
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInjectable in situ gelling methylcellulose-based hydrogels for bone tissue regeneration†
Lorenzo
Bonetti
 a,
Silvia
Borsacchi
*bc,
Alessandra
Soriente
d,
Alberto
Boccali
a,
Lucia
Calucci
bc,
Maria Grazia
Raucci
d and
Lina
Altomare
*ae
a,
Silvia
Borsacchi
*bc,
Alessandra
Soriente
d,
Alberto
Boccali
a,
Lucia
Calucci
bc,
Maria Grazia
Raucci
d and
Lina
Altomare
*ae
aDepartment of Chemistry, Materials, and Chemical Engineering “G. Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: lina.altomare@polimi.it
bInstitute of Chemistry of Organometallic Compounds (ICCOM), Italian National Research Council (CNR), Via G. Moruzzi 1, 56124 Pisa, Italy. E-mail: silvia.borsacchi@pi.iccom.cnr.it
cCenter for Instrument Sharing of the University of Pisa (CISUP), Lungarno Pacinotti 43/44, 56126 Pisa, Italy
dInstitute for Polymers, Composites and Biomaterials (IPCB), Italian National Research Council, Viale J.F. Kennedy 54, Mostra d’Oltremare Pad 20, 80125 Napoli, Italy
eNational Interuniversity Consortium for Materials Science and Technology (INSTM), Via Giuseppe Giusti 9, 50121 Firenze, Italy
First published on 17th April 2024
Abstract
Injectable bone substitutes (IBSs) represent a compelling choice for bone tissue regeneration, as they can be exploited to optimally fill complex bone defects in a minimally invasive manner. In this context, in situ gelling methylcellulose (MC) hydrogels may be engineered to be free-flowing injectable solutions at room temperature and gels upon exposure to body temperature. Moreover, incorporating a suitable inorganic phase can further enhance the mechanical properties of MC hydrogels and promote mineralization, thus assisting early cell adhesion to the hydrogel and effectively guiding bone tissue regeneration. In this work, thermo-responsive IBSs were designed selecting MC as the organic matrix and calcium phosphate (CaP) or CaP modified with graphene oxide (CaPGO) as the inorganic component. The resulting biocomposites displayed a transition temperature around body temperature, preserved injectability even after loading with the inorganic components, and exhibited adequate retention on an ex vivo calf femoral bone defect model. The addition of CaP and CaPGO promoted the in vitro mineralization process already 14 days after immersion in simulated body fluid. Interestingly, combined X-ray diffraction and solid state nuclear magnetic resonance characterizations revealed that the formed biomimetic phase was constituted by crystalline hydroxyapatite and amorphous calcium phosphate. In vitro biological characterization revealed the beneficial impact of CaP and CaPGO, indicating their potential in promoting cell adhesion, proliferation and osteogenic differentiation. Remarkably, the addition of GO, which is very attractive for its bioactive properties, did not negatively affect the injectability of the hydrogel nor the mineralization process, but had a positive impact on cell growth and osteogenic differentiation on both pre-differentiated and undifferentiated cells. Overall, the proposed formulations represent potential candidates for use as IBSs for application in bone regeneration both under physiological and pathological conditions.
1. Introduction
Methylcellulose (MC) hydrogels are well-known biomaterials that have attracted significant attention in the fields of tissue engineering and regenerative medicine.1 MC hydrogels exhibit lower critical solution temperature (LCST) behavior, undergoing a reversible thermo-responsive gelation process around their transition temperature (Tt or Tgel). Upon heating, MC hydrogels show a transition from a sol (at T < Tt) to a gel state (at T > Tt), as a result of MC self-assembly into stiff fibrils that percolate into a fibrillar network. When the generated fibrils cool, they unbind and the MC gel returns to its original solution state.2–6 MC hydrogels display peculiar features due to the weak nature of the interactions involved in the gelation process, including reversibility of the sol–gel transition and decreased cytotoxicity (due to the lack of chemical crosslinkers),7 and low cost. Moreover, the possibility of easily tuning the Tt of MC hydrogels makes them even more attractive. In this regard, it is known that pristine MC hydrogels display a Tt in the 55–60 °C temperature range (i.e., 2% w/v MC solution), far enough from physiological temperature to make them non-responsive to body temperature. However, Tt can be simply modified by acting on different parameters in the preparation of the hydrogels: the degree of substitution, MC concentration, and the presence of salts or additives (e.g., other polymers, sugars) in solution are among the main players in this tuning.1,2Injectable hydrogels have been largely investigated in several reviews,8–10 displaying how different types of hydrogels may be prepared and how their properties (e.g., gelling time, mechanical properties) can be modified according to the final application. In general, injectable hydrogels must be flowable and easily extrudable through needles or catheters, but this implies that they may be affected by poor mechanical properties. Moreover, only a few studies11,12 address the issue of injectability, and the findings are controversial and challenging to compare, mainly due to variations in injection conditions (e.g., injection rate, needle dimensions).
In this panorama, in situ gelling – or in situ forming – MC hydrogels may be engineered to be free-flowing injectable solutions at room temperature (T < Tt) and gels upon exposure to body temperature (T ≥ Tt). Due to in situ gelation, MC hydrogels have specific benefits over standard injectable hydrogel formulations, including the potential to be minimally invasive, fill irregularly shaped voids, and ensure shape stability and excellent mechanical properties following injection.
In the field of bone tissue engineering, the addition of an inorganic phase (e.g., calcium phosphates, CaPs, or bioglasses) can further improve the mechanical properties of MC hydrogels, also promoting mineralization, a property of paramount importance to assist early cell adhesion to the hydrogel and successfully guide bone tissue regeneration.1,10
In this scenario, MC hydrogels have been explored as injectable bone substitutes (IBSs) and two main approaches have been reported to produce MC-based IBSs: CaP loading and CaP nucleation and growth into MC-based hydrogels. In the first approach, CaPs were added directly into the MC hydrogel. Following this approach, Oğuz et al.13 proposed IBSs prepared from MC (8% (w/v)), gelatin (2.5% (w/v)), and sodium citrate (3% (w/v)) loaded with different amounts of CaPs (0–50 wt%), obtaining injectable formulations with improved mechanical performance (five-fold increase in compressive strength compared to pristine MC hydrogels) and capable of forming a gel around 37 °C. Interestingly, the limit for injectability was achieved for 50% CaP loading. The same hydrogel formulation, with the addition of graphene oxide (GO), was also investigated for the bioactive potential of GO.14 In the second approach, precursor salts (i.e., CaCl2 and Na2HPO4), added to the MC solutions, supported both the in situ nucleation of CaPs and the tuning of the Tt of the resulting MC hydrogels. Following this approach, Kim and co-workers11,15,16 obtained CaP-loaded MC hydrogels via an in situ, one-pot synthesis process. The obtained hydrogels displayed Tt lower than body temperature (28–29 °C) and in vitro biocompatibility. The in vivo study in a rabbit calvarial defect model indicated a superior regeneration rate of new mature bone in the MC-CaP composite hydrogel compared with the pristine MC hydrogel. Interestingly, injectability tests revealed a maximum injection force lower than 30 N at room temperature, representing the upper limit for manual injection of an IBS.11 Overall, these studies reveal that MC-based hydrogels are promising materials for filling bone tissue deficiencies and assisting bone tissue regeneration.
The aim of the present work was to develop novel IBSs selecting: (i) MC as the organic matrix, to provide injectable and in situ gelling hydrogels; (ii) calcium phosphate (CaP) or CaP modified with graphene oxide (CaPGO) as the inorganic component, to mimic the native environment of bone tissue and offer additional biological properties in the case of CaPGO. These biocomposites were designed to preserve the gels injectability even after loading with the inorganic phases, as well as to promote the formation of bone substitute phases upon mineralization. The prepared hydrogels were first characterized from a rheological point of view to assess their thermo-responsive behavior and their viscoelastic properties prior to and after gelation. Then, injectability tests were carried out to measure the force needed to extrude them through an 18 G needle and their retention capacity was evaluated on an ex vivo calf femoral bone defect model. Moreover, the in vitro mineralization of the hydrogels was investigated by performing X-Ray Diffraction (XRD) and Solid State NMR (SSNMR) experiments on samples taken at selected mineralization times. XRD allowed crystalline phases to be identified, while by exploiting SSNMR it was possible to detect and quantify crystalline, nanocrystalline, and amorphous phases. The possibility of easily observing 31P and 1H nuclei, whose NMR properties (e.g., chemical shift, dipolar interactions, relaxation times) are directly related to the structural and dynamic features of their local environment, makes SSNMR a very powerful investigation technique for bone-related materials, unique in the panorama of MC-based injectable IBSs. In this work, by combining XRD and SSNMR, the phosphate-containing phases formed during the in vitro mineralization process of CaP- and CaPGO-loaded MC gels have been identified and quantified, characterizing their time evolution and the effect of the presence of GO. Lastly, an in vitro biological characterization was carried out, investigating the impact of MC-based formulations on cell proliferation and osteogenic differentiation.
2. Experimental section
2.1. Materials
Unless stated, all reagents were purchased from Merck (Merck Life Science S.r.l., Italy) and used without further purification.2.2. Hydrogel preparation
MC hydrogels were prepared according to previously reported procedures.17–19 Briefly, MC (2% w/v) was added to a hot (55 °C) Na2SO4 water solution (150 mM) under stirring. This formulation was selected on the basis of both our previous studies on MC hydrogels1,17,20–22 and a composition optimization investigation centred on the swelling behavior and gelation temperature of MC hydrogels prepared with a Na2SO4 concentration ranging from 100 to 200 mM (S1, ESI†). The obtained solution was cooled to room temperature under stirring, then stored in the refrigerator (4 °C) overnight to allow complete MC hydration. To prepare the biocomposites, CaP and CaPGO powders were produced using sol–gel technology as previously reported,12,23 starting from calcium nitrate tetrahydrate [Ca(NO3)2·4H2O] and di-ammonium hydrogen phosphate [(NH4)2HPO4], as precursors for Ca2+ and PO43−, respectively, with a Ca/P molar ratio in the range of 1.6–1.7. By including GO (1.5 wt%, Nanesa s.r.l.) during the in situ sol–gel synthesis of CaP, CaPGO was obtained. The addition of CaP and CaPGO powders (1% (w/v)) to the gel was carried out with two syringes, one loaded with the MC hydrogel and the other with the powder suspension in deionised (DI) water. This set-up was studied to allow working and mixing under sterile conditions. The two syringes were connected with a three-way Luer Stopcock and mixing was achieved through 50 passes between the syringes, until a uniform gel was obtained.12 From this point on, the samples analysed will be referred to as MC, MC-CaP, and MC-CaPGO.2.3. Rheological tests
A thorough rheological characterization was performed using a rotational rheometer (MCR 302, Anton Paar GmbH, Graz, Austria) equipped with a parallel plate geometry (∅: 25 mm, gap = 1 mm).Amplitude sweep tests were carried out on each formulation to identify the linear viscoelastic region (LVR) by applying an oscillatory strain (γ) in the 0.01–10% range, setting a frequency (ν) of 1 Hz, at 20 and 40 °C.
Then, to identify the transition temperature (Tgel) of the composite MC gels, temperature sweep tests were carried out in the 20–50 °C range, with a heating ramp of 2 °C min−1, γ = 1%, ν = 1 Hz. The transition temperatures were identified from the storage (G′) and loss moduli (G′′) as the crossover point (G′ = G′′).24
Time sweep tests were carried out to identify the gelation time (tgel) of the gels. Before starting the test, the temperature was set at 20 °C; then, the temperature was impulsively increased to 37 °C and the viscoelastic properties of the materials were measured in a time window of 300 s, γ = 1%, ν = 1 Hz. The tgel was identified from the crossover point (G′ = G′′).
Lastly, the viscosity of the samples was measured via shear rate sweep tests, by applying a shear rate (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ) ramp in the 0.1–1000 s−1 range at 20 °C.
) ramp in the 0.1–1000 s−1 range at 20 °C.
2.4. Injectability tests
Injectability tests were carried out on a uniaxial tensile testing machine (MTS 1/MH, 5 kN load cell, MTS System, Eden Prairie, MN, USA) in compression configuration. The hydrogels were loaded into 5 mL syringes fitted with 18 G needles and positioned in a previously reported custom experimental set-up.12 The measurements were carried out at room temperature (20 °C). A pre-load of 0.15 N was set, then the syringe piston was compressed at a rate of 0.6 mm s−1 and the force applied on it was recorded. The resulting force value was then multiplied by a factor of 1.1 to take into account the resistance of biological tissues toward injection.112.5. Ex vivo tilt tests
Calf femurs were purchased from a local butcher. Bone slices (15 mm in thickness) were obtained from calf femoral heads using a die grinder (Dremel® 3000) equipped with a 32 mm cut-off wheel. A hemispherical hole, 6 mm in diameter, was then drilled into the cancellous bone of each slice using the die grinder equipped with a 3.2 mm tungsten carbide cutter ball tip. Subsequently, each cavity was filled with 500 μL of MC, MC-CaP, or MC-CaPGO. The samples were then incubated at either 20 or 37 °C for 3 h, followed by tilt tests.20 In the test, the samples were inverted (180° rotation) and any flow of the MC formulations from the bone cavity was noted.2.6. In vitro mineralization tests
Mineralization tests were performed by immersing the hydrogels in simulated body fluid (SBF), prepared according to the protocol described by Kokubo et al.25 For the evaluation of mineralization, aliquots (0.5 mL) of composite hydrogels were added in dialysis tubes (10 mm width, Mw cut-off = 14 kDa), submerged in 10 mL of SBF inside 15 mL Falcon tubes, and kept under stirring (37 °C, 50 rpm) in a shaking incubator (SKI 4, Argo Lab, Giorgio Bormac S.r.l. Carpi MO, Italy). The SBF was renewed every 48 h and, at selected time points (7, 14, 28 days), the specimens were withdrawn and washed 3 times (24 hours total washing time) with DI water inside the dialysis tubes to remove soluble inorganic species.For the XRD and SSNMR experiments, the washed specimens were first frozen at −20 °C, then freeze-dried (−40 °C, <0.5 mbar). Hereafter, these samples will be indicated as MC_xd, MC-CaP_xd, and MC-CaPGO_xd, where x is the mineralization time (in days) and 0 days refers to samples that were not immersed in SBF.
2.7. In vitro biological characterization
MC hydrogels were prepared using a filter sterilized Na2SO4 solution (0.22 μm pore size filter) and UV light sterilized MC, CaP, and CaPGO powders (2 h).To create a suitable microenvironment for cell growth within the formulations (MC, MC-CaP, and MC-CaPGO), DMEM powder was added. Subsequently, cells (7F2: 5 × 104; hMSC: 2 × 104) were resuspended in 20 μL of FBS and inoculated into 200 μL of each formulation at room temperature in non-adherent 24-well plates. The plates were then incubated (37 °C, 5% CO2, 95% humidity) to induce the sol–gel transition. Following this, 2 mL of medium was added to each well and incubated for different time intervals.
The in vitro biocompatibility of the MC-based formulations was evaluated using the Alamar Blue assay (Life Technologies, Italy) at 3, 7, and 14 days, according to the manufacturer's protocol. Fluorescence (λex = 560 nm; λem = 590 nm) was measured using a spectrophotometer (Victor X3, PerkinElmer, Italy).
First, alkaline phosphatase (ALP) activity within the gels was examined at 3, 7, and 14 days of culture. To quantify the ALP levels, cells were lysed using a cell lysis buffer, and the ALP activity was measured using the SensoLyte® pNPP Alkaline Phosphatase Assay kit (AnaSpec, DBA, Italy) following the manufacturer's instructions. Absorbance readings were taken in a 96-well plate at 405 nm using a spectrophotometer (Victor X3, PerkinElmer, Italy). The nanomolar (nM) ALP concentration was then calculated based on a standard curve. Next, to normalize the ALP values, the amount of double-stranded DNA (dsDNA) was quantified using a PicoGreen™ dsDNA quantification kit (Life Technologies, Italy). Fluorescence readings were recorded at 502/523 nm using a spectrophotometer (Victor X3, PerkinElmer, Italy). A calibration curve of a λ-dsDNA standard in a buffer of 10 mM Tris, 1 mM EDTA, pH = 7.5 was used to quantify dsDNA. The results of the ALP activity were reported as the nanomolar concentration of ALP normalized to the micrograms of total DNA content (nM ALP/μg DNA).
Additionally, osteocalcin levels, produced by hMSCs within the gels at 7 and 14 days, were analyzed using an ELISA kit (Elabscience, Italy) following the manufacturer's protocol. The optical density (OD value) was determined at 450 nm (Victor X3, PerkinElmer, Italy). The results are expressed as nanograms of osteocalcin on micrograms of total DNA content (ng OCN/μg DNA).
2.8. Statistical data analysis
Where possible, tests were run in triplicate (n = 3). Data are expressed as mean (M) ± standard deviation (SD). To compare the data, t-tests or one-way ANOVA with Tukey's post hoc tests were used, considering a significance level of α = 0.05.3. Results and discussion
3.1. Rheological, injectability, and retention properties of MC hydrogels
In order to test the applicability of the MC hydrogels loaded with CaP or CaPGO as injectable bone substitutes, it is fundamental to characterize their rheological and injectability properties. Toward this aim, storage and loss moduli were measured on MC, MC-CaP, and MC-CaPGO in amplitude, temperature, and time sweep tests, determining the viscoelastic behavior at room and physiological temperature, as well as the gelation temperature and time. Moreover, the viscosity of the samples was measured via shear rate sweep tests and compared with the outcomes of the injectability tests, carried out under the operational conditions used in surgery.Shear strain tests were conducted to determine the Linear Viscoelastic Region (LVR) of MC, MC-CaP, and MC-CaPGO samples (S2, ESI†).20,21 As an outcome, a strain value γ = 1% was selected for all the subsequent tests.
To quantitatively evaluate Tgel of the three formulations, temperature sweep tests were carried out between 20 and 50 °C, applying a heating ramp of 2 °C min−1 (Fig. 1A1–A3). The Tgel was determined from the crossover point between the conservative and the loss modulus (i.e., G′ = G′′), where a transition from the sol phase (G′ < G′′) to the gel phase (G′ > G′′) occurs. A similar trend is detected in all the temperature sweep curves: for T < Tgel, a slight decrease of the viscoelastic parameters occurs, due to partial disentanglement of the MC chains upon shear stress; for T > Tgel, an increase of G′ occurs due to the aggregation of MC into stiff fibrils and percolation into a fibrillar network (i.e., sol–gel transition).11,15,17,18 The addition of CaP and CaPGO seems not to impact on the Tgel, which is 34–35 °C for the composites and 36 °C for MC (Table 1).
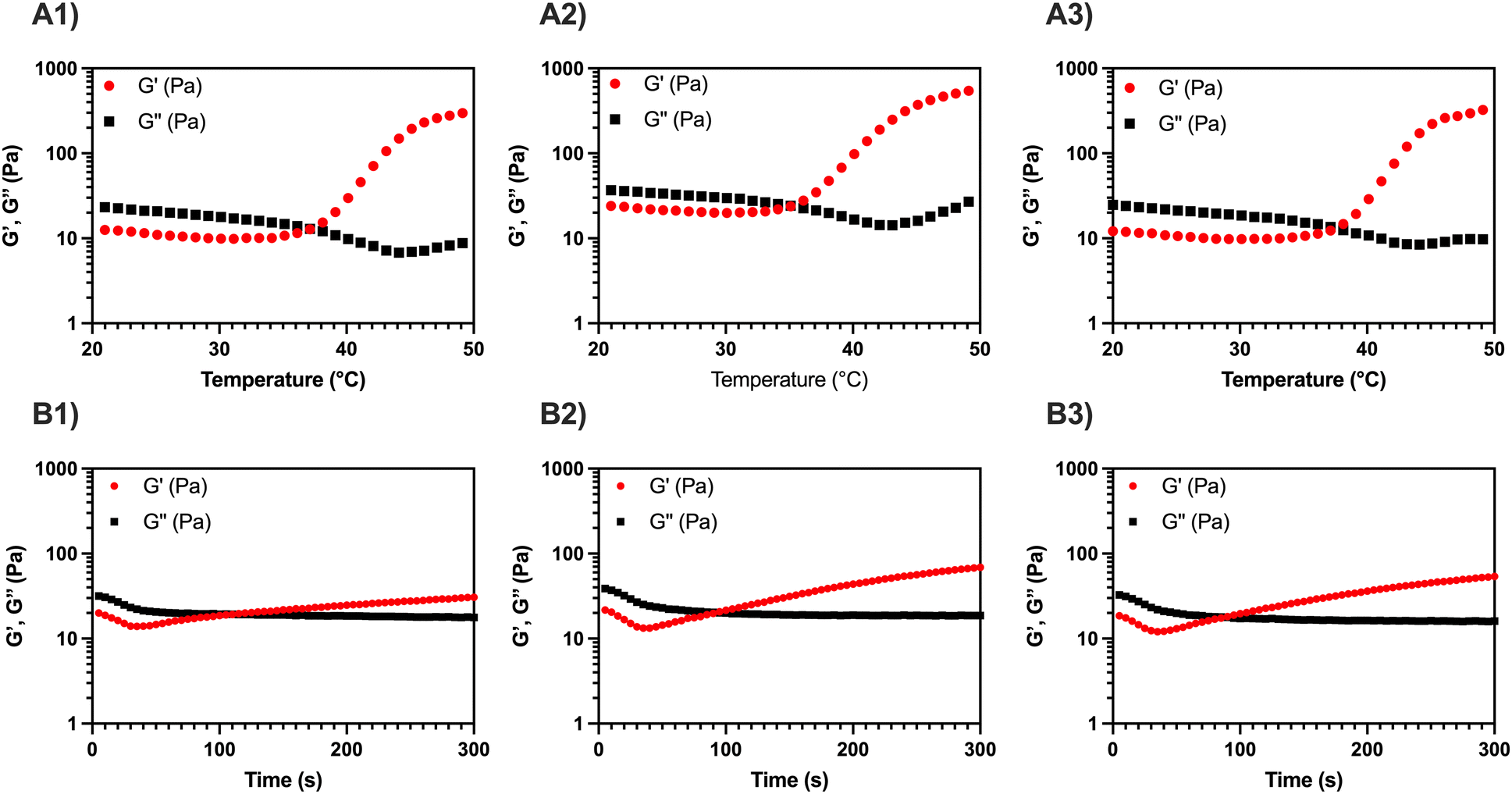 | ||
| Fig. 1 Representative temperature sweep tests on (A1) MC, (A2) MC-CaP, and (A3) MC-CaPGO formulations. The tests were carried out in the 20–50 °C range (2 °C min−1 ramp), γ = 1%, ν = 1 Hz. Representative time sweep tests on (B1) MC, (B2) MC-CaP, and (B3) MC-CaPGO formulations. The tests were carried out in a time window of 300 s, γ = 1%, ν = 1 Hz. | ||
| MC | MC-CaP | MC-CaPGO | |
|---|---|---|---|
| T gel (°C) | 35.8 ± 2.3 | 34.6 ± 0.7 | 34.5 ± 3.7 |
| t gel (s) | 98.8 ± 20.2 | 86.7 ± 2.9 | 85.0 ± 5.0 |
Then, a time sweep test was performed to quantitatively assess the gelation time, tgel, of the three formulations (Fig. 1B1–B3). To replicate the injection process in the human body, the temperature was first set at 20 °C (operation room temperature) and then rapidly raised to 37 °C. In this regard, the standard ISO 5833/1-1999-E identifies an in situ gelling filler as clinically suitable when its tgel is lower than 15 min.27 All three tested formulations displayed a gelation time of about 90 s, without significant differences induced by the addition of the inorganic powders (Table 1). On the other hand, a reduction in the setting time for MC hydrogels containing GO has been reported by Oğuz et al.,28 but this variation can be ascribed both to the presence of free GO in the hydrogel formulation and to the higher amount of GO in the gel (used to enhance both biological response and rheological characteristics) compared to the present work.
During in situ gelling, thermo-responsive systems are generally formulated to be free-flowing (i.e., injectable) at room temperature (T < Tgel), and readily gel upon exposure to body temperature (T ≥ Tgel).1 All the MC-based formulations here reported, displaying a Tgel < 37 °C and a tgel ≪ 15 min, lend themselves well as injectable fillers.
Moreover, the viscosity of MC, MC-CaP, and MC-CaPGO was analyzed in rotational mode, applying a shear rate in the 0.1–1000 s−1 range (Fig. 2). All the samples exhibit shear-thinning behavior in this range, characterized by a significant decrease in viscosity as the shear rate increases. This behavior, characteristic of non-Newtonian fluids, indicates the disruption of the MC hydrogel network,12 and is of pivotal importance in the design of injectable hydrogels.1,29
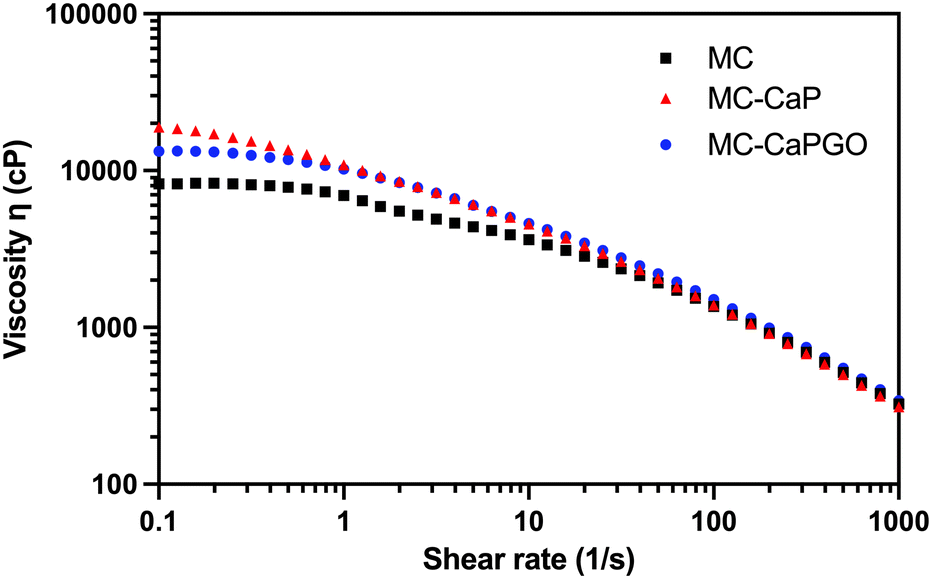 | ||
| Fig. 2 Representative flow curves for MC, MC-CaP, and MC-CaPGO formulations. The tests were carried out at 20 °C, applying a shear rate ramp from 0.1 to 1000 s−1. | ||
Following the rheological characterization, injectability tests were carried out on MC, MC-CaP, and MC-CaPGO hydrogels by loading them into 5 mL sterile syringes equipped with an 18 G needle. The tests were carried out at 20 °C, setting the crosshead speed of the MTS apparatus at 0.6 mm s−1 to mimic the operational conditions during surgery.12,30,31 The obtained curves (Fig. 3), expressed as force vs. extension, display the same trend for all samples: after an initial exponential increase of the force value, associated to the extrusion of the hydrogel throughout the needle, a force plateau (Fmean) is reached. No significant differences were detected among the three formulations, which display Fmean values of about 11 N (Table 2), in accordance with similar hydrogel formulations.11,12 Interestingly, the Fmean values settle far below 30 N, commonly recognized as the upper force limit for manual injection of injectable materials.15,32,33 Moreover, the loading of CaP and CaPGO powders into MC hydrogel did not lead to any obstruction of the needle, thus indicating that the proposed formulations can be effortlessly injected in vivo at a reasonable rate.
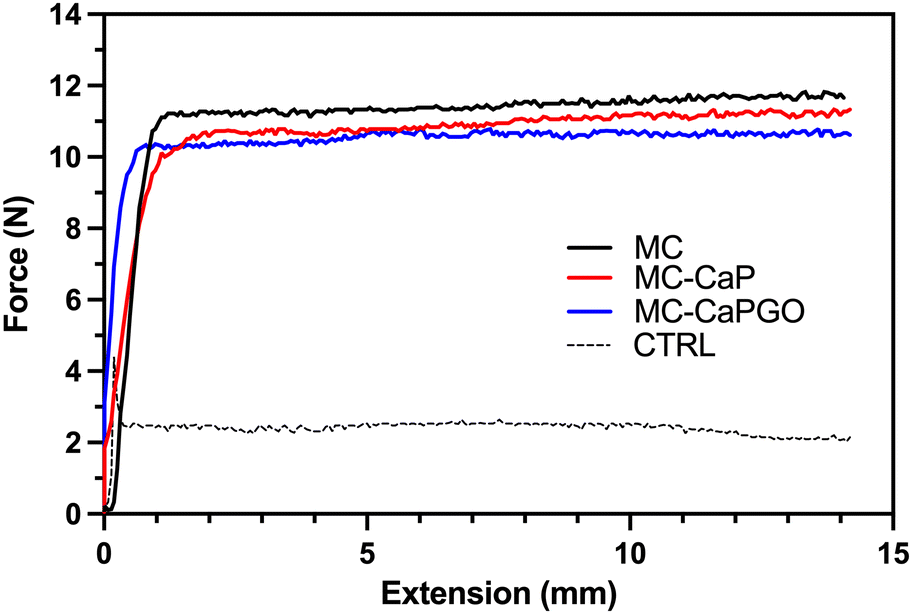 | ||
| Fig. 3 Representative injectability tests for MC, MC-CaP, and MC-CaPGO formulations. CTRL = empty syringe. The hydrogel formulations were extruded through an 18 G needle at 20 °C at a crosshead speed of 0.6 mm s−1, measuring the force needed for the process. | ||
| MC | MC-CaP | MC-CaPGO | CTRL | |
|---|---|---|---|---|
| F mean (N) | 11.7 ± 0.2 | 11.0 ± 0.1 | 10.6 ± 0.1 | 2.2 ± 0.4 |
The retention capacity of the developed hydrogel formulations (MC, MC-CaP, MC-CaPGO) was evaluated on an ex vivo calf femoral bone defect model. Bone defects were filled with the hydrogel formulations and incubated (20 or 37 °C, 3 h). Bone samples were then tilted to observe the flow of the MC formulations from the bone cavity (Fig. 4 and Videos S1–S6, ESI†). All the formulations incubated at 20 °C rapidly flowed after inversion (Fig. 4A1–A3), as a consequence of the low viscosity in the sol state.20 Conversely, the increase of temperature (37 °C) prevented any flow of the MC formulations from the bone defects (Fig. 4B1–B3), suggesting optimal retention in the defect site induced by the sol–gel transition.20
 | ||
| Fig. 4 Ex vivo tilt tests, displaying the flowability of the formulations as a function of temperature. Tests carried out at 20 °C on (A1) MC, (A2) MC-CaP, and (A3) MC-CaPGO; tests carried out at 37 °C on (B1) MC, (B2) MC-CaP, and (B3) MC-CaPGO. | ||
Considering the results of the rheological, injectability and retention tests, it is evident that all the developed formulations have several benefits over conventional hydrogels and scaffolds, including the ability to fill irregularly shaped cavities (such as bone deformities) and guarantee continuity (mechanical and physicochemical features) in the host tissue, and may be implanted using minimally invasive procedures. The addition of CaP and CaPGO maintains the gelation and injectability properties of the hydrogels at values suitable for IBSs. Moreover, CaP and CaPGO can promote the in vivo mineralization processes, leading to the deposition of a new bone matrix in the defect area.1,11,13,15,16,34
3.2. In vitro mineralization
To evaluate the effectiveness of CaP and CaPGO in enhancing the hydrogels’ bioactivity in bone tissue regeneration, MC, MC-CaP and MC-CaPGO were subjected to a mineralization process in SBF up to 28 days, mimicking the mineralization process that occurs in native bone tissue.The phosphate-containing phases formed in the mineralization process were identified and characterized in detail by means of XRD and SSNMR experiments, carried out on samples taken at selected mineralization times, washed, and freeze-dried (samples indicated as MC_xd, MC-CaP_xd, and MC-CaPGO_xd, where x is the mineralization time in days). While the XRD analysis allows crystalline phases to be detected, SSNMR can also reveal the presence of amorphous phases and provide a quantitative determination of all the phases.
The XRD analysis of MC_xd samples (Fig. 5A) revealed that the peaks corresponding to MC (green dotted lines) remain consistent up to 28 days in SBF. No evidence of hydroxyapatite (Ca10(PO4)6(OH)2) formation was observed, indicating that MC itself does not undergo mineralization. In the diffractograms of MC-CaP_xd (Fig. 5B) and MC-CaPGO_xd (Fig. 5C), beside the MC peaks, signals of monetite (CaHPO4) are observed (red dotted lines) at the initial stages of mineralization, owing to the presence of monetite in CaP and CaPGO (vide infra). The characteristic diffraction pattern of hydroxyapatite12,35 at 2θ angles of 10.6°, 25.9°, 31.8°, 32.8°, 34.0°, 39.6°, 46.7°, 49.5°, and 53.2° (blue dotted lines) becomes detectable already after 7 days of SBF immersion (data not shown). For longer immersion times (up to 28 days), the hydroxyapatite peak intensity progressively increases, while the peaks related to the monetite phase almost disappear. This indicates that the presence of CaP induces the formation of hydroxyapatite, replicating the mineralization process observed in native bone tissue.35
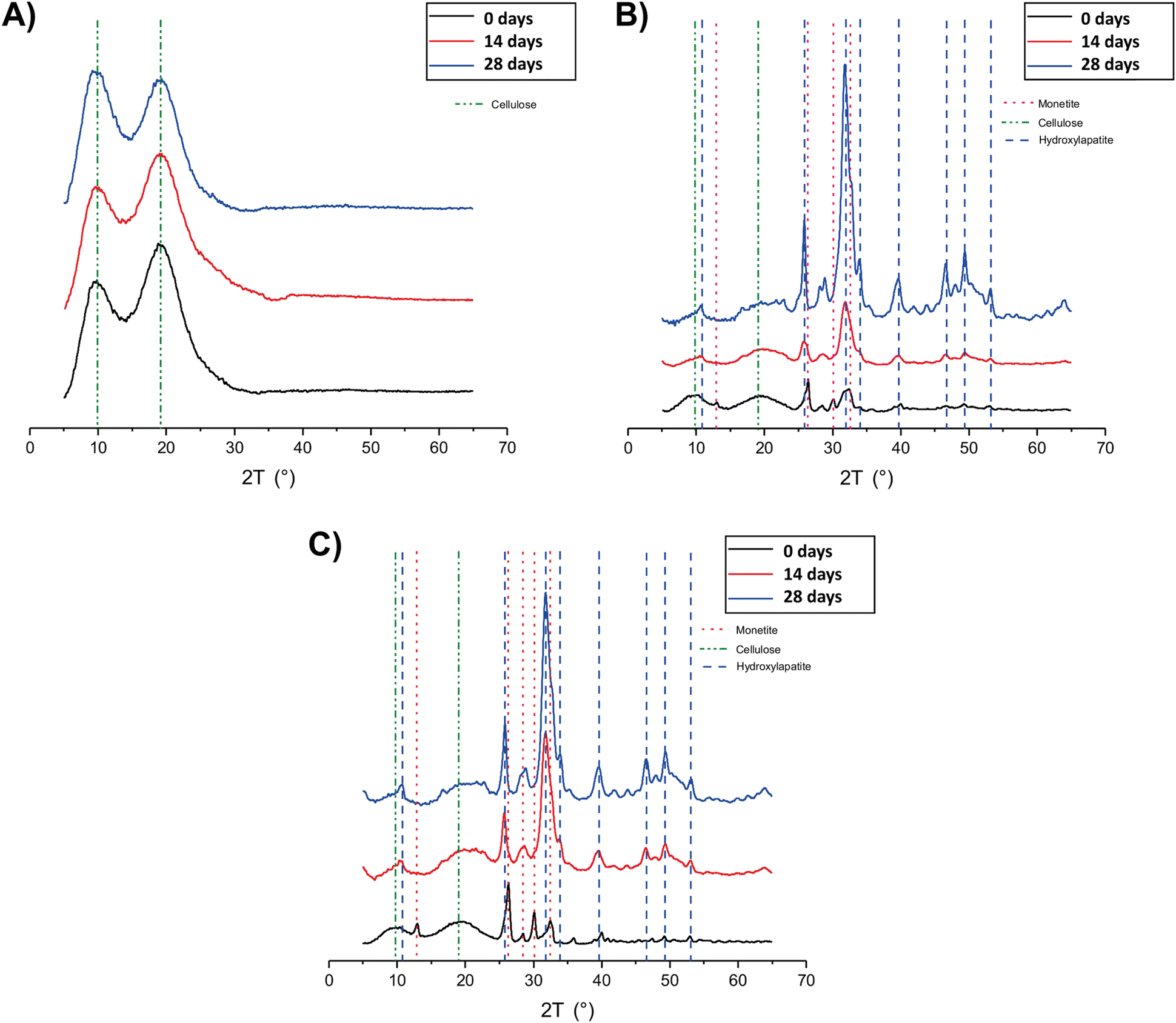 | ||
| Fig. 5 XRD patterns of (A) MC_xd, (B) MC-CaP_xd, and (C) MC-CaPGO_xd formulations at selected time points. | ||
The XRD patterns of MC-CaPGO_xd samples resemble those of MC-CaP_xd, suggesting that the presence of GO does not influence the mineralization process. Similar observations have been reported by Oğuz et al.,13 who showed that the addition of GO to a MC hydrogel did not cause changes to the XRD pattern.
A more complete picture of the phosphate-containing phases formed during the mineralization of the hydrogels was obtained from 31P and 1H SSNMR experiments. Indeed, SSNMR played a significant role in the progress of knowledge of the complex chemical structure of bone and bone-related materials, as well as in understanding the mineralization processes of biomaterials.36–44
The 31P MAS spectra of MC_xd samples do not show any signals, confirming that mineralization does not occur in pristine MC hydrogels. In Fig. 6 the quantitative 31P DE-MAS spectra of MC-CaP_xd samples, with x = 0, 14, 28, are shown. The spectrum of the non-mineralized sample, MC-CaP_0d, shows peaks at about −1 and 0.5 ppm, ascribable to monetite arising from the CaP powder (S3, ESI†), and one intense peak at 3.3 ppm, due to hydroxyapatite.41,45–48 A spectral deconvolution performed through a fitting procedure allowed us to recognize that the signal at 3.3 ppm results from the superimposition of two peaks (both with the same chemical shift): the first one with a linewidth of 270 Hz and a Lorentzian lineshape, already observed in the spectrum of CaP (Fig. S4, ESI†) and ascribable to crystalline hydroxyapatite; the second one with a linewidth of 820 Hz and a Gaussian lineshape, ascribable to an amorphous calcium phosphate phase (ACP), already observed to form on the pore walls of CaO–SiO2–P2O5 mesoporous bioactive glasses soaked in SBF.48–50 It is worth mentioning that the observed composite signal could also be ascribed to nanocrystalline hydroxyapatite, which is reported to be constituted by a core of crystalline hydroxyapatite and a surface layer of ACP.37 The formation of ACP, not observed in CaP (S3, ESI†), is clearly favored by the environment created by the hydrogel and it is of particular interest for its biomimetic tissue-bonding ability. Moreover, the comparison of the 31P DE-MAS spectrum of MC-CaP_0d with that of CaP (Fig. S4, ESI†) also shows that the hydrogel strongly favors the formation of hydroxyapatite instead of monetite.
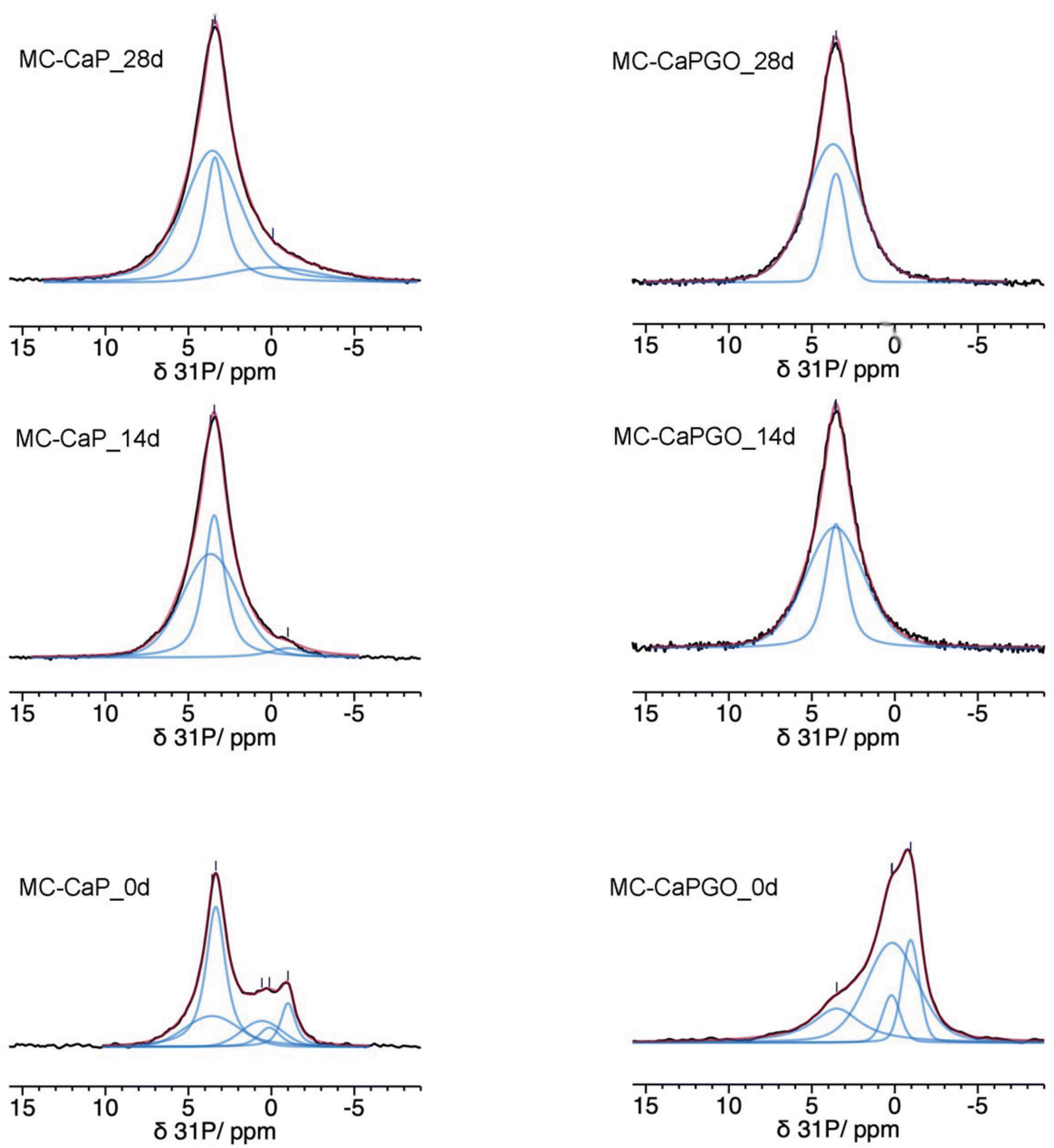 | ||
| Fig. 6 31P quantitative DE-MAS NMR spectra (black) of the indicated samples. The calculated spectra are shown as red traces, while the single peaks are in blue. | ||
A comparison of the normalized total spectral areas of MC-CaP_0d, MC-CaP_14d and MC-CaP_28d (Table 3) shows that soaking the MC-CaP hydrogel in SBF results in a progressive increase of the total amount of phosphate-containing phases, indicating that mineralization proceeds. In fact, the number of detected 31P nuclei doubles in the first 14 days and then further slightly increases up to 28 days. From the relative intensities of the peaks present in the spectra of the three samples, it is possible to infer that, after 14 days in SBF, the content of monetite is substantially reduced and ACP and hydroxyapatite are the main phases present. From 14 to 28 days, hydroxyapatite does not increase, while ACP still grows. The relative percentages of ACP and crystalline hydroxyapatite, in terms of P atoms, obtained from the areas of the respective signals, change from 41/59 in MC-CaP_0d to 60/40 in MC-CaP_14d and 71/29 in MC-CaP_28d, indicating that the mineralization preferentially results in the formation of ACP.
| Sample | Total P | P of monetite | P of hydroxyapatite | P of ACP |
|---|---|---|---|---|
| MC-CaP_0d | 1 | 0.27 | 0.43 | 0.30 |
| MC-CaP_14d | 2 | 0.10 | 0.76 | 1.14 |
| MC-CaP_28d | 2.5 | 0 | 0.73 | 1.80 |
| MC-CaPGO_0d | 1.3 | 1 | 0 | 0.30 |
| MC-CaPGO_14d | 1.9 | 0 | 0.59 | 1.31 |
| MC-CaPGO_28d | 2.4 | 0 | 0.50 | 1.90 |
Useful information is also provided by 1H MAS spectra (Fig. 7). For all MC-CaP_xd samples, a narrow peak is observed at about 0.6 ppm, which can be ascribed to –OH hydrogens of hydroxyapatite,45,48 observed also in CaP (Fig. S5, ESI†). The most intense signal is centered at a chemical shift of 4.3 ppm, and it displays a narrow and a broad component. This signal is also present in the spectrum of MC_0d; thus, it must be partly ascribed to hydrogens of MC. However, the intensity of the broad component of this signal visibly increases moving from MC_0d to MC-CaP_0d and also increases with mineralization time, suggesting that hydrogens of ACP also contribute to this signal.
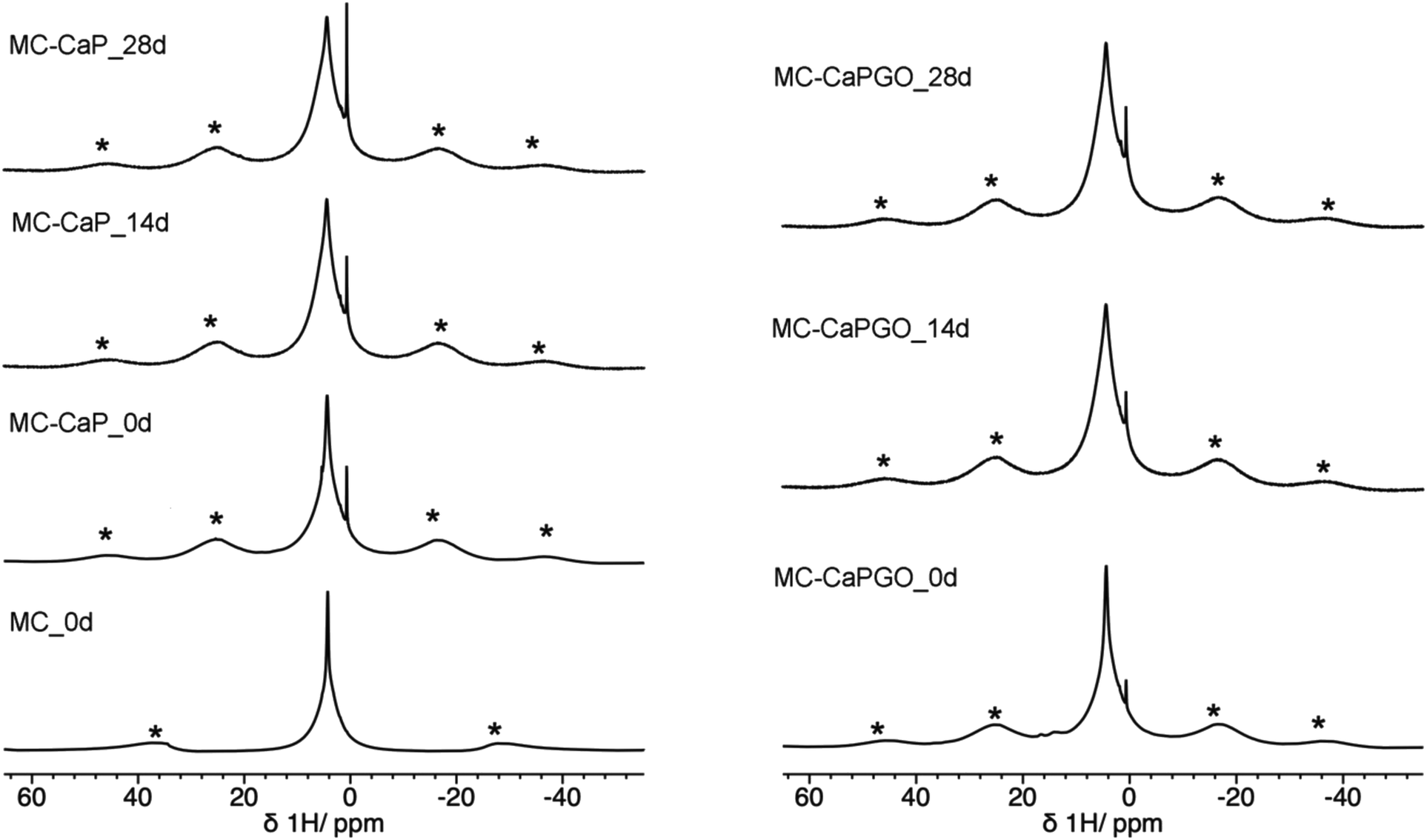 | ||
| Fig. 7 1H MAS spectra of MC_0d (MAS frequency of 15 kHz) and of MC-CaP_xd and MC-CaPGO_xd samples. Asterisks indicate spinning sidebands of the broad peak at 4.3 ppm. | ||
In this context it is interesting to compare the 31P DE-MAS spectrum of MC-CaP_28d with CP-MAS ones, in which signals of P atoms in spatial proximity to hydrogens are enhanced (Fig. 8). In agreement with the literature,48 the CP-MAS spectrum recorded with a short contact time of 0.5 ms, which favors the signals of 31P nuclei within a few Å from 1H nuclei, highlights the broad signal of ACP, indicating that this phase contains many more hydrogens than hydroxyapatite. However, by recording the CP-MAS spectrum with a longer contact time (2 ms), the 1H to 31P magnetization transfer is allowed to proceed also for 31P nuclei at larger distances from 1H ones. In fact, in the CP spectrum with contact time of 2 ms the narrower signal of hydroxyapatite can also be recognized. It must be mentioned that the CP spectra are much less intense than the quantitative DE-MAS spectrum, indicating that only a part of ACP is hydrogen rich. On the basis of the literature45,48 these hydrogen atoms can be ascribed to structural water molecules present in ACP.
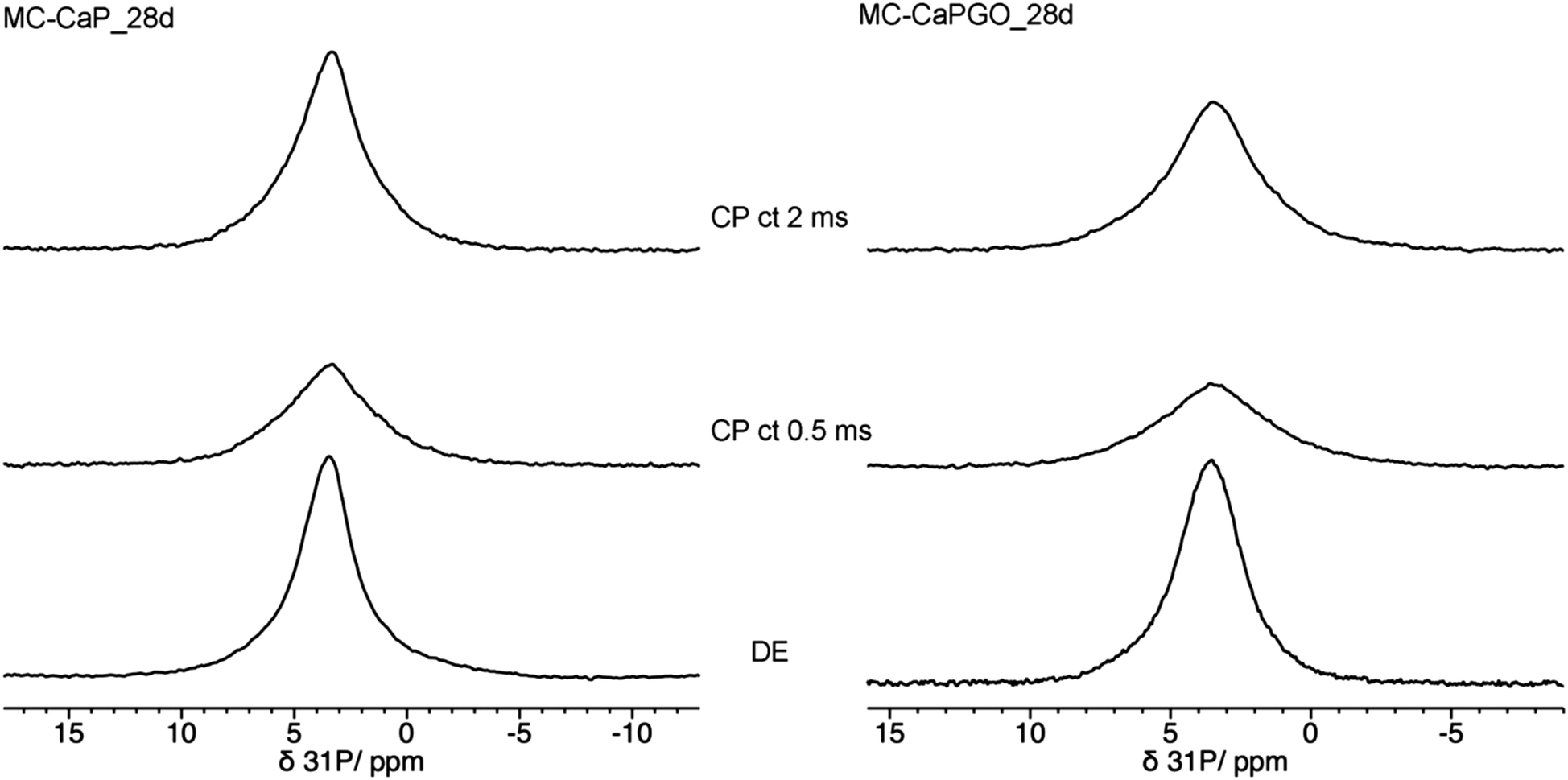 | ||
| Fig. 8 31P MAS NMR spectra of (left) MC-CaP_28d and (right) MC-CaPGO_28d, recorded with, from bottom to top, DE experiment and CP experiments with contact times (ct) of 0.5 ms and 2 ms. | ||
In order to investigate the effect of GO on the hydrogels’ mineralization, 31P and 1H SSNMR experiments were carried out on MC-CaPGO_xd samples with x = 0, 14, and 28. The 31P DE-MAS spectrum of the non-mineralized sample, MC-CaPGO_0d, (Fig. 6) shows that monetite is the major phase and crystalline hydroxyapatite is not present, similarly to what observed for CaPGO (Fig. S4, ESI†). On the other hand, the broad peak ascribable to ACP is observed. After 14 days in SBF, the monetite signal disappears and the narrower peak of hydroxyapatite appears, in agreement with the XRD results. After 28 days of mineralization the ACP/hydroxyapatite ratio, in terms of P atoms, is 79/21, slightly higher than in MC-CaP_28d, indicating a certain antagonistic effect of GO toward the growth of hydroxyapatite, already evident from the comparison between the 31P spectra of CaP and CaPGO (Fig. S4, ESI†). Analogously to what observed for MC-CaP_28d, the 1H–31P CP-MAS spectrum of MC-CaPGO_28d with a contact time of 0.5 ms highlights the signal of ACP (Fig. 8). The 1H MAS spectra of MC-CaP_GO_xd samples (Fig. 7) are very similar to those of the corresponding MC-CaP_xd samples, showing an intensity increase of the broad signal at 4.3 ppm with the progress of mineralization, ascribable to an increase of ACP.
3.3. In vitro biological properties
The impact of bioactivated gels, i.e., gels incorporating CaP and CaPGO nanoparticles, was evaluated in vitro by investigating cell adhesion, proliferation and osteogenic differentiation. Previous literature has firmly established that CaP nanoparticles, synthesized via the sol–gel approach, exhibit notable bioactivity and osteogenic potential even under basal culture conditions.51 Moreover, Raucci et al.23 reported that biomineralized GO, synthesized using the sol–gel method at room temperature, demonstrates significant effects on cellular behavior, particularly in terms of early and late-stage osteogenic activities.The chemical composition of the GO surface enables robust interactions with calcium phosphates, as evidenced by its active functionalities for cellular processes.52 These unique properties of GO, combined with the bioactivity of CaP nanoparticles, suggest a synergistic effect in promoting cell adhesion and proliferation while facilitating osteogenic differentiation. These findings not only underscore the potential of CaP and CaPGO nanoparticles in tissue engineering, but also highlight the importance of understanding their interaction for optimizing biomaterial design in bone tissue regeneration applications.
In this study, we performed experiments using murine osteoblast cells (7F2) and human mesenchymal stem cells (hMSCs) to assess the effects of the developed formulations (MC, MC-CaP, MC-CaPGO) on two distinct states of cellular differentiation, as pre-differentiated in the case of 7F2 cells and undifferentiated in the case of hMSCs. By examining these specific cell types at different stages of differentiation, we aimed at gaining a comprehensive understanding of how the bioactivated MC-based hydrogels influence cellular behaviors.
First, the bioactivated formulations (MC-CaP, MC-CaPGO) were evaluated in terms of cell viability normalized to the MC hydrogel control. Considering 7F2 cells, a decline in cell viability for MC-CaP and MC-CaPGO compared to the MC control was observed at day 7 (Fig. 9A), paralleling the transition of cells into a differentiation state (as revealed below, Fig. 10A). This transition causes a decrease in cell proliferation and subsequently reduces their metabolic activity.53 Considering hMSCs encapsulated in MC-CaP and MC-CaPGO, an early decrease in cell viability compared to MC control occurred at day 3 (Fig. 9B). This response in undifferentiated cells may be attributed to the time required for cells to adapt to the bioactive environment. Remarkably, for both cell types, the incorporation of bioactive materials had no long-term adverse effect on cell proliferation.
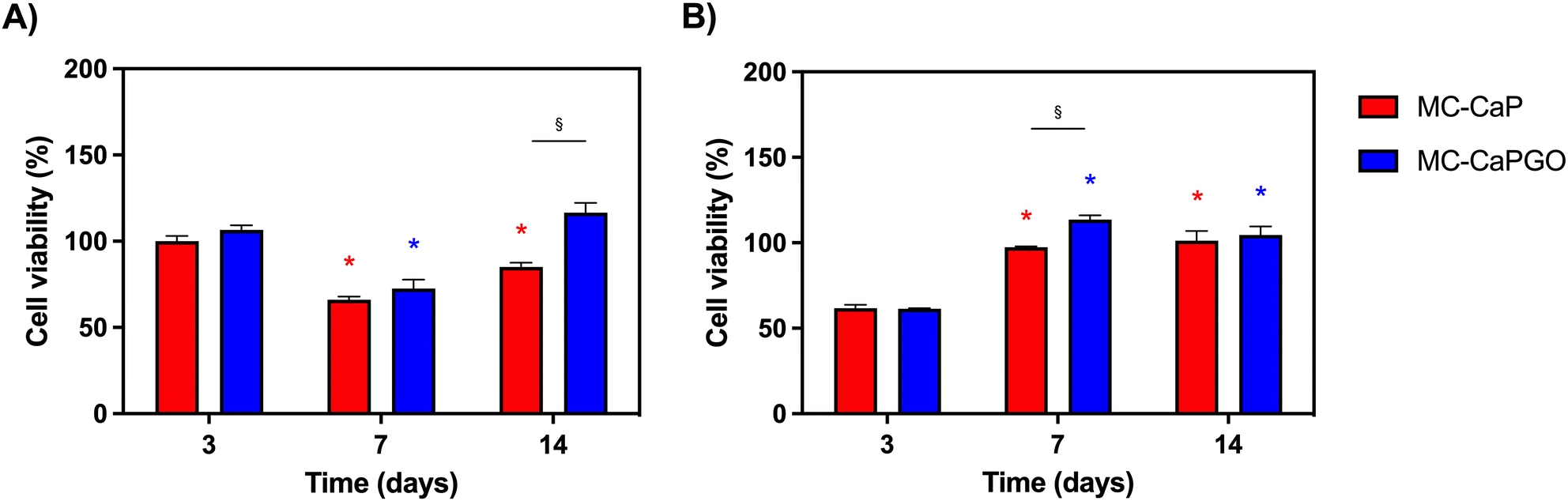 | ||
| Fig. 9 Cell viability normalized with respect to MC for (A) 7F2 cells and (B) hMSCs within hydrogel formulations at 3, 7, and 14 days. *p < 0.05 between the same formulation type (vs. time point 3 days); §p < 0.05 between different formulations (MC-CaP and MC-CaPGO) at the same time point. | ||
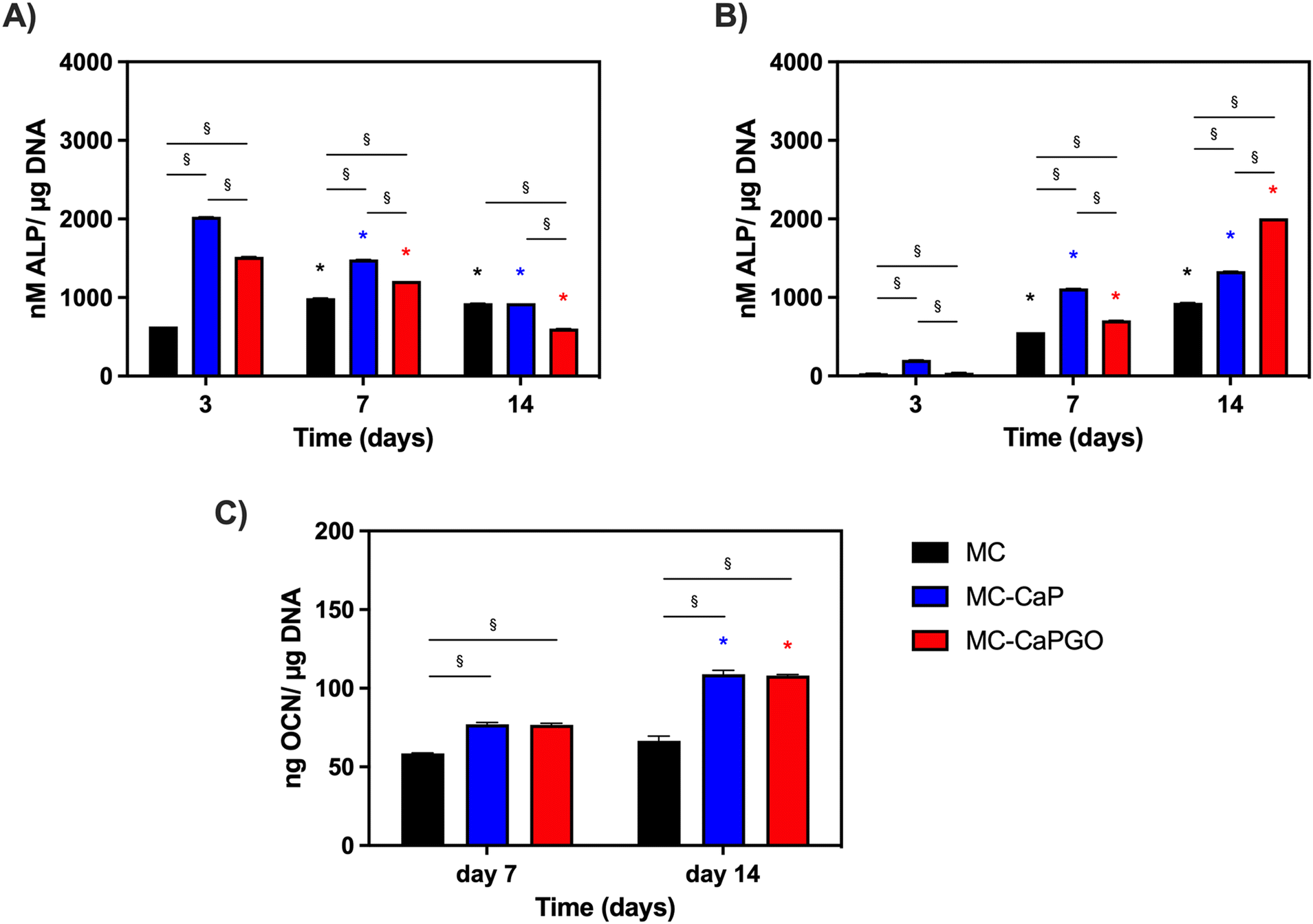 | ||
| Fig. 10 In vitro osteogenic differentiation: (A) ALP expression of 7F2 cells after 3, 7, and 14 days of cell culture for MC-based hydrogels. *p < 0.05 between the same formulation type (vs. time point 3 days); §p < 0.05 between different formulations (MC, MC-CaP, and MC-CaPGO) at the same time point. (B) ALP expression of hMSCs after 3, 7 and 14 days of cell culture for MC-based hydrogels, *p < 0.05 between the same formulation type (vs. time point 3 days); §p < 0.05 between different formulations (MC, MC-CaP, and MC-CaPGO) at the same time point. (C) OCN levels of hMSCs at 7 and 14 days for MC, MC-CaP, and MC-CaGO. *p < 0.05 between the same formulation type (7 vs. 14 days); §p < 0.05 between different formulations (MC, MC-CaP, and MC-CaPGO) at the same time point. | ||
The effect of GO in the bioactive formulations was then investigated. By comparing MC-CaP and MC-CaPGO formulations for 7F2 cells, no significant differences in cell viability were observed at day 3 (Fig. 9A). Increased viability was observed for the MC-CaPGO formulation (vs. MC-CaP) at day 7. Interestingly, this difference was even more noticeable at day 14 (Fig. 9A). By comparing MC-CaP and MC-CaPGO formulations for hMSCs, the highest viability levels were instead observed for MC-CaPGO at day 7 (Fig. 9B). These results underscore the beneficial impact of incorporating GO in the hydrogel formulation, indicating its potential to enhance cell proliferation. Overall, the improved cellular response observed with MC-CaPGO suggests a conductive environment for cell adhesion and proliferation.
Furthermore, the impact of bioactivated hydrogels on osteogenic differentiation was assessed in basal medium by measuring the expression of alkaline phosphatase (ALP) and osteocalcin (OCN) as markers of early and late osteogenesis, respectively.
The results depicted in Fig. 10A and B illustrate distinct patterns of ALP expression in 7F2 cells and hMSCs over time. Notably, at day 3, the effects of the bioactivated hydrogels (MC-CaP and MC-CaPGO) were already evident for 7F2 cells. Interestingly, ALP levels decreased throughout the culture period (Fig. 10A). The response differed for hMSCs, which are undifferentiated cells with osteogenic potential at later stages. For this second cell type, higher ALP levels were observed around day 7 and by day 14 it was evident that CaPGO exhibited a more pronounced effect compared to MC-CaP (Fig. 10B).
Lastly, we focused the investigation on hMSCs to assess the impact of bioactivated formulations on osteocalcin levels (OCN) normalized to μg of DNA. This decision was based on the significance of hMSCs in bone tissue engineering and the importance of OCN as a key marker of their osteogenic differentiation. Therefore, analyzing OCN levels in hMSCs offers valuable insights into their reaction to the bioactivated formulations. The results (Fig. 10C) demonstrated that at day 7 MC-CaP and MC-CaPGO show a slight increase of OCN level compared to MC control. However, the highest levels of OCN expression were recorded at day 14. This aligns with previous studies suggesting that CaP nanoparticles and GO incorporation into hydrogels enhance osteogenic differentiation markers.54,55
Overall, these findings underscore the potential of bioactivated MC-based hydrogels in promoting osteogenic differentiation, as evidenced by the enhanced expression of ALP and OCN. The different responses observed between 7F2 cells and hMSCs, attributed to their different states of differentiation, highlight the importance of considering cell type-specific responses in the development of biomaterials for tissue engineering applications.
4. Conclusions
The thorough characterization presented in this work demonstrates that the proposed biocomposites based on MC, CaP, and CaPGO lend themselves well as potential thermo-responsive IBSs, thanks to their fast gelation (∼90 s) at body temperature (37 °C), ease of injectability, and retention at the bone defect site. Thanks to these properties, the developed formulations are superior to conventional hydrogels and scaffolds, since they can be implanted via minimally invasive procedures, filling irregularly shaped cavities often occurring after trauma or surgery, and guaranteeing continuity into the host tissues. The addition of CaP and CaPGO led to a significant mineralization as revealed by the formation of a biomimetic phase, which was found to be constituted by crystalline hydroxyapatite and amorphous ACP through a detailed SSNMR characterization. The in vitro biological characterization revealed the favorable impact of CaP and CaPGO incorporation in promoting cell proliferation and osteogenic differentiation. Remarkably, the addition of GO, which is very attractive for its bioactive potential, did not negatively affect the injectability of the hydrogel nor the mineralization process, but had a positive impact in providing a conducive environment for cell growth and osteogenic differentiation on both pre-differentiated and undifferentiated cells.Overall, the proposed formulations have the potential to be directly employed for the ultimate purpose without any modifications. In conclusion, further in vitro and in vivo assessments could open the floodgates to the development of novel, injectable formulations with potential as IBSs in bone regeneration both under physiological and pathological conditions.
Author contributions
Conceptualization, methodology, formal analysis, investigation, writing – original draft, writing – review and editing, and visualization, L. B.; conceptualization, resources, methodology, investigation, writing – original draft, and writing – review and editing, S. B. and L. C.; methodology, investigation, writing – review and editing, A. S.; methodology and investigation, A. B.; methodology, writing – review and editing, M. G. R.; conceptualization, resources, writing – original draft, writing – review and editing, supervision, L. A.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Ministero dell’Università e della Ricerca (MUR), grant number PRIN ‘ACTION’ 2017SZ5WZB_002.Notes and references
- L. Bonetti, L. De Nardo and S. Farè, Tissue Eng., Part B, 2021, 27, 486–513 CrossRef CAS PubMed.
- M. L. Coughlin, L. Liberman, S. P. Ertem, J. Edmund, F. S. Bates and T. P. Lodge, Prog. Polym. Sci., 2021, 112, 101324 CrossRef CAS.
- S. Morozova, Polym. Int., 2020, 69, 125–130 CrossRef CAS.
- P. W. Schmidt, S. Morozova, S. P. Ertem, M. L. Coughlin, I. Davidovich, Y. Talmon, T. M. Reineke, F. S. Bates and T. P. Lodge, Macromolecules, 2020, 53, 398–405 CrossRef CAS.
- J. R. Lott, J. W. McAllister, M. Wasbrough, R. L. Sammler, F. S. Bates and T. P. Lodge, Macromolecules, 2013, 46, 9760–9771 CrossRef CAS.
- J. R. Lott, J. W. McAllister, S. A. Arvidson, F. S. Bates and T. P. Lodge, Biomacromolecules, 2013, 14, 2484–2488 CrossRef CAS PubMed.
- L. Bonetti, L. De Nardo and S. Farè, Soft Matter, 2023, 19, 7869–7884 RSC.
- P. Kondiah, Y. Choonara, P. Kondiah, T. Marimuthu, P. Kumar, L. Du Toit and V. Pillay, Molecules, 2016, 21, 1580 CrossRef PubMed.
- A. Mellati and J. Akhtari, Res. Mol. Med., 2018, 6(4), 1–14 CAS.
- G. A. N. Atia, H. K. Shalaby, N. G. Ali, S. M. Morsy, M. M. Ghobashy, H. A. N. Attia, P. Barai, N. Nady, A. S. Kodous and H. R. Barai, Pharmaceuticals, 2023, 16, 702 CrossRef CAS PubMed.
- M. H. Kim, B. S. Kim, H. Park, J. Lee and W. H. Park, Int. J. Biol. Macromol., 2018, 109, 57–64 CrossRef CAS.
- A. Fiorati, C. Linciano, C. Galante, M. G. Raucci and L. Altomare, Materials, 2021, 14, 4511 CrossRef CAS PubMed.
- Ö. Demir Oğuz and D. Ege, Materials, 2018, 11, 604 CrossRef.
- M. Maleki, R. Zarezadeh, M. Nouri, A. R. Sadigh, F. Pouremamali, Z. Asemi, H. S. Kafil, F. Alemi and B. Yousefi, Biomol. Concepts, 2020, 11, 182–200 CAS.
- M. H. Kim, H. Park and W. H. Park, Carbohydr. Polym., 2018, 191, 176–182 CrossRef CAS PubMed.
- H. Park, M. H. Kim, Y. I. Yoon and W. H. Park, Carbohydr. Polym., 2017, 157, 775–783 CrossRef CAS PubMed.
- L. Bonetti, L. De Nardo and S. Farè, Gels, 2021, 7, 141 CrossRef CAS PubMed.
- L. Bonetti, L. D. Nardo, F. Variola and S. Farè, Soft Matter, 2020, 16, 5577–5587 RSC.
- L. Bonetti, L. De Nardo, F. Variola and S. Farè, Mater. Lett., 2020, 274, 128011 CrossRef CAS.
- L. Altomare, A. Cochis, A. Carletta, L. Rimondini and S. Farè, J. Mater. Sci.: Mater. Med., 2016, 27, 95 CrossRef PubMed.
- A. Cochis, L. Bonetti, R. Sorrentino, N. Contessi Negrini, F. Grassi, M. Leigheb, L. Rimondini and S. Farè, Materials, 2018, 11, 579 CrossRef PubMed.
- L. Bonetti, A. Fiorati, A. D’Agostino, C. M. Pelacani, R. Chiesa, S. Farè and L. De Nardo, Gels, 2022, 8, 298 CrossRef CAS.
- M. G. Raucci, D. Giugliano, A. Longo, S. Zeppetelli, G. Carotenuto and L. Ambrosio, J. Tissue Eng. Regener. Med., 2017, 11, 2204–2216 CrossRef CAS.
- J. Desbrières, M. Hirrien and S. B. Ross-Murphy, Polymer, 2000, 41, 2451–2461 CrossRef.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Solid State Nucl. Magn. Reson., 2008, 33, 41–56 CrossRef CAS.
- G. T. Gold, D. M. Varma, D. Harbottle, M. S. Gupta, S. S. Stalling, P. J. Taub and S. B. Nicoll, J. Biomed. Mater. Res., Part A, 2014, 102, 4536–4544 Search PubMed.
- Ö. D. Oğuz and D. Ege, MRS Commun., 2019, 9, 1174–1180 CrossRef.
- D. J. Overstreet, D. Dutta, S. E. Stabenfeldt and B. L. Vernon, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 881–903 CrossRef CAS.
- S. Yasmeen, M. K. Lo, S. Bajracharya and M. Roldo, Langmuir, 2014, 30, 12977–12985 CrossRef CAS PubMed.
- V. Burckbuchler, G. Mekhloufi, A. P. Giteau, J. L. Grossiord, S. Huille and F. Agnely, Eur. J. Pharm. Biopharm., 2010, 76, 351–356 CrossRef CAS PubMed.
- C. Chung and J. A. Burdick, Adv. Drug Delivery Rev., 2008, 60, 243–262 CrossRef CAS PubMed.
- E. B. Hunziker, Osteoarthritis Cartilage, 2002, 10, 432–463 CrossRef CAS.
- S. Ghanaati, M. Barbeck, U. Hilbig, C. Hoffmann, R. E. Unger, R. A. Sader, F. Peters and C. J. Kirkpatrick, Acta Biomater., 2011, 7, 4018–4028 CrossRef CAS PubMed.
- C. C. Lopes, W. A. Pinheiro, D. Navarro Da Rocha, J. G. Neves, A. B. Correr, J. R. M. Ferreira, R. M. Barbosa, J. R. F. Soares, J. L. Santos and M. H. Prado Da Silva, Ceram. Int., 2021, 47, 7653–7665 CrossRef CAS.
- A. Kaflak-Hachulska, A. Samoson and W. Kolodziejski, Calcif. Tissue Int., 2003, 73, 476–486 CrossRef CAS.
- M. Edén, Materialia, 2021, 17, 101107 CrossRef.
- Y.-Y. Hu, X. P. Liu, X. Ma, A. Rawal, T. Prozorov, M. Akinc, S. K. Mallapragada and K. Schmidt-Rohr, Chem. Mater., 2011, 23, 2481–2490 CrossRef CAS.
- R. Gelli, M. Tonelli, F. Martini, L. Calucci, S. Borsacchi and F. Ridi, Constr. Build. Mater., 2022, 348, 128686 CrossRef CAS.
- Y. Coppel, Y. Prigent and G. Grégoire, Acta Biomater., 2021, 120, 156–166 CrossRef CAS.
- Y. Yu, H. Guo, M. Pujari-Palmer, B. Stevensson, J. Grins, H. Engqvist and M. Edén, Ceram. Int., 2019, 45, 20642–20655 CrossRef CAS.
- E. E. Wilson, A. Awonusi, M. D. Morris, D. H. Kohn, M. M. J. Tecklenburg and L. W. Beck, Biophys. J., 2006, 90, 3722–3731 CrossRef CAS PubMed.
- S. Hayakawa, K. Tsuru, C. Ohtsuki and A. Osaka, J. Am. Ceram. Soc., 2004, 82, 2155–2160 CrossRef.
- C. Gervais, C. Bonhomme and D. Laurencin, Solid State Nucl. Magn. Reson., 2020, 107, 101663 CrossRef CAS PubMed.
- Y. Yu, B. Stevensson, M. Pujari-Palmer, H. Guo, H. Engqvist and M. Edén, Int. J. Mol. Sci., 2019, 20, 6356 CrossRef CAS PubMed.
- W. P. Rothwell, J. S. Waugh and J. P. Yesinowski, J. Am. Chem. Soc., 1980, 102, 2637–2643 CrossRef CAS.
- S. Hayakawa, K. Tsuru, H. Iida, C. Ohtsuki and A. Osaka, J. Ceram. Soc. Jpn., 1996, 104, 1000–1003 CrossRef CAS.
- R. Mathew, P. N. Gunawidjaja, I. Izquierdo-Barba, K. Jansson, A. García, D. Arcos, M. Vallet-Regí and M. Edén, J. Phys. Chem. C, 2011, 115, 20572–20582 CrossRef CAS.
- P. N. Gunawidjaja, A. Y. H. Lo, I. Izquierdo-Barba, A. García, D. Arcos, B. Stevensson, J. Grins, M. Vallet-Regí and M. Edén, J. Phys. Chem. C, 2010, 114, 19345–19356 CrossRef CAS.
- E. Leonova, I. Izquierdo-Barba, D. Arcos, A. López-Noriega, N. Hedin, M. Vallet-Regí and M. Edén, J. Phys. Chem. C, 2008, 112, 5552–5562 CrossRef CAS.
- B. Yuan, M. G. Raucci, Y. Fan, X. Zhu, X. Yang, X. Zhang, M. Santin and L. Ambrosio, J. Mater. Chem. B, 2018, 6, 7974–7984 RSC.
- A. Raslan, L. Saenz Del Burgo, J. Ciriza and J. L. Pedraz, Int. J. Pharm., 2020, 580, 119226 CrossRef CAS PubMed.
- M. E. Frohbergh, A. Katsman, G. P. Botta, P. Lazarovici, C. L. Schauer, U. G. K. Wegst and P. I. Lelkes, Biomaterials, 2012, 33, 9167–9178 CrossRef CAS.
- J. Su, Z. Du, L. Xiao, F. Wei, Y. Yang, M. Li, Y. Qiu, J. Liu, J. Chen and Y. Xiao, Mater. Sci. Eng., C, 2020, 113, 110983 CrossRef CAS.
- A. Şelaru, H. Herman, G. M. Vlăsceanu, S. Dinescu, S. Gharbia, C. Baltă, M. Roşu, C. V. Mihali, M. Ioniţă, A. Serafim, H. Iovu, A. Hermenean and M. Costache, IJMS, 2022, 23, 491 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02414h |
| This journal is © The Royal Society of Chemistry 2024 |