
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in the direct electro-conversion of captured CO2 into valuable products
Kezia
Langie†
ab,
Gwangsu
Bak†
c,
Ung
Lee
 ade,
Dong Ki
Lee
aef,
Chan Woo
Lee
b,
Yun Jeong
Hwang
*cg and
Da Hye
Won
*adh
ade,
Dong Ki
Lee
aef,
Chan Woo
Lee
b,
Yun Jeong
Hwang
*cg and
Da Hye
Won
*adh
aClean Energy Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: dahye0803@kist.re.kr
bDepartment of Chemistry, Kookmin University, Seoul 02707, Republic of Korea
cDepartment of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: yjhwang1@snu.ac.kr
dDivision of Energy and Environmental Technology, KIST School, Korea University of Science and Technology (UST), Seoul 02792, Republic of Korea
eGraduate School of Energy and Environment (Green School), Korea University, Seoul 02841, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Yonsei-KIST Convergence Research Institute, Yonsei University, Seoul 03722, Republic of Korea
gCenter for Nanoparticle Research, Institute for Basic Science (IBS), Seoul 08826, Republic of Korea
hKHU-KIST Department of Converging Science and Technology, Kyung Hee University, Seoul 02477, Republic of Korea
First published on 3rd April 2024
Abstract
The direct electrochemical conversion of captured CO2 (capt-eCO2R) into valuable chemicals has recently emerged as a promising carbon capture and utilisation technology that will contribute to achieving net-zero carbon emissions. Conventional electrochemical CO2 (eCO2R) typically uses pure CO2 gas as a reactant; thus, this system requires substantial energy and capital allocation across the entire process, from the initial CO2 capture to the post-CO2 conditioning for product separation. The capt-eCO2R addresses these limitations and presents a compelling economic advantage by integrating the CO2 capture and direct electro-conversion of captured CO2 in the form of carbamate and (bi)carbonate without a CO2 conditioning process. The capt-eCO2R is still in the early stages of development and is not as mature as the conventional eCO2R; thus, several challenges remain to be addressed to improve system performance. This review provides a comprehensive overview of the capt-eCO2R system, including various system configurations, suitable catalysts, and strategies to enhance performance within captured media. The reaction mechanisms depend on the form of captured CO2; therefore, we categorised them according to the type of CO2 absorbent. The outlook, ongoing challenges, and strategies for future development are also presented.
1. Introduction
The concentration of CO2 in the atmosphere has increased over hundreds of years owing to anthropological activities, reaching 413.2 ppm in 2020, which represents an increase higher than the average growth rate observed over the past decade (2.5 ppm per year).1 Carbon capture, utilisation, and storage (CCUS) technology has attracted considerable interest as a means to overcome the climate crisis caused by CO2.2–4 Electrochemical CO2 reduction (eCO2R) to produce value-added chemicals is one of the most significant CCUS technologies owing to its high potential to achieve net-zero carbon emission through integration with renewable energy sources. Although eCO2R has a relatively low level of technological readiness, recent developments have markedly improved its practical feasibility.Among the various components of the eCO2R system, a state-of-the-art electrolyser or reactor that directly supplies gaseous CO2 to the catalyst layer through a gas diffusion electrode (GDE) considerably improves the scale and selectivity of the reaction (Fig. 1a), leading to record-breaking current densities ranging from hundreds of mA cm−2 to several A cm−2.5,6 In addition, the conventional eCO2R achieves a high Faradaic efficiency (FE) of more than 90% and 80% in the case of C1 (i.e. CO and formate)7,8 and C2+ (i.e. ethylene, ethanol acetate, n-propanol, etc.)9,10 compounds, respectively. Nevertheless, techno-economics analysis (TEA) indicates that the economic viability of the eCO2R system is limited considering the current levels of technology and the market price of the target chemicals.11,12 The energy-intensive upstream utilisation of CO2 as a reactant gas is one of the factors undermining the technological competitiveness of eCO2R.13 Expensive, energy-intensive processes such as CO2 absorption, desorption, and CO2 compression are required to supply high-purity CO2 from flue gas or even direct air capture (DAC). For instance, CO2 capture using a commercially available CO2 absorbent, such as monoethanolamine, requires up to 4.3 GJ tCO2−1 for amine regeneration.14,15 This drawback compromises the environmental sustainability of the technology. To implement a profitable and environmentally sustainable eCO2R system, an advanced system linking low-concentration CO2 capture and conversion is critical.
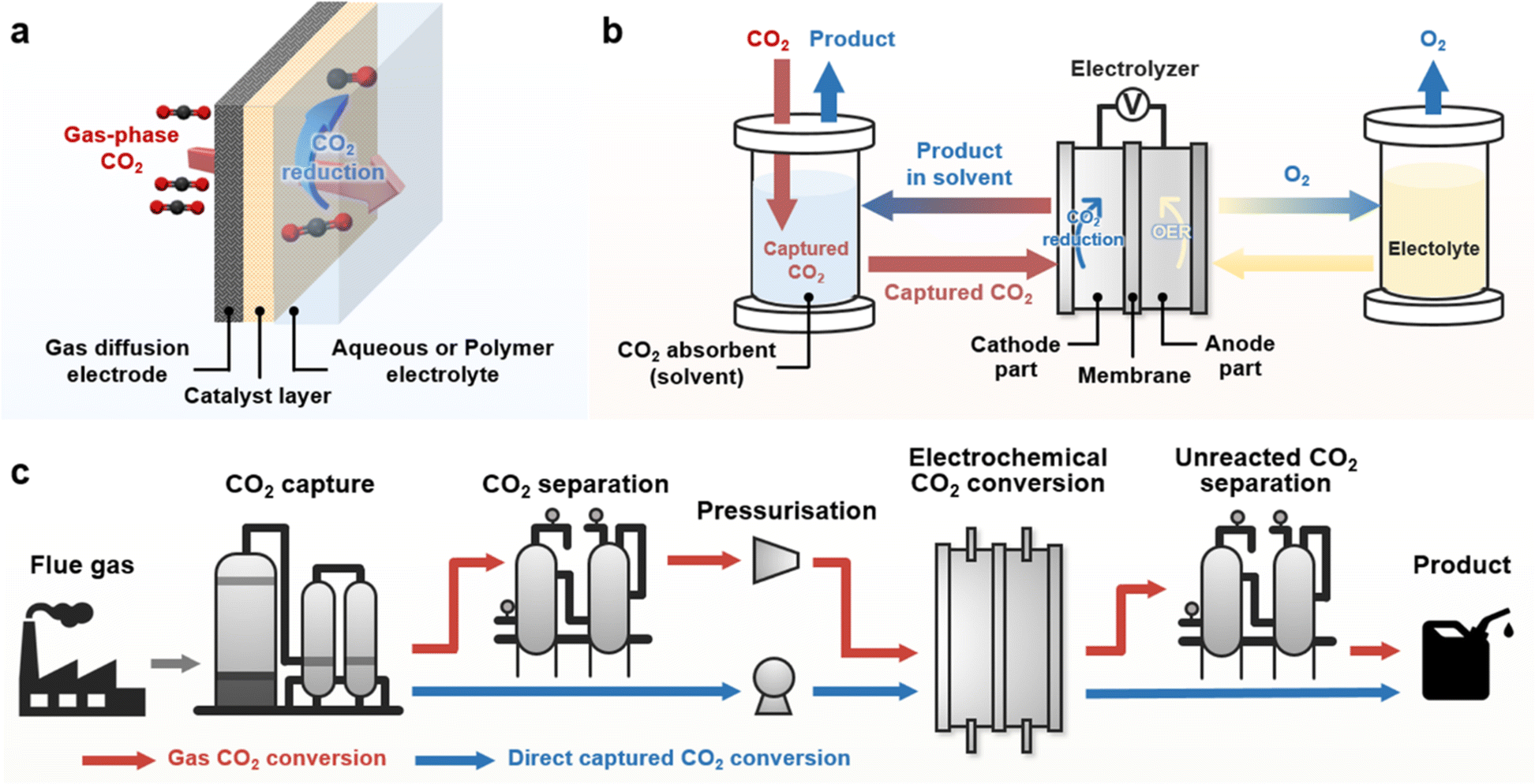 | ||
| Fig. 1 (a) Schematic of the GDE electrolyser for eCO2R. (b) System for capt-eCO2R. (c) Comparison of the systematic pathway of conventional gas-eCO2 (red line) and capt-eCO2R (blue line). | ||
The direct electrochemical conversion of captured CO2 (capt-eCO2R) has recently been recognised as a state-of-the-art eCO2R system that simplifies the system configuration (Fig. 1b). Rather than relying on gaseous CO2 as a reactant, this system directly utilises captured CO2 in CO2 absorbent and thus integrates the CO2 capture and electrochemical conversion processes. Unreacted CO2 gas is absorbed in the reaction media (i.e., CO2 absorbent), allowing concentrated gas products to be obtained from the capt-eCO2R. In contrast, the conventional gas-fed eCO2R system (gas-eCO2R) necessitates an additional process to separate the gas products from the unreacted CO2 gas. Thus, in principle, systems that directly convert captured CO2 conversion do not require energy-intensive processes such as CO2 conditioning (CO2 desorption and compression) and product separation, which are essential in gas-eCO2R (Fig. 1c).16 Based on these intrinsic advantages, a few studies have compared the life cycle analysis (LCA) and TEA of capt-eCO2R systems with those of gas-eCO2R systems. Langie et al. compared the LCA and TEA of the direct electrochemical conversion of triethylamine (TREA) captured CO2 with those of several syngas production CCU systems such as the reverse water gas shift reaction and conventional gas-eCO2R.17 The capt-eCO2R system using TREA recorded the most favourable operating expenditure (OPEX) and lowest break-even syngas price. Moreover, evaluation of the global warming potential (GWP) through global sensitivity analysis showed that the capt-eCO2R system using TREA can achieve near net-zero CO2 emissions, making the capt-eCO2R system industrially feasible, environmentally sustainable, and economically viable. Despite these advantages, the performance of capt-eCO2R is rather low; however, LCA/TEA analyses suggest that improving the current density at lower overpotentials may make the system more compelling. A similar direction of development was proposed by Gao et al., who found that lower capital expenses could be achieved with capt-eCO2R, but a carbon-negative system still requires high current densities of over 0.15 A cm−2.18 Therefore, to implement a profitable and environmentally sustainable system, improving technological maturity through technical development and a greater theoretical understanding of the capt-eCO2R is urgently required.
In this study, we investigated various capt-eCO2R systems that employ different types of electrolysers and absorbents. The solvent in such systems acts as both a CO2 absorbent and an electrolyte, thus determining the form of the captured CO2 (e.g., carbamate, bicarbonate/carbonate, etc.). Furthermore, because the CO2 reactant is captured in a liquid solvent, system components such as electrodes, membranes, electrolytes, electrolysers, and other parameters should differ from those of the conventional gas-eCO2R system. Therefore, this review has a particular focus on the role of the system elements, the reaction mechanism of the capt-eCO2R, and strategies to improve performance based on the form of the captured CO2 and the type of solvent. A perspective on an advanced future capt-eCO2R system with industrial applications is also presented.
2. Direct conversion of captured CO2
The capt-eCO2R system achieves the electrochemical conversion of captured CO2 in a liquid electrolyte without additional feeding of gaseous CO2 (Fig. 1b). In this system, carbamates or bicarbonate/carbonates, the dominant forms of captured CO2 in the aqueous absorbent, are utilised directly as reactants. The formation of carbamate or bicarbonate is determined by the type of absorbent used. The capture abilities and forms of major CO2 absorbents are outlined in Table 1. Primary and secondary amines capture one CO2 molecule per two amine molecules, primarily as a carbamate. Theoretically, tertiary amines can capture one CO2 molecule per one amine molecule, but the reported low CO2 capture ability appears to be due to a slow CO2 absorption rate. Potassium hydroxide (KOH) captures one CO2 molecule per one molecule, but salt formation hinders the recording of the theoretical CO2 capture ability. Considering that the optimal CO2 absorbent concentrations for primary/secondary amines and tertiary/KOH are 30 wt% and 3 M, respectively, the concentrations of carbamate and bicarbonate/carbonate would be similar regardless of the types of CO2 absorbents used. Therefore, in this review, we categorised the previous studies based on the forms of the captured CO2 (Fig. 2). As carbamate and (bi)carbonate electrolytes have different reaction mechanisms, their utilisation requires different strategies and approaches.| CO2 capture agent | Capture ability | Concentration | Capture condition | Capture form | Reference | |
|---|---|---|---|---|---|---|
| a The capture ability is evaluated by feeding 99% CO2. b The capture ability is evaluated by feeding 15% CO2 and 85% N2 concentration. c The capture ability is evaluated by feeding 5% CO2 concentration in flue gas. | ||||||
| Metal hydroxide | KOH | ∼0.745 molCO2 molKOH−1 | 0.412 mol L−1 | 35 °C, 4 bara | Bicarbonate | 19 |
| Primary amine | Monoethanolamine | 0.58 molCO2 molamine−1 | 30 wt% | 40 °C, 1 barb | Carbamate | 20 |
| Secondary amine | Diethanolamine | 0.53 molCO2 molamine−1 | 30 wt% | 40 °C, 1 barb | Carbamate | 20 |
| Tertiary amine | Triethanolamine | 0.39 molCO2 molamine−1 | 30 wt% | 40 °C, 1 barb | Bicarbonate | 20 |
| Triethylamine | ∼0.193 molCO2 molamine−1 | 3 M | R. T. ambientc | 17 | ||
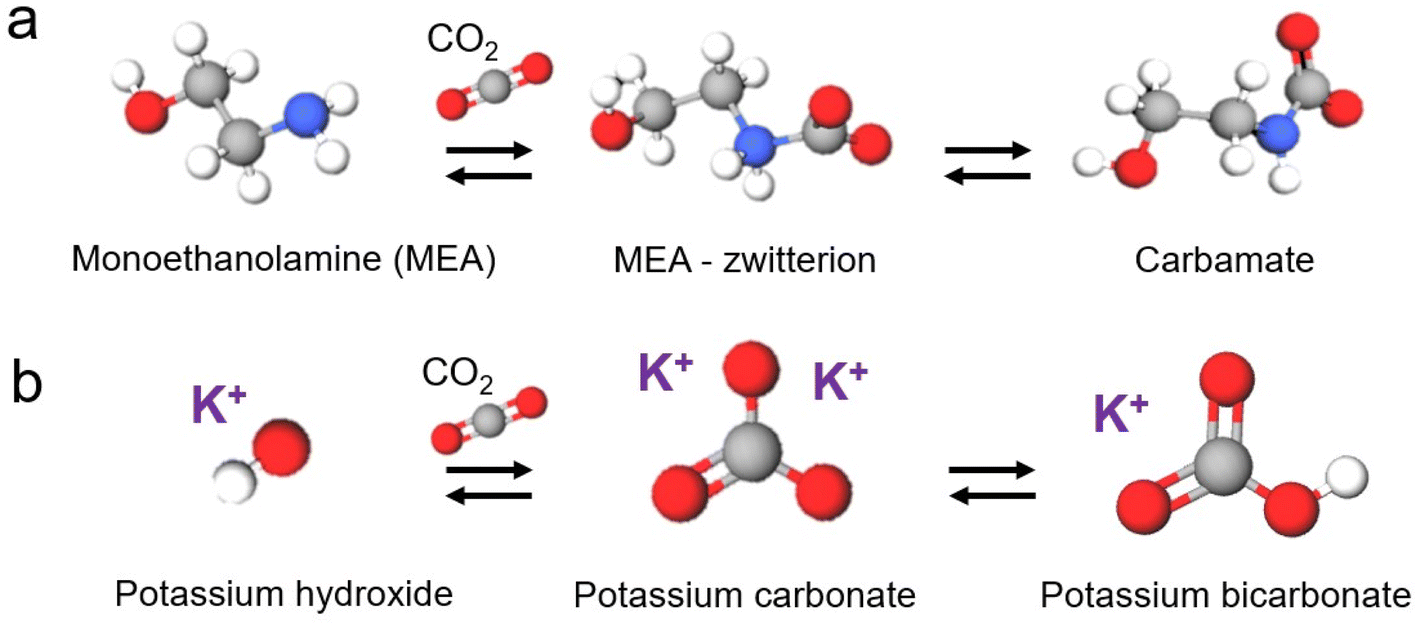 | ||
| Fig. 2 Reaction mechanism of the direct conversion of CO2 into (a) carbamate and (b) bicarbonate. | ||
2.1 CO2 captured in carbamate form
Many primary and secondary amines, such as monoethanolamine, diethanolamine (DEA), 2-amino-2-methyl-1-propanol (AMP), and their mixtures, are commercially available CO2 absorbent, primarily capturing CO2 as carbamates (RNHCOO−). Therefore, CO2 captured by these types of amines can be easily integrated into established industrial processes; however, the utilisation of carbamate can prove challenging owing to the very strong C–N bond, which necessitates the use of an energy-intensive amine regeneration process.21,22 Furthermore, the negatively charged moiety of the carbamate results in electrostatic repulsion between the cathode and carbamate. Several approaches have been developed to overcome the limitations imposed by the intrinsic properties of carbamate.2,23,24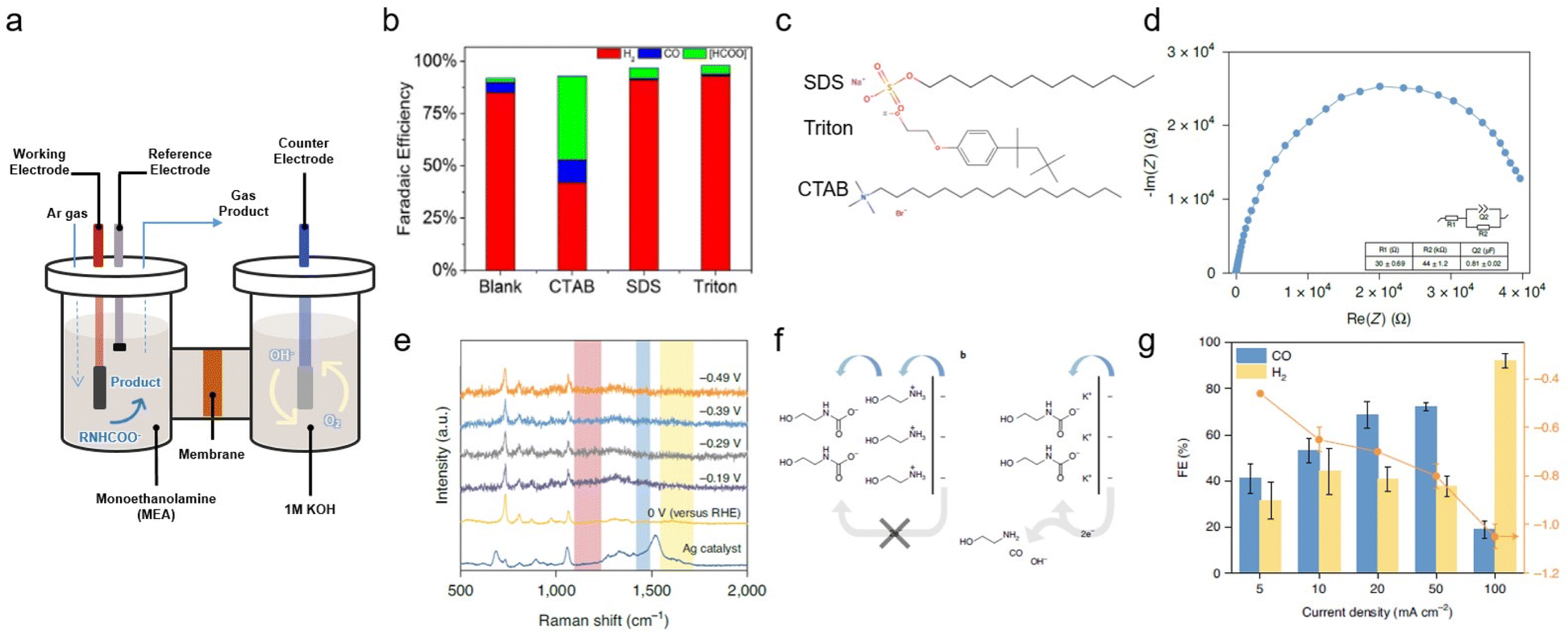 | ||
| Fig. 3 (a) Schematic of an H-type cell. (b) FE of products obtained with a smooth indium plate at a potential of −0.8 V vs. RHE in monoethanolamine containing 0.1% (w/w) of various surfactants. (c) Structures of the examined surfactants. Copyright 2017,25 Wiley. (d) EIS of the monoethanolamine/KCl electrolyte collected at open circuit potential, approximately −0.145 V vs. Ag/AgCl. Inset: the equivalent circuit and fit values of each component. R1 and R2 are the series and charge transfer resistances, respectively; Q2 is the constant phase element. The error of each component denotes the standard deviation of three independent measurements. (e) In situ surface-enhanced Raman spectra of the monoethanolamine/KCl electrolyte. (f) Proposed interfacial structure near the electrode surface. (g) Product distribution of monoethanolamine-CO2 conversion to H2 and CO at different applied current densities, ranging from 5 mA cm−2 to 100 mA cm−2 in a flow cell system. Copyright 2020,24 Springer Nature. | ||
| F.E. (%) | Catalyst | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In | Sn | Bi | Pb | Pd | Ag | Cu | Zn | |||||||||||||||||
| −VRHE | 0.8 | 1.1 | 1.3 | 0.8 | 1.1 | 1.3 | 0.8 | 1.1 | 1.3 | 0.8 | 1.1 | 1.3 | 0.1 | 0.5 | 0.8 | 0.8 | 1.1 | 1.3 | 0.8 | 1.1 | 1.3 | 0.8 | 1.1 | 1.3 |
| H2 | 41.9 | 42.0 | 44.3 | 68.6 | 78.5 | 93.4 | 69.5 | 87.1 | 93.4 | 79.6 | 79.2 | 85.1 | 91.6 | 87.3 | 96.0 | 62.8 | 84.7 | 89.5 | 79.7 | 98.1 | 91.0 | 103.0 | 91.4 | 102.0 |
| CO | 17.0 | 10.7 | 11.2 | 9.0 | 3.6 | 2.6 | 7.0 | 4.9 | 2.6 | 1.9 | 3.0 | 3.5 | — | — | — | 33.4 | 15.9 | 9.2 | 1.7 | — | — | 3.7 | 2.9 | 0.5 |
| HCOOH | 45.4 | 39.4 | 36.5 | 19.0 | 16.4 | 2.0 | 24.3 | 7.1 | 3.9 | 8.5 | 8.7 | 6.1 | 4.0 | 4.1 | 0.1 | 2.0 | 2.8 | 1.7 | 19.1 | 0.5 | 0.1 | 5.4 | 2.0 | 2.0 |
| Total | 104.3 | 92.1 | 92.0 | 96.6 | 98.5 | 98.0 | 100.8 | 99.1 | 99.9 | 90.0 | 90.9 | 94.7 | 95.6 | 91.4 | 96.1 | 100.8 | 103.4 | 100.4 | 100.5 | 98.6 | 91.1 | 112.1 | 96.3 | 104.5 |
A similar monoethanolamine utilisation system was developed by Lee et al.; however, this system also exhibited low capt-eCO2R performance.24 In CO2-saturated monoethanolamine solution, a Ag catalyst showed a CO FE of just 5% in the potential range from −0.46 to −0.78 V. Electrochemical impedance spectroscopy (EIS) and in situ Raman analysis of capt-eCO2R in monoethanolamine solution showed the formation of an electrochemical double layer (EDL) composed of ethanol–ammonium ions and carbamate. Due to the cationic ethanol–ammonium ion forming an inner Helmholtz layer owing to the negatively biased electrode surface, electron transfer must first pass through the ethanol–ammonium ion before reaching carbamate, which consequently inhibits the facile conversion of carbamate (Fig. 3f). Various alkali cations, such as Li+, Na+, K+, Rb+, and Cs+, were added to the electrolyte to systematically tune the EDL. Among the various alkali cations, K+ significantly improved the capt-eCO2R performance, by facilitating the adjustment of EDL, as evidenced by data from EIS and in situ Raman analysis (Fig. 3d and e). The addition of Li+ and Na+ resulted in the production of less CO than the pure monoethanolamine solution. This trend correlates with the ionic radius of the cations owing to differences in water solvation. Increasing the reaction temperature to facilitate cleavage of the C–N bond can also improve the performance of the system. The current density at 60 °C was 15 times higher than that at room temperature. By adjusting the EDL with a high concentration of alkali salt, 2 M KCl, and circulating the catholyte at 60 °C in the flow cell, CO production remarkably increased to 72% at 50 mA cm−2 and −0.8 V versus reversible hydrogen electrode (RHE) by directly utilising monoethanolamine captured CO2 (Fig. 3g).
To understand the capt-eCO2R system, its catalytic activity was investigated in various absorbent media. Atomically dispersed nickel-doped carbon (Ni–N/C), a promising capt-eCO2R catalyst, was applied in CO2-captured 5 M monoethanolamine solutions (Fig. 4a and b).26 The Ni–N/C catalyst has demonstrated high CO production selectivity comparable to that of a Ag catalyst, while the atomically dispersed active sites efficiently suppress the HER. Using Ni–N/C in an H-type electrolyser using CO2-captured 5 M monoethanolamine solutions, they achieved a CO partial current density three times higher than that obtained with a commercial Ag electrode (cAg). Furthermore, the introduction of a zero-gap membrane electrode assembly (MEA) equipped with a bipolar membrane (BPM) (Fig. 4c) improved the system efficiency, with Ni–N/C resulting in a 64.9% CO FE at −50 mA cm−2, 2.5 times higher than that obtained using the cAg (Fig. 4d). To understand the catalytic performance of the Ni–N/C catalyst, the reaction mechanisms were investigated by adjusting the carbamate concentration (from 1 M to 5 M monoethanolamine) (Fig. 4e). Interestingly, the reaction rate was unaffected by the carbamate concentration, indicating a zeroth-order dependence on the carbamate concentration in this range. This implied that the CO2 released from the carbamate, rather than the carbamate itself, is the main reactant in the capt-eCO2R. Temperatures higher than 60 °C typically favoured H2 generation and thus reduced CO selectivity (Fig. 4f and g). The similarity between the capt-eCO2R pathway and the conventional gas-eCO2R pathway leads to the high performance of the Ni–N/C catalyst in the CO2-capturing amine-based solvent, which is attributed to its outstanding intrinsic activity. The performances of Ni–N/C and cAg in the capt-eCO2R were further investigated in different CO2 capturing media, such as 1 M 3-amino-1-propanol, 2-(methlyamino)ethanol, AMP, DEA, and KHCO3, in order to investigate the effect of bulky cations. Considering that alkali metal cations with a smaller effective size have a higher surface density and thus stabilise the CO2-intermediate, the Ni–N/C exhibited remarkable CO FE performance irrespective of the type of electrolyte. Additionally, the CO FE of cAg decreases significantly as the cation size increases. The weak cation sensitivity of Ni–N/C was further demonstrated in M2CO3 electrolytes containing various alkali metal cations (M = Li+, Na+, K+, and Cs+) (Fig. 4h and i). This unique feature of Ni–N/C may be derived from its potential for zero charge (PZC), which is higher than that of cAg. The high PZC of Ni–N/C was thought to increase the surface charge density, which in turn reduces the impact of the steric bulk of the cation on surface intermediate stabilisation, thereby showing universal performance. This further highlights the importance of charge density at the EDL in improving the capt-eCO2R, in addition to its inherent catalytic activity.
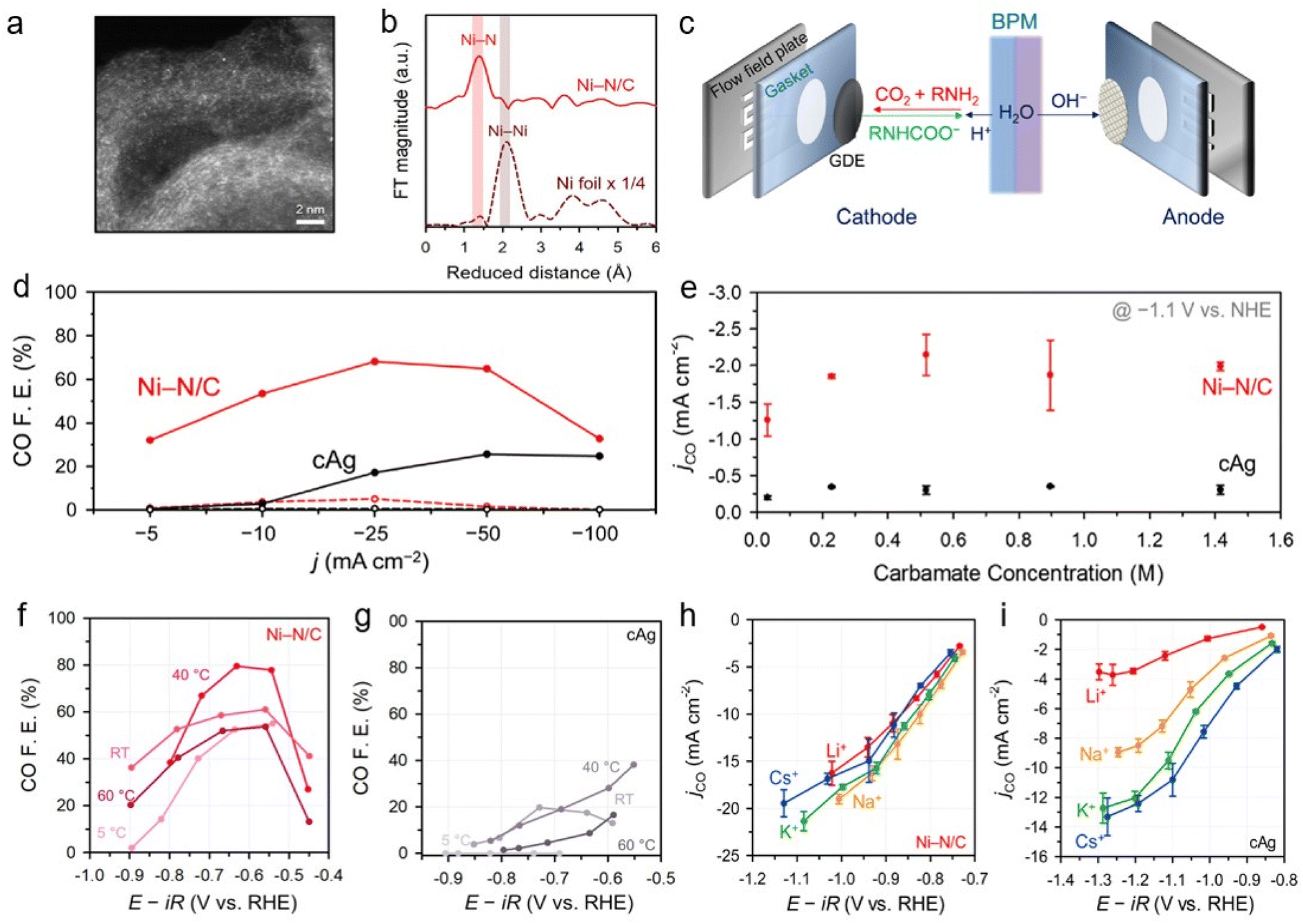 | ||
| Fig. 4 Physical characterisation of Ni–N/C (a) high-angle annular dark-field scanning transmission electron microscopy image. (b) Extended X-ray absorption fine structure spectra of Ni–N/C and Ni-foil. (c) Schematic of capt-eCO2R in MEA cell with BPM, anode and cathode. (d) Ni–N/C and cAg performance in membrane electrode assembly using 5 M monoethanolamine. (e) jco of Ni–N/C and cAg with respect to carbamate concentration. CO FE of (f) Ni–N/C and cAg (g) at various temperatures. Cation effect and sensitivity of Ni–N/C (h) and cAg (i) by occupying conventional gas-eCO2R. Copyright 2020,26 Royal Society of Chemistry. | ||
Modification of the catalyst morphology is an effective strategy for improving performance and has been applied to the capt-eCO2R system. Hossain et al. prepared Cu, Ag, and Au nanodendrites and conducted the capt-eCO2R in CO2-captured 0.05 M ethanolamine solutions (Fig. 5a–c).27 The nanodendrites exhibited a high electrochemically active surface area with a large double-layer capacitance, and consequently showed a higher eCO2R current density than a smooth surface. The Au nanodendrites outperformed the other electrodes, showing the highest current efficiency and lowest onset potential. These properties may arise from the synergistic effect between the dendritic structure and the intrinsic activity of Au, which is a well-known catalyst for CO production. The as-synthesised Au nanodendrites showed the most efficient eCO2R with a high current density in the linear sweeping voltammetry studies and recorded the lowest Tafel slope (Fig. 5d and e). A comparison of the eCO2R performance of nanodendrites in CO2-saturated NaHCO3 and CO2-saturated ethanolamine demonstrated that ethanolamine is a promising absorbent for the capt-eCO2R system. Ethanolamine plays an important role in achieving high current density and catalytic activity because the protonated NH3+ group of ethanolamine increases CO2 solubility.28,29 Furthermore, as-synthesised Au nanodendrites exhibited a significant performance in capt-eCO2R in ethanolamine, demonstrating its potential as a catalyst in this system (Fig. 5f).
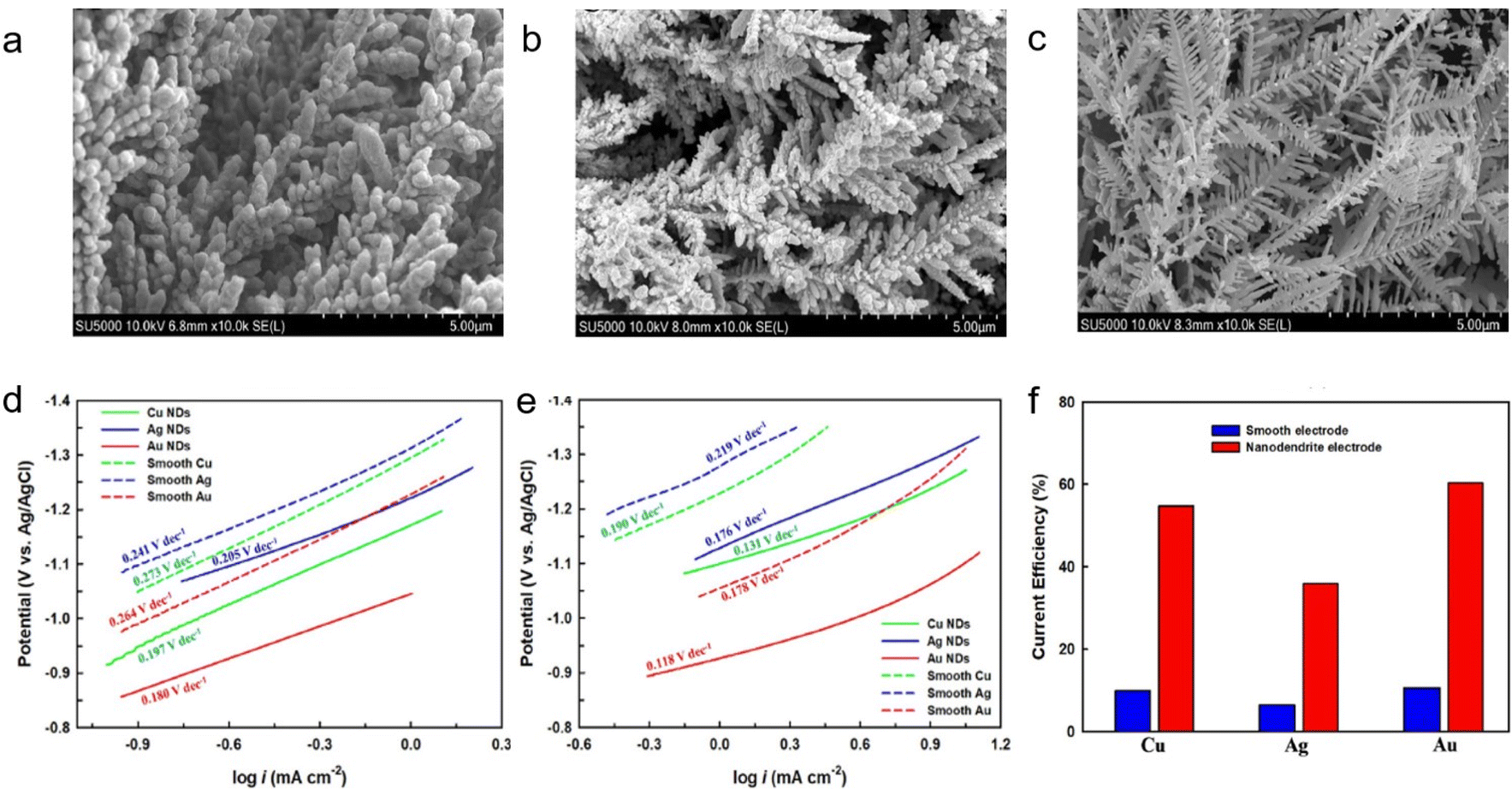 | ||
| Fig. 5 SEM images of as prepared (a) Cu, (b) Ag, and (c) Au nanodendrites. Tafel slope of various electrodes in CO2-saturated solutions of (d) 0.1 M NaHCO3 and (e) 0.05 mol fraction ethanolamine solution. (f) Current efficiency of smooth and nanodendritic electrodes in CO2-saturated 0.05 mol fraction ethanolamine solution at −1.0 V (vs. Ag/AgCl). Copyright 2021,27 Wiley. | ||
Recent studies have identified the active species in the reduction of CO2 in monoethanolamine-containing aqueous electrolytes.30 The carbamate moiety is not the only active site in aqueous monoethanolamine, and bicarbonate formation occurred as the partial pressure of CO2 increased, as detected by 13C NMR. This bicarbonate was formed via carbamate hydrolysis and was confirmed by the reduction in pH as the bicarbonate concentration and CO2 pressure increased. Though the reduction of CO2 to CO in monoethanolamine is limited by the adsorption of dissolved CO2, the addition of proton-donating ammonium cations influences the HER. Further study is required to achieve a better understanding of the active state of monoethanolamine during CO2 reduction.
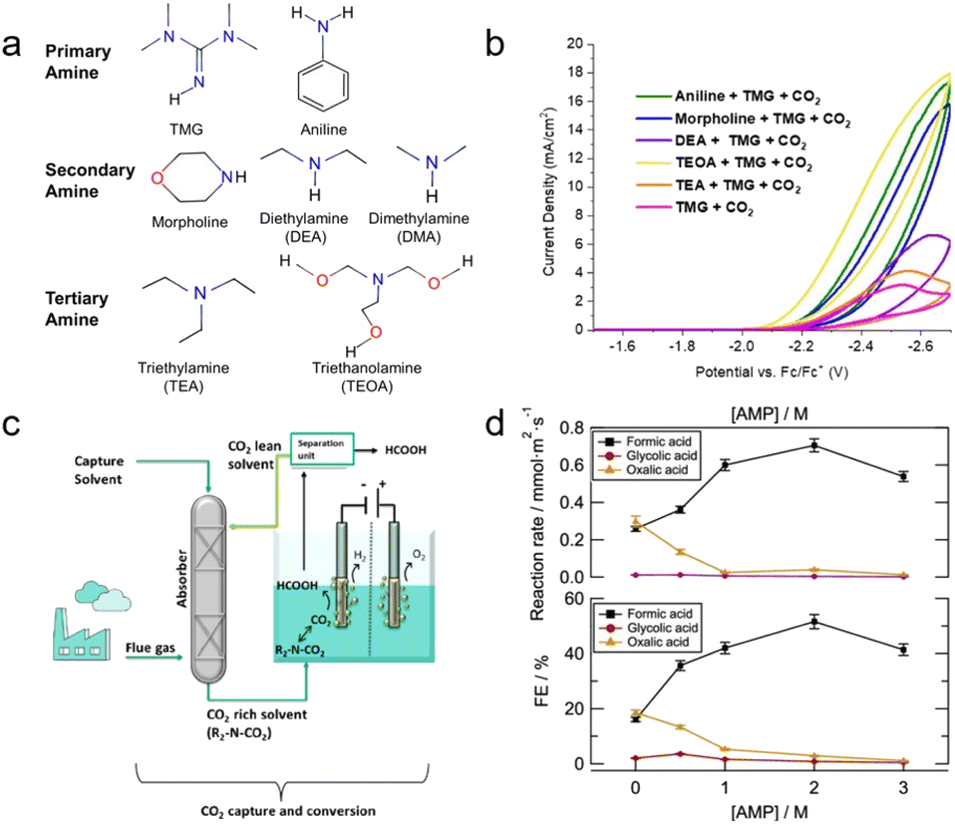 | ||
| Fig. 6 (a) Primary, secondary, and tertiary amines tested in this experiment. (b) CV responses of various amines in the presence of CO2 and 0.1 M TMG at a scan rate of 0.1 V s−1, scanning negative. Copyright 2020,31 The Electrochemical Society. (c) The proposed CO2 capture and conversion system. (d) AMP concentration effect of reaction rate and FE of formate, glycolate, and oxalate during electrolysis at −2.5 V vs. Ag/AgCl at 75 °C. Copyright 2021,32 American Chemical Society. | ||
Pérez-Gallent et al. developed an integrated system for CO2 capture and conversion by combining chemical and physical absorption solvents as electrolytes and increasing the temperature of the system in a mixed solution of AMP and propylene carbonate (PC).32 The system is functioned by combining these two solvents, where AMP is CO2 capturing media and PC is the physical solvent. This method involves heating an electrochemical reactor to liberate the captured CO2 (Fig. 6c). The concentration of AMP affected the reduction of CO2 at an optimal concentration of 2 M. Further increasing the concentration of AMP to 3 M results in a highly viscous solution that limits the conductivity and mass transfer properties of the solvent (Fig. 6d). Increasing the temperature also affects the performance of the system,24,26 which will be discussed in detail in Section 3.3. A Pb catalyst produced formic acid with a FE of up to 45% and a reaction rate of 0.56 mmol m−2 s−1. Recent research has indicated that AMP predominantly captures CO2 in the carbamate form, which undergoes hydrolysis in an aqueous solution to form bicarbonate.33,34 This phenomenon enhances the efficacy of AMP relative to other primary amines. Bicarbonate conversion is further discussed in Section 2.2.
2.2 (Bi)carbonate conversion
Metal hydroxides such as NaOH and KOH chemically absorb CO2 to form bicarbonates or carbonates depending on the pH of the solution. Such absorbents offer several advantages over amine absorbents, including lower volatility and absence of odour; however, their industrial application is limited by the drawbacks of salt formation during CO2 conditioning, high regeneration energy, and corrosivity arising from their high alkalinity.35–43 Despite these drawbacks, metal hydroxides are currently suitable CO2 absorbents for DAC systems, owing to their high selective absorption capacity in low-concentration CO2 environments. Moreover, the application of metal hydroxides to the capt-eCO2R system can address the high regeneration energy issue because the high-temperature (>700 °C)44 calcination process to release captured CO2 is replaced by electrochemical conversion of bicarbonate/carbonate. Therefore, capt-eCO2R systems using metal hydroxides are promising systems for achieving net-zero CCUS and carbon-negative direct air capture and utilisation (DACU).Tertiary amines absorbents chemically absorb CO2 in the form of bicarbonate. Unlike primary and secondary amines, tertiary amines do not directly bond with CO2 but act as base catalysts in the CO2 capture process (Fig. 7). Compared to primary and secondary amines, the C–H bonds of tertiary amines give an energetical advantage over the C–N bonds of primary and secondary amines in the desorption process. Additionally, tertiary amines generally have a higher capacity because one tertiary amine molecule captures one CO2 molecule, whereas two molecules of primary and secondary amines are required to capture one CO2 molecule as mentioned in Section 2. The performance of tertiary amine-based systems is expected to depend on the amine type, given that metal hydroxides provide the alkali cations required for electrochemical conversion reactions.
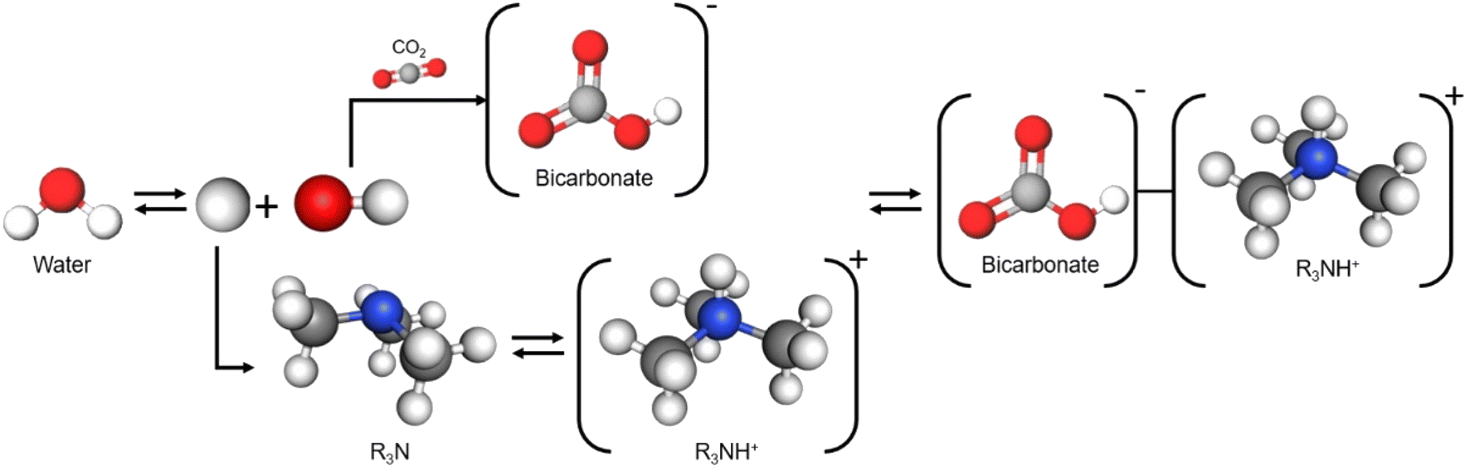 | ||
| Fig. 7 Representation of the process of CO2 capture by tertiary amines. | ||
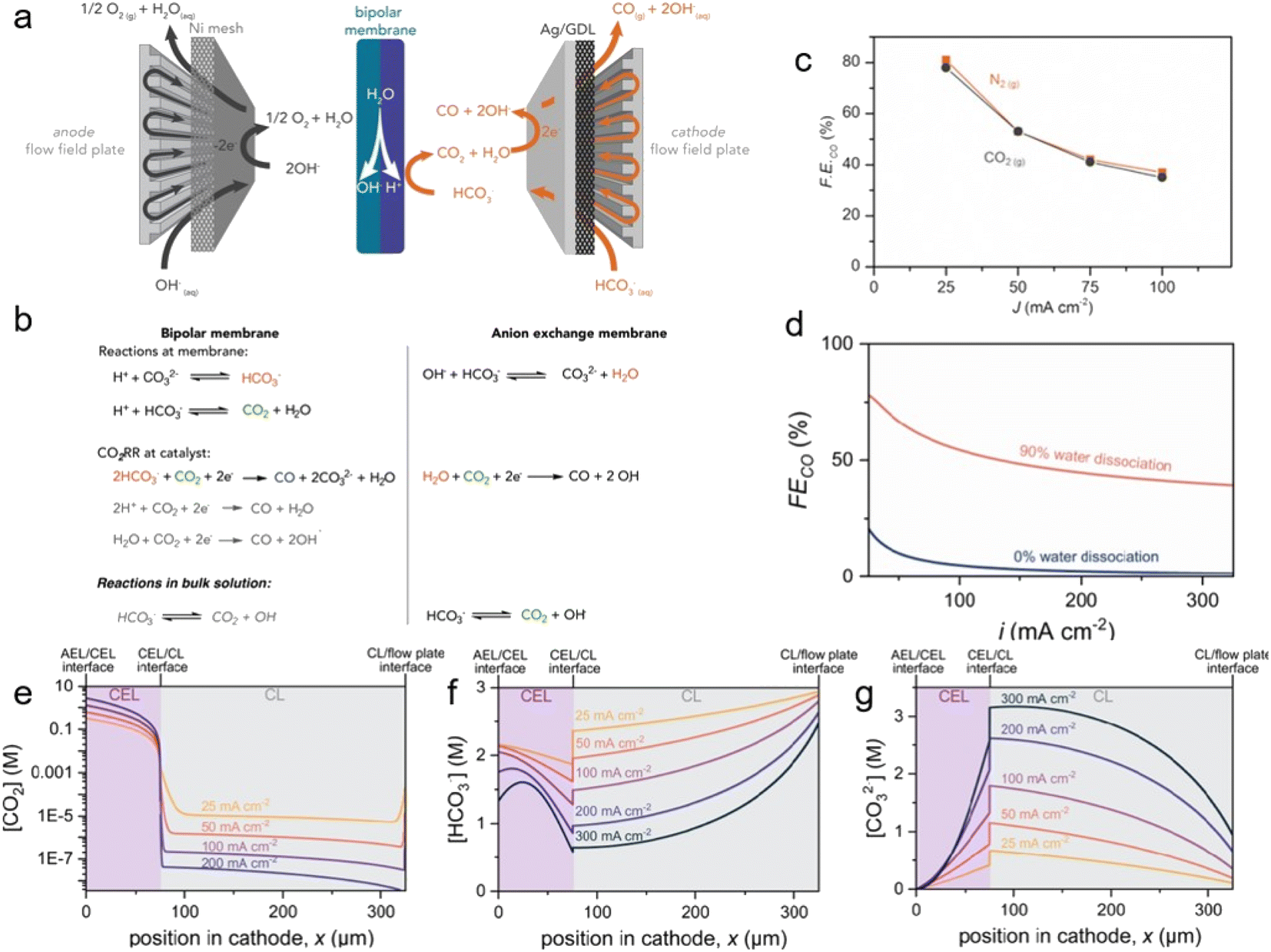 | ||
| Fig. 8 (a) Schematic of the MEA electrolyser with BPM. (b) The proposed dominant reactions that occur using each membrane. (c) CO FE between 25 and 100 mA cm−2 in a 3.0 M KHCO3 solution. Copyright 2019,47 Elsevier. (d) CO FE as a function of the water dissociation performance of BPM in the bicarbonate electrolyser cathode. Modeling results of (e) CO2, (f) HCO3−, (g) CO32− concentrations at various current densities. Copyright 2022,48 American Chemical Society. | ||
The bicarbonate conversion products were diversified by using different catalyst materials. Similar to conventional gas-eCO2R, the major products are determined by the type of catalyst. CO is primarily produced using Ag catalysts. Zhang et al. used a cathode consisting of porous Ag foam that showed high activity of 59% CO FE at −100 mA cm−2 in 3 M KHCO3 (Fig. 9b).49 Kim et al. reported that Ni–N/C showed significantly higher selectivity (91.0%) than cAg catalyst (28.8%) at −0.65 VRHE in 1 M KHCO3 (Fig. 9c).26 In addition, Li et al. noted that the H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO ratio of syngas, the feedstock for the Fischer–Tropsch reaction, varied from 2.5 : 1 to 7 : 1, and they produced a 3:1 ratio of syngas from CO2 captured 2 M KOH using a Ag catalyst.50 In particular, they also showed operational stability of 145 hours in the system. As a study on the production of formate, Bonet Navarro et al. used a bulk Sn catalyst to obtain 18% and 47% formate FE in 1.5 M KHCO3 in the case of CO2 pre-saturated and CO2 pre-saturated 1.5 M KHCO3, respectively, at −1.6 V vs. Ag/AgCl.51 In addition, Li et al. reported that electrodeposited Bi on carbon paper produced formate with 64% FE at −100 mA cm−2 and 27% FE at −400 mA cm−2 in 3 M KHCO3 (Fig. 9a).52 Lees et al. also demonstrated that Cu foam produced methane with a partial current density of −120 ± 10 mA cm−2 in 34 ± 7% yield from capt-eCO2R in 3 M KHCO3 with 3 mM CTAB.53 1D continuum modeling suggests that multi-carbon products are formed if the H+ supply from the membrane is smoother and the catalyst surface becomes more acidic. To study the multi-carbon products, Lee et al. composed a bilayer with one layer consisting of a mixture of Ag catalyst, Nafion, and poly(tetrafluoroethylene) (PTFE) and the other comprising a mixture of Cu catalyst and Sustainion.54 A flow reactor equipped with this bilayer recorded 41.6 ± 0.39% C2+ FE at −100 mA cm−2 in 3 M KHCO3 (Fig. 9d).
:CO ratio of syngas, the feedstock for the Fischer–Tropsch reaction, varied from 2.5 : 1 to 7 : 1, and they produced a 3:1 ratio of syngas from CO2 captured 2 M KOH using a Ag catalyst.50 In particular, they also showed operational stability of 145 hours in the system. As a study on the production of formate, Bonet Navarro et al. used a bulk Sn catalyst to obtain 18% and 47% formate FE in 1.5 M KHCO3 in the case of CO2 pre-saturated and CO2 pre-saturated 1.5 M KHCO3, respectively, at −1.6 V vs. Ag/AgCl.51 In addition, Li et al. reported that electrodeposited Bi on carbon paper produced formate with 64% FE at −100 mA cm−2 and 27% FE at −400 mA cm−2 in 3 M KHCO3 (Fig. 9a).52 Lees et al. also demonstrated that Cu foam produced methane with a partial current density of −120 ± 10 mA cm−2 in 34 ± 7% yield from capt-eCO2R in 3 M KHCO3 with 3 mM CTAB.53 1D continuum modeling suggests that multi-carbon products are formed if the H+ supply from the membrane is smoother and the catalyst surface becomes more acidic. To study the multi-carbon products, Lee et al. composed a bilayer with one layer consisting of a mixture of Ag catalyst, Nafion, and poly(tetrafluoroethylene) (PTFE) and the other comprising a mixture of Cu catalyst and Sustainion.54 A flow reactor equipped with this bilayer recorded 41.6 ± 0.39% C2+ FE at −100 mA cm−2 in 3 M KHCO3 (Fig. 9d).
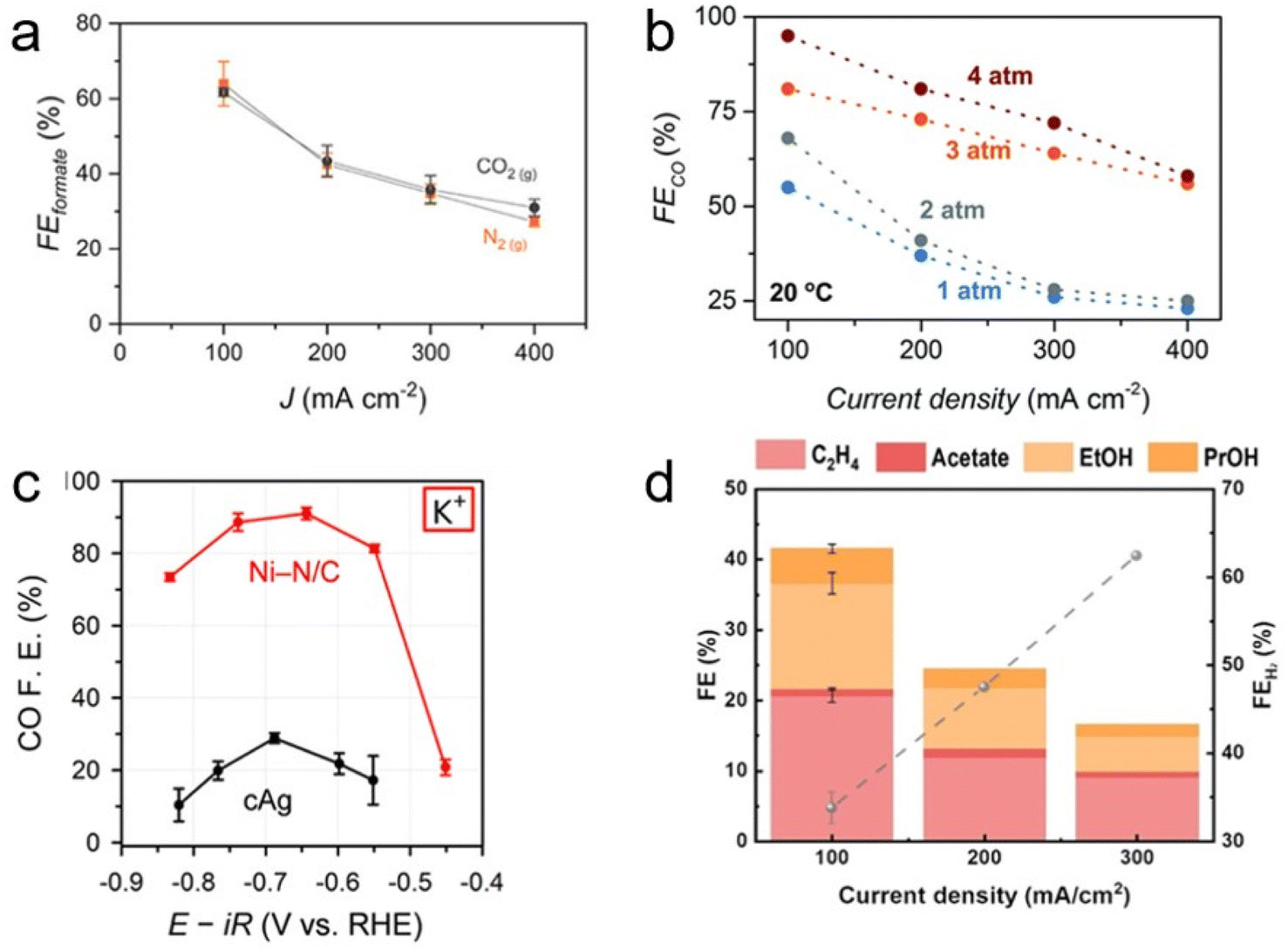 | ||
| Fig. 9 (a) Formate FE as a function of current density in 3.0 M KHCO3 solution purged with CO2 (g) or N2 (g). Copyright 2020,52 American Chemical Society. (b) CO FE as a function of pressure at different current densities. Copyright 2022,49 Royal Society of Chemistry. (c) CO FE of Ni–N/C and cAg in 1 M KHCO3. Copyright 2022,26 Royal Society of Chemistry. (d) FE of products as a function of current density. Copyright 2022,54 Wiley. | ||
Another study targeting multi-carbon products found that controlling the microenvironment was crucial, in addition to catalyst design. Lee et al. investigated instantaneous CO2 detachment by a pH equilibrium reaction of the absorbed CO2 species according to distances between the membrane and the cathode.55 The use of a 0.5 M H2SO4 anolyte and Nafion as a cation exchange membrane (CEM) with various thicknesses of interposers enabled the construction of well-defined spacing between 135 and 540 μm (Fig. 10a–c). In this configuration, high C2+ FE of 38% at a distance of 135 μm and a current density of −300 mA cm−2 were obtained in the presence of Cu nanoparticle catalysts (Fig. 10d). Also noteworthy, the CEM lowered the overpotential of the system by facilitating proton transfer to the cathodic side owing to the concentration gradient (Fig. 10e). To enhance C2+ production, cobalt phthalocyanines on carbon nanotubes (CoPc-CNTs) was applied to the Cu to produce CO from CO2 with high turnover frequencies, resulting in a higher C2+ FE of 47% at −300 mA cm−2 (Fig. 10f). These efficient systems highlight the importance of adjusting the mass transport to improve current density.
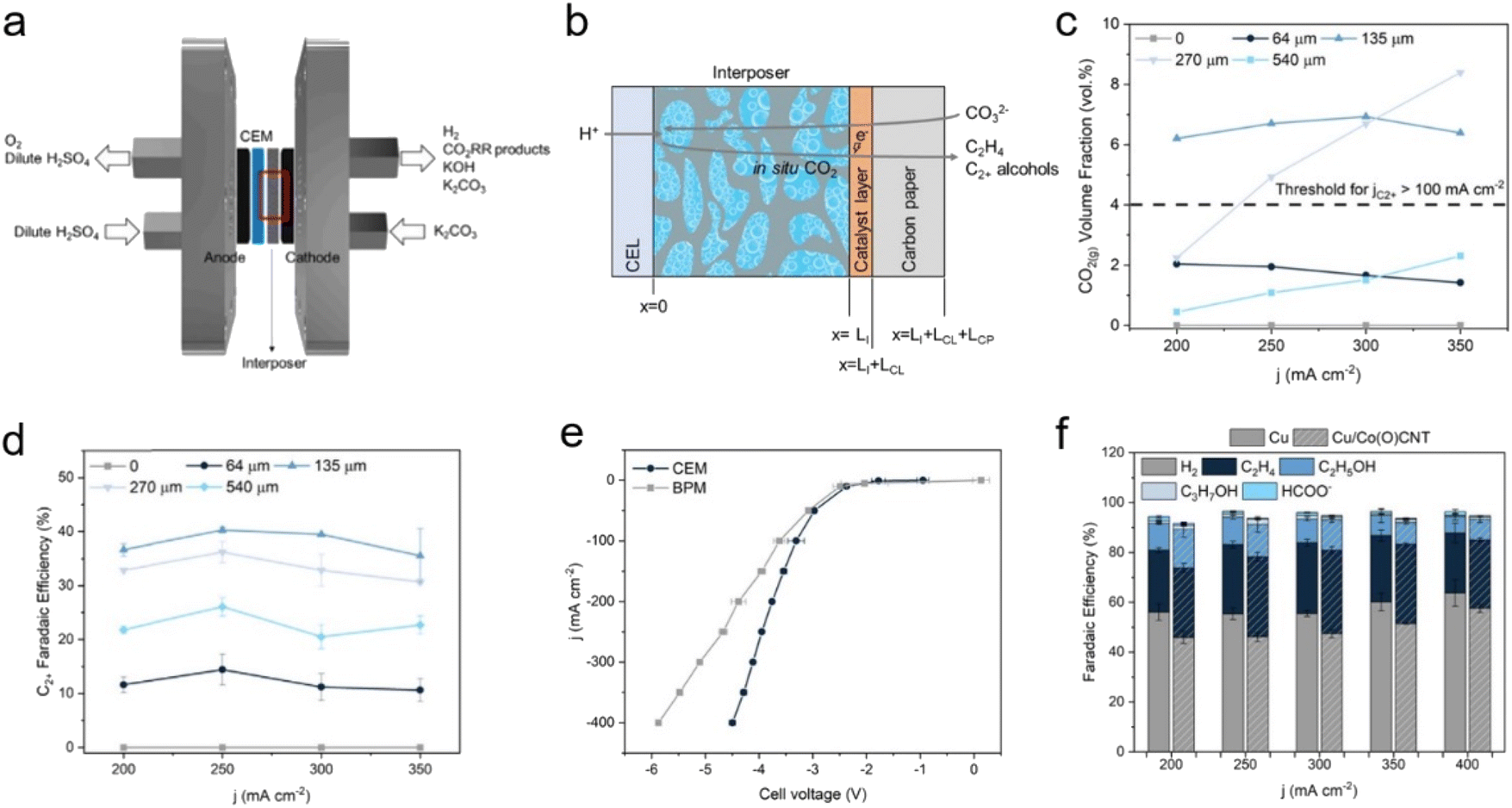 | ||
| Fig. 10 (a) Schematic of CO32−-fed electrolyser. (b) Representation of the cation-exchange layer, interposer, catalyst layer, and carbon paper. (c) CO2 (g) volume fraction under different spacing conditions and current densities. (d) C2+ FE of CO32− fed electrolyser with Cu electrocatalyst. (e) Full cell j–V curve with Cu electrocatalyst with a CEM (Nafion) and a BPM in 1.5 M K2CO3 solution with 135 μm interposer. (f) Product distribution of Cu electrocatalyst and Cu/CoPc-CNTs catalyst at applied current densities ranging from 200 to 400 mA cm−2. Copyright 2023,55 Elsevier. | ||
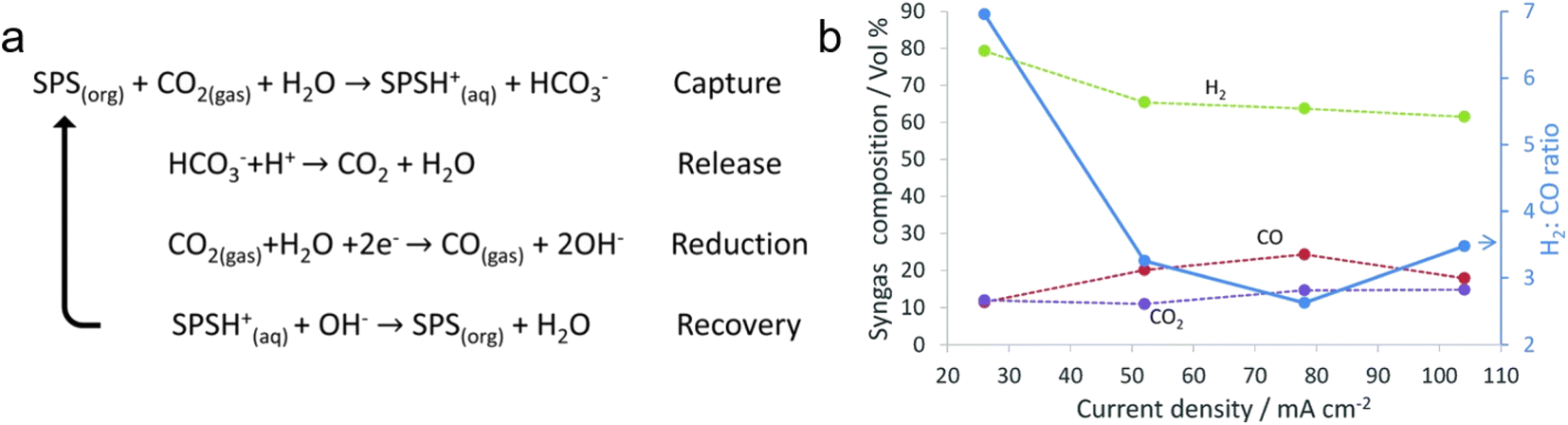 | ||
| Fig. 11 (a) A proposed amine cycle. (b) H2:CO ratio of the syngas obtained at different current densities. Copyright 2018,56 Royal Society of Chemistry. | ||
Langie et al. developed a unique bicarbonate conversion system known as a reaction swing absorption (RSA) using TREA as a CO2 absorbent.17 The tertiary amine TREA was used in place of a metal hydroxide owing to its high selectivity towards bicarbonate and industrially applicable CO2 absorption rate. TREA demonstrated a CO2 absorption rate of 84.5% at a flow rate of 0.8 m3 h−1 with 3% CO2, and 95.1% at 0.5 m3 h−1 with 3% CO2, measured in an absorption tower with a height of 3 m (Fig. 12a and b). Various membranes, substrates, and cathode catalysts were evaluated to identify the viable configuration for the utilisation of TREA-captured CO2. The optimal catalyst for syngas consisted of hydrophilic nano-coral structured Ag with carbon (coral Ag/C), which achieved a CO FE of 68.7% at −20 mA cm−2 and 29.2% at −200 mA cm−2 in a BPM equipped MEA electrolyser (Fig. 12c). Remarkably, the CO2 concentration in the outlet gas is nearly zero, highlighting the effective absorption of TREA and its suitability as a CO2 absorbent for the capt-eCO2R system. Also, they have reported up to 70 hours of stable performance in TREA capt-eCO2R through the use of optimized catalyst. The feasibility of the RSA system compares favourably with traditional CO2-to-syngas technologies, such as the reverse water–gas shift reaction and gas-eCO2R, according to TEA and LCA. RSA is in the early stages of development and thus incurs higher capital expenditure (CAPEX) (Fig. 12d); however, it offers the lowest OPEX and break-even prices among the evaluated CCU processes (Fig. 12e). In the optimistic scenario, with potential improvements in CO FE and reduced voltage, the break-even price of RSA could drop to as low as $0.65 per kg of syngas, significantly lowering both the CAPEX and OPEX. The RSA also showed the highest net present value (NPV), indicating a substantial positive increase in profitability (Fig. 12f). LCA showed that RSA had the lowest GWP in all tested scenarios, potentially being as low as 0.27 kg CO2 eq. kg−1 of syngas, moving towards net-zero CO2 emissions (Fig. 12g). These findings highlighted the economic and environmental advantages of bicarbonate systems. Recent achievements in bicarbonate conversion are summarised in Table 3.
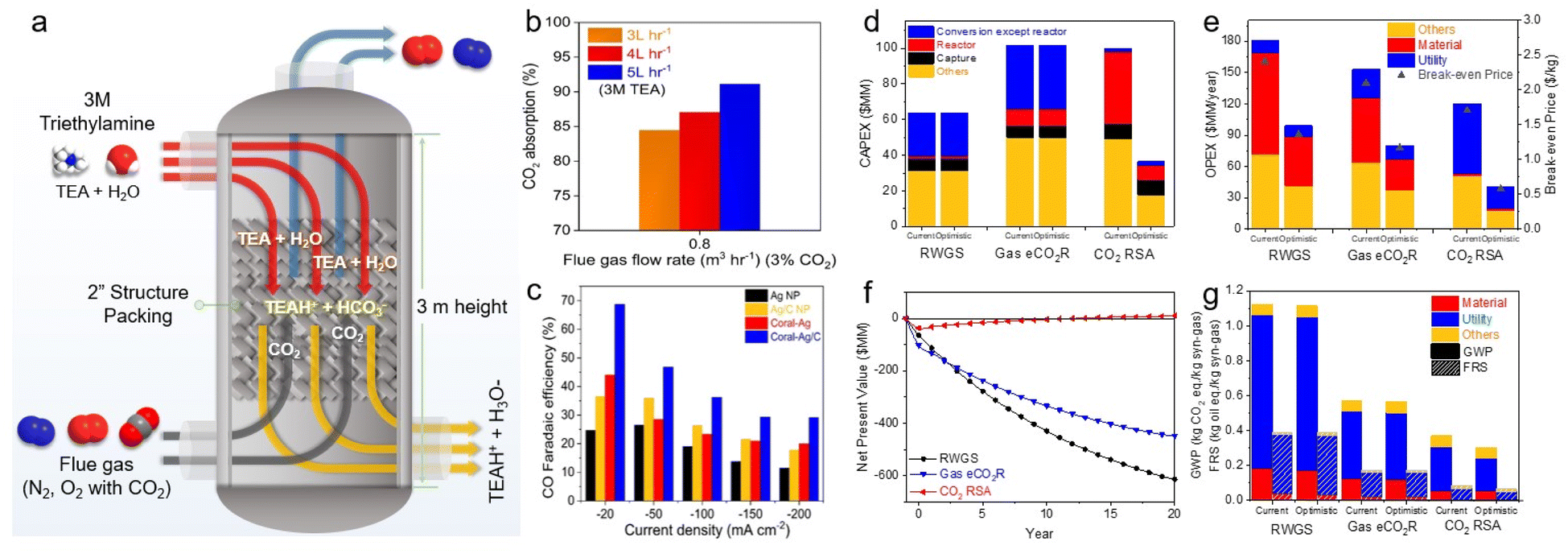 | ||
| Fig. 12 (a) Schematic of the CO2 absorption column used in the capture experiments. (b) CO2 absorption rates at various TREA flow rates at fixed gas flow rate and CO2 concentration. (c) CO FE of Ag nanoparticle, Ag/C, coral-Ag, and coral Ag/C in 3 M TREA. (d) CAPEX of three processes. (e) OPEX and break-even price of three processes. (f) LCA of the current and optimistic scenarios of three processes. (f) NPV of an optimistic scenario with a selling price of $0.8 per kg syngas. (g) LCA of the current and optimistic scenarios of three processes. Copyright 2022,17 Springer Nature. | ||
| Catalyst | Catholyte | Product | Faradaic efficiency (%) | Current density (mA cm−2) | Temperature | Pressure | Reference |
|---|---|---|---|---|---|---|---|
| Ag | 1.25 M CHP–H2CO3 | CO | 30 | −104 | 25 °C | 40 psig | 56 |
| Coral Ag/C | 3 M TREA | CO | 30 | −200 | R.T. | Ambient | 17 |
| Ag foam | 1.5 M CsHCO3 | CO | 80 | −100 | R.T. | Ambient | 58 |
| Ag | 3 M KHCO3 | CO | 82 ± 2 | −100 | R.T. | Ambient | 59 |
| Ag foam | 3 M KHCO3 | CO | 95 | −100 | R.T. | 4 atm | 49 |
| Bi | 3 M KHCO3 | HCOO− | 62 ± 1 | −100 | R.T. | Ambient | 52 |
| Cu foam | 3 M KHCO3 | CH4 | 30 | −400 | R.T. | Ambient | 53 |
| Cu/Ag bilayer | 3 M KHCO3 | C2+ | 41.6 ± 0.39 | −100 | R.T. | Ambient | 54 |
| Cu/CoPc-CNTs | 1.5 M K2CO3 | C2+ | 47 | −300 | R.T. | Ambient | 55 |
To investigate the dependence of the electrochemical activity on the type of tertiary amine, Hosseini et al. analysed solutions of monoethanolamine, diethylenetriamine (DETA), diisopropylamine (DIPA), and aminoethylpiperazine (AEP) with and without the addition of bicarbonate using CV.57 Of these; only DIPA showed a significant catalytic effect on bicarbonate reduction, then the mechanism of carbonate reduction in DIPA was explored electrochemically by scan rate control experiments and EIS analysis.
3 Strategy for improve performance of capt-eCO2R
Recent studies that aimed to identify the active characteristics of primary amine-based systems have proposed that the release or dissolution of CO2 may constitute an active component rather than the carbamate itself. Increasing the ratio of free CO2 to the captured absorbent is important to promote more efficient utilisation regardless of the absorbent type. Similar strategies have therefore been employed to improve the performance of capt-eCO2R system, regardless of the type of CO2 absorbent.3.1 Control the wettability of electrodes
In the case of an MEA cell or flow cell for the gas-eCO2R, where reactions occur at a triple-phase boundary, various strategies have been applied to reduce the involvement of water on the solid electrode surface to inhibit the HER and flooding.60–62 For example, porous carbon paper treated with hydrophobic materials, such as PTFE and microporous layers (MPL), is preferred in the gas-eCO2R system because it alleviates flooding and enables gaseous CO2 to access the catalyst layer. In contrast, the capt-eCO2R requires different mass transport approaches to achieve feasibility because the reactant is delivered in the liquid phase rather than the gas phase. Hydrophobic treatment of the carbon substrate inhibits intimate contact between the liquid reactants and the catalyst layer and may therefore have the opposite effect.Lees et al. compared the performance of Ag catalysts on carbon papers under different PTFE and MPL conditions.59 Placing the Ag catalyst on carbon paper without PTFE and MPL achieved the highest performance (Fig. 13a and b), indicating that a well-wetted environment facilitates the approach of the reactants to the electrode in the capt-eCO2R. Based on this observation, Zhang et al. used porous Ag foam as an electrode, which showed significantly higher performance than the control carbon cloth as mentioned in Section 2.2.1.49 Langie et al. also constructed an MEA cell using carbon papers that were not treated with PTFE or MPL.17 To enhance the hydrophilicity of the catalyst and improve CO production, the coral Ag/C was prepared through electrochemical oxidation and reduction processes. The performance of coral Ag/C was compared with Ag nanoparticles, coral Ag, and Ag/C to determine the origin of the high performance associated with the coral Ag/C. They found that the hydrophilicity of the catalyst material, through the transition from Ag nanoparticles to coral Ag and Ag/C structures, along with the measurement of the contact angle with water, contributes significantly to the performance improvement (Fig. 12c and 13c). Additionally, the CO production performance of various carbon papers, both treated and untreated with PTFE, showed a consistent trend where hydrophobic carbon paper resulted in reduced CO production (Fig. 13d).
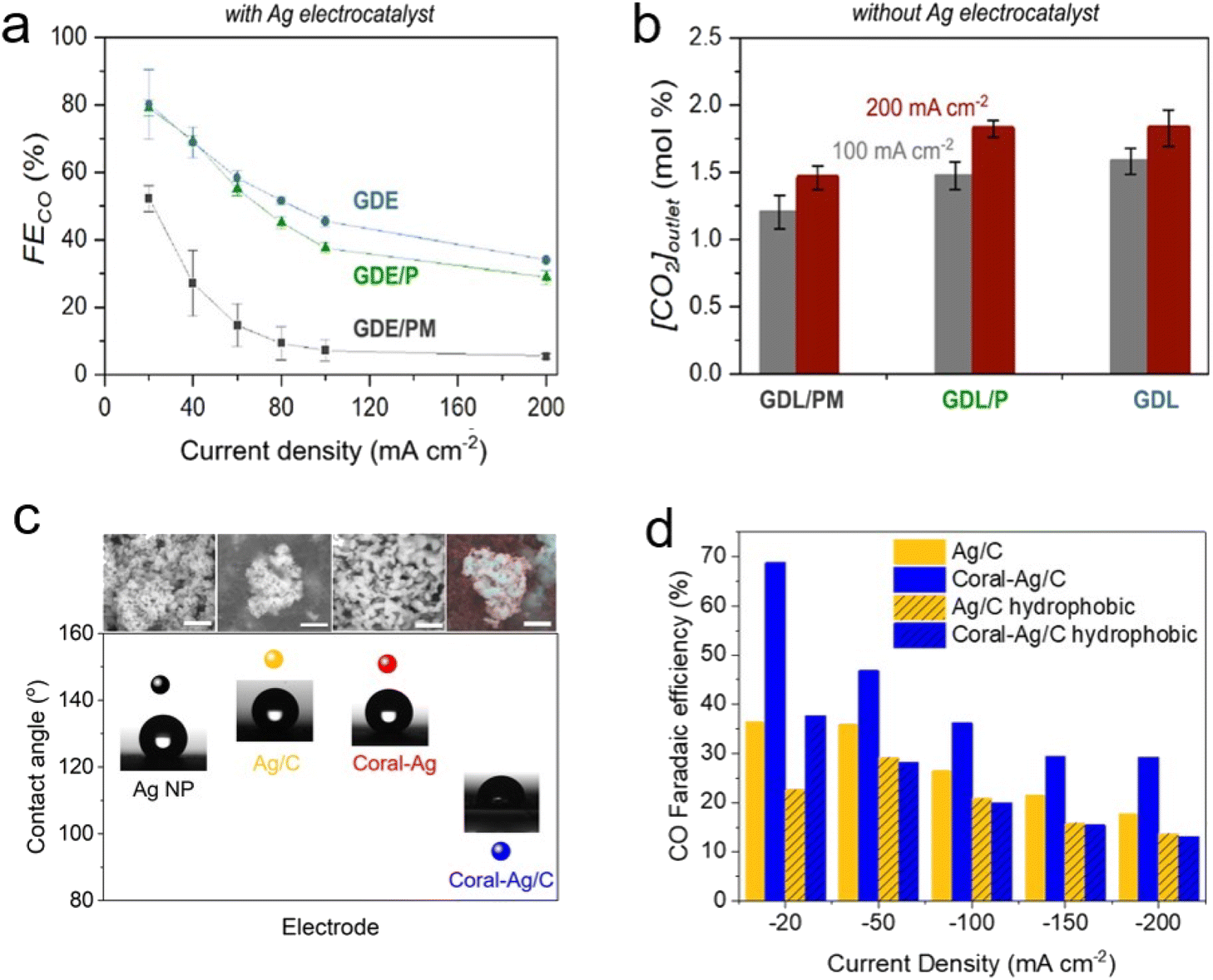 | ||
| Fig. 13 (a) CO FE of three different electrode conditions; GDE with neither PTFE nor an MPL, GDE/P containing PTFE but with an MPL, and GDE/PM containing both PTFE and an MPL. (b) The concentration of CO2 in the outlet gas under the three electrode conditions. Copyright 2020,59 American Chemical Society. (c) SEM images (scale: 500 nm) and contact angles of all Ag electrodes. (d) CO FE of prepared hydrophobic electrodes in the RSA system displayed with those of Ag/C and coral Ag/C. Copyright 2022,17 Springer Nature. | ||
3.2 Addition of various additives
CO2 absorbents such as amino-alcohol solutions can interfere with the catalyst surface in the capt-eCO2R system. As explained in Section 2.1.1, Lee et al. observed an increase in performance by adding alkali cation, especially K+, to the monoethanolamine solution, and observed the effect of alkali cation on the composition of electrode surface EDL through Raman spectroscopy and EIS analysis.24 These results demonstrate that while bulky ammonium cations inhibit reaction rates by preventing the direct transfer of electrons from the cathode to the reactants, this can be overcome by adding alkali cations.Fink et al. confirmed a cation effect similar to that observed in conventional gas-eCO2R by adjusting the type and concentration of alkali cations in MHCO3 (M = Li, Na, K, Cs) solutions.58 Furthermore, the influence of the cation on the chemical desorption and electrochemical conversion of CO2 was investigated. The amount of in situ CO2 was similar regardless of the identity of the cation; however, larger cations increased the CO selectivity, consistent with the known trend of the cation effect. They concluded that solvated alkali cations affect the stabilisation of the reaction intermediate, similar to the gas-eCO2R.
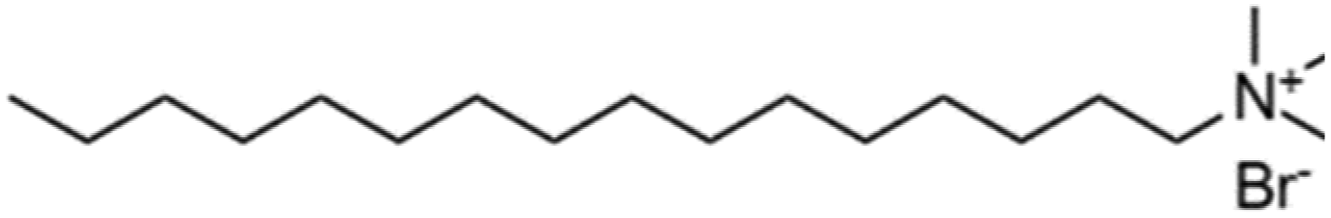
The organic chains of cationic surfactants are known to inhibit the HER, which competes with the eCO2R; thus, cationic surfactants have been used as alternative additives to metal cations. Banerjee et al. reported that a cationic surfactant suppressed the HER in the gas-eCO2R by adsorbing onto the surface of the cathode and thereby preventing the approach of water or hydrated cations in the electrolyte.63 Several similar studies using surfactants have been conducted in the capt-eCO2R. Lees et al. added 3 mM CTAB to 3 M KHCO3 to convert captured CO2 to methane and compared this method to that using a solution without CTAB using a porous copper electrode.53 The selectivity of the conversion reaction increased from nearly 0% to approximately 30%. In another study, Gutiérrez-Sánchez et al. compared the influence of four different surfactants, CTAB, CKC, SDS, and CTAC, on the formate production performance (Table 4) using a Sn wire as a working electrode.64 In 2 M KHCO3, the 1000 μM of CKC addition resulted in the highest activity, increasing the FE from 12% to 66% at −0.9 VRHE. The aromatic ring of CKC was hypothesised to increase resistance to H+, thereby improving the FE by more than 20% relative to CTAB, which exhibited the second-best performance.
| Surfactant | Abbreviation | Charge | Counter ion | Structure |
|---|---|---|---|---|
| Hexadecyl trimethylammonium bromide | CTAB | + | Br− |
|
| Benzyldimethyl hexadecylammonium chloride | CKC | + | Cl− |
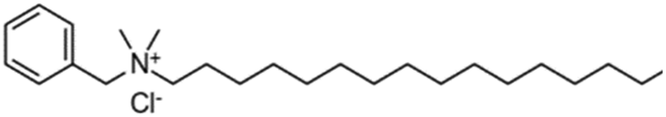
|
| Sodium dodecyl sulphate | SDS | − | Na+ |
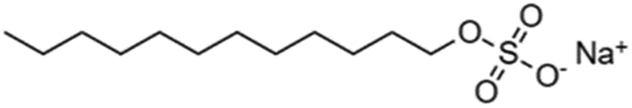
|
| Hexadecyltrimethylammonium chloride | CTAC | + | Cl− |
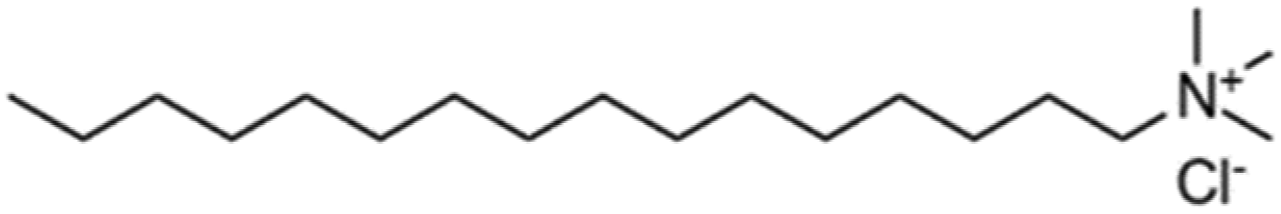
|
3.3 Temperature and pressure
During capt-eCO2R, desorption of CO2 from the absorption medium is expected to be necessary for electrochemical conversion into a product. Optimising the physical conditions, such as temperature and pressure, should improve the capt-eCO2R performance by facilitating the breaking of chemical bonds between the absorbent and CO2; however, the additional energy required for this process must be considered to ensure overall energy efficiency.The optimal temperatures vary according to the components and target products of the capt-eCO2R because temperature affects multiple properties, including the desorption–absorption equilibrium, catalytic reaction rates, and mass transfer. Lee et al. adjusted the temperatures of a flow cell system with a Ag catalyst as a cathode.24 The current density at 60 °C was 15 times higher than at room temperature (RT) and CO FE also increases with temperature (Fig. 14a and b). The additional thermal energy facilitated cleavage of the C–N bond between the amine and CO2, and accelerated the transfer of reactants. Kim et al. studied the effect of temperature in capt-eCO2R at RT, 40 °C, and 60 °C and found that the capt-eCO2R demonstrated peak efficiency at 40 °C (Fig. 14c and d).26 A higher temperature of 60 °C had positive effects on the desorption of CO2 but also simultaneously accelerated HER; thus, 40 °C was determined to be the optimal temperature for the capt-eCO2R. In addition, Pérez-Gallent et al. estimated the CO2 conversion rate, along with the amounts of liberated and converted CO2 at 15 °C, 45 °C, and 75 °C with a Pb electrode in 1 M AMP and PC solution.32 The highest CO2 liberation and conversion amounts were achieved at 75 °C, demonstrating 8 times higher performance than at 15 °C (Fig. 14f and g). Additionally, the highest CO2 conversion rate was observed at 45 °C (Fig. 14e). While additional thermal energy assists in the desorption and reactions of CO2, the rate of CO2 conversion has an optimal temperature of 45 °C.
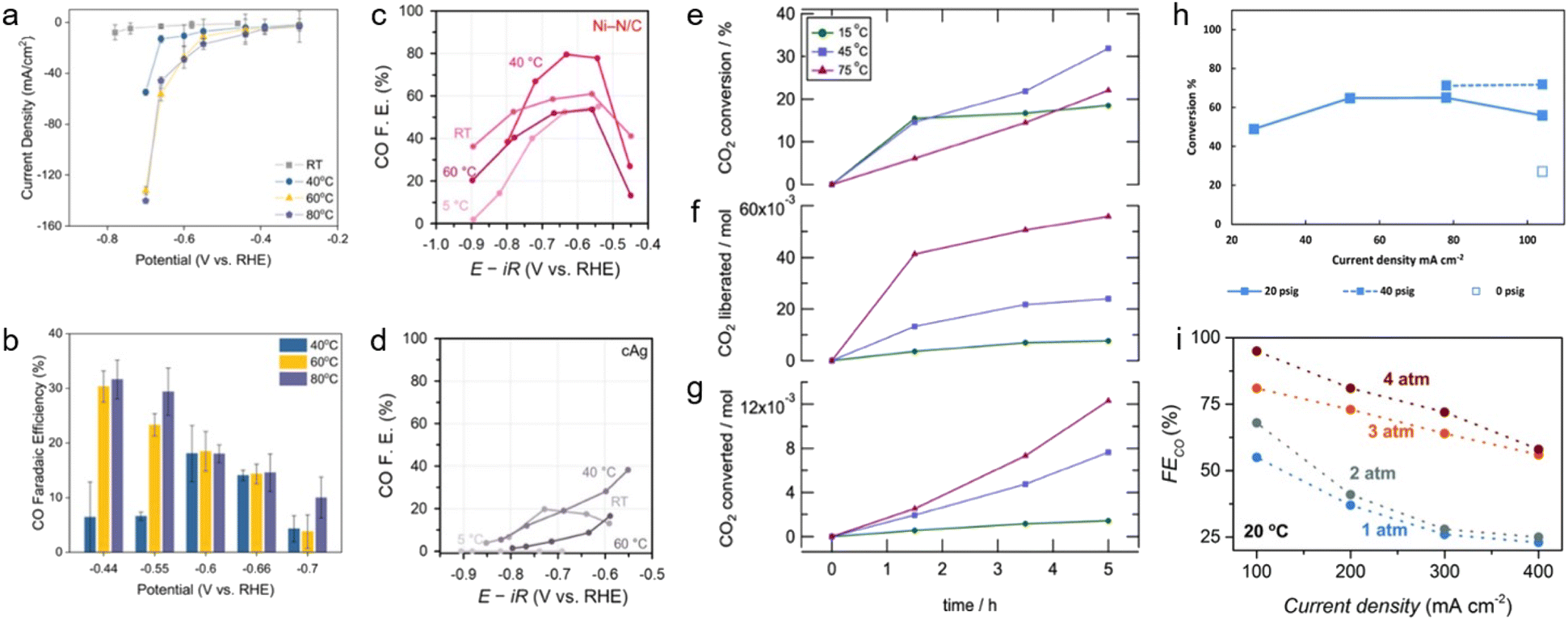 | ||
| Fig. 14 (a) j–V curve, (b) CO FE of 2 M monoethanolamine with 2 M KCl electrolyte at different temperatures. Copyright 2020,24 Springer Nature. CO FE of (c) Ni–N/C, and (d) cAg at different temperatures. Copyright 2022,26 Royal Society of Chemistry. (e) CO2 conversion, (f) liberated CO2, and (g) converted CO2 on a Pb electrode in a 1 M AMP in PC solution as functions of time and temperature. Copyright 2021,32 American Chemical Society. (h) Conversion profiles of CO2 released from the CP–H2CO3 solution at different current densities and pressures. Copyright 2018,56 Royal Society of Chemistry. (i) FECO as a function of pressure at different current densities of Ag foam in 3 M KHCO3. Copyright 2022,49 Royal Society of Chemistry. | ||
Pressure also significantly affects the solubility and reaction rates of gaseous molecules. To understand the effect of pressure effect on capt-eCO2R, Diaz et al. measured the CO2 conversion ratio at various pressures of 0, 20, and 40 psig at a current density of −104 mA cm−2 in a CHP–H2CO3 mixed solution.56 The highest CO2 conversion of 71.4% was obtained at 40 psig. High pressure facilitated the redissolution of unreacted CO2 and its participation in the reaction (Fig. 14h). Zhang et al. improved the performance on an MEA cell with a porous Ag foam cathode at a constant current density of −100 mA cm−2, by tuning the pressure and temperature.49 Increasing the temperature from 20 °C to 70 °C increased the CO FE from 59% to 78%, while increasing the pressure from 1 atm to 4 atm increased the CO FE from 55% to 95% (Fig. 14i). Temperature may affect CO2 desorption and mass transfer, whereas increasing the pressure increases the kinetic supply and reaction rate of CO2.
3.4 Other modification
The electrode surface was modified to use the flue gas directly via physical separation rather than chemical separation. Al-Attas et al. fabricated a permselective GDE composed of a sputtered Ag catalyst on one side and a metal–organic framework (MOF) mixed with Nafion on the other.65 Owing to its strong ability to physisorb CO2, the MOF called Calgary framework-20 (CALF-20) was used to increase CO2 permselectivity among the multiple gases that make up the flue gas. With the introduction of 10% CO2 into the N2-based gas stream, the permselective GDE showed 95% CO FE while the Ag/PTFE electrode showed only 58% CO FE at −1.32 VRHE. CALF-20 showed superior performance in suppressing the oxygen reduction reaction with an oxygen-containing gas stream and improved the wet stability with a wet gas stream.Kim et al. demonstrated that the activity of a given electrode is strongly dependent on the electrolyte composition because it can overcome the interference of the bulky cations typically present in CO2 absorbents.26 Developing catalyst materials affords control over the EDL and enables the manipulation of PZC of the electrode, which determines the electric field required for ion accumulation under electrocatalytic reaction conditions and tailored the catalyst to be insensitive to the cation species, as explained in Section 2.1.1. Developing catalyst materials is a promising strategy to enhance performance because it can be implemented into existing systems and circumvents the undesirable side effects of additives such as salt formation.
4. Perspective
The capt-eCO2R system is a promising system that complements the weaknesses of both CO2 capture and CO2 conversion systems. In the field of CO2 capture, capt-eCO2R systems eliminate the requirement for energy-intensive solvent regeneration, gas conditioning, and compression. Concurrently, because the majority of unreacted CO2 remains within the capture medium, eCO2R systems significantly improve the economic feasibility of eCO2R by simplifying or eliminating product separation and recycling process. Although the capt-eCO2R seems a seamless integration of CO2 capture and CO2 conversion, coupled studies considering both reactions thoroughly are essential for developing an advanced, high-performance system.In the CO2 capture process, the captured CO2 is regenerated by environmental factors such as temperature and pressure. The capt-eCO2R exploits the proton conductivity of the capture medium such that CO2 can be regenerated and activated by pH changes or applied potentials. To date, various CO2 absorbents, such as primary, secondary, and tertiary amine, as well as metal hydroxides, have been preferentially screened as well-known CO2 capture materials, while mixed absorbents or additives have been applied to improve the performance of capt-eCO2R systems. However, in the capt-eCO2R system, the CO2 absorbent acts as both a CO2 absorbent and an electrolyte. Given the active participation of the electrolyte-like cation effect observed in the capt-eCO2R system, further research is required to elucidate the role of the absorbent and identify or develop an ideal absorbent for the capt-eCO2R. In the screening of a promising CO2 absorbent for the capt-eCO2R, we propose to consider multiple properties that specifically address the binding energy and electroconductivity, such as pKa, molecular structure and size, solvation effect, and charge distribution. An understanding of the effects of these parameters is expected to afford control over the strength of CO2 binding, adsorption, and desorption of the reactants and products, as well as interactions with the catalyst surface. Beyond empirical approaches for selecting a CO2 absorbent, further studies are required to understand the key parameters and their respective impacts.
The electrolyser configuration must also be redesigned to enhance the circulatory efficiency of CO2 absorption, desorption, and conversion. Recent studies commonly employ MEA-type electrolysers for capt-eCO2R systems; however, because MEA electrolysers are designed for use with gas-phase CO2 rather than liquid reactants, the system configuration must be adjusted to ensure their optimal applicability in capt-eCO2R systems. Strongly hydrophobic substrates appear to limit the interaction between the liquid reactant and the catalyst, indicating that superior performance may be achieved using hydrophilic substrates (Section 3.1). This discrepancy may arise from the current designs of the flow path and substrate location, which are optimised for gas delivery. Consequently, a new generation of electrolysers more suitable for liquid reactants is required. In addition, most studies employed carbon substrates, while other types of substrates remain relatively unexplored.
The selection and development of membranes should be a major focus of future work to improve the performance of the MEA configuration. Current capt-eCO2R systems typically use BPM as a membrane owing to its high H+ and OH− reflux to each cathode and anode, respectively, which provides sufficient acidity at the cathode side to enable the release and conversion of CO2. The BPM would have been a good choice to ensure good separation, because most previous studies conducted the cathodic and anodic reactions under different electrolyte conditions. However, the structure of the BPM overlaps with those of the CEM and AEM, resulting in a high resistance due to its thickness, where consequently limits the current density that the BPM can provide to approximately 600 mA cm−2.66 Recent progress in enhancing the performance of the BPM can ensure the longevity of the capt-eCO2R; however, a newly designed or configured electrolyser that does not rely on a BPM should also be considered. Recently, a novel CEM-based system with an interposer was introduced to increase the concentration of in situ CO2 at the desired local pH at the catalyst; however, this configuration still requires a high overpotential owing to the presence of an interposer.55 A new membrane optimised for the capt-eCO2R system should therefore be developed to generate a higher current density and low overpotential.
A thorough understanding of the overall reaction mechanisms could also provide a breakthrough in the development of this system. Opinions regarding the identity of the active species in the capt-eCO2R differ. Some studies have suggested that captured CO2 is reduced directly; however, (bi)carbonate/carbamate are generally considered to be preferentially regenerated to in situ CO2 that is then reduced on the catalyst surface. Further reductions to afford the desired products are expected to be similar to those in the gas-eCO2R system; however, this remains to be conclusively demonstrated. Hence, a comprehensive investigation of these mechanisms requires direct observations by operando/in situ spectroscopy, alongside thermodynamic calculations and multiphysics modeling. Fundamental research in this area can provide valuable insights that inform the development of effective strategies and identify promising approaches.
In the current state of research, significant emphasis has been placed on advancing system development and catalyst engineering to enhance the performance of capt-eCO2R. Given the relatively early stage of development in this field, only a few publications reported on the long-term stability of their system comprehensively.17,50 To achieve a long-term stable capt-eCO2R system, we may encounter challenges similar to those in conventional gas-eCO2R systems, such as catalyst degradation, membrane stability, and electrolyte changes during prolonged operation. Furthermore, depending on the CO2 absorbent used, different degradation phenomena and their degrees may be expected, necessitating a thorough investigation of the effects of the CO2 absorbent. For instance, with a KOH solution, salt formation on the catalyst surface and flow path is probably unavoidable during extended operation.38,40,42 Amine-type absorbents may alleviate this issue, but some amines can potentially damage the catalyst and carbon substrate due to their corrosiveness. Moreover, it is essential to consider that catalysts can degrade due to fluctuations in reactant concentration and physical delamination under a liquid reactant circulating system. Therefore, observing the phenomenon of prolonged reactions should be a primary focus, followed by comprehensive studies to address it. All components of the electrochemical system, including the membrane, cathode, anode, and electrolyte, are crucial areas that require further study for long-term stability.
This technology is still in the early stages of research and development; thus, its performance is currently inferior to that of gas-eCO2R technologies. Nevertheless, significant performance improvements are expected over time as the development of the aforementioned component technologies continues to mature, driven by empirical insights gained through gas-CO2R research. In addition, the simplicity of this system will afford economic and environmental benefits that far outstrip those of existing CCUS technologies.
Author contributions
K. L. and G. B. contributed equally to this work, collected and analysed data, and wrote the manuscript. U. L., D. K. L., and C. W. L. contributed to the investigation of recent research trends and wrote the perspective part. Y. J. H. and D. H. W. supervised this study.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by “DACU project” (No. RS-2023-00259920), “Carbon to X Project” (Project No. 2020M3H7A1098229), and NRF-2022R1A2B5B02001380 from the National Research Foundation of Korea (NRF) grant, funded by Ministry of Science and ICT, Republic of Korea, and a KIST institutional project.References
- WMO, WMO Greenhouse Gas Bulletin (GHG Bulletin) – No. 17: the State of Greenhouse Gases in the Atmosphere Based on Global Observations through 2020, WMO, 2021 Search PubMed.
- O. Gutiérrez-Sánchez, B. Bohlen, N. Daems, M. Bulut, D. Pant and T. Breugelmans, ChemElectroChem, 2022, 9, e202101540 CrossRef.
- A. Dubey and A. Arora, J. Cleaner Prod., 2022, 373, 133932 CrossRef CAS.
- A. S. Reis Machado and M. Nunes da Ponte, Curr. Opin. Green Sustainable Chem., 2018, 11, 86–90 CrossRef.
- G. Y. Duan, X. Q. Li, G. R. Ding, L. J. Han, B. H. Xu and S. J. Zhang, Angew Chem. Int. Ed. Engl., 2022, 61, e202110657 CrossRef CAS PubMed.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- T. Jaster, A. Gawel, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U. P. Apfel, iScience, 2022, 25, 104010 CrossRef CAS PubMed.
- P. Yue, Q. Fu, J. Li, X. Zhu and Q. Liao, Green Chem., 2022, 24, 2927–2936 RSC.
- J. Fernández-González, M. Rumayor, A. Domínguez-Ramos and Á. Irabien, Int. J. Greenhouse Gas Control, 2022, 114, 103549 CrossRef.
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- R. Veneman, Adsorptive System for Post-combustion CO2 Capture, University of Twente, Enschede, 2015 Search PubMed.
- F. Yulia, R. Sofianita, K. Prayogo and N. Nasruddin, Case Stud. Therm. Eng., 2021, 26, 101093 CrossRef.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- K. M. G. Langie, K. Tak, C. Kim, H. W. Lee, K. Park, D. Kim, W. Jung, C. W. Lee, H.-S. Oh, D. K. Lee, J. H. Koh, B. K. Min, D. H. Won and U. Lee, Nat. Commun., 2022, 13, 7482 CrossRef CAS PubMed.
- N. S. J. Gao, C. Quiroz-Arita, L. A. Diaz and T. E. Lister, J. CO2 Util., 2021, 43, 101365 CrossRef CAS.
- Z. Rastegar and A. Ghaemi, J. Heat Mass Transf. Res., 2021, 58, 365–381 CrossRef.
- N. El Hadri, D. V. Quang, E. L. V. Goetheer and M. R. M. Abu Zahra, Appl. Energy, 2017, 185, 1433–1449 CrossRef CAS.
- D. Fernandes, W. Conway, R. Burns, G. Lawrance, M. Maeder and G. Puxty, J. Chem. Thermodyn., 2012, 54, 183–191 CrossRef CAS.
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC.
- B. Yoon, D. C. Calabro, L. S. Baugh, S. Raman and G. S. Hwang, J. Environ. Chem. Eng., 2022, 10, 108987 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2020, 6, 46–53 CrossRef.
- L. Chen, F. Li, Y. Zhang, C. L. Bentley, M. Horne, A. M. Bond and J. Zhang, ChemSusChem, 2017, 10, 4109–4118 CrossRef CAS PubMed.
- J. H. Kim, H. Jang, G. Bak, W. Choi, H. Yun, E. Lee, D. Kim, J. Kim, S. Y. Lee and Y. J. Hwang, Energy Environ. Sci., 2022, 15, 4301–4312 RSC.
- M. N. Hossain, S. Ahmad, I. S. da Silva and H. B. Kraatz, Chemistry, 2021, 27, 1346–1355 CrossRef CAS PubMed.
- Y. Qiu, H. Zhong, W. Xu, T. Zhang, X. Li and H. Zhang, J. Mater. Chem. A, 2019, 7, 5453–5462 RSC.
- M. Abdinejad, Z. Mirza, X.-a. Zhang and H.-B. Kraatz, ACS Sustain. Chem. Eng., 2020, 8, 1715–1720 CrossRef CAS.
- G. Leverick, E. M. Bernhardt, A. I. Ismail, J. H. Law, A. Arifutzzaman, M. K. Aroua and B. M. Gallant, ACS Catal., 2023, 13, 12322–12337 CrossRef CAS.
- M. Bhattacharya, S. Sebghati, Y. M. Vercella and C. T. Saouma, J. Electrochem. Soc., 2020, 167, 086507 CrossRef CAS.
- E. Pérez-Gallent, C. Vankani, C. Sánchez-Martínez, A. Anastasopol and E. Goetheer, Ind. Eng. Chem. Res., 2021, 60, 4269–4278 CrossRef.
- Y. Matsuzaki, H. Yamada, F. A. Chowdhury, S. Yamamoto and K. Goto, Ind. Eng. Chem. Res., 2019, 58, 3549–3554 CrossRef CAS.
- H. M. Stowe and G. S. Hwang, Phys. Chem. Chem. Phys., 2017, 19, 32116–32124 RSC.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, e1803111 CrossRef PubMed.
- J. Zhang, W. Luo and A. Züttel, J. Mater. Chem. A, 2019, 7, 26285–26292 RSC.
- E. J. Dufek, T. E. Lister and M. E. McIlwain, Electrochem. Solid-State Lett., 2012, 15, B48 CrossRef CAS.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O'Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- P. Jeanty, C. Scherer, E. Magori, K. Wiesner-Fleischer, O. Hinrichsen and M. Fleischer, J. CO2 Util., 2018, 24, 454–462 CrossRef CAS.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew Chem. Int. Ed. Engl., 2020, 59, 14736–14746 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2019, 3, 75–82 CrossRef.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- Y. Hori and S. Suzuki, J. Electrochem. Soc., 1983, 130, 2387 CrossRef CAS.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 CrossRef CAS.
- E. W. Lees, J. C. Bui, D. Song, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 834–842 CrossRef CAS.
- Z. Zhang, E. W. Lees, F. Habibzadeh, D. A. Salvatore, S. Ren, G. L. Simpson, D. G. Wheeler, A. Liu and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 705–713 RSC.
- Y. G. C. Li, G. Lee, T. G. Yuan, Y. Wang, D. H. Nam, Z. Y. Wang, F. P. G. de Arquer, Y. Lum, C. T. Dinh, O. Voznyy and E. H. Sargent, ACS Energy Lett., 2019, 4, 1427–1431 CrossRef CAS.
- A. Bonet Navarro, A. Nogalska and R. Garcia-Valls, Electrochem, 2021, 2, 64–70 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 CrossRef CAS.
- E. W. Lees, A. Liu, J. C. Bui, S. Ren, A. Z. Weber and C. P. Berlinguette, ACS Energy Lett., 2022, 7, 1712–1718 CrossRef CAS.
- J. Lee, H. Liu and W. Li, ChemSusChem, 2022, 15, e202201329 CrossRef CAS PubMed.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O'Brien, Z. Chen, D. Sinton and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- L. A. Diaz, N. Gao, B. Adhikari, T. E. Lister, E. J. Dufek and A. D. Wilson, Green Chem., 2018, 20, 620–626 RSC.
- S. Hosseini, H. Moghaddas, S. Masoudi Soltani, M. K. Aroua, S. Kheawhom and R. Yusoff, J. Environ. Chem. Eng., 2018, 6, 6335–6343 CrossRef CAS.
- A. G. Fink, E. W. Lees, Z. Zhang, S. Ren, R. S. Delima and C. P. Berlinguette, ChemElectroChem, 2021, 8, 2094–2100 CrossRef CAS.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 CrossRef CAS.
- M. D. Gálvez-Vázquez, P. Moreno-García, H. Xu, Y. H. Hou, H. F. Hu, I. Z. Montiel, A. V. Rudnev, S. Alinejad, V. Grozovski, B. J. Wiley, M. Arenz and P. Broekmann, ACS Catal., 2020, 10, 13096–13108 CrossRef.
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2017, 3, 193–198 CrossRef.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- S. Banerjee, X. Han and V. S. Thoi, ACS Catal., 2019, 9, 5631–5637 CrossRef CAS.
- O. Gutiérrez-Sánchez, N. Daems, W. Offermans, Y. Y. Birdja, M. Bulut, D. Pant and T. Breugelmans, J. CO2 Util., 2021, 48, 101521 CrossRef.
- T. Al-Attas, S. NabilK, A. S. Zeraati, H. S. Shiran, T. Alkayyali, M. Zargartalebi, T. Tran, N. N. Marei, M. A. Al Bari, H. Lin, S. Roy, P. M. Ajayan, D. Sinton, G. Shimizu and M. G. Kibria, ACS Energy Lett., 2022, 8, 107–115 CrossRef.
- C. Shen, R. Wycisk and P. N. Pintauro, Energy Environ. Sci., 2017, 10, 1435–1442 RSC.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |