
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical passivation of 2D transition metal dichalcogenides: strategies, mechanisms, and prospects for optoelectronic applications
Zhaojun
Li
 *a,
Hope
Bretscher
b and
Akshay
Rao
c
*a,
Hope
Bretscher
b and
Akshay
Rao
c
aSolid State Physics, Department of Materials Science and Engineering, Uppsala University, 75103 Uppsala, Sweden. E-mail: zhaojun.li@angstrom.uu.se
bThe Max Planck Institute for the Structure and Dynamics of Matter, 22761, Hamburg, Germany
cCavendish Laboratory, University of Cambridge, JJ Thomson Avenue, CB3 0HE, Cambridge, UK
First published on 19th April 2024
Abstract
The interest in obtaining high-quality monolayer transition metal dichalcogenides (TMDs) for optoelectronic device applications has been growing dramatically. However, the prevalence of defects and unwanted doping in these materials remain challenges, as they both limit optical properties and device performance. Surface chemical treatments of monolayer TMDs have been effective in improving their photoluminescence yield and charge transport properties. In this scenario, a systematic understanding of the underlying mechanism of chemical treatments will lead to a rational design of passivation strategies in future research, ultimately taking a step toward practical optoelectronic applications. We will therefore describe in this mini-review the strategies, progress, mechanisms, and prospects of chemical treatments to passivate and improve the optoelectronic properties of TMDs.
1. Introduction
Since the successful exfoliation of monolayer graphene, research attention on various 2D materials has grown significantly.1–5 Among 2D materials, transition metal dichalcogenides (TMDs), with the chemical structure MX2 (M = Mo, W; X = S, Se, Te), have been of great interest for optoelectronic applications due to their electronic and optical properties such as direct bandgaps, strong light–matter interactions, mechanical flexibility, and chemical and thermal stability to highlight a few.6–11 Many proof-of-concept 2D TMD-based optoelectronic devices have been demonstrated.9,12–18 However, in general, monolayer TMDs are imperfect and often contain various intrinsic and extrinsic defects.19 Despite the remarkable potential of TMDs and the progress towards various optoelectronic applications, many challenges in improving their intrinsic qualities and limiting performance losses due to trion formation and defects persist.In contrast to bulk TMD materials, excitons (electron–hole pairs) are strongly confined to the monolayer plane and also experience reduced screening due to the change in the dielectric environment.20 A strong Coulomb interaction results in tightly bound excitons with binding energies of several hundred meV, which dominate the optical and charge transport properties of 2D TMDs.21–23 Quasiparticles, the so-called trions, formed by excitons and induced free charges through defects or adsorbates, have also been identified even at room temperature due to their strong electrostatic interactions and exhibit binding energies of tens of meV.24 Background carrier concentrations can therefore play an important role in the recombination pathways and affect the optical and electronic properties of 2D TMDs. Additionally, oxygen and other adsorbates can alter the properties or degrade the qualities of 2D TMDs.25,26 Substrate-supported TMD monolayers also suffer from strain variations introduced by the roughness of substrates, which modifies their electronic properties.27 More importantly, due to their reduced dimensionality, 2D TMDs are predisposed to form atomic defects, such as vacancies, self-interstitials, grain boundaries, etc.19,28–32 The electrons and holes in TMDs can be trapped in defect-resulting potentials, leading to localized excitons and non-radiative recombination pathways, which strongly influences the optical and electronic properties of TMDs.32–34
To this end, extensive efforts have been devoted to exploring approaches for preparing and improving the quality of semiconducting 2D TMD materials. The fact that 2D materials are essentially all surfaces provides a unique opportunity for controlling and tuning their optical and electronic properties. 2D TMD layers are extremely sensitive to all influences of the surrounding environment, and their properties can therefore be easily modified by external variables. Substrate engineering like using a thin flake of hexagonal boron nitride (h-BN) as an interfacial layer reduces the structural damage and the associated interface states, which leads to cleaner optical and electronic properties.35–37 Strain engineering has also emerged as a powerful strategy for tuning the optical bandgaps of TMD materials.38,39 Phase engineering has been employed to enhance the electronic properties of TMDs.40,41 Additionally, the surface chemical strategy, a versatile and non-destructive method, is one of the most effective approaches to tailor the properties of 2D TMD materials for practical device applications.42 Compared to other approaches, chemical treatments are advantageous as they can be made compatible with other processing steps required for scaling-up device fabrication. However, designing treatments compatible with industry-scale device processing will require a precise understanding of the mechanisms behind known chemical treatments. Thus, setting up rational selection rules for chemicals to increase the potential of 2D TMDs in practical optoelectronic applications is of crucial importance. Previous review articles have discussed tuning optoelectronic properties by strain or substrate engineering and the observed effects of various chemical treatments.42–45 However, to the best of our knowledge, no reviews addressed both the effects and mechanisms behind these effects of surface chemical treatments on 2D TMDs. To this end, this mini-review focuses on the recent efforts using various surface chemical treatments to achieve high quality optoelectronic applications, paying particular focus on the mechanisms behind such treatments.
In this mini-review, we start with an overview of the major studies over the past few years on chemical treatments that improve the semiconducting quality of 2D transition metal disulfides (TMDSs) including MoS2 and WS2 (Section 2). More specifically, Section 2.1 covers the characterization approaches of TMDs utilizing photoluminescence (PL) and electron mobility as the main quality indicators, as well as discrepancies among the reports in the literature, and Section 2.2 discusses the mechanisms of each chemical treatment including different proposed mechanisms. Given that the understanding of how several chemical treatments work has evolved over the past few years, we think the discussion of discrepancies between different works is important to clarify the chemical selection rules and guide the further design of chemical treatment strategies for defect passivation and property control. In Section 3, we will then turn our focus to the major studies and accompanying mechanisms of chemical treatments improving the quality of MoSe2 and WSe2. In recent years, there have also been numerous studies on the preparation of high-quality 2D MoTe2, and it was theoretically predicted that Te vacancies can open a bandgap, which could be tuned by lattice strain or external forces.46,47 However, little research has been done up to this point on chemical treatments of MoTe2, thus discussions of MoTe2 and WTe2 are not included in this mini-review.48 Finally, we present our concluding remarks including issues addressed by chemical treatments, challenges facing the chemical treatment strategy development, and an outlook of future research directions in this area.
2. Chemical passivation of MoS2 and WS2
The intrinsic doping of transition metal disulfides (TMDSs) may lead to the formation of positively or negatively charged excitons (trions) that redshift and broaden the PL spectra. Control of the carrier density is effective to modulate the optical properties of monolayer TMDs induced by the many-body bound effect.Defects attenuate properties and device performance. In light-harvesting devices, defects can be detrimental if they assist carrier recombination and reduce mean free paths of carriers, consequently diminishing device performance. The predominant defect species and their percentages in 2D TMD monolayers synthesized via different methods or via mechanically exfoliated (ME) from bulk crystals will be different, and hence lead to different chemical treatment requirements. This might also be one of the factors that has caused discrepancy between the mechanisms disclosed in different studies. A large concentration of defects in ME MoS2 monolayers observed in transmission electron microscopy (TEM) and scanning tunneling microscopy (STM) was thought to be sulfur vacancies (SVs) that possess the lowest formation energy, and in contrast to atom dislocations observed at the grain boundaries in chemical vapor deposition (CVD)-grown MoS2.49–51 Hong et al. reported that antisite defects with Mo replacing the S atom are dominant point defects in physical vapour deposition (PVD)-grown ML MoS2, while SVs are predominant in ME and CVD-grown samples.49 The defect density in the CVD-grown ML WS2 interior is reported to be ∼0.33 nm−2 through atomically resolved scanning electron microscopy by Terrones and co-workers in 2017, which is four orders of magnitude higher than in mechanically exfoliated WS2.52 In addition, the density of sulfur vacancies near the edges is around three times higher than in the interior in the CVD-grown WS2. Similar defect distribution in monolayer CVD-grown MoS2 was reported by Schuck and co-workers.53 On the other hand, Su et al. showed CVD-grown rhombic monolayer MoS2 with PL intensity eight times stronger than CVD-grown triangular samples, indicating a low density of defects in rhombic monolayer MoS2. This was attributed to SV passivation by oxygen atoms which is predicted through density functional theory (DFT) simulations to remove in-gap states.54 Moreover, there is still debate on the origin of defects existing in TMDs. A previous study has shown that oxygen substitutions can be the dominant defects instead of sulfur vacancies and that differentiating between them is not possible using high-resolution TEM alone.55 Therefore, we have carefully included synthesis methods of TMDs for different chemical passivation studies.
2.1 Characterization
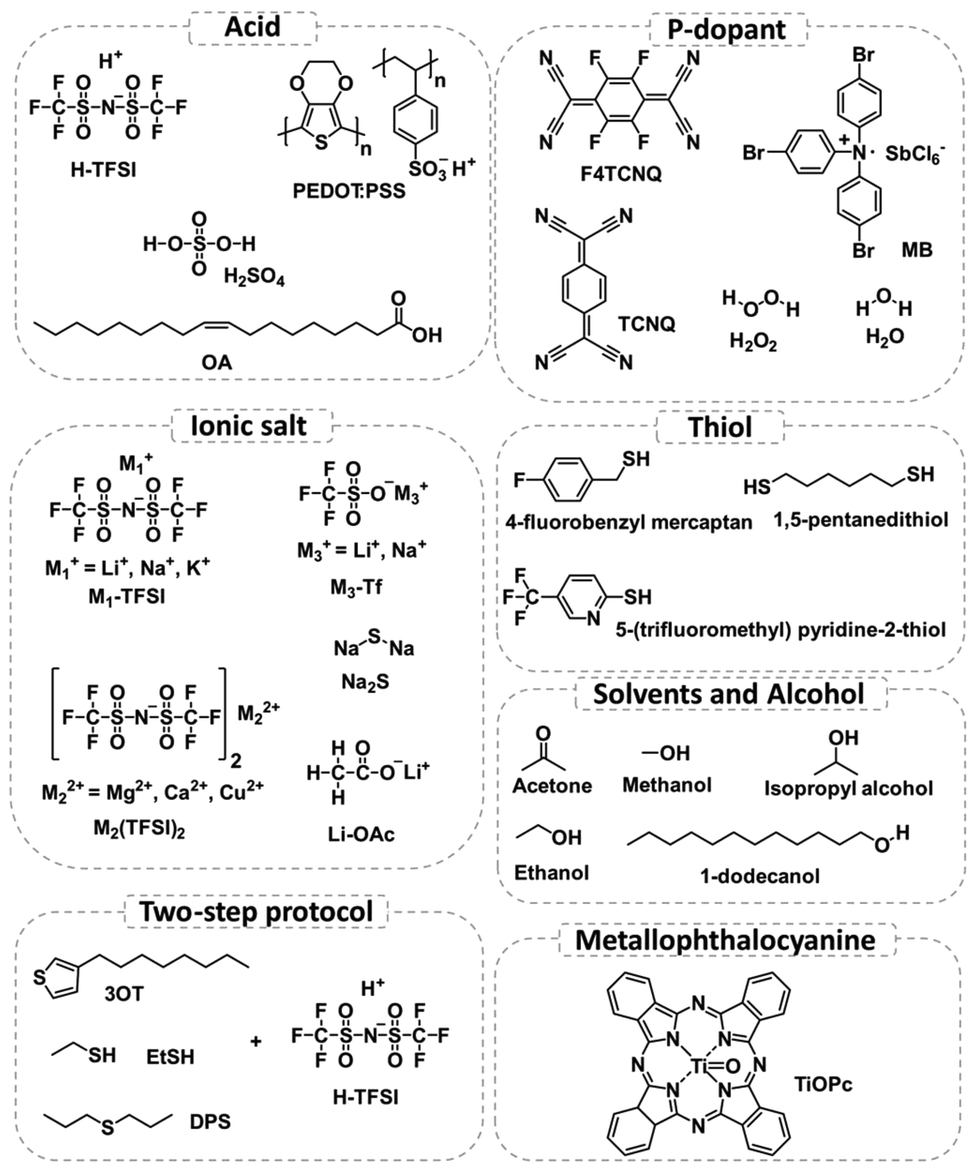 | ||
| Fig. 1 Structures of chemicals used in previous studies for the PL enhancement of 2D TMDs. This figure has been adapted from ref. 94 with permission from Spring Nature, copyright 2021. | ||
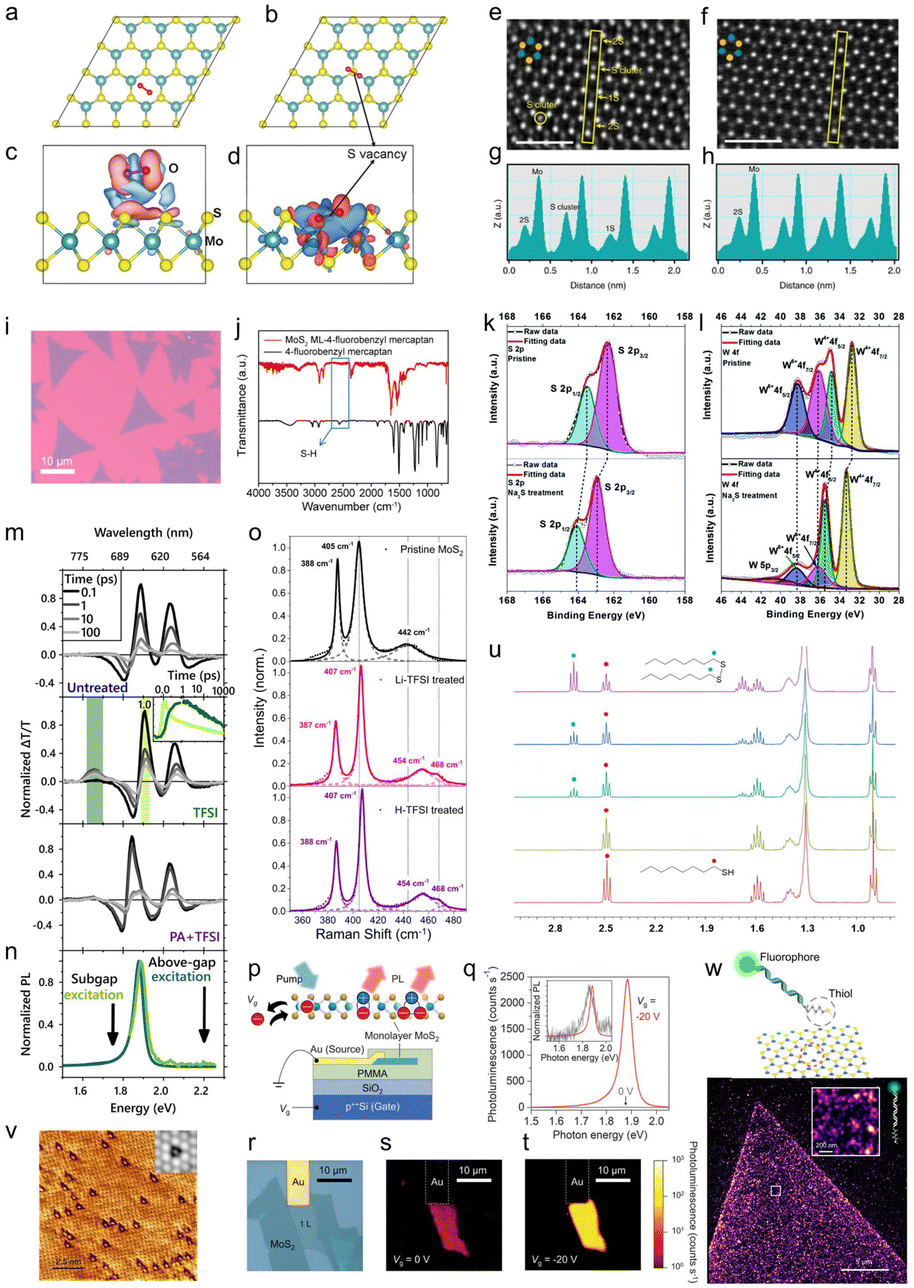 | ||
| Fig. 2 Summary of varied techniques employed to illustrate the chemical treatment mechanisms. DFT simulation on the adsorption of O2 on the 2D MoS2 surface. Relaxed configuration and charge density difference of an O2 molecule physisorbed on perfect monolayer MoS2 (a and c) and chemisorbed on defective monolayer MoS2 containing a monosulfur vacancy (b and d). The positive and negative charges are shown in red and blue, respectively. Reproduced from ref. 64 with permission from American Chemical Society, copyright 2014. (e–h) PSS-induced SVSH mechanism supported by HAADF STEM images. HAADF images obtained before (e) and after (f) PSS-induced SVSH, together with the Z-contrast mapping obtained before (g) and after (h) PSS-induced SVSH in the areas marked with yellow rectangles, revealing that the SVs (1S) are healed spontaneously by the sulfur adatom clusters on the MoS2 surface through a PSS-induced hydrogenation process. The cyan and yellow dots indicate the Mo and S atoms, respectively. Scale bar, 1 nm. Adopted from ref. 72 with permission from Springer Nature, copyright 2017. (i and j) Functionalization of the CVD-grown MoS2 monolayer with thiol molecules supported by FTIR measurement. (i) Typical optical microscopy image of CVD-grown MoS2 monolayers on a sapphire substrate. (j) FTIR spectra of 4-fluorobenzyl mercaptan-functionalized MoS2 MLs (red) in comparison with those of the free 4-fluorobenzyl mercaptan ligand (black). Reproduced from ref. 73 with permission from American Chemical Society, copyright 2014. (k and l) High-resolution XPS spectra of CVD-grown WS2. (k) XPS spectra of S 2p and (l) XPS spectra of W 4f before and after chemical treatment with Na2S solution (0.05 M). Reproduced from ref. 74 with permission from the Royal Society of Chemistry, copyright 2018. (m and n) Defect passivation of ME MoS2 supported by optical spectroscopy techniques. (m) Pump–probe spectra of MoS2 untreated (top), H-TFSI-treated (middle), and thiol + H-TFSI-treated (bottom) MoS2. H-TFSI treatment results in a prominent sub-gap bleach associated with sulfur vacancy defects. The inset shows the normalized kinetics taken at the A exciton bleach (light green) and defect peak (dark green) in the TFSI-treated sample, illustrating the transfer from the band edge to the sub-gap defect state. (n) PL emission with sub-gap excitation of H-TFSI-treated MoS2 occurs at the same energy as PL emitted by the above-bandgap excitation of H-TFSI-treated MoS2. Reproduced from ref. 83 with permission from American Chemical Society, copyright 2021, licensed under CC-BY 4.0. (o) Chemical adsorption supported by Raman measurements. Raman spectra of untreated, H-TFSI-treated, and Li-TFSI-treated monolayer MoS2. The decomposed Lorentzian peak fitting of each spectrum is presented as a short, dashed line and the cumulative fitting is presented as a solid line. The positions of A1g and 2LA modes of untreated MoS2 and the A2u mode of MoS2 with an adatom (Li, for example) are illustrated in each spectrum with a short black dashed line for direct comparison. The value of each peak position is also stated in the spectra. Reproduced from ref. 94 with permission from Springer Nature, copyright 2021. (p–t) Schematics of the device and the gate dependence of photoluminescence in MoS2. (p) Schematic showing control of different quasiparticles by the gate voltage Vg and generation rate G. (q) PL spectra of the MoS2 monolayer device under gate voltages of Vg = −20 and 0 V at a generation rate of G = 1018 cm−2 s−1. Inset: normalized PL spectra. (r) Top-view optical micrograph of a MoS2 device. (s and t) PL images of the device at (r) Vg = 0 V and (s) Vg = −20 V. Reproduced from ref. 103 with permission from AAAS, copyright 2019. (u) 1H NMR (400 MHz, CD3OD) spectra of pure 1-octanethiol after heating 1-octanethiol in CD3OD under reflux (in air) for 24 h and a 1-octanethiol/exfoliated 2H-MoS2 reaction mixture (in air) after 4 h, 6 h, and 24 h (from top to bottom). Highlighted triplet resonances: 1-octanethiol, δ = 2.49 ppm, red dot; dioctyl disulfide, δ = 2.68 ppm, blue dot. Other signals are associated with either 1-octanethiol or the disulfide derivative thereof. Reproduced from ref. 113 with permission from American Chemical Society, copyright 2018. (v) Higher-resolution STM image displaying the incorporation of O atoms (bright spots) into S vacancies (dark triangles). Inset: simulated STM image based on DFT calculations of an O-saturated S vacancy site in the 2D MoS2 crystals. Reproduced from ref. 118 with permission from Springer Nature, copyright 2018. (w) 2D-PAINT image acquired using fluorescent probes consisting of a fluorophore head, dsDNA of 70 bp as a linker molecule, and a thiol tail (FAM-dsDNA70bp-SH). The zoomed-in image reveals a high density of binding on MoS2. Reproduced from ref. 115 with permission from American Chemical Society, copyright 2021, licensed under CC-BY-NC-ND 4.0. | ||
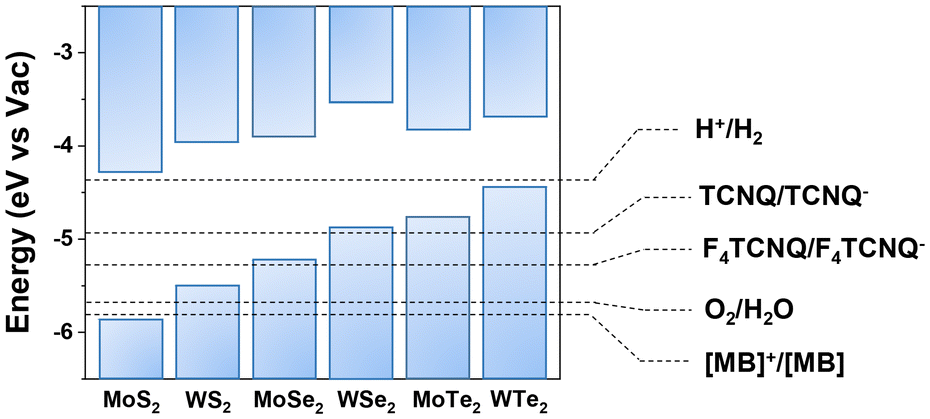 | ||
| Fig. 3 Calculated band alignment of MoS2, WS2, MoSe2, WSe2, MoTe2, and WTe2 along with electrochemical redox potentials of the p-dopants from the literature. This figure has been adapted from ref. 42 with permission from the Royal Society of Chemistry, copyright 2018. | ||
In 2015, Javey and co-workers demonstrated a near-unity PLQY with no change in the overall spectral shape for ME MoS2 monolayers on oxide substrates through a chemical treatment by the nonoxidizing organic superacid bis-(trifluoromethane)sulfonimide (H-TFSI).67 They also observed that the PL lifetime of MoS2 was lengthened from roughly 250 ps to 10 ns after the H-TFSI treatment. Later on, Javey and co-workers reported an encapsulation approach with an amorphous fluoropolymer CYTOP and a subsequent H-TFSI treatment, which yielded a near-unity PLQY in both ME MoS2 and WS2 monolayers with excellent stability against post-processing.68 They proposed that the strong protonating nature of the superacid removed the contaminants on the surface and suppressed defect-mediated nonradiative recombination in the monolayers. Goodman et al. reported that the deep trapped dark exciton states which were associated with native structural defects were responsible for the long PL lifetime of H-TFSI-treated MoS2, and the H-TFSI treatment reduced nonradiative recombination through these states.69 The exact mechanism of H-TFSI treatment is, however, not fully understood, which has been investigated by a few research groups. This will be further discussed in detail in Section 2.2.
In 2017, Atallah et al. reported that charged defects in CVD-grown MoS2 monolayers could be electrostatically passivated by ionic liquids (ILs) with a grounded metal contact, leading to up to two orders of magnitude increase in the PL yield.70 Similarly, Park et al. showed PL enhancement and defect passivation of CVD-grown ML MoS2 with a ML of titanyl phthalocyanine (TiOPc).71 Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) was reported to passivate the SVs in CVD-grown MoS2 by a sulfur adatom cluster through a hydrogenation process confirmed by the high-angle annular-dark-field (HAADF) scanning transmission electron microscopy (STEM) images (Fig. 2e–h) and X-ray photoelectron spectroscopy (XPS) measurements.72 In that study, a high-performance lateral monolayer MoS2 homojunction with an excellent photoresponsivity of ∼308 mA W−1 and outstanding air stability after two months was achieved employing that strategy. In that study, the electron concentration of MoS2 after the treatment decreased by 643 times, and led to a work function increase of ∼150 meV and an enhanced PL intensity. Jin and co-workers demonstrated the passivation of SVs in both CVD-grown and ME MoS2 monolayers via various thiol molecules, which led to an enhanced PL intensity.73 The authors used thiol molecules with F-containing ligands as markers, and the functionalized products were characterized with XPS and Fourier transform infrared (FTIR) spectroscopy (Fig. 2i and j). The reported mechanism of this chemical treatment is discussed in Section 2.2.2. Yao et al. immersed a CVD-grown WS2 monolayer into sodium sulfide (Na2S) solution and achieved enhanced PL emission with WO3−x defect passivation validated by XPS measurements (Fig. 2k and l).74 In their study, the inhomogeneous PL emission in the inner and edge regions of the pristine WS2 monolayer was attributed to the different charge populations and defect states across the monolayer area, which was clarified by the STEM images showing both SVs and W vacancies. The authors also observed a redshift of the PL spectra of ML WS2 after the Na2S treatment, which was due to the increased formation of trions and biexcitons evidenced by steady-state low temperature and laser power-dependent PL measurements.74 Recently, Zhang and co-workers reported that the PLQY of MoS2 quantum sheets produced by silica-assisted ball milling (BE) and sonication-assisted solvent exfoliation was enhanced to 18.5% via a heat treatment method based on the polar solvent tetrahydrofuran (THF).75 They showed increased radiative relaxation by time-resolved PL (TRPL) measurements and their XPS and high resolution transmission electron microscopy (HRTEM) results suggested that S and Mo formed an oxidized state and the passivation/deformation was from the edge inwards.
In 2019, Rao and co-workers reported a greatly enhanced PL intensity of ME WS2 monolayers via oleic acid (OA, shown in Fig. 1) treatment comparable to that of H-TFSI-treated monolayers, and simultaneously improved the mobility in WS2-based FET devices due to defect passivation.82 Later, they reported a generalizable SV passivation protocol using a passivating agent (thiol, thiophene, or sulfide, Fig. 1), followed by the H-TFSI treatment. This two-step chemical treatment simultaneously achieved improved mobility and an increase in the PL intensity of both MoS2 and WS2 monolayers.83 The detailed mechanism of this chemical treatment is discussed together with the H-TFSI treatment in Section 2.2.1. In 2021, Samorì and co-workers used π-conjugated dithiolated molecules to bridge adjacent MS2 flakes produced by liquid exfoliation. This led to an enhanced field-effect mobility (∼10−2 cm2 V−1 s−1) and ION/IOFF (∼104), along with the fastest switching time (∼18 ms), showing the importance of chemical treatment for the development of high-performance and printed electronics based on solution-processed TMDs.84,85
2.2 Mechanisms of chemical treatments
Cation intercalation has been proposed as another mechanism of chemical treatments. The intercalated cations can result in p-type doping to the monolayer TMDs and reduce the substrate influence simultaneously. In 2017, Yu et al. demonstrated that the PL of the CVD as-grown and transferred WS2 monolayer was enhanced due to the intercalation of small cations (H+ and Li+) between the monolayers and underlying substrates, which was achieved by simply immersing substrate-supported monolayers into a certain chemical solution.96 The intercalation was evidenced by an increase in the atomic force microscopy (AFM)-measured height of the as-grown monolayers after the chemical treatment. Through a series of steady-state PL measurements, they also concluded that intercalation was less likely to occur in TMD monolayers that interacted with substrates more strongly, like for the as-grown monolayers or monolayers on 2D material substrates (h-BN, for example).
The mechanism of H-TFSI treatment is a topic of specific focus since this chemical treatment has received the most attention in the past few years following the report of Javey and co-workers. In 2017, Kim and co-workers found that the H-TFSI treatment had a minimal effect on the inner region of triangular CVD-grown WS2 monolayers, whereas the PL of WS2 in the edge regions was enhanced up to 25 times.97 They concluded that H-TFSI p-doped the sample, and reduced defects which they assumed were distributed unequally throughout the sample, thus leading to spatially heterogeneous effects. Subsequently, Kim and co-workers reported that SVs in CVD-grown MoS2 were directly repaired by the extrinsic sulfur atoms produced from the dissociation of H-TFSI, evidenced through a correlative combination of optical characterization, atomic-scale STEM, and DFT calculations.98 The detailed mechanism of the H-TFSI treatment proposed by the authors is shown in Fig. 4a, where the H-TFSI molecule initially released SO2, after which a SV was passivated, resulting in the dissociation of an O2 molecule from the SO2 after SV passivation. The authors also observed that the PL peak position of MoS2 blueshifted by ∼5 nm and the A1g Raman mode blueshifted by 0.6 cm−1 after the H-TFSI treatment, suggesting a p-type doping effect. On the other hand, Kiriya et al. compared the effect of H-TFSI treatments in various solvents on the PL intensity of the ME MoS2 monolayer with H2SO4 and Li2SO4 in water and concluded that the proton is a key factor in enhancing the PL intensity of MoS2.99 Relatedly, Lu et al. reported that the effectiveness of H-TFSI depended critically on the charge state and protons donated by H-TFSI.100 According to their DFT simulations, three H atoms symmetrically adsorbed around the SV site in its −1 charge state, which could remove all gap states (Fig. 4b). Schwermann et al. revealed through first-principles calculations that H-TFSI transferred oxygen to the surface of monolayer MoS2, yielding a defect-free electronic band structure like that of pristine MoS2.101 The authors also pointed out that there were similar reactions with H2O2, O2, and H2SO4 (but not H2O) treatments, which was supported by simulations and steady-state PL measurements. Molas et al. observed that the H-TFSI treatment resulted in progressive quenching of defect-related emission in ME MoS2 monolayers at low temperatures, again concluding the defect-passivating effect of H-TFSI treatment.102
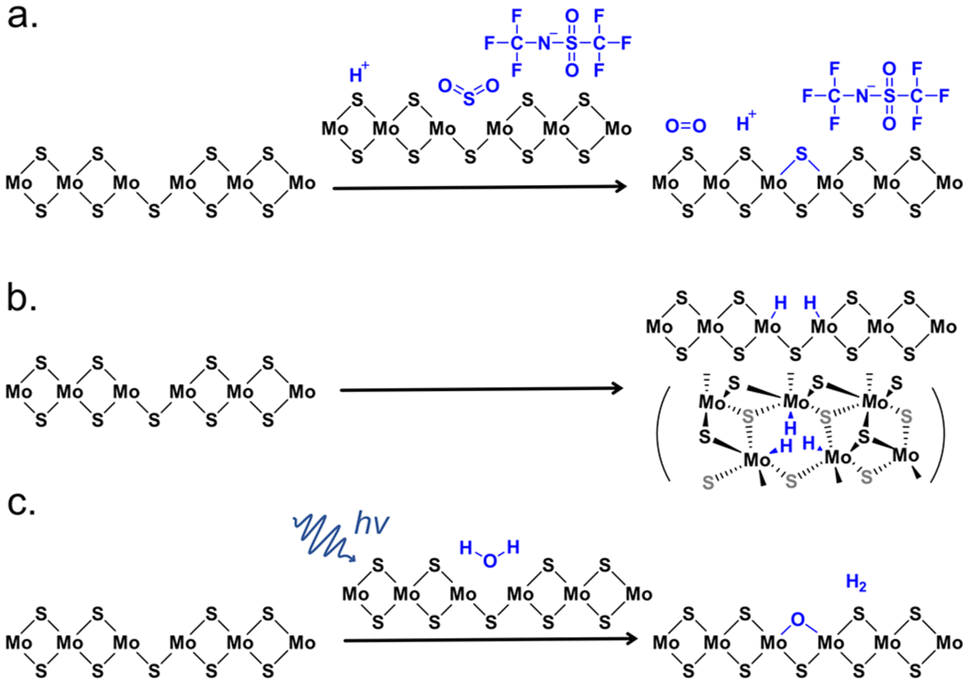 | ||
| Fig. 4 Reported mechanisms behind H-TFSI and H2O treatments for monolayer MoS2 defect passivation. (a) Illustration of the reaction between H-TFSI and monolayer MoS2 through the SV sites. (b) Illustration of the interaction between H-TFSI and monolayer MoS2 through the SV sites. (c) Illustration of photon-mediated passivation with a H2O molecule on the surface of MoS2. Adapted from ref. 121 with permission from American Chemical Society, copyright 2019. | ||
Countering the growing body of claims that H-TFSI passivates defects through some mechanisms, in 2019, Javey and co-workers showed a near-unity PL QY of pristine ME MoS2 and WS2 monolayers through electrostatic doping and revealed that the underlying mechanism of the H-TFSI treatment is p-type doping without defect passivation further justified by the TRPL measurements, where the H-TFSI treatment led to similar decay kinetics compared to electrostatic doping (Fig. 2p–t).103 Their work implied that all neutral excitons in 2D TMDSs radiatively recombined even in the presence of native defects. On the other hand, Pain and co-workers reported the PL enhancement of 2D TMDSs with superacid analogues and pointed out that acidity and the inclusion of sulfur and oxygen from H-TFSI did not necessarily play roles in defect passivation.104 Rao and co-workers observed a much longer PL lifetime (1–20 ns) upon H-TFSI treatment compared to the ME pristine MoS2 monolayer which fell below the instrument response of 100 ps, indicating a trap-mediated exciton recombination process after the H-TFSI treatment.83 In addition, the authors experimentally observed sub-gap trap sites (originating from SVs) in H-TFSI-treated MoS2, which appeared as a positive feature at 730 nm in ultrafast pump–probe spectra. This sub-gap defect bleach grew simultaneously with the initial A exciton decay, confirming a transfer in population from the A excitons to the defect states. They also reported a decrease in carrier mobility by over two orders of magnitude in H-TFSI-treated FETs compared to untreated devices. The authors concluded that even though H-TFSI treatment increased the PLQY, the SVs were still present and significantly limited the quality of the TMD material. In addition, they have conducted a two-step chemical treatment, a passivating agent (thiol, thiophene, or sulfide) followed by the H-TFSI treatment, which achieved an enhanced PL intensity and shortened emission lifetime compared to the H-TFSI-treated-only sample.83 In contrast to the H-TFSI-only treatment, the sub-gap bleach was greatly reduced in the two-step treatment, suggesting the passivation of SV sites (Fig. 2m and n). The understanding of the mechanism behind H-TFSI treatment is then the key to further designing chemical treatments to passivate the defects of 2D TMDs.
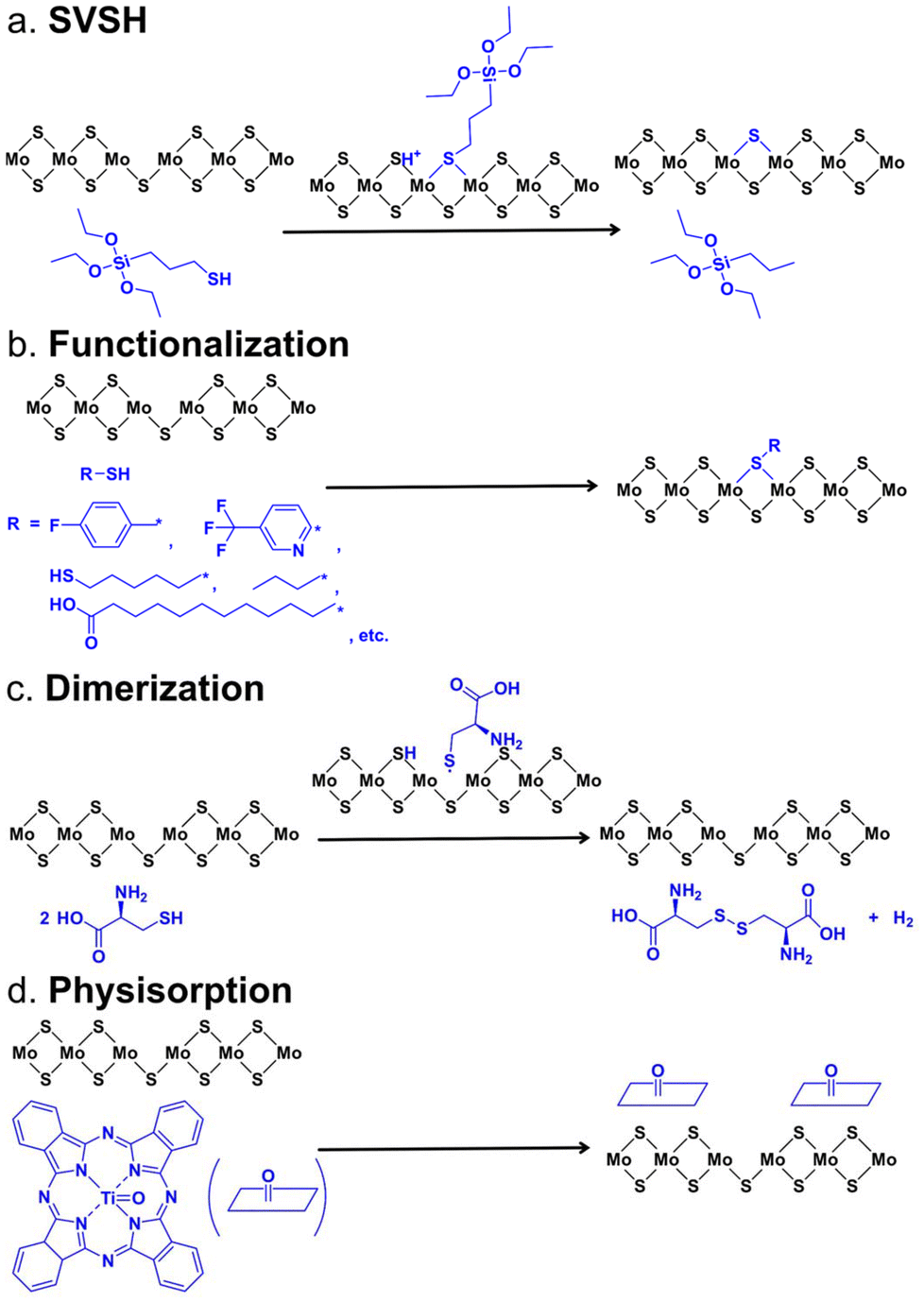 | ||
| Fig. 5 Schematics of the reaction kinetics that have been reported for passivating the SVs. (a) Reaction between the MPS molecules and monolayer MoS2 through the sulfur vacancy self-healing (SVSH) mechanism. (b) Reaction between various thiol molecules and monolayer MoS2 through the functionalization mechanism. (c) Reaction between the thiol molecule and monolayer MoS2 through the dimerization mechanism. (d) Interaction between the TiOPc molecule and monolayer MoS2 through the physisorption mechanism. | ||
The SV passivation with thiol chemistry has also been explored by multiple groups, yet both the resulting products and reaction mechanisms remain controversial. One reported mechanism of SV passivation is that thiol molecules conjugated to the TMD surface with the S–H bond cleaved rather than physisorption or chemisorption, as illustrated in Fig. 5b.73,86,108–110 This was proposed as the functionalization mechanism and was often visualized by FTIR measurements in which the S–H band from thiol molecules was found at 2563 cm−1, but absent after conjugation with MoS2. Similarly, Cho et al. reported that alkanethiol molecules passivated the SVs through chemisorption at the SV sites of few-layer MoS2, evidenced by a shift of the characteristic peak position in XPS after the treatment.111 On the other hand, McDonald and co-workers proposed that TMDs facilitated the oxidation of organic thiols to disulfides, which were physisorbed on the 2D TMD surfaces through electrostatic interactions, rather than coordinate at SVs.112 This was proposed as the dimerization mechanism. The disulfide products were evidenced by diffuse reflectance infrared Fourier transform (DRIFT) measurements. In this scenario, thiols initially donated a hydrogen atom to the TMD. The formed thiyl radicals yielded disulfides and the H[MoS2] released hydrogen gas. The proposed mechanism is illustrated in Fig. 5c. Subsequently, McDonald and co-workers quantitatively monitored the consumption of 1-octanethiol in the presence of liquid exfoliated MoS2 nanosheets using 1H nuclear magnetic resonance (NMR) spectroscopy and further confirmed the dimerization mechanism they proposed previously where MoS2 facilitated the oxidation of thiols to disulfides (Fig. 2u).113 In 2017, Wang and co-workers investigated the reaction mechanisms between the defective MoS2 monolayer and thiol molecules employing potential energy surface calculations and kinetic studies.114 They concluded that the reactions were dominated by two competing mechanisms, dimerization or SVSH, and the dominant pathway was largely determined by the polarization of thiol molecules and the temperature. It is also worth noting that the authors predicted that the Mo–H bond was formed in the dimerization mechanism which is different from the other report about the same mechanism.113,114 In 2021, Zhang et al. directly monitored the interaction between the fluorescent thiol and SVs in a metal–organic chemical vapor deposition (MOCVD)-grown MoS2 monolayer via 2D point accumulation for imaging in a nanoscale topography (PAINT) strategy (Fig. 2w) and revealed a hydroxide-assisted transition from the reversible interaction (physisorption) to covalent binding by deprotonation of the thiol while increasing the pH.115
The SVs could also be passivated by the formation of a van der Waals interface, proposed as a physisorption mechanism like the use of ML TiOPc on the MoS2 surface.71 Park et al. revealed a van der Waals interaction via scanning tunneling microscopy (STM) and DFT modeling, in which a negative charge transfer from MoS2 to TiOPc removes defect states.71 As illustrated in Fig. 5d, it was hypothesized that a thermally stable TiOPc ML was formed on the MoS2 surface, which did not induce physical reconstructions of defects. Similarly, Ahn et al. reported that MPcs passivated SVs in ME MoS2, evidenced by the weakened PL peak at 1.79 eV (associated with excitons bound to defects) after the treatment in low-temperature PL measurements.116,117
Besides organic thiol molecules and MPcs, there were other approaches reported for passivating defects in 2D TMDSs. Tapasztó and co-workers reported that the defects in MoS2 resulted from O2 oxidation, where O2 spontaneously incorporated into the basal plane of monolayer MoS2 during ambient exposure (Fig. 2v). The substitutional oxidation of MoS2 could be fully recovered to pristine via annealing the oxygen-substituted MoS2 under a H2S atmosphere at 200 °C, which was evidenced by STM images and DFT calculations.118 On the other hand, there were a few research groups that reported that the chemisorption of O2 could passivate the SVs of TMDSs and result in a defect-free electronic band structure similar to that of perfect monolayer TMDSs.64,101,119,120 Sivaram et al. proposed that H2O could passivate SVs in the CVD-grown MoS2, but the reaction required photo-generated excitons to overcome a large absorption barrier (Fig. 4c).121 The H2O molecule was physisorbed on the surface of MoS2 using an empty antibonding orbital, then the exciton-mediated dissociation of H2O resulted in the O atom bonded to the SV with a valency of −2, and the H2 molecule desorbed from the MoS2 surface.
3. Chemical passivation of MoSe2 and WSe2
3.1 Characterization
To date, few general chemical treatments enhance the quality of both sulfur-based and selenide-based 2D TMD materials, which can be attributed to their different intrinsic doping levels and defects.81,91 For instance, 2D WSe2 is known to be a p-type semiconductor while MoSe2 is known to be an n-type semiconductor due to its intrinsic defects.122,123 In 2016, Javey and co-workers reported the effects of H-TFSI treatments on the PLQY of MoS2, WS2, MoSe2, and WSe2, and suggested that only the defects in sulfur-based 2D TMD materials were amenable to the H-TFSI treatment.124 Later, they reported that ME MoS2 and WSe2 monolayers were hardly doped, and the electrostatic doping was, therefore, not able to enhance their emission.103 This work bore some differences from a related study by Yu et al., who reported a PL increase of the CVD as-grown WSe2 and MoSe2 monolayers after the H-TFSI treatment.96 On the other hand, Ahn et al. reported that both n-type dopant zinc phthalocyanine (ZnPc) and p-type dopant zinc hexadecafluoro phthalocyanine (F16ZnPc) resulted in quenching of PL from CVD-grown few-layer MoSe2.116 In 2021, Rao and co-workers demonstrated that the OA treatment on ME MoSe2 monolayers enhanced the PL yield of MoSe2 by an average of 58-fold, while also improving the spectral uniformity of brightness and reducing the emission linewidth.125 The authors revealed trap-free neutral exciton movement in OA-treated MoSe2 monolayers evidenced by steady-state excitation intensity-dependent PL and TRPL studies and thus postulated that the defect passivation scheme of the OA treatment was related to selenium vacancy passivation through oleate coordination to Mo dangling bonds without distinguishable structural changes. Recently, Deng and co-workers reported MoSe2 monolayers with lateral size >1 mm with a uniformly high PLQY using 1-dodecanol encapsulation, which both passivated the chalcogen vacancies and suppressed substrate quenching of the excitons.1263.2 Mechanisms of chemical passivation
Han et al. reported that the HBr treatment enhanced the PL intensity of CVD-grown monolayer MoSe2 more than 30 times through p-doping and defect healing.127 The p-doping effect was validated by the intensity increase and frequency upshift of A1g mode in Raman spectroscopy. Undesired oxidized Se4+ and bridging Se22− defects were removed, which was visualized by the peak shift and full width at half-maximum (FWHM) decrease of Mo(IV) 3d, as well as the elemental ratio increase of the anion to Mo in XPS spectra. The possible mechanism of HBr treatment proposed by the authors is illustrated in Fig. 6.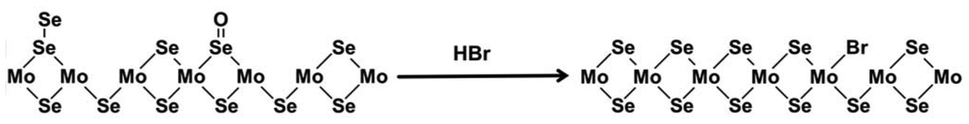 | ||
| Fig. 6 Illustration of the chemical reaction on the surface of MoSe2 during the HBr treatment. This figure has been adapted from ref. 127 with permission from American Chemical Society, copyright 2015. | ||
A few other molecules and treatment protocols have also been reported for passivating defects. Guo et al. proposed that the Se vacancies in MoSe2 could be well passivated with halogen atoms (except F) using first-principles calculation, through a p-doping process.128 Mahjouri-Samani et al. reported that the vaporization of selenium in a vacuum using a pulsed laser repaired Se vacancies in the synthesized MoSe2.129 Lu et al. demonstrated the passivation of Se vacancies by oxygen through a focused laser treatment in air on CVD-grown WSe2, which was verified by an enhancement in the PL intensity, an improvement of the photoconductivity (∼150 times), an increase of the W oxidation ratio in XPS, as well as an increase in thickness in AFM images.130 Ahn et al. reported that metallophthalocyanines (MPcs) exhibited defect-healing effects on the surface of the CVD-grown MoSe2 monolayer, evidenced by the temperature-dependent blueshift of the band gap, the narrower PL bandwidth, and the suppression of mid-gap defect-induced absorption in ultrafast pump–probe spectroscopy.116
Moreover, Javey and co-workers reported a PLQY of ∼60% in CVD-grown WSe2 monolayers after undergoing a solvent evaporation-mediated decoupling (SEMD) process, which was also higher than that in ME WSe2 monolayers by an order of magnitude.131 They attributed the enhanced PLQY to reduced nonradiative recombination due to the release of built-in strain by decoupling the grown WSe2 monolayer during the SEMD process validated by electron diffraction, in situ PL imaging, TEM, and TRPL measurements. Similarly, Chen et al. demonstrated that the PLQYs of both MoS2 and MoSe2 were enhanced by the solvent with a moderate volatilization rate like ethanol.132
4. Conclusions and perspectives
This mini-review provides an overview of the state-of-the-art chemical treatments and related mechanisms on TMDs. The low quality of 2D TMD materials has been a veritable bottleneck to the incorporation of 2D TMDs in practical optoelectronic devices. Thus, we focus more on strategies to improve the intrinsic quality of TMDs by passivating atomic defects or reducing their inherent doping with surface chemical treatments and discuss the discrepancies of reported related mechanisms. The photoluminescence of monolayer TMDs and the mobility of FET devices built on 2D TMDs are utilized as the two most important quality factors to evaluate the effect of chemical treatments. The PL and mobility of resulting FET devices have been hugely improved with varied surface chemical treatments in most cases. The mechanisms behind the chemical treatments have also been theoretically and experimentally explored for further development of chemical treatment approaches. We have thus tried to present the research to date on chemical treatments and the mechanisms behind them in this mini-review, and in some cases, the literature is converging on a unified picture. However, disagreement and discrepancies across different treatments and mechanisms remain. This must be resolved to move forward. A summary of the reported data for the effect of chemical treatments on the PL of TMDs can be found in Table 1. Since it is technically very difficult to measure the PLQY from 100 μm2 monolayer flakes, the shape and position of the steady-state PL spectra also serve as a quality indicator of 2D TMDs both the PL intensity and PL shape of the same monolayer flakes before and after chemical treatment should be measured.133 Moreover, as previously described, some of the discrepancies in disclosed mechanisms may be ascribed to the lack of comparison of various chemical treatments on TMD monolayers with the same synthesis method and under the same measurement conditions, as well as to the precision limit in characterization tools. Therefore, more attention should be paid to the synthesis of materials, and the experiments utilized for characterization should be carefully chosen and scrutinized to determine what can provide the most useful information for determining mechanisms. For instance, electrical transport measurements were utilized to support the defects passivation mechanism with enhanced mobility after chemical treatments, whereas the resulting mobilities were often limited by the contact between the monolayers and the electrodes, and chemicals like H-TFSI could corrode the contact, which led to an unfair comparison among various chemicals. From our perspective, an essential tool to characterize the effect of chemical treatments on 2D TMD materials is ultrafast spectroscopy, which can reveal the carrier dynamics associated with defects without the additional complications of contacts and electrodes. However, this should not be performed in isolation, as the variation of exciton dynamics associated with defects and doping renders multiple processes with a variety of time scales that can also overlap.134 Advanced microscopy techniques such as cavity-enhanced extinction microscopy and single-molecule localization microscopy coupled with fluorescence labelling are reported to give new insights into the defects’ properties of 2D materials.115,135 Therefore, we contend that the most robust approach is to combine various experimental approaches to construct a hypothesis.| Material (monolayer) | Exfoliation/synthesis method | Untreated | Treated | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PL peak (eV) | QY (%) | PL lifetime (ns) | Chemical treatment | PL peak (eV) | I | QY (%) | PL lifetime (ns) | |||
| a PL intensity enhancement factor (times). | ||||||||||
| MoS2 | ME | 1.85 | — | — | F4TCNQ | 1.88 | 10 | — | — | 59 and 60 |
| 1.87 | — | — | H2O2 | 1.89 | 27.4 | 12 | — | 61 | ||
| 1.84 | — | — | O2, H2O (after annealing) | 1.88 | 100 | — | — | 62 | ||
| 1.79 | — | — | O2 (after annealing) | 1.83 | 30 | — | — | 64 | ||
| 1.88 | 1 | 0.3 | H-TFSI | 1.88 | 190 | 95 | 10.8 | 67 | ||
| 1.88 | 0.1–1 | — | CYTOP + H-TFSI | 1.88 | >100 | 55–100 | 15 | 68 | ||
| 1.88 | — | <0.1 | Thiols + H-TFSI | 1.88 | 275 | — | 2.5 | 83 | ||
| 1.86 | — | — | MB | 1.88 | 2 | — | — | 91 | ||
| 1.87 | — | <0.1 | Li-TFSI | 1.88 | 50 | — | 0.32 | 94 | ||
| ∼1.89 | — | — | H2SO4/H2O2 | ∼1.91 | 20 | — | — | 101 | ||
| CVD | 1.89 | — | — | IL | 1.89 | 100 | — | — | 70 | |
| 1.84 | — | — | TiOPc | 1.85 | 3 | — | — | 71 | ||
| 1.86 | — | — | PEDOT:PSS | 1.88 | 2 | — | — | 72 | ||
| 1.87 | — | 0.11 | Methanol | 1.86 | 2.2 | — | 0.15 | 81 | ||
| ∼1.85 | — | — | H-TFSI/Li-TFSI | ∼1.91 | >80 | — | — | 96 | ||
| 1.89 | 0.1 | — | H-TFSI | 1.90 | 10 | 1.5–15 | — | 98 | ||
| 1.85 | — | — | H2O + hv | 1.85 | 200 | — | — | 121 | ||
| CVD/ME | ∼1.81 | — | — | Thiols | ∼1.82 | ∼2 | — | — | 73 | |
| BE + LPE | — | <1 | 6.08 | THF + heating | ∼2.66 | 64 | 18.5 | 4.67 | 75 | |
| WS2 | ME | 1.96 | — | — | F4TCNQ | 2.02 | 3 | — | — | 63 |
| 2.01 | — | 0.06 | OA | 2.02 | 40 | — | 0.25 | 82 | ||
| CVD | 2.01 | — | — | Na2S | 1.96 | 25 | — | — | 74 | |
| ∼1.88 | — | — | H-TFSI/Li-TFSI | ∼2.0 | >60 | — | — | 96 | ||
| 1.94 | — | — | H-TFSI | 2.01 | 25 | — | — | 97 | ||
| MoSe2 | CVD | ∼1.57 | — | — | H-TFSI/Li-TFSI | ∼1.58 | >10 | — | — | 96 |
| 1.53 | — | — | HBr | 1.55 | 30 | — | — | 127 | ||
| 1.53 | 2.5 | 0.48 | Ethanol | 1.55 | 3.5 | 16 | 1.67 | 132 | ||
| ME | 1.56 | — | 1.07 | OA | 1.58 | 61 | — | 3.3 | 125 | |
| WSe2 | CVD | 1.56 | 1 | 1 | Acetone | 1.65 | ∼15 | 60 | 4.1 | 131 |
Other challenges must be addressed to take full advantage of 2D TMDs in practical optoelectronic applications. For instance, another area we identify for specific future work and possible commercialization is the improvement of liquid exfoliation (LE) of 2D TMDs.85,136 Although the LE methodology and the quality of liquid-exfoliated TMD nanosheets have been developed and improved constantly, the PLQY of liquid-exfoliated TMD nanosheets remains an outstanding challenge, which limits the scalability of device application to a large extent.44,137–140 Chemical treatment development towards being compatible with the liquid exfoliation process could be extremely fruitful for possible applications in flexible optoelectronics.
Author contributions
Z. Li conceived the idea and wrote the paper with input from H. Bretscher and A. Rao. All authors contributed to the organization of the content.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. is grateful for the invitation to this contribution from the editor office. We acknowledge funding from the Swedish Research Council, Vetenskapsrådet 2018-06610, and ÅForsk Foundation nr. 22-390.References
- K. S. Novoselov, V. I. Fal'Ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- C. Tan, X. Cao, X. J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- T. D. Thanh, N. D. Chuong, H. Van Hien, T. Kshetri, L. H. Tuan, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2018, 96, 51–85 CrossRef CAS.
- X. Li, L. Tao, Z. Chen, H. Fang, X. Li, X. Wang, J. Bin Xu and H. Zhu, Appl. Phys. Rev., 2017, 4, 021306 Search PubMed.
- A. Hirsch and F. Hauke, Angew. Chem., Int. Ed., 2018, 57, 4338–4354 CrossRef CAS PubMed.
- F. Yan, Z. Wei, X. Wei, Q. Lv, W. Zhu and K. Wang, Small Methods, 2018, 2, 1–14 Search PubMed.
- K. F. Mak and J. Shan, Nat. Photonics, 2016, 10, 216–226 CrossRef CAS.
- A. Krasnok, S. Lepeshov and A. Alú, Opt. Express, 2018, 26, 15972 CrossRef CAS PubMed.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, ACS Nano, 2014, 8, 1102–1120 CrossRef CAS PubMed.
- W. Zheng, Y. Jiang, X. Hu, H. Li, Z. Zeng, X. Wang and A. Pan, Adv. Opt. Mater., 2018, 6, 1–29 Search PubMed.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 2–5 CrossRef PubMed.
- J. Shim, H. Y. Park, D. H. Kang, J. O. Kim, S. H. Jo, Y. Park and J. H. Park, Adv. Electron. Mater., 2017, 3, 1600364 CrossRef.
- J. Kim, S. Zhang, S. Hou, B. Lee, G. Wei and S. R. Forrest, ACS Photonics, 2021, 8, 1152–1158 CrossRef CAS.
- J. W. T. Seo, J. Zhu, V. K. Sangwan, E. B. Secor, S. G. Wallace and M. C. Hersam, ACS Appl. Mater. Interfaces, 2019, 11, 5675–5681 CrossRef CAS PubMed.
- Y. Cui, Z. Zhou, T. Li, K. Wang, J. Li and Z. Wei, Adv. Funct. Mater., 2019, 29, 1–33 Search PubMed.
- Y. Liu, X. Duan, H. J. Shin, S. Park, Y. Huang and X. Duan, Nature, 2021, 591, 43–53 CrossRef CAS PubMed.
- Z. Cheng, R. Cao, K. Wei, Y. Yao, X. Liu, J. Kang, J. Dong, Z. Shi, H. Zhang and X. Zhang, Adv. Sci., 2021, 8, 1–22 CAS.
- K. Nassiri Nazif, A. Daus, J. Hong, N. Lee, S. Vaziri, A. Kumar, F. Nitta, M. E. Chen, S. Kananian, R. Islam, K.-H. Kim, J.-H. Park, A. S. Y. Poon, M. L. Brongersma, E. Pop and K. C. Saraswat, Nat. Commun., 2021, 12, 7034 CrossRef CAS PubMed.
- D. Rhodes, S. H. Chae, R. Ribeiro-Palau and J. Hone, Nat. Mater., 2019, 18, 541–549 CrossRef CAS PubMed.
- A. Chernikov, T. C. Berkelbach, H. M. Hill, A. Rigosi, Y. Li, O. B. Aslan, D. R. Reichman, M. S. Hybertsen and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 1–5 CrossRef PubMed.
- Y. You, X. X. Zhang, T. C. Berkelbach, M. S. Hybertsen, D. R. Reichman and T. F. Heinz, Nat. Phys., 2015, 11, 477–481 Search PubMed.
- Z. Ye, T. Cao, K. O'Brien, H. Zhu, X. Yin, Y. Wang, S. G. Louie and X. Zhang, Nature, 2014, 513, 214–218 CrossRef CAS PubMed.
- M. M. Ugeda, A. J. Bradley, S. F. Shi, F. H. Da Jornada, Y. Zhang, D. Y. Qiu, W. Ruan, S. K. Mo, Z. Hussain, Z. X. Shen, F. Wang, S. G. Louie and M. F. Crommie, Nat. Mater., 2014, 13, 1091–1095 CrossRef CAS PubMed.
- K. F. Mak, K. He, C. Lee, G. H. Lee, J. Hone, T. F. Heinz and J. Shan, Nat. Mater., 2013, 12, 207–211 CrossRef CAS PubMed.
- J. Pető, T. Ollár, P. Vancsó, Z. I. Popov, G. Z. Magda, G. Dobrik, C. Hwang, P. B. Sorokin and L. Tapasztó, Nat. Chem., 2018, 10, 1246–1251 CrossRef PubMed.
- N. Kang, H. P. Paudel, M. N. Leuenberger, L. Tetard and S. I. Khondaker, J. Phys. Chem. C, 2014, 118, 21258–21263 CrossRef CAS.
- B. G. Shin, G. H. Han, S. J. Yun, H. M. Oh, J. J. Bae, Y. J. Song, C. Y. Park and Y. H. Lee, Adv. Mater., 2016, 28, 9378–9384 CrossRef CAS PubMed.
- X. Zou and B. I. Yakobson, Acc. Chem. Res., 2015, 48, 73–80 CrossRef CAS PubMed.
- S. Haldar, H. Vovusha, M. K. Yadav, O. Eriksson and B. Sanyal, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 1–12 CrossRef.
- H. Liu, N. Han and J. Zhao, RSC Adv., 2015, 5, 17572–17581 RSC.
- J. A. Robinson and B. Schuler, Appl. Phys. Lett., 2021, 119, 140501 CrossRef CAS.
- S. M. Hus and A. P. Li, Prog. Surf. Sci., 2017, 92, 176–201 CrossRef CAS.
- S. Y. Seo, D. H. Yang, G. Moon, O. F. N. Okello, M. Y. Park, S. H. Lee, S. Y. Choi and M. H. Jo, Nano Lett., 2021, 21, 3341–3354 CrossRef CAS PubMed.
- Q. Zhang, A. T. S. Wee, Q. Liang, X. Zhao and M. Liu, ACS Nano, 2021, 15, 2165–2181 CrossRef PubMed.
- Y. Yu, Y. Yu, C. Xu, Y. Q. Cai, L. Su, Y. Zhang, Y. W. Zhang, K. Gundogdu and L. Cao, Adv. Funct. Mater., 2016, 26, 4733–4739 CrossRef CAS.
- O. A. Ajayi, J. V. Ardelean, G. D. Shepard, J. Wang, A. Antony, T. Taniguchi, K. Watanabe, T. F. Heinz, S. Strauf, X. Y. Zhu and J. C. Hone, 2D Mater., 2016, 26, 4733–4739 CrossRef.
- F. Cadiz, E. Courtade, C. Robert, G. Wang, Y. Shen, H. Cai, T. Taniguchi, K. Watanabe, H. Carrere, D. Lagarde, M. Manca, T. Amand, P. Renucci, S. Tongay, X. Marie and B. Urbaszek, Phys. Rev. X, 2017, 7, 1–12 Search PubMed.
- Z. Liu, M. Amani, S. Najmaei, Q. Xu, X. Zou, W. Zhou, T. Yu, C. Qiu, A. G. Birdwell, F. J. Crowne, R. Vajtai, B. I. Yakobson, Z. Xia, M. Dubey, P. M. Ajayan and J. Lou, Nat. Commun., 2014, 5, 5246 CrossRef PubMed.
- Z. Li, Y. Lv, L. Ren, J. Li, L. Kong, Y. Zeng, Q. Tao, R. Wu, H. Ma, B. Zhao, D. Wang, W. Dang, K. Chen, L. Liao, X. Duan, X. Duan and Y. Liu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- H. H. Huang, X. Fan, D. J. Singh and W. T. Zheng, Nanoscale, 2020, 12, 1247–1268 RSC.
- R. Wang, Y. Yu, S. Zhou, H. Li, H. Wong, Z. Luo, L. Gan and T. Zhai, Adv. Funct. Mater., 2018, 28, 1802473 CrossRef.
- S. Bertolazzi, M. Gobbi, Y. Zhao, C. Backes and P. Samorì, Chem. Soc. Rev., 2018, 47, 6845–6888 RSC.
- K. Cho, J. Pak, S. Chung and T. Lee, ACS Nano, 2019, 13, 9713–9734 CrossRef CAS PubMed.
- S. Ippolito, A. Ciesielski and P. Samorì, Chem. Commun., 2019, 55, 8900–8914 RSC.
- L. Huang, A. Krasnok, A. Alu, Y. Yu, D. Neshev and A. E. Miroshnichenko, Rep. Prog. Phys., 2022, 85, 046401 CrossRef CAS PubMed.
- X. Dai, Z. Yang, A. Li, J. Yang and F. Ouyang, Superlattices Microstruct., 2019, 130, 528–538 CrossRef CAS.
- J. Zhou, F. Liu, J. Lin, X. Huang, J. Xia, B. Zhang, Q. Zeng, H. Wang, C. Zhu, L. Niu, X. Wang, W. Fu, P. Yu, T. R. Chang, C. H. Hsu, D. Wu, H. T. Jeng, Y. Huang, H. Lin, Z. Shen, C. Yang, L. Lu, K. Suenaga, W. Zhou, S. T. Pantelides, G. Liu and Z. Liu, Adv. Mater., 2017, 29, 1603471 CrossRef PubMed.
- D. Qu, X. Liu, M. Huang, C. Lee, F. Ahmed, H. Kim, R. S. Ruoff, J. Hone and W. J. Yoo, Adv. Mater., 2017, 29, 1–11 Search PubMed.
- J. Hong, Z. Hu, M. Probert, K. Li, D. Lv, X. Yang, L. Gu, N. Mao, Q. Feng, L. Xie, J. Zhang, D. Wu, Z. Zhang, C. Jin, W. Ji, X. Zhang, J. Yuan and Z. Zhang, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- X. Zou, Y. Liu and B. I. Yakobson, Nano Lett., 2013, 13, 253–258 CrossRef CAS PubMed.
- S. Najmaei, Z. Liu, W. Zhou, X. Zou, G. Shi, S. Lei, B. I. Yakobson, J. C. Idrobo, P. M. Ajayan and J. Lou, Nat. Mater., 2013, 12, 754–759 CrossRef CAS PubMed.
- V. Carozo, Y. Wang, K. Fujisawa, B. R. Carvalho, A. McCreary, S. Feng, Z. Lin, C. Zhou, N. Perea-López, A. L. Elías, B. Kabius, V. H. Crespi and M. Terrones, Sci. Adv., 2017, 3, 1–10 Search PubMed.
- W. Bao, N. J. Borys, C. Ko, J. Suh, W. Fan, A. Thron, Y. Zhang, A. Buyanin, J. Zhang, S. Cabrini, P. D. Ashby, A. Weber-Bargioni, S. Tongay, S. Aloni, D. F. Ogletree, J. Wu, M. B. Salmeron and P. J. Schuck, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- W. Su, L. Jin, X. Qu, D. Huo and L. Yang, Phys. Chem. Chem. Phys., 2016, 18, 14001–14006 RSC.
- S. Barja, S. Refaely-Abramson, B. Schuler, D. Y. Qiu, A. Pulkin, S. Wickenburg, H. Ryu, M. M. Ugeda, C. Kastl, C. Chen, C. Hwang, A. Schwartzberg, S. Aloni, S. K. Mo, D. Frank Ogletree, M. F. Crommie, O. V. Yazyev, S. G. Louie, J. B. Neaton and A. Weber-Bargioni, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- J. D. Lin, C. Han, F. Wang, R. Wang, D. Xiang, S. Qin, X. A. Zhang, L. Wang, H. Zhang, A. T. S. Wee and W. Chen, ACS Nano, 2014, 8, 5323–5329 CrossRef CAS PubMed.
- M. Tebyetekerwa, J. Zhang, Z. Xu, T. N. Truong, Z. Yin, Y. Lu, S. Ramakrishna, D. Macdonald and H. T. Nguyen, ACS Nano, 2020, 14, 14579–14604 CrossRef CAS PubMed.
- Y. Zhu, J. Lim, Z. Zhang, Y. Wang, S. Sarkar, H. Ramsden, Y. Li, H. Yan, D. Phuyal, N. Gauriot, A. Rao, R. L. Z. Hoye, G. Eda and M. Chhowalla, ACS Nano, 2023, 17, 13545–13553 CrossRef CAS PubMed.
- S. Mouri, Y. Miyauchi and K. Matsuda, Nano Lett., 2013, 13, 5944–5948 CrossRef CAS PubMed.
- S. Mouri, Y. Miyauchi and K. Matsuda, Appl. Phys. Express, 2016, 9, 1–4 Search PubMed.
- W. Su, H. Dou, J. Li, D. Huo, N. Dai and L. Yang, RSC Adv., 2015, 5, 82924–82929 RSC.
- S. Tongay, J. Zhou, C. Ataca, J. Liu, J. S. Kang, T. S. Matthews, L. You, J. Li, J. C. Grossman and J. Wu, Nano Lett., 2013, 13, 2831–2836 CrossRef CAS PubMed.
- N. Peimyoo, W. Yang, J. Shang, X. Shen, Y. Wang and T. Yu, ACS Nano, 2014, 8, 11320–11329 CrossRef CAS PubMed.
- H. Nan, Z. Wang, W. Wang, Z. Liang, Y. Lu, Q. Chen, D. He, P. Tan, F. Miao, X. Wang, J. Wang and Z. Ni, ACS Nano, 2014, 8, 5738–5745 CrossRef CAS PubMed.
- L. Sun, X. Zhang, F. Liu, Y. Shen, X. Fan, S. Zheng, J. T. L. Thong, Z. Liu, S. A. Yang and H. Y. Yang, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- L. Xu, L. Zhao, Y. Wang, M. Zou, Q. Zhang and A. Cao, Nano Res., 2019, 12, 1619–1624 CrossRef CAS.
- M. Amani, D. H. Lien, D. Kiriya, J. Xiao, A. Azcatl, J. Noh, S. R. Madhvapathy, R. Addou, K. C. Santosh, M. Dubey, K. Cho, R. M. Wallace, S. C. Lee, J. H. He, J. W. Ager, X. Zhang, E. Yablonovitch and A. Javey, Science, 2015, 350, 1065–1068 CrossRef CAS PubMed.
- H. Kim, D. H. Lien, M. Amani, J. W. Ager and A. Javey, ACS Nano, 2017, 11, 5179–5185 CrossRef CAS PubMed.
- A. J. Goodman, A. P. Willard and W. A. Tisdale, Phys. Rev. B, 2017, 96, 1–6 CrossRef.
- T. L. Atallah, J. Wang, M. Bosch, D. Seo, R. A. Burke, O. Moneer, J. Zhu, M. Theibault, L. E. Brus, J. Hone and X. Y. Zhu, J. Phys. Chem. Lett., 2017, 8, 2148–2152 CrossRef CAS PubMed.
- J. H. Park, A. Sanne, Y. Guo, M. Amani, K. Zhang, H. C. P. Movva, J. A. Robinson, A. Javey, J. Robertson, S. K. Banerjee and A. C. Kummel, Sci. Adv., 2017, 3, 1–7 Search PubMed.
- X. Zhang, Q. Liao, S. Liu, Z. Kang, Z. Zhang, J. Du, F. Li, S. Zhang, J. Xiao, B. Liu, Y. Ou, X. Liu, L. Gu and Y. Zhang, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- Q. Ding, K. J. Czech, Y. Zhao, J. Zhai, R. J. Hamers, J. C. Wright and S. Jin, ACS Appl. Mater. Interfaces, 2017, 9, 12734–12742 CrossRef CAS PubMed.
- H. Yao, L. Liu, Z. Wang, H. Li, L. Chen, M. E. Pam, W. Chen, H. Y. Yang, W. Zhang and Y. Shi, Nanoscale, 2018, 10, 6105–6112 RSC.
- Z. Li, Z. Chen, L. Xiao, X. Zhou, C. Zhao and Y. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 15487–15495 CrossRef CAS PubMed.
- M. Y. Chan, K. Komatsu, S. L. Li, Y. Xu, P. Darmawan, H. Kuramochi, S. Nakaharai, A. Aparecido-Ferreira, K. Watanabe, T. Taniguchi and K. Tsukagoshi, Nanoscale, 2013, 5, 9572–9576 RSC.
- S. Najmaei, X. Zou, D. Er, J. Li, Z. Jin, W. Gao, Q. Zhang, S. Park, L. Ge, S. Lei, J. Kono, V. B. Shenoy, B. I. Yakobson, A. George, P. M. Ajayan and J. Lou, Nano Lett., 2014, 14, 1354–1361 CrossRef CAS PubMed.
- D. Lim, E. S. Kannan, I. Lee, S. Rathi, L. Li, Y. Lee, M. A. Khan, M. Kang, J. Park and G. H. Kim, Nanotechnology, 2016, 27, 225201 CrossRef PubMed.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- Z. Yu, Y. Pan, Y. Shen, Z. Wang, Z. Y. Ong, T. Xu, R. Xin, L. Pan, B. Wang, L. Sun, J. Wang, G. Zhang, Y. W. Zhang, Y. Shi and X. Wang, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- G. P. Neupane, M. D. Tran, S. J. Yun, H. Kim, C. Seo, J. Lee, G. H. Han, A. K. Sood and J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 11950–11958 CrossRef CAS PubMed.
- A. O. A. Tanoh, J. Alexander-Webber, J. Xiao, G. Delport, C. A. Williams, H. Bretscher, N. Gauriot, J. Allardice, R. Pandya, Y. Fan, Z. Li, S. Vignolini, S. D. Stranks, S. Hofmann and A. Rao, Nano Lett., 2019, 19, 6299–6307 CrossRef CAS PubMed.
- H. Bretscher, Z. Li, J. Xiao, D. Y. Qiu, S. Refaely-Abramson, J. A. Alexander-Webber, A. Tanoh, Y. Fan, G. Delport, C. A. Williams, S. D. Stranks, S. Hofmann, J. B. Neaton, S. G. Louie and A. Rao, ACS Nano, 2021, 15, 8780–8789 CrossRef CAS PubMed.
- S. Ippolito, A. G. Kelly, R. Furlan de Oliveira, M. A. Stoeckel, D. Iglesias, A. Roy, C. Downing, Z. Bian, L. Lombardi, Y. A. Samad, V. Nicolosi, A. C. Ferrari, J. N. Coleman and P. Samorì, Nat. Nanotechnol., 2021, 16, 592–598 CrossRef CAS PubMed.
- S. Ippolito, F. Urban, W. Zheng, O. Mazzarisi, C. Valentini, A. G. Kelly, S. M. Gali, M. Bonn, D. Beljonne, F. Corberi, J. N. Coleman, H. I. Wang and P. Samorì, Adv. Mater., 2023, 2211157 CrossRef CAS PubMed.
- E. P. Nguyen, B. J. Carey, J. Z. Ou, J. Van Embden, E. Della Gaspera, A. F. Chrimes, M. J. S. Spencer, S. Zhuiykov, K. Kalantar-Zadeh and T. Daeneke, Adv. Mater., 2015, 27, 6225–6229 CrossRef CAS PubMed.
- D. Pierucci, H. Henck, Z. Ben Aziza, C. H. Naylor, A. Balan, J. E. Rault, M. G. Silly, Y. J. Dappe, F. Bertran, P. Le Fèvre, F. Sirotti, A. T. C. Johnson and A. Ouerghi, ACS Nano, 2017, 11, 1755–1761 CrossRef CAS PubMed.
- D. M. Sim, M. Kim, S. Yim, M. J. Choi, J. Choi, S. Yoo and Y. S. Jung, ACS Nano, 2015, 9, 12115–12123 CrossRef CAS PubMed.
- S. H. Amsterdam, T. K. Stanev, L. Wang, Q. Zhou, S. Irgen-Gioro, S. Padgaonkar, A. A. Murthy, V. K. Sangwan, V. P. Dravid, E. A. Weiss, P. Darancet, M. K. Y. Chan, M. C. Hersam, N. P. Stern and T. J. Marks, J. Am. Chem. Soc., 2021, 143, 17153–17161 CrossRef CAS PubMed.
- A. Tarasov, S. Zhang, M. Y. Tsai, P. M. Campbell, S. Graham, S. Barlow, S. R. Marder and E. M. Vogel, Adv. Mater., 2015, 27, 1175–1181 CrossRef CAS PubMed.
- S. Zhang, H. M. Hill, K. Moudgil, C. A. Richter, A. R. Hight Walker, S. Barlow, S. R. Marder, C. A. Hacker and S. J. Pookpanratana, Adv. Mater., 2015, 27, 02991 Search PubMed.
- B. Birmingham, J. Yuan, M. Filez, D. Fu, J. Hu, J. Lou, M. O. Scully, B. M. Weckhuysen and Z. Zhang, J. Phys. Chem. C, 2019, 123, 15738–15743 CrossRef CAS.
- Y. Wang, A. Slassi, M. A. Stoeckel, S. Bertolazzi, J. Cornil, D. Beljonne and P. Samorì, J. Phys. Chem. Lett., 2019, 10, 540–547 CrossRef CAS PubMed.
- Z. Li, H. Bretscher, Y. Zhang, G. Delport, J. Xiao, A. Lee, S. D. Stranks and A. Rao, Nat. Commun., 2021, 12, 6044 CrossRef CAS PubMed.
- Z. Li, D. Li, H. Wang, X. Xu, L. Pi, P. Chen, T. Zhai and X. Zhou, ACS Nano, 2022, 16, 4884–4891 CrossRef CAS PubMed.
- Y. Yu, G. Li, L. Huang, A. Barrette, Y. Q. Cai, Y. Yu, K. Gundogdu, Y. W. Zhang and L. Cao, ACS Nano, 2017, 11, 9390–9396 CrossRef CAS PubMed.
- K. P. Dhakal, S. Roy, S. J. Yun, G. Ghimire, C. Seo and J. Kim, J. Mater. Chem. C, 2017, 5, 6820–6827 RSC.
- S. Roy, W. Choi, S. Jeon, D. H. Kim, H. Kim, S. J. Yun, Y. Lee, J. Lee, Y. M. Kim and J. Kim, Nano Lett., 2018, 18, 4523–4530 CrossRef CAS PubMed.
- D. Kiriya, Y. Hijikata, J. Pirillo, R. Kitaura, A. Murai, A. Ashida, T. Yoshimura and N. Fujimura, Langmuir, 2018, 34, 10243–10249 CrossRef CAS PubMed.
- H. Lu, A. Kummel and J. Robertson, APL Mater., 2018, 6, 066104 CrossRef.
- C. Schwermann, T. Stiehm, P. Tonndorf, R. Schneider, R. Schmidt, J. Kern, S. Michaelis De Vasconcellos, R. Bratschitsch and N. L. Doltsinis, Phys. Chem. Chem. Phys., 2018, 20, 16918–16923 RSC.
- M. R. Molas, K. Gołasa, Ł. Bala, K. Nogajewski, M. Bartos, M. Potemski and A. Babiński, Sci. Rep., 2019, 9, 1–7 CrossRef CAS PubMed.
- D. H. Lien, S. Z. Uddin, M. Yeh, M. Amani, H. Kim, J. W. Ager, E. Yablonovitch and A. Javey, Science, 2019, 364, 468–471 CrossRef CAS PubMed.
- S. L. Pain, N. E. Grant and J. D. Murphy, ACS Nano, 2022, 16, 1260–1270 CrossRef CAS PubMed.
- C. G. Wiegenstein and K. H. Schulz, J. Phys. Chem. B, 1999, 103, 6913–6918 CrossRef CAS.
- M. Makarova, Y. Okawa and M. Aono, J. Phys. Chem. C, 2012, 116, 22411–22416 CrossRef CAS.
- J. Y. Noh, H. Kim and Y. S. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 1–12 CrossRef.
- S. S. Chou, M. De, J. Kim, S. Byun, C. Dykstra, J. Yu, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2013, 135, 4584–4587 CrossRef CAS PubMed.
- J. S. Kim, H. W. Yoo, H. O. Choi and H. T. Jung, Nano Lett., 2014, 14, 5941–5947 CrossRef CAS PubMed.
- S. Karunakaran, S. Pandit, B. Basu and M. De, J. Am. Chem. Soc., 2018, 140, 12634–12644 CrossRef CAS PubMed.
- K. Cho, M. Min, T. Y. Kim, H. Jeong, J. Pak, J. K. Kim, J. Jang, S. J. Yun, Y. H. Lee, W. K. Hong and T. Lee, ACS Nano, 2015, 9, 8044–8053 CrossRef CAS PubMed.
- X. Chen, N. C. Berner, C. Backes, G. S. Duesberg and A. R. McDonald, Angew. Chem., Int. Ed., 2016, 55, 5803–5808 CrossRef CAS PubMed.
- X. Chen, C. McGlynn and A. R. McDonald, Chem. Mater., 2018, 30, 6978–6982 CrossRef CAS.
- Q. Li, Y. Zhao, C. Ling, S. Yuan, Q. Chen and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 10501–10505 CrossRef CAS PubMed.
- M. Zhang, M. Lihter, T. H. Chen, M. Macha, A. Rayabharam, K. Banjac, Y. Zhao, Z. Wang, J. Zhang, J. Comtet, N. R. Aluru, M. Lingenfelder, A. Kis and A. Radenovic, ACS Nano, 2021, 15, 7168–7178 CrossRef CAS PubMed.
- H. Ahn, Y. C. Huang, C. W. Lin, Y. L. Chiu, E. C. Lin, Y. Y. Lai and Y. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 29145–29152 CrossRef CAS PubMed.
- S. Tongay, J. Suh, C. Ataca, W. Fan, A. Luce, J. S. Kang, J. Liu, C. Ko, R. Raghunathanan, J. Zhou, F. Ogletree, J. Li, J. C. Grossman and J. Wu, Sci. Rep., 2013, 3, 1–5 Search PubMed.
- J. Pető, T. Ollár, P. Vancsó, Z. I. Popov, G. Z. Magda, G. Dobrik, C. Hwang, P. B. Sorokin and L. Tapasztó, Nat. Chem., 2018, 10, 1246–1251 CrossRef PubMed.
- B. Akdim, R. Pachter and S. Mou, Nanotechnology, 2016, 27, 185701 CrossRef PubMed.
- Y. Liu, P. Stradins and S. Wei, Angew. Chem., 2016, 128, 977–980 CrossRef.
- S. V. Sivaram, A. T. Hanbicki, M. R. Rosenberger, G. G. Jernigan, H. J. Chuang, K. M. McCreary and B. T. Jonker, ACS Appl. Mater. Interfaces, 2019, 11, 16147–16155 CrossRef CAS PubMed.
- M. Y. Tsai, S. Zhang, P. M. Campbell, R. R. Dasari, X. Ba, A. Tarasov, S. Graham, S. Barlow, S. R. Marder and E. M. Vogel, Chem. Mater., 2017, 29, 7296–7304 CrossRef CAS.
- L. C. Upadhyayula, J. J. Loferski, A. Wold, W. Giriat and R. Kershaw, J. Appl. Phys., 1968, 39, 4736–4740 CrossRef CAS.
- M. Amani, P. Taheri, R. Addou, G. H. Ahn, D. Kiriya, D. H. Lien, J. W. Ager, R. M. Wallace and A. Javey, Nano Lett., 2016, 16, 2786–2791 CrossRef CAS PubMed.
- A. O. A. Tanoh, J. Alexander-Webber, Y. Fan, N. Gauriot, J. Xiao, R. Pandya, Z. Li, S. Hofmann and A. Rao, Nanoscale Adv., 2021, 3, 4216–4225 RSC.
- Q. Li, A. Alfrey, J. Hu, N. Lydick, E. Paik, B. Liu, H. Sun, Y. Lu, R. Wang, S. Forrest and H. Deng, Nat. Commun., 2023, 14, 1837 CrossRef CAS PubMed.
- H. V. Han, A. Y. Lu, L. S. Lu, J. K. Huang, H. Li, C. L. Hsu, Y. C. Lin, M. H. Chiu, K. Suenaga, C. W. Chu, H. C. Kuo, W. H. Chang, L. J. Li and Y. Shi, ACS Nano, 2016, 10, 1454–1461 CrossRef CAS PubMed.
- Y. Guo, Y. Ji, H. Dong, L. Wang and Y. Li, AIP Adv., 2019, 9, 025202 CrossRef.
- M. Mahjouri-Samani, L. Liang, A. Oyedele, Y. S. Kim, M. Tian, N. Cross, K. Wang, M. W. Lin, A. Boulesbaa, C. M. Rouleau, A. A. Puretzky, K. Xiao, M. Yoon, G. Eres, G. Duscher, B. G. Sumpter and D. B. Geohegan, Nano Lett., 2016, 16, 5213–5220 CrossRef CAS PubMed.
- J. Lu, A. Carvalho, X. K. Chan, H. Liu, B. Liu, E. S. Tok, K. P. Loh, A. H. Castro Neto and C. H. Sow, Nano Lett., 2015, 15, 3524–3532 CrossRef CAS PubMed.
- H. Kim, G. H. Ahn, J. Cho, M. Amani, J. P. Mastandrea, C. K. Groschner, D. H. Lien, Y. Zhao, J. W. Ager, M. C. Scott, D. C. Chrzan and A. Javey, Sci. Adv., 2019, 5, 1–8 CAS.
- K. Chen, S. Deng, E. Chen, S. Wen, T. Ouyang, X. Wang, R. Zhan, J. Cai, X. Wan and H. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 44814–44823 CrossRef CAS PubMed.
- M. Tebyetekerwa, J. Zhang, Z. Xu, T. N. Truong, Z. Yin, Y. Lu, S. Ramakrishna, D. Macdonald and H. T. Nguyen, ACS Nano, 2020, 14, 14579–14604 CrossRef CAS PubMed.
- L. Gao, Z. Hu, J. Lu, H. Liu and Z. Ni, Phys. Chem. Chem. Phys., 2021, 23, 8222–8235 RSC.
- F. Sigger, I. Amersdorffer, A. Hötger, M. Nutz, J. Kiemle, T. Taniguchi, K. Watanabe, M. Förg, J. Noe, J. J. Finley, A. Högele, A. W. Holleitner, T. Hümmer, D. Hunger and C. Kastl, J. Phys. Chem. Lett., 2022, 13, 10291–10296 CrossRef CAS PubMed.
- Z. Li, F. Rashvand, H. Bretscher, B. M. Szydłowska, J. Xiao, C. Backes and A. Rao, J. Phys. Chem. C, 2022, 126, 21681–21688 CrossRef CAS PubMed.
- C. Backes, B. M. Szydłowska, A. Harvey, S. Yuan, V. Vega-Mayoral, B. R. Davies, P. L. Zhao, D. Hanlon, E. J. G. Santos, M. I. Katsnelson, W. J. Blau, C. Gadermaier and J. N. Coleman, ACS Nano, 2016, 10, 1589–1601 CrossRef CAS PubMed.
- S. Witomska, T. Leydecker, A. Ciesielski and P. Samorì, Adv. Funct. Mater., 2019, 29, 1–23 CrossRef.
- F. Bonaccorso, A. Bartolotta, J. N. Coleman and C. Backes, Adv. Mater., 2016, 28, 6136–6166 CrossRef CAS PubMed.
- A. G. Kelly, V. Vega-Mayoral, J. B. Boland and J. N. Coleman, 2D Mater., 2019, 6, 045036 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |