
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of a hybrid PrPfr state in the dark reversion of a bathy phytochrome indicates inter-dimer allostery
Sayan
Prodhan
 a,
Petra
Mészáros
a,
Szabolcs
Bódizs
a,
Yalin
Zhou
a,
Michał
Maj
b and
Sebastian
Westenhoff
*a
a,
Petra
Mészáros
a,
Szabolcs
Bódizs
a,
Yalin
Zhou
a,
Michał
Maj
b and
Sebastian
Westenhoff
*a
aDepartment of Chemistry – BMC, Biochemistry, Uppsala University, 75123 Uppsala, Sweden. E-mail: sebastian.westenhoff@kemi.uu.se
bDepartment of Chemistry—Ångström Laboratory, Uppsala University, Uppsala, Sweden
First published on 4th September 2025
Abstract
Phytochromes are photosensor proteins found in plants, fungi, and bacteria. They photoswitch between red light absorbing (Pr) and far-red light absorbing (Pfr) states. Thermal reversion in the dark, however, is an equally important factor in controlling their signaling levels. Phytochromes are generally dimeric proteins, therefore, mixed PrPfr states are possible. These states have been implicated in dark reversion studies of plant phytochromes, but not in bacterial phytochromes. Here, we investigate the dark reversion kinetics of the 'bathy' phytochrome from P. aeruginosa (PaBphP) using UV-Vis absorption spectroscopy. A single set of time-resolved spectra does not conclusively reveal the presence of a mixed PrPfr state, as both a direct Pr → Pfr model or a sequential Pr → PrPfr → Pfr model fit the spectral kinetics. However, a systematic analysis of dark reversion kinetics with varying Pr/Pfr ratios can only be satisfactorily fit by the sequential model, which indicates the presence of an intermediate PrPfr state. A newly designed monomeric variant of PaBphP provides strong support for this interpretation. Temperature-dependent kinetics revealed similarly low activation energies for the dark reversion processes of both proteins, consistent with a previously proposed keto–enol tautomerization preceding dark reversion. Interestingly, our results suggest allosteric regulation of dark reversion across the dimer, which we propose to be a contributing factor in phytochrome signaling.
1 Introduction
Many organisms rely on photoreception to adapt to changing environmental conditions.1,2 Among various light-sensing systems, phytochromes are red light-detecting photoreceptors that regulate physiological responses in plants, fungi, and bacteria.3–5 In plants, they control essential processes like seed germination, chloroplast development, and flowering,6–8 while in bacteria, they regulate pigmentation, motility, and biofilm formation.9–11 Therefore, the phytochrome photocycle is extensively studied for biotechnological applications, as well as for the engineering of optogenetic tools and near-infrared fluorescent markers used in microscopy and cell biology.12,13Phytochromes are typically homodimeric proteins with a modular structure.14 Each monomer consists of an N-terminal photosensory module (PSM), responsible for light sensing, and a C-terminal output module (OPM), which mediates signal transduction. In some cases, additional N-terminal extensions may also contribute to the effector function of the protein. The PSM is composed of three functionally distinct domains: PAS (Per-ARNT-Sim),15 GAF (cGMP phosphodiesterase/adenylate cyclase/FhlA),16 and PHY (phytochrome-specific domain), and contains a bilin cofactor that serves as the chromophore.17
Phytochromes interconvert between two structurally distinct states: the red light-absorbing Pr state (λmax ≈ 700 nm, for bacterial phytochromes) and the far-red-light-absorbing Pfr state (λmax ≈ 750 nm). These states are associated with distinct biochemical properties and, consequently, different signaling activities.18,19 Based on the relative free energies of these states, phytochromes are classified as either canonical, the most common type with a thermally stable Pr state, or reverse-activated (bathy), in which the Pfr state is thermodynamically favored (Fig. 1(a)).20–22 In the absence of light, phytochromes thermally revert to their resting state.
 | ||
| Fig. 1 The phytochrome photoswitches between Pr and Pfr states, and chromatography confirms the oligomeric states of the two protein samples. (a) Conformational differences in the biliverdin structure distinguish the Pfr (≈750 nm) and Pr (≈700 nm) states, which are reflected in their steady-state absorption spectra. (b) Preparatory SEC analysis of FL-PaBphP-D and PSM-PaBphP-M, revealing their oligomerization state. The shaded region indicates the fraction collected for FL-PaBphP-D. For PSM-PaBphP-M, the collection zone was around the peak maximum. (c) Analytical SEC of FL-PaBphP-D in Pr and Pfr state confirms that the protein is predominantly dimeric. The slight shift in the retention time between the states likely reflects conformational changes. | ||
Canonical and bathy phytochromes have been extensively studied using spectroscopy and structural biology methods. Most of these studies focus on understanding the sequence of events during photoactivation.21,23–27 They show that upon photoactivation, the chromophore undergoes isomerization around the C15![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C16 bond, followed by a series of structural changes through intermediate states (i.e., Lumi-F/R and Meta-F/R).28–30 Crystal, NMR, and cryo-EM structures of phytochromes in both the Pr and Pfr states have been solved.17,31–35 Of particular interest are two recent crystal and cryo-EM structures, which reveal phytochromes in mixed Pr/Pfr heterodimers.36,37 However, it remains unclear how these intermediate states are formed.
C16 bond, followed by a series of structural changes through intermediate states (i.e., Lumi-F/R and Meta-F/R).28–30 Crystal, NMR, and cryo-EM structures of phytochromes in both the Pr and Pfr states have been solved.17,31–35 Of particular interest are two recent crystal and cryo-EM structures, which reveal phytochromes in mixed Pr/Pfr heterodimers.36,37 However, it remains unclear how these intermediate states are formed.
The dark reversion has been studied in both plant and bacterial phytochromes,19,24,27,28,33,35,38–48 with studies on plant phytochromes providing the most detailed insights, including differences in activation energy across various temperature ranges,38 the presence of PrPfr hybrid states,46 and temperature-dependent kinetics that allow phytochromes to function as temperature sensors.19,42 Plants also have multiple isoforms of phytochromes, which contribute to varying dark reversion rates, allowing for more complex adaptation to different environmental conditions.41
In bacterial phytochromes, the dark reversion process is less well understood. While dark reversion is commonly used as a standard assay in structural and biochemical studies of phytochromes, only a few detailed studies have been conducted.43–45,49–51 However, a hybrid PrPfr state has not been observed as part of the dark reversion process in bacterial phytochromes yet.
In this study, we systematically investigate the dark reversion kinetics of a bathy phytochrome from Pseudomonas aeruginosa (PaBphP). By comparing the wild-type full-length dimer (FL-PaBphP-D) with a newly designed monomeric variant (PSM-PaBphP-M), we provide clear evidence for the presence of PrPfr states as intermediates in the dark reversion process for the wild-type protein. This finding suggests a novel allosteric mechanism between the two protomers.
2 Materials and methods
2.1 Recombinant protein production
The plasmid used for the recombinant expression of the full-length P. aeruginosa phytochrome contained the PaBphP insert (GenBank: AAG07504.1) with a C-terminal His6-tag and a pET28a(+) backbone (Novagen). The plasmid used for the recombinant expression of the P. aeruginosa phytochrome PAS-GAF-PHY mutant fragment contained the PaBphP insert (GenBank: AAG07504.1) with a C-terminal His6-tag and a pET24a(+) plasmid backbone (GenScript). To facilitate biliverdin incorporation during expression, all the phytochrome constructs were co-expressed with a heme oxygenase plasmid, kindly provided by Prof. Janne A. Ihalainen. The plasmids were transformed into BL21(DE3) Escherichia coli strain for expression.Cells were grown in LB medium supplemented with 50 μg mL−1 kanamycin and 34.5 μg mL−1 chloramphenicol at 37 °C until reaching an OD6oo of 0.8. Expression of PaBphP and heme oxygenase was induced by the addition of 1 mM IPTG and 1 mM 5-aminolevulinic acid. Protein expression was carried out overnight at 18 °C.
2.2 Protein purification
Cells were re-suspended in 10 mL of lysis buffer per gram of pellets (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5% glycerol), supplemented with one tablet of EDTA-free protease inhibitor (cOmplete Protease Inhibitor Cocktail, Roche) and 10 U mL−1 DNase I. Lysis was carried out by sonication on ice at 70% amplitude for 5 min, with 5 s pulses on and 10 s pulses off. The supernatant was collected after centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 35 min (JA 14.50 rotor, Beckman) and was briefly incubated with a molar excess of biliverdin hydrochloride. The lysate was purified by Ni-NTA affinity chromatography using a HisTrap HP 5 mL column (Cytiva). The bound protein was washed with wash buffer I (50 mM Tris, 50 mM NaCl, pH 8.0) and wash buffer II (50 mM Tris, 1 M NaCl, pH 8.0), and eluted with elution buffer (50 mM Tris, 50 mM NaCl, 300 mM imidazole, pH 8.0). This was followed by size-exclusion chromatography using a HiLoad 16/600 Superdex 200 pg column (Cytiva) in wash buffer I. The purified protein was then flash-frozen for storage. There was no further buffer exchange when conducting the spectroscopic measurements. The buffer used for the final spectroscopic measurements is wash buffer I (50 mM Tris, 50 mM NaCl, pH 8.0).
000g for 35 min (JA 14.50 rotor, Beckman) and was briefly incubated with a molar excess of biliverdin hydrochloride. The lysate was purified by Ni-NTA affinity chromatography using a HisTrap HP 5 mL column (Cytiva). The bound protein was washed with wash buffer I (50 mM Tris, 50 mM NaCl, pH 8.0) and wash buffer II (50 mM Tris, 1 M NaCl, pH 8.0), and eluted with elution buffer (50 mM Tris, 50 mM NaCl, 300 mM imidazole, pH 8.0). This was followed by size-exclusion chromatography using a HiLoad 16/600 Superdex 200 pg column (Cytiva) in wash buffer I. The purified protein was then flash-frozen for storage. There was no further buffer exchange when conducting the spectroscopic measurements. The buffer used for the final spectroscopic measurements is wash buffer I (50 mM Tris, 50 mM NaCl, pH 8.0).
2.3 Design of monomeric variant
The mutant plasmid was designed using only the PSM of the protein, excluding the histidine kinase module, to promote monomerization with minimal mutational changes. Mutation sites were selected based on amino acid properties and spatial proximity (within 4 Å). I294E and V298E mutations were introduced into the wild-type sequence to replace hydrophobic residues with charged glutamate residues (Fig. 2(a)). The mutant plasmid was generated using GenScript's site-directed mutagenesis service. | ||
| Fig. 2 Design and structural prediction of truncated monomeric mutant PaBphP construct. (a) Locations of I294E and V298E mutation sites in the wild-type, truncated structure, highlighting their spatial proximity. (b) AlphaFold prediction of the wild-type truncated sequence as a dimer, showing proper dimerization and a conformation comparable to the crystal structure. (c) AlphaFold prediction of the I294E/V298E mutant in its monomeric form, showing proper folding of the monomeric unit. | ||
2.4 UV-Vis absorption spectroscopy
Steady-state UV-Vis absorption measurements were performed using a Shimadzu UV-1900i spectrophotometer equipped with LabSolutions UV-Vis software (Version 1.13). For dark reversion kinetics, 100 sequential UV-Vis spectra scans were recorded at 45-second intervals with a delay of 4 seconds between diode illumination of the phytochrome samples and the start of each measurement. We verified that the choice of sampling rate did not influence the kinetics. For time-dependent diode illumination experiments, samples were exposed to a 780 nm light-emitting diode (LED) for durations ranging from 6 to 60 seconds. Temperature-dependent measurements were conducted by irradiating the phytochrome samples with a 780 nm LED for 120 seconds at temperatures ranging from 10 °C to 35 °C.2.5 Calculation of Pr/Pfr fraction in diode illuminated phytochrome
To determine the relative amounts of Pr and Pfr in the illuminated samples, we first acquired absorption spectra for the pure Pr and pure Pfr states. The pure Pfr spectrum was recorded after incubating the sample in the dark for approximately three hours. The pure Pr spectrum was recorded after illuminating the sample with a 780 nm LED for 120 seconds. The concentrations of Pr and Pfr in the illuminated sample were determined by fitting the initial spectrum obtained after illumination to the following equation:| I(λ) = X·IPfr(λ) + (1 − X)·IPr(λ) | (1) |
2.6 Global kinetic analysis
The time-resolved absorption data were globally fitted using two kinetic schemes: a direct model and a sequential model (further detailed in eqn (3)–(8)). For both models, the analysis relied on a set of basis spectra representing the pure chemical species. The basis spectrum for the initial PrPr state was defined as the first spectrum measured after illumination, while the basis spectrum for the final PfrPfr state was defined as the last spectrum of the kinetic series. The spectrum for the intermediate species, PrPfr, was assumed to be the average of these initial and final basis spectra (Fig. 3). | ||
| Fig. 3 Basis spectra used for the global fitting of FL-PaBphP-D using sequential model. | ||
The initial conditions for the kinetic models were set according to the type of experiment. For temperature-dependent dark reversion measurements, we used maximal illumination times (120 seconds) and therefore the system was assumed to start in a pure PrPr state, with initial fractional populations of PrPr, PrPfr, and PfrPfr set to 1, 0, and 0, respectively. For illumination-dependent measurements, the initial spectrum of each kinetic trace was first fitted as a linear combination of pure Pr and Pfr spectra. The resulting φ (eqn (9)) were then used to calculate the starting fractions of PrPr, PrPfr, and PfrPfr (eqn (10)–(12), see Fig. 5(c)), which served as the initial conditions for the subsequent global fit.
To ensure a realistic assessment of parameter uncertainty, we employed the profile-likelihood method to calculate confidence intervals, as it systematically maps the true error landscape and is robust to parameter correlations. The boundary of the confidence interval is defined by a threshold value for the residual sum of squares (RSS), determined by varying one parameter while optimizing all others. Any set of parameters having an RSS value above this threshold is considered statistically inconsistent with the data. The threshold is calculated using the following formula:
 | (2) |
3 Results
3.1 Design of monomeric variant
To study the dark reversion kinetics as a function of the oligomeric state of PaBphP, we prepared a truncated, monomeric variant of the protein. Mutations I294E and V298E were introduced into the PSM to disrupt dimer formation by weakening hydrophobic interactions and introducing electrostatic repulsion (Fig. 2(a)). This mutational strategy was motivated by previous studies on the canonical phytochrome from Deinococcus radiodurans, which showed that glutamate substitutions at L311 and L314 and a cysteine mutation at F145 positions in DrBphP effectively disrupted dimerization and produced stable monomers.45,52 Our AlphaFold predictions supported that these mutations would disrupt the dimer interface while maintaining proper monomer folding. Structural models of both the wild-type and mutant truncated proteins were generated using the GenBank sequence AAG07504.1, in both dimeric and monomeric forms. While the wild-type sequence was reliably predicted to form a stable dimer (Fig. 2(b)), AlphaFold did not predict a stable dimeric structure for the mutant variant. However, it successfully predicted a stable monomeric fold for the mutant (Fig. 2(c)), indicating that the introduced mutations disrupted dimerization without compromising the native folding.3.2 Size-exclusion chromatography
To confirm the oligomeric states of the full-length protein (FL-PaBphP-D) and the truncated mutant (PSM-PaBphP-M), we performed size-exclusion chromatography (SEC). Preparative SEC (Fig. 1(b)) showed that FL-PaBphP-D (blue trace) predominantly forms dimers, indicated by a major peak at lower elution volumes (68–72 mL). A minor peak at 7–8 mL suggests a small monomeric population. The shaded region marks the fraction collected for further analysis. In contrast, PSM-PaBphP-M (red trace) showed a single peak around 88 mL, consistent with monomeric species. Analytical SEC of the FL-PaBphP-D fraction, performed both in the dark and under continuous 780 nm illumination, confirmed the dimeric state in both Pr and Pfr forms, with elution volumes of 1.24–1.26 mL. A small shoulder at 1.5 mL may correspond to a minor monomeric fraction (Fig. 1(c)). A slight shift in the dimer peak between states likely reflects conformational differences between Pr and Pfr that influence the protein's shape. We conclude that PSM-PaBphP-M is monomeric, whereas the wild-type FL-PaBphP-D is predominantly dimeric (>90%).3.3 Dark reversion
To measure dark reversion kinetics, all phytochrome samples were illuminated for 120 seconds with LED light at 780 nm to populate the Pr state. Fig. 4(a) and (d) show the time-dependent spectral evolution from the initially photoconverted Pr state back to the Pfr state. Under these conditions, FL-PaBphP-D and PSM-PaBphP-M display qualitatively similar spectral features over time. | ||
| Fig. 4 Both, the sequential and direct model fit the dark reversion kinetics of FL-PaBphP-D and PSM-PaBphP-M reasonably well. UV-Vis spectra recorded over time during dark reversion for (a) FL-PaBphP-D, and (d) PSM-PaBphP-M. Kinetic traces at 700 and 750 nm with global fitting by direct and sequential model for (b) FL-PaBphP-D and (e) PSM-PaBphP-M. Dashed lines represent the global fit with a direct model, while solid lines show the fit using a sequential model. Residual of global fitting at 700 and 750 nm with both direct and sequential model for (c) FL-PaBphP-D and (f) PSM-PaBphP-M. The temperature for these experiments is maintained at 20 °C. | ||
The dark reversion kinetics of the protein is analyzed using two kinetic models to elucidate the underlying mechanism: a direct transition and a sequential model involving an intermediate state. We first describe the direct transition model:
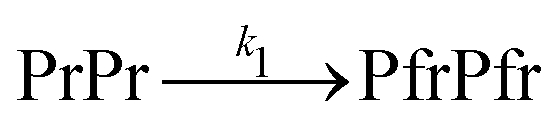 | (3) |
| c1(λ, t) = c(1)0(λ)·exp(−k1·t) | (4) |
 | (5) |
| c1(λ, t) = c(1)0(λ)·exp(−k1·t), | (6) |
 | (7) |
| c3(λ, t) = 1 − c1(λ, t) − c2(λ, t), | (8) |
In the first experiment, dark reversion kinetics are measured starting from a state with a maximal Pr population. Global fitting of kinetic data for FL-PaBphP-D and PSM-PaBphP-M is performed using direct (eqn (3)) and sequential (eqn (5)) models. The resulting kinetic fits at 700 and 750 nm are shown in Fig. 4(b) and (e). The sequential model yields a better fit than the direct model for FL-PaBphP-D as judged from the residual plot (Fig. 4(c)), whereas for PSM-PaBphP-M the trends in the residuals (Fig. 4(f)) are indistinguishable However, the effect is small and we do not consider this difference as sufficient evidence to favor one model over the other.
To differentiate between the models, we investigated the dependence of the refined rate constants on the initial photostationary state. To achieve varying initial photostationary states, we altered the duration of the illumination at 780 nm from 6 to 60 seconds. This variation in illumination time modified the initial fraction of the Pr state at the onset of the dark reversion measurement (Fig. 5(a) and (b)). The resulting photostationary states are defined as the fraction of Pr relative to the total phytochrome population, as given by:46
 | (9) |
| PrPr = φ2 | (10) |
| PrPfr = 2(1 − φ)φ, | (11) |
| PfrPfr = (1 − φ)2. | (12) |
 | ||
| Fig. 5 Preparation of different initial concentrations of Pr and Pfr and their global kinetic fits at 20 °C. Spectra at various photostationary states for (a) PSM-PaBphP-M and (b) FL-PaBphP-D, generated by variation of the diode illumination time as indicated in the panels. (c) Refined starting fractions of PrPr, PrPfr, and PfrPfr states using eqn (9)–(12) for FL-PaBphP-D. (d) Variation of dark reversion rate constants with φ, obtained from global fitting with the direct step model for FL-PaBphP-D and PSM-PaBphP-M. (e) Variation of fast (k1) and slow (k2) dark reversion rate constants with φ from the fits of the sequential model for FL-PaBphP-D. (f) Resulting simulated relative concentration of PrPr, PrPfr, and PfrPfr over time during temperature-dependent dark reversion at φ = 1. | ||
After global fitting of all data sets, we now analyze the rate-refined constants as a function of the initial photostationary states. Using the direct model (eqn (3) and (4)), we observed that the rate constants (k1) remain independent of the initial photostationary states for PSM-PaBphP-M, as shown in Fig. 5(d). This is expected as the rate constant of dark reversion should not change with the population of the initial state. In contrast, for the FL-PaBphP-D, the refined rates vary significantly with φ (Fig. 5(d)). This dependence indicates that the direct transition model does not adequately describe the reaction, as true rate constants should be independent of initial concentrations.
When examining the rates of the sequential model as a function of φ, we find that k1 and k2 remain constant for φ > 0.5 (Fig. 5(e) and Table 1). At the lowest excitation fraction (φ = 0.3), k1 appears as an outlier. This is likely due to the low abundance of PrPr under this illumination condition, which limits the reliable determination of k1. The analysis strongly supports the applicability of the sequential model to the dark reversion of the dimeric protein, since only for this model the rate constants are independent of the initial population distribution.
| φ | FL-PaBphP-D | |
|---|---|---|
| k 1 (s−1) | k 2 (s−1) | |
| 0.30 | 0.012 | 0.0011 |
| 0.55 | 0.0051 | 0.0012 |
| 0.64 | 0.0054 | 0.0012 |
| 0.70 | 0.0059 | 0.0012 |
| 0.80 | 0.0055 | 0.0013 |
| 0.85 | 0.0056 | 0.0013 |
| 0.92 | 0.0057 | 0.0013 |
| 0.98 | 0.0057 | 0.0013 |
We note here that although the change in k1 rate constants from the direct and sequential models for the dimeric variant fall within a similar numerical range, they exhibit distinctly different trends. The rate constant from the direct model shows a systematic 61.3% increase across the φ values (Fig. 5d), whereas the value from the sequential model remains stable, fluctuating slightly around its mean (Fig. 5e). The monomeric variant further confirms this notion because the same dependence determined for the monomeric variant does not show an increase with φ, but fluctuations around a mean value (Fig. 5d).
Having established the presence of a PrPfr intermediate state in the dark reversion of the dimeric variant, we extended our investigation by probing the temperature dependence of dark reversion kinetics. Spectra for both proteins were recorded across a temperature range of 10 °C to 35 °C. Each measurement was initiated from the pure Pr (PrPr) state at maximum photoexcitation. The initial spectra recorded immediately after illumination are consistent across all temperatures, confirming a uniform starting population of the Pr state. To extract the temperature-dependent rate constants, we globally fit the kinetic data using the sequential model (eqn (5)) for FL-PaBphP-D and the direct model (eqn (3)) for PSM-PaBphP-M.
The resulting temperature dependence of the rate constants is shown in Fig. 6a and b and tabulated in Table 2. For both proteins, the kinetic rate constants increase significantly with temperature, indicating that thermal energy facilitates dark reversion. To quantify this effect, the activation energy (Ea) for each kinetic step was determined using the Arrhenius equation. For FL-PaBphP-D, the activation energies for k1 and k2 are 3.18 ± 1.92 and 6.16 ± 2.94 kJ mol−1, respectively. These values lead to the unexpected conclusion that the photochemical state of the sister protomer in the dimer influences the activation energy of the reaction. In contrast, PSM-PaBphP-M shows a significantly higher activation energy of 15.24 ± 0.83 kJ mol−1 for k1, suggesting that even the quaternary structure of the protein impacts the activation energy.
 | ||
Fig. 6 Arrhenius analysis shows notably low activation energies for the dark reversion of PaBphP. Logarithmic variation of (a) fast (k1) and slow (k2) dark reversion rate constants with the inverse of temperature for FL-PaBphP-D and (b) logarithmic variation of fast (k1) dark reversion rate constants with the inverse of temperature for PSM-PaBphP-M. Activation energy is calculated using Arrhenius equation  . . | ||
| Temperature (°C) | FL-PaBphP-D | PSM-PaBphP-M | |
|---|---|---|---|
| k 1 (s−1) | k 2 (s−1) | k (s−1) | |
| 10 | — | — | 0.0009 |
| 15 | — | — | 0.0011 |
| 16 | 0.0055 | 0.0011 | — |
| 20 | 0.0057 | 0.0013 | 0.0013 |
| 25 | 0.0059 | 0.0012 | 0.0014 |
| 27 | 0.0058 | 0.0012 | — |
| 30 | 0.0063 | 0.0012 | 0.0015 |
| 34 | 0.0063 | 0.0014 | — |
| 35 | 0.0057 | 0.0013 | 0.0017 |
Our kinetic analysis shows that the transition rate (k1) from PrPr to PrPfr is about 2–3 times faster than the transition rate (k2) from PrPfr to PfrPfr. This suggests that a significant portion of the PrPfr intermediate state is formed during the intermediate phase of dark reversion in FL-PaBphP-D (Fig. 5(f)). More importantly, this shows that substantial allosteric interactions may occur between the two protomers in the dimer, as the dark reversion time differs depending on whether both monomers are in the Pr state or if one is in the Pfr state. This points to an as-yet undescribed interaction between the two protomers in the dimer.
4 Discussion
Our kinetic analysis of PaBphP provides the first direct demonstration of a PrPfr heterodimer intermediate during the thermal reversion of bacterial phytochromes. By analyzing kinetic models in a series of experiments with varying initial concentrations of Pr and Pfr, we show that a two-step mechanism accurately describes the dark reversion pathway. Only this model yields rate constants that are independent of the initial Pr:Pfr ratio.
This finding mirrors observations made for plant phytochromes, where dark reversion proceeds via a PrPfr heterodimer before returning to the Pr state.39,46PaBphP being a bathy phytochrome exhibits a similar sequential interconversion pathway, but in the reverse direction, from Pr to Pfr.
To our knowledge and somewhat surprisingly, earlier studies on bacterial phytochromes have not considered the possibility of a PrPfr intermediate state.21,22,33,35,44 This may be due to the fact that, in canonical phytochromes, the initial state is typically a mixture of Pr and Pfr, making it difficult to assign a specific dark recovery pathway. In contrast, bathy phytochromes allow for the generation of nearly pure Pr states through far-red illumination, enabling the identification of a PrPfr intermediate state.
It is established that the Pfr form of PaBphP represents the biochemical “off” state.35,44 Huber et al. qualitatively correlated biochemical kinase activity, measured via radionucleotide assays, with the decaying Pr/Pfr ratio during dark reversion.44 However, their study did not consider the potential presence of a mixed PrPfr heterodimer. This raises the question of whether the PrPfr state is also biochemically active and whether it might even substitute for PrPr as the functional state. Notably, in plant phytochromes, the hybrid PrPfr state is known to play a distinct functional role compared to the pure PrPr or PfrPfr states.39,48 A definitive assignment of the PrPfr state's function would require a quantitative analysis of the radionucleotide assays, a challenging task, as acknowledged by Huber et al. Nonetheless, their assays show high biochemical activity after prolonged far-red illumination,44 a condition in which the PrPr population is significantly higher than that of PrPfr (see Fig. 5(c) at φ = 1). We believe that PrPr is indeed kinase-active. However, this line of reasoning does not rule out the possibility that the PrPfr state also exhibits biochemical activity.
The unusually low activation barriers observed for the PaBphP protein are difficult to explain solely by a steric model of C15C16 double bond isomerization. An alternative chemical mechanism may be involved. In fact, keto–enol tautomerization of the D-ring in its Z-isomerized (Pr) state has been proposed to precede isomerization of the C15–C16 bond.43 The enol form lowers the bond order of the C15–C16 bond, which would significantly reduce the energy of the transition state and could account for the unusually low activation energy observed in our measurements.
Interestingly, we observe distinct rates and activation energies for the two steps of dark reversion (Fig. 6), suggesting that the state of the sister protomer within the dimer influences the chromophore's reversion reaction. This strongly indicates an allosteric interaction. Supporting this, the monomeric variant, which lacks a dimer interface, exhibits an even higher activation energy for dark reversion (Fig. 6(b)). A similar observation has been made for plant phytochromes, suggesting that the two-step mechanism is tightly regulated by the protein and likely optimized through evolution.40 The observed differences in thermal reversion kinetics likely arise from these protomer–protomer interactions altering the chromophore pocket. We propose that these structural modulations influence the transient keto–enol tautomerization of the chromophore—the presumed rate-limiting step—thus accounting for the different measured activation energies.
So far, allostery in bacterial phytochromes has primarily been discussed in the context of linking chromophore photoactivation to the regulation of biochemical output. However, the allosteric interaction observed here happens between the sister protomers within the dimer, suggesting the existence of a previously unidentified signaling pathway between the two protein subunits. Recent NMR investigations of the D. radiodurans phytochrome support this idea, revealing chemical shift changes that propagate from the chromophore to the dimer interface within the PAS/GAF domains.53 Further structural studies are needed to uncover the molecular basis of this signaling route.
5 Conclusion
In this study, we have successfully demonstrated the transient accumulation of a mixed PrPfr hybrid state in the bacterial phytochrome PaBphP during the dark reversion process. Our kinetic analysis indicates that both subunits within the dimeric phytochrome can exist in different photoisomeric states simultaneously during dark reversion, and that the hybrid state is transiently populated at high concentration. We also observe that a monomeric variant of PaBphP follows a direct Pr-to-Pfr reaction scheme, which further supports the assignment of the PrPfr intermediate as a feature arising from the dimeric nature of the wild-type protein. Furthermore, the temperature-dependent kinetics reveal low activation energies for the dark reversion processes in both the dimeric and monomeric PaBphP, consistent with a previously proposed keto–enol tautomerization as a crucial step preceding dark reversion. The distinct kinetic behavior observed in the dimeric PaBphP, specifically, the transient accumulation of the PrPfr hybrid state and the difference in activation energy, suggests the presence of allosteric regulation between the two subunits. We propose that this plays an important role in controlling the signaling output of bacterial phytochromes.Author contributions
Sayan Prodhan: conceptualization, spectroscopic investigation, data curation, formal analysis, kinetic model determination, verification, and writing – original draft and review & editing. Petra Mészáros: protein production and purification, design of monomeric variant, writing, review & editing. Szabolcs Bódizs: protein production and purification, review & editing. Yalin Zhou: kinetic model determination, formal analysis. Michał Maj: conceptualization, supervision, and writing – review & editing. Sebastian Westenhoff: conceptualization, supervision, funding acquisition, project administration, resources, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
All essential data supporting the findings of this study are available within the article. The data files are deposited at Figshare.Acknowledgements
The author acknowledge Göran Gustaffson foundations and Carl-Trygger Foundation for supporting this work. MM acknowledges the Swedish Research Council (Grant No. VR 2020-05403), the Swedish Society for Medical Research (S20-0156), and Harald and Greta Jeanssons Foundation Grant (J2021-0114). We thank Dr Heikki Takala (University of Jyväskylä) for initial design of the mutation sites for generating the monomeric variant of PSM-PaBphP.Notes and references
- P. Hegemann, Annurev. Arplant, 2008, 59, 167–189 CAS.
- A. Möglich, X. Yang, R. A. Ayers and K. Moffat, Annurev. Arplant, 2010, 61, 21–47 Search PubMed.
- N. C. Rockwell, Y.-S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS.
- H. Wang and H. Wang, Mol. Plant, 2015, 8, 540–551 CrossRef CAS PubMed.
- T. Lamparter, N. Krauß and P. Scheerer, Photochem. Photobiol., 2017, 93, 642–655 CrossRef CAS PubMed.
- J. J. Casal and R. A. Sánchez, Seed Sci. Res., 1998, 8, 317–329 CrossRef CAS.
- E. Monte, J. M. Tepperman, B. Al-Sady, K. A. Kaczorowski, J. M. Alonso, J. R. Ecker, X. Li, Y. Zhang and P. H. Quail, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16091–16098 CrossRef CAS PubMed.
- M. Endo, Y. Tanigawa, T. Murakami, T. Araki and A. Nagatani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18017–18022 CrossRef CAS.
- J. A. Kyndt, T. E. Meyer and M. A. Cusanovich, Photochem. Photobiol. Sci., 2004, 3, 519–530 CrossRef CAS.
- L. Wu, R. S. McGrane and G. A. Beattie, mBio, 2013, 4, 10–1128 CrossRef.
- R. K. Verma, A. Biswas, A. Kakkar, S. K. Lomada, B. B. Pradhan and S. Chatterjee, Cell Rep., 2020, 32(13), 108202 CrossRef CAS PubMed.
- K. G. Chernov, T. A. Redchuk, E. S. Omelina and V. V. Verkhusha, Chem. Rev., 2017, 117, 6423–6446 CrossRef CAS.
- A. A. Kaberniuk, A. A. Shemetov and V. V. Verkhusha, Nat. Methods, 2016, 13, 591–597 CrossRef CAS PubMed.
- A. T. Ulijasz and R. D. Vierstra, Curr. Opin. Plant Biol., 2011, 14, 498–506 CrossRef CAS PubMed.
- A. Möglich, R. A. Ayers and K. Moffat, Structure, 2009, 17, 1282–1294 CrossRef.
- L. Aravind and C. P. Ponting, Trends Biochem. Sci., 1997, 22, 458–459 CrossRef CAS PubMed.
- L.-O. Essen, J. Mailliet and J. Hughes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14709–14714 CrossRef CAS.
- E. S. Burgie and R. D. Vierstra, Plant Cell, 2014, 26, 4568–4583 CrossRef CAS PubMed.
- M. Legris, C. Klose, E. S. Burgie, C. C. R. Rojas, M. Neme, A. Hiltbrunner, P. A. Wigge, E. Schäfer, R. D. Vierstra and J. J. Casal, Science, 2016, 354, 897–900 CrossRef CAS PubMed.
- G. Rottwinkel, I. Oberpichler and T. Lamparter, J. Bacteriol. Res., 2010, 192, 5124–5133 CrossRef CAS.
- F. Velázquez Escobar, D. Buhrke, N. Michael, L. Sauthof, S. Wilkening, N. N. Tavraz, J. Salewski, N. Frankenberg-Dinkel, M. A. Mroginski and P. Scheerer, et al. , Photochem. Photobiol., 2017, 93, 724–732 CrossRef.
- M. F. López, M. Dahl, F. V. Escobar, H. R. Bonomi, A. Kraskov, N. Michael, M. A. Mroginski, P. Scheerer and P. Hildebrandt, Phys. Chem. Chem. Phys., 2022, 24, 11967–11978 RSC.
- S. Westenhoff, P. Meszaros and M. Schmidt, Curr. Opin. Struct. Biol., 2022, 77, 102481 CrossRef CAS PubMed.
- A. Björling, O. Berntsson, H. Lehtivuori, H. Takala, A. J. Hughes, M. Panman, M. Hoernke, S. Niebling, L. Henry, R. Henning, I. Kosheleva, V. Chukharev, N. V. Tkachenko, A. Menzel, G. Newby, D. Khakhulin, M. Wulff, J. A. Ihalainen and S. Westenhoff, Sci. Adv., 2016, 2, e1600920 CrossRef.
- M. Chenchiliyan, J. Kübel, S. A. Ooi, G. Salvadori, B. Mennucci, S. Westenhoff and M. Maj, J. Chem. Phys., 2023, 158, 085103 CrossRef CAS PubMed.
- S. G. Sokolovski, E. A. Zherebtsov, R. K. Kar, D. Golonka, R. Stabel, N. B. Chichkov, A. Gorodetsky, I. Schapiro, A. Möglich and E. U. Rafailov, Biophys. J., 2021, 120, 964–974 CrossRef CAS PubMed.
- Y. Yang, T. Stensitzki, L. Sauthof, A. Schmidt, P. Piwowarski, F. Velazquez Escobar, N. Michael, A. D. Nguyen, M. Szczepek, F. N. Brünig, R. R. Netz, M. A. Mroginski, S. Adam, F. Bartl, I. Schapiro, P. Hildebrandt, P. Scheerer and K. Heyne, Nat. Chem., 2022, 14, 823–830 CrossRef CAS PubMed.
- H. Takala, P. Edlund, J. A. Ihalainen and S. Westenhoff, Photochem. Photobiol. Sci., 2020, 19, 1488–1510 CrossRef CAS PubMed.
- X. Yang, Z. Ren, J. Kuk and K. Moffat, Nature, 2011, 479, 428–432 CrossRef CAS PubMed.
- X. Yang, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15639–15644 CrossRef CAS PubMed.
- J. R. Wagner, J. S. Brunzelle, K. T. Forest and R. D. Vierstra, Nature, 2005, 438, 325–331 CrossRef CAS PubMed.
- X. Yang, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14715–14720 CrossRef CAS PubMed.
- W. Y. Wahlgren, E. Claesson, I. Tuure, S. Trillo-Muyo, S. Bódizs, J. A. Ihalainen, H. Takala and S. Westenhoff, Nat. Commun., 2022, 13, 7673 CrossRef CAS PubMed.
- H. Li, E. S. Burgie, Z. T. K. Gannam, H. Li and R. D. Vierstra, Nature, 2022, 604, 127–133 CrossRef CAS PubMed.
- S. Bódizs, P. Mészáros, L. Grunewald, H. Takala and S. Westenhoff, Structure, 2024, 32, 1952–1962.e3 CrossRef PubMed.
- D. Buhrke, G. Gourinchas, M. Müller, N. Michael, P. Hildebrandt and A. Winkler, J. Biol. Chem., 2020, 295, 539–551 CrossRef CAS PubMed.
- T. N. Malla, C. Hernandez, S. Muniyappan, D. Menendez, D. Bizhga, J. H. Mendez, P. Schwander, E. A. Stojković and M. Schmidt, Sci. Adv., 2024, 10, eadq0653 CrossRef CAS PubMed.
- E. Schäfer and W. Schmidt, Planta, 1974, 116, 257–266 CrossRef.
- J. Brockmann, S. Rieble, N. Kazarinova-Fukshansky, M. Seyfried and E. Schäfer, Plant, Cell Environ., 1987, 10, 105–111 CrossRef CAS.
- L. Hennig and E. Schäfer, J. Biol. Chem., 2001, 276, 7913–7918 CrossRef CAS PubMed.
- E. S. Burgie, Z. T. K. Gannam, K. E. McLoughlin, C. D. Sherman, A. S. Holehouse, R. J. Stankey and R. D. Vierstra, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2105649118 CrossRef CAS.
- J.-H. Jung, M. Domijan, C. Klose, S. Biswas, D. Ezer, M. Gao, A. K. Khattak, M. S. Box, V. Charoensawan, S. Cortijo, M. Kumar, A. Grant, J. C. W. Locke, E. Schäfer, K. E. Jaeger and P. A. Wigge, Science, 2016, 354, 886–889 CrossRef CAS PubMed.
- F. Velazquez Escobar, P. Piwowarski, J. Salewski, N. Michael, M. Fernandez Lopez, A. Rupp, B. M. Qureshi, P. Scheerer, F. Bartl, N. Frankenberg-Dinkel, F. Siebert, M. Andrea Mroginski and P. Hildebrandt, Nat. Chem., 2015, 7, 423–430 CrossRef CAS PubMed.
- C. Huber, M. Strack, I. Schultheiß, J. Pielage, X. Mechler, J. Hornbogen, R. Diller and N. Frankenberg-Dinkel, J. Biol. Chem., 2024, 300, 107148 CrossRef CAS PubMed.
- H. Takala, A. Björling, M. Linna, S. Westenhoff and J. A. Ihalainen, J. Biol. Chem., 2015, 290, 16383–16392 CrossRef CAS PubMed.
- W. Schmidt and E. Schäfer, Planta, 1974, 116, 267–272 CrossRef CAS PubMed.
- M. Medzihradszky, J. Bindics, É. Ádám, A. Viczián, É. Klement, S. Lorrain, P. Gyula, Z. Mérai, C. Fankhauser, K. F. Medzihradszky, T. Kunkel, E. Schöfer and F. Nagy, The Plant Cell, 2013, 25, 535–544 CrossRef CAS PubMed.
- C. Klose, F. Venezia, A. Hussong, S. Kircher, E. Schäfer and C. Fleck, Nat. Plants, 2015, 1, 15090 CrossRef CAS PubMed.
- G. Merga, M. F. Lopez, P. Fischer, P. Piwowarski, Z. Nogacz, A. Kraskov, D. Buhrke, F. V. Escobar, N. Michael, F. Siebert, P. Scheerer, F. Bartl and P. Hildebrandt, Phys. Chem. Chem. Phys., 2021, 23, 18197–18205 RSC.
- K. D. Piatkevich, F. V. Subach and V. V. Verkhusha, Nat. Commun., 2013, 4, 2153 CrossRef PubMed.
- A. Kraskov, A. D. Nguyen, J. Goerling, D. Buhrke, F. Velazquez Escobar, M. Fernandez Lopez, N. Michael, L. Sauthof, A. Schmidt, P. Piwowarski, Y. Yang, T. Stensitzki, S. Adam, F. Bartl, I. Schapiro, K. Heyne, F. Siebert, P. Scheerer, M. A. Mroginski and P. Hildebrandt, Biochemistry, 2020, 59, 1023–1037 CrossRef CAS PubMed.
- M. E. Auldridge, K. A. Satyshur, D. M. Anstrom and K. T. Forest, J. Biol. Chem., 2012, 287, 7000–7009 CrossRef CAS PubMed.
- L. Isaksson, E. Gustavsson, C. Persson, U. Brath, L. Vrhovac, G. Karlsson, V. Orekhov and S. Westenhoff, Structure, 2020, 29, 151–160.e3 CrossRef PubMed.
| This journal is © the Owner Societies 2025 |