the fluorinating agent would preferentially approach from the upper side of the enolate, leading to the observed α-(S)-fluorinated product (Fig. 6).
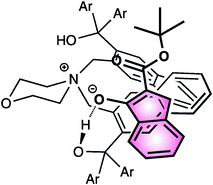 | ||
| Fig. 6 | ||
Unlike the α-fluorination reaction, protocols for the α-chlorination do not make use of phase transfer catalysis. Melchiorre, Bartoli et al. reported a methodology for α-chlorination of 1,3-dicarbonyl compounds with O-benzoylquinidine 22 as the catalyst and a polychlorinated quinolinone as the halogen source (Scheme 25).53
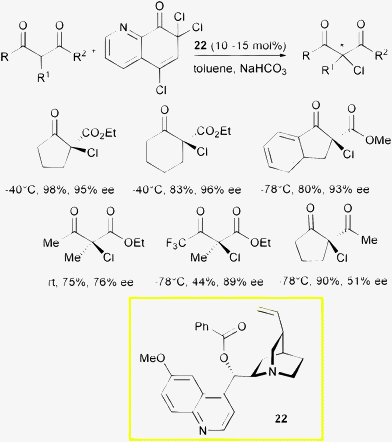 | ||
| Scheme 25 | ||
Trichloroquinolinone proved to be an effective source of chlorine in the enantioselective enolate halogenation, good to high levels of enantioinduction, but only moderate to good yield were obtained working at low temperature with an inorganic base. While cyclic β-ketoesters provided chlorinated products in excellent optical purity, good enantioselectivity but decreased reactivity were found for acyclic products. Challenging β-diketones were also chlorinated but with modest enantiocontrol. Temperature and reaction conditions were modulated according to the substrate structure.
The addition of one equivalent of NaHCO3, responsible for reaction rate acceleration, serves to a rapid deprotonation of the ammonium ion 23, which is tightly associated with the aromatic phenolate anion 24, released after the halogenation process (Fig. 7).
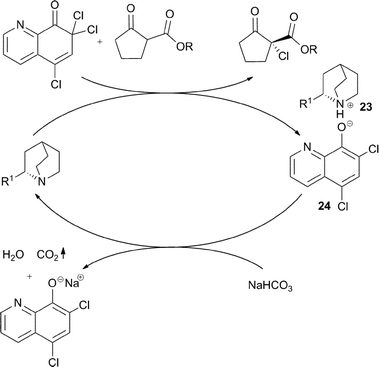 | ||
| Fig. 7 | ||
The faster regeneration of the chiral active catalyst, would help to minimize a racemic chlorination path. Recently, Feng and co-workers disclosed a chiral N,N′-dioxide with great potential as ligand in metal-catalyzed asymmetric processes and also as an organocatalyst.54 They developed a new catalytic system based on N,N′-dioxide 25 derived from pipecolic acid with easily available and low cost NCS as the chlorine source (Scheme 26).55
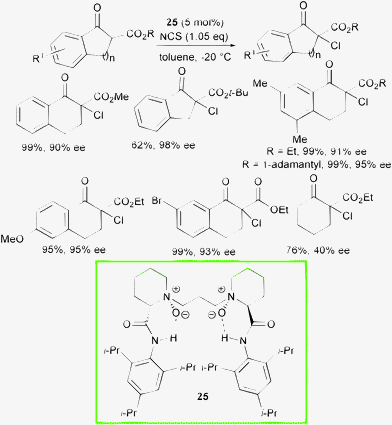 | ||
| Scheme 26 | ||
This process allowed differently substituted chlorinated products to be obtained in generally excellent yield and enantioselectivity. Electron-withdrawing and electron-donating substituents on the aromatic ring were well-tolerated and bulky ester groups resulted in increased enantioselectivity. Unfortunately, simple cyclic β-ketoesters afforded modest results, whereas acyclic β-ketoesters furnished less than 10% conversion to the product.
Díaz-de-Villegas, Gálvez et al. developed and screened several chiral amino diol derivatives as modular finely tunable organocatalysts.56
Enantioselective α-chlorination of cyclic β-ketoesters was performed with the most effective catalyst 26 and NCS as the halogen source (Scheme 27).
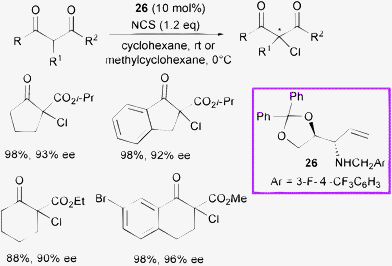 | ||
| Scheme 27 | ||
A wide exploration of the reaction conditions and scope of the process was carried out on five- and six-membered cyclic β-ketoesters. Although the latter were less reactive, high conversion and high levels of enantioselectivity were observed.
Rare examples of the enantioselective α-bromination and α-iodination of β-ketoesters have been reported. The introduction of bromine onto β-ketoesters was described by Melchiorre, Bartoli et al. as an extension of the α-chlorination process with the corresponding trihaloquinolinone and compound 22 as the catalyst (Scheme 28).53
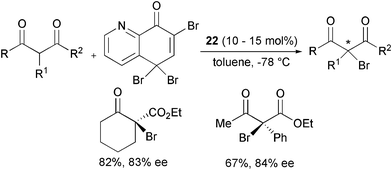 | ||
| Scheme 28 | ||
The products were obtained in good yield and enantioselecticity.
Feng et al. compared the efficiency of different N-halosuccinimides as a source of chlorine, bromine and iodine in the α-halogenation of β-ketoesters with catalyst 25 (Scheme 29).55 While the yields of the products were excellent, the enantioselectivity drastically dropped from chlorine to iodine.
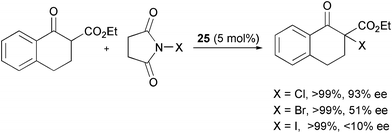 | ||
| Scheme 29 | ||
α-Carbon–phosphorus bond formation
Jørgensen and co-authors have very recently disclosed the first example of enantioselective C–P bond formation by means of a phosphine electrophile.57 This approach is complementary to the methodology illustrated in Scheme 22, where the phosphorus containing moiety is installed in the nucleophile. α-Alkyl substituted cyanoacetates were reacted with diaryl phosphine chlorides, commercially available (DHQD)2PYR 18 as the catalyst and proton sponge (1,8-bis(dimethylamino)naphthalene) as acid scavenger.Indeed, through a one-pot procedure the corresponding enantiomerically enriched quaternary α-phosphino β-aminoacid esters were directly obtained in satisfactory yield and high enantioselectivity (Scheme 30).
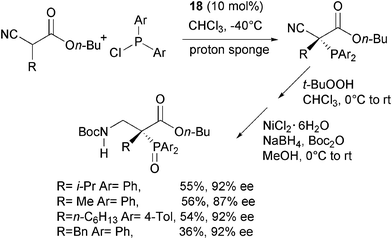 | ||
| Scheme 30 | ||
From a mechanistic point of view, useful insights came from the 31P-NMR analyses of the mixtures containing diarylphosphine chlorides with quinuclidine or catalyst 18 and α-alkyl substituted cyanoacetates. Novel signals appeared and then decreased in the course of the reaction, suggesting the formation of an intermediate. According to these and other studies, a catalytic cycle has been suggested where catalyst 18 gives rise to a nucleophilic substitution to generate the intermediate 27, containing a more electrophilic phosphorus group (Scheme 31).
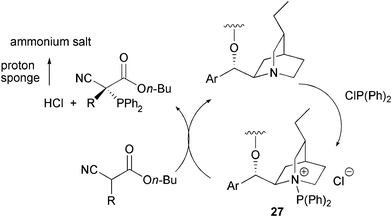 | ||
| Scheme 31 | ||
A base promoted nucleophilic displacement of the catalyst by the α-alkyl substituted cyanoacetate and the electrophilic intermediate 27 occurs to give the product and the HCl formed is neutralized by proton sponge.
Conclusions and outlooks
The development of asymmetric methodologies for the formation of carbon–heteroatom bearing quaternary stereocenters, by reacting relatively acidic α-substituted 1,3-dicarbonyl and related compounds with different electrophiles has been a fertile area of organocatalysis over the last years. A great variety of organic promoters, from mono- to multifunctional molecules and phase transfer catalysts, can be successfully used to install the desired carbon–heteroatom bond in cyclic active methines with generally high enantiocontrol. Current limitations concern the low reactivity and modest enantioselectivity achieved in the case of the acyclic counterparts, which will require the design and employment of more active organocatalysts to promote the process. It is expected that differently substituted activated methines will be investigated as other suitable substrates for these processes. Finally, the search for more effective and novel electrophilic heteroatom sources should also be pursued.Acknowledgements
Italian Ministry of University and Research (MIUR) is acknowledged for financial support.References
- (a) For reviews, see: B. List, Acc. Chem. Res., 2004, 37, 548 Search PubMed; (b) M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001 RSC; (c) G. Guillena and D. J. Ramón, Tetrahedron: Asymmetry, 2006, 17, 1465 CrossRef CAS; (d) T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917 Search PubMed.
- (a) For recent reviews, see: Asymmetric Organocatalysis, ed. A. Berkessel and H Gröger, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) H. Pellissier, Tetrahedron, 2007, 63, 9267 CrossRef CAS; (d) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS; (e) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS; (f) Asymmetric Organocatalysis: Topics in Current Chemistry, ed. B. List, Springer-Verlag,Berlin-Heidelberg, 2009, vol. 291 Search PubMed.
- (a) For a recent review, see: G. Guillena and D. J. Ramón, Curr. Org. Chem., 2011, 15, 296 Search PubMed.
- (a) For reviews, see: A. Mielgo and C. Palomo, Chem.–Asian J., 2008, 3, 922 Search PubMed; (b) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC; (c) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (d) L.-W. Xu, L. Li and Z.-H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS.
- (a) For reviews, see: S. J. Connon, Chem. Commun., 2008, 2499 Search PubMed; (b) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632 RSC.
- (a) For recent reviews on catalytic asymmetric synthesis of quaternary stereocenters, see: J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473 Search PubMed; (b) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (c) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS.
- A. M. R. Smith and K. K. Hii, Chem. Rev., 2011, 111, 1637 CrossRef CAS.
- (a) G. Guillena and D. J. Ramón, Tetrahedron: Asymmetry, 2006, 17, 1465 CrossRef CAS; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS; (c) P. M. Pihko, I. Majander and A. Erkkilä, Top. Curr. Chem., 2009, 291, 29 Search PubMed.
- (a) For recent reviews, see: M. Andrei and K. Undheim, Tetrahedron: Asymmetry, 2004, 15, 53 Search PubMed; (b) J. Watts, A. Benn, N. Flinn, T. Monk, M. Ramjee, P. Ray, Y. Wang and M. Quibell, Bioorg. Med. Chem., 2004, 12, 2903 Search PubMed; (c) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2007, 18, 569 CrossRef CAS; (d) C. Cativiela and M. Ordóńez, Tetrahedron: Asymmetry, 2009, 20, 1 CrossRef CAS; (e) J. Michaux, G. Niel and J.-M. Campagne, Chem. Soc. Rev., 2009, 38, 2093 RSC.
- (a) G. C. Barrett, in Amino Acids, Peptides and Proteins; The Royal Society of Chemistry, 1998, vol. 29 Search PubMed; (b) D. D. Schoepp, D. E. Jane and J. A. Monn, Neuropharmacology, 1999, 38, 1431 CrossRef CAS; (c) J. Venkatraman, S. C. Shankaramma and P. Balaram, Chem. Rev., 2001, 101, 3131 CrossRef CAS.
- (a) For reviews, see: M. T. Reetz, Chem. Rev., 1999, 99, 1121 Search PubMed; (b) H.-S. Lee and S. H. Kang, Synlett, 2004, 1673 CrossRef CAS; (c) C. A. de Parrodi and E. Juaristi, Synlett, 2006, 2699 CrossRef.
- (a) For reviews, see: K. Kacprzak and J. Gawronski, Synthesis, 2001, 961 Search PubMed; (b) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229 CrossRef CAS; (c) H. Li, Y. Chen and L. Deng, in Privileged Chiral Ligands and Catalyst, ed. Q.-L. Zhou, Wiley-VCH, Weiheim, 2011, p. 361 Search PubMed.
- P. M. Pihko and A. Pohjakallio, Synlett, 2004, 2115.
- S. Saaby, M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 8120 CrossRef CAS.
- (a) For reviews, see: S.-K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 Search PubMed; (b) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496 CrossRef CAS.
- X. Liu, H. Li and L. Deng, Org. Lett., 2005, 7, 167 CrossRef CAS.
- X. Liu, B. Sun and L. Deng, Synlett, 2009, 1685 CAS.
- (a) T. Ishikawa and T. Isobe, Chem.–Eur. J., 2002, 8, 552 CrossRef CAS; (b) T. Ishikawa and T. Isobe, J. Synth. Org. Chem. Jpn., 2003, 61, 58 Search PubMed; (c) D. Leow and C.-H. Tan, Chem.–Asian J., 2009, 4, 488 CrossRef CAS.
- M. Terada, M. Nakano and H. Ube, J. Am. Chem. Soc., 2006, 128, 16044 CrossRef CAS.
- M. Terada, D. Tsushima and M. Nakano, Adv. Synth. Catal., 2009, 351, 2817 CrossRef CAS.
- X. Xu, T. Yabuta, P. Yuan and Y. Takemoto, Synlett, 2006, 137.
- Phase-Transfer Catalysis: Fundamentals, Applications, and Industrial Perspectives, ed. C. M. Starks, C. L. Liotta, M. Halpern, Chapman & Hall, New York, 1994, p. 132 Search PubMed.
- (a) R. He, X. Wang, T. Hashimoto and K. Maruoka, Angew. Chem., Int. Ed., 2008, 47, 9466 CrossRef CAS; (b) R. He and K. Maruoka, Synthesis, 2009, 2289 Search PubMed.
- (a) T. Negoro, M. Murata, S. Ueda, B. Fujitani, Y. Ono, A. Kuromiya, K. Suzuki and J.-I. Matsumoto, J. Med. Chem., 1998, 41, 4118 CrossRef CAS; (b) T. Mashiko, N. Kumagai and M. Shibasaki, Org. Lett., 2008, 10, 2725 Search PubMed.
- Q. Lan, X. Wang, R. He, C. Ding and K. Maruoka, Tetrahedron Lett., 2009, 50, 3280 Search PubMed.
- (a) For selected reviews on asymmetric phase transfer catalysis, see: M. J. O'Donnell, in Catalytic Asymmetric Synthesis, ed I. Ojima, Verlag-Chemie, New York, 1993, ch. 8 Search PubMed; (b) S. Shirakawa and K. Maruoka, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New York, 3rd edn, 2010, p. 95 Search PubMed.
- S. H. Jung and D. Y. Kim, Tetrahedron Lett., 2008, 49, 5527 Search PubMed.
- J. Y. Mang and D. Y. Kim, Bull. Korean Chem. Soc., 2008, 29, 2091 Search PubMed.
- (a) D. O'Hagan and H. S. Rzepa, Chem. Commun., 1997, 645 Search PubMed; (b) G. Thomas, Medicinal Chemistry: An Introduction, Wiley, New York, 2000 Search PubMed; (c) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- (a) For reviews see: K. Mikami, Y. Itoh and M. Yamanaka, Chem. Rev., 2004, 104, 1 Search PubMed; (b) S. France, A. Weatherwax and T. Lectka, Eur. J. Org. Chem., 2005, 475 CrossRef CAS.
- S. M. Kim, J. H. Lee and D. Y. Kim, Synlett, 2008, 2659 Search PubMed.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS.
- Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CAS.
- H. Konishi, T. Y. Lam, J. P. Malerich and V. H. Rawal, Org. Lett., 2010, 12, 2028 CrossRef CAS.
- X. Han, F. Zhong and Y. Lu, Adv. Synth. Catal., 2010, 352, 2778 Search PubMed.
- Z.-H. Zhang, X.-Q. Dong, H.-Y. Tao and C.-J. Wang, Arkivoc, 2011, 137 Search PubMed.
- For a review see: J. Christoffers, A. Baro and T. Werner, Adv. Synth. Catal., 2004, 346, 143 Search PubMed.
- M. R. Acocella, O. G. Mancheño, M. Bella and K. A. Jørgensen, J. Org. Chem., 2004, 69, 8165 CrossRef CAS.
- M. Lu, D. Zhu, Y. Lu, X. Zeng, B. Tan, Z. Xu and G. Zhong, J. Am. Chem. Soc., 2009, 131, 4562 CrossRef CAS.
- B. Gong, Q. Meng, T. Su, M. Lian, Q. Wang and Z. Gao, Synlett, 2009, 2659 CAS.
- M. Lian, Z. Li, J. Du, Q. Meng and Z. Gao, Eur. J. Org. Chem., 2010, 6525 CrossRef CAS.
- (a) D. A. Evans, K. R. Campos, J. S. Tedrow, F. E. Michael and M. R. Gagne, J. Am. Chem. Soc., 2000, 122, 7905 CrossRef CAS; (b) A. M. Masdeu-Bulto, M. Dieguez, E. Martin and M. Gomez, Coord. Chem. Rev., 2003, 242, 159 CrossRef CAS.
- S. Sobhani, D. Fielenbach, M. Marigo, T. C. Wabnitz and K. A. Jørgensen, Chem.–Eur. J., 2005, 11, 5689 CrossRef CAS.
- For a review on α,α-L-diaryl prolinols as organocatalysts, see: A. Lattanzi, Chem. Commun., 2009, 1452 Search PubMed.
- L. Fang, A. Lin, H. Hu and C. Zhu, Chem.–Eur. J., 2009, 15, 7039 CrossRef CAS.
- V. P. Kukhar and H. R. Hudson, in Aminophosphonic and Aminophosphinic Acids, Chemistry and Biological Activity, John Wiley & Sons, New York, 2000, p. 634 Search PubMed.
- A. Lin, L. Fang, X. Zhu, C. Zhu and Y. Cheng, Adv. Synth. Catal., 2011, 353, 545 Search PubMed.
- (a) N. De Kimpe and R. Verhé, The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines, John Wiley & Sons, New York, 1988 Search PubMed; (b) C. Czekelius and C. C. Tzschucke, Synthesis, 2010, 543 Search PubMed.
- (a) For recent reviews on asymmetric halogenation reactions, see: H. Ibrahim and A. Togni, Chem. Commun., 2004, 1147 Search PubMed; (b) M. Oestreich, Angew. Chem., Int. Ed., 2005, 44, 2324 CrossRef CAS.
- D. Y. Kim and E. J. Park, Org. Lett., 2002, 4, 545 CrossRef CAS.
- X. Wang, Q. Lan, S. Shirakawa and K. Maruoka, Chem. Commun., 2010, 46, 321 RSC.
- (a) For recent reviews, see: M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539 Search PubMed; (b) M. Terada, Curr. Org. Chem., 2011, 15, 2227 Search PubMed; (c) A. Zamfir, S. Schenker, M. Freund and S. B. Tsogoeva, Org. Biomol. Chem., 2010, 5262 Search PubMed.
- G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, P. Melchiorre and L. Sambri, Angew. Chem., Int. Ed., 2005, 44, 6219 CrossRef CAS.
- (a) Z. P. Yu, X. H. Liu, Z. H. Dong, M. S. Xie and X. M. Feng, Angew. Chem., Int. Ed., 2008, 47, 1308 CrossRef CAS; (b) K. Zheng, J. Shi, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2008, 130, 15770 CrossRef CAS; (c) X. Zhou, D. J. Shang, Q. Zhang, L. L. Lin, X. H. Liu and X. M. Feng, Org. Lett., 2009, 11, 1401 CrossRef CAS.
- Y. Cai, W. Wang, K. Shen, J. Wang, X. Hu, L. Lin, X. Liu and X. Feng, Chem. Commun., 2010, 46, 1250 RSC.
- P. Etayo, R. Badorrey, M. D. Dìaz-de-Villegas and J. A. Gàlvez, Adv. Synth. Catal., 2010, 352, 3329 Search PubMed.
- M. Nielsen, C. Borch Jacobsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 3211 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |