
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complementarity of two proteomic data analysis tools in the identification of drug-metabolising enzymes and transporters in human liver†
Areti-Maria
Vasilogianni‡
 ab,
Sarah
Alrubia‡
ac,
Eman
El-Khateeb‡
ade,
Zubida M.
Al-Majdoub
a,
Narciso
Couto
a,
Brahim
Achour
af,
Amin
Rostami-Hodjegan
ae and
Jill
Barber
*a
ab,
Sarah
Alrubia‡
ac,
Eman
El-Khateeb‡
ade,
Zubida M.
Al-Majdoub
a,
Narciso
Couto
a,
Brahim
Achour
af,
Amin
Rostami-Hodjegan
ae and
Jill
Barber
*a
aCentre for Applied Pharmacokinetic Research, School of Health Sciences, University of Manchester, Stopford Building, Oxford Road, Manchester, M13 9PT, UK. E-mail: Jill.Barber@manchester.ac.uk
bDMPK, Oncology R&D, AstraZeneca, Cambridge, UK
cPharmaceutical Chemistry Department, College of Pharmacy, King Saud University, Riyadh, Saudi Arabia
dClinical Pharmacy Department, Faculty of Pharmacy, Tanta University, Tanta, Egypt
eCertara Inc (Simcyp Division), 1 Concourse Way, Sheffield, UK
fDepartment of Biomedical and Pharmaceutical Sciences, College of Pharmacy, University of Rhode Island, Kingston, Rhode Island, USA
First published on 13th November 2023
Abstract
Several software packages are available for the analysis of proteomic LC-MS/MS data, including commercial (e.g. Mascot/Progenesis LC-MS) and open access software (e.g. MaxQuant). In this study, Progenesis and MaxQuant were used to analyse the same data set from human liver microsomes (n = 23). Comparison focussed on the total number of peptides and proteins identified by the two packages. For the peptides exclusively identified by each software package, distribution of peptide length, hydrophobicity, molecular weight, isoelectric point and score were compared. Using standard cut-off peptide scores, we found an average of only 65% overlap in detected peptides, with surprisingly little consistency in the characteristics of peptides exclusively detected by each package. Generally, MaxQuant detected more peptides than Progenesis, and the additional peptides were longer and had relatively lower scores. Progenesis-specific peptides tended to be more hydrophilic and basic relative to peptides detected only by MaxQuant. At the protein level, we focussed on drug-metabolising enzymes (DMEs) and transporters, by comparing the number of unique peptides detected by the two packages for these specific proteins of interest, and their abundance. The abundance of DMEs and SLC transporters showed good correlation between the two software tools, but ABC showed less consistency. In conclusion, in order to maximise the use of MS datasets, we recommend processing with more than one software package. Together, Progenesis and MaxQuant provided excellent coverage, with a core of common peptides identified in a very robust way.
1. Introduction
Recent years have witnessed increased use of mass spectrometry-based proteomics for the identification and quantification of pharmacologically relevant proteins in different populations.1–6 This powerful analytical technique allows characterisation of complex biological matrices (such as enriched fractions, cell culture lysates, tissue extracts, and biopsies) as well as quantification of specific proteins of special interest.7,8 A wide variety of mass spectrometry-based strategies are available, taking advantage of the technique's selectivity, sensitivity and ability to detect many proteins simultaneously.9,10Drug-metabolizing enzymes (DMEs), such as cytochrome P450 (CYP450)11–19 and uridine 5′-diphospho-glucuronosyltransferase (UGT) enzymes, have received particular attention owing to their role in determining the kinetics of the majority of drugs on the market.20 However, even with recent advances in technology, measuring UGT enzymes remains challenging because of their membrane topology and high sequence homology.21 Similarly, transporters are difficult to quantify because of low abundance and membrane localization, and therefore their characterization requires enrichment of plasma membrane fractions and the use of highly sensitive instrumentation.22
The increased activity in this area, however, has highlighted inter-laboratory and inter-methodological variation in quantification.6 There is no simple relationship between the size of a mass spectrometry signal and the concentration of analyte. Worse, the LC-MS/MS workflow does not normally sample every available peptide but selects the most intense signals at any time point. Quantification of DMEs and transporters is important – it provides numbers used in silico to represent patients in virtual clinical trials.23–25 The community therefore assembled in September 2018 to address best practice in proteomic analysis and quantification methods, resulting in a white paper.26
Differences in quantification can arise from differences in sample preparation,27,28 quantification methodology,28,29 including whether measurement is targeted or untargeted,6,30,31 LC-MS/MS parameters and instrumentation, even when the sample is the same. In practice, we are not especially interested in measuring the same sample because biological differences between samples are the main subject of our investigations. Multivariate statistical techniques, such as principal components analysis (PCA), have been used to discern biological and technical variation within groups of samples27,32,33 but are of limited utility in assessing cross-laboratory measurements.
At this stage, strategies for overcoming these differences would inevitably involve many replicate analyses, which are at best costly, and often impossible where samples are small and of human origin. There is, however, less excuse for differences in quantification resulting from data analysis. The commonly used data analysis tools, required to convert RAW data files into quantification of proteins, have different algorithms that can generate variable results, and one useful idea is to assess their complementarity. Comparative reports for different data analysis tools have been generated (Table 1) with varied conclusions. A single 2012 study sought to compare data processing using complex samples from animal retinas, concluding that the total number of proteins identified by MaxQuant and Progenesis is highly comparable, with 74% overlap.34 Another study using five different data analysis tools to identify potato and human synthetic peptides concluded that MaxQuant achieved the highest peptide coverage based on charge-state merging, while Progenesis was the best based on the obtained original data, as a result of all alignment features and normalization before LC-MS/MS.35 Comparison of different tools using a plant-derived standard proteins mix demonstrated high variability in protein abundance measured by the different tools, suggesting caution should be applied with discovery proteomics data.34 Finally, a study using Universal Proteomics Standard Set and yeast concluded that Progenesis performed consistently well in differential expression analysis and produced few missing intensity values, whereas data filtering or imputation methods improved the performance of commonly used software for proteomics including MaxQuant, Proteios, PEAKS, and OpenMS.7
| Study | Sample | Compared software | Analysis technique (instrument) | Outcomes compared |
|---|---|---|---|---|
| DDA: data dependent acquisition; SRM: selected reaction monitoring; SILAC: stable isotope labelling by amino acids in cell culture. | ||||
| Merl et al. 201242 | Retinal cells (healthy animals) | Progenesis | Label free versus SILAC (Orbitrap) | Quantification accuracy |
| MaxQuant | Dynamic range | |||
| Sensitivity | ||||
| Chawade et al. 201543 | Synthetic peptides (potato and human) | Progenesis | DDA and SRM (Orbitrap XL ETD) | Peptide coverage |
| MaxQuant | F1-score (harmonic mean of precision and sensitivity) | |||
| Proteios | Mean accuracy (proportion of true positive and negative identifications) | |||
| Skyline | Number of unique peptides | |||
| Anubis | ||||
| Välikangas et al. 201744 | Universal proteomics standard set and yeast Saccharomyces cerevisiae | Progenesis | DDA (Orbitrab Velos) | The number of proteins quantified. |
| MaxQuant | The extent of missing data | |||
| Proteios | ||||
| PEAKS | ||||
| OpenMS | ||||
| Al Shweiki et al. 201745 | Standard proteins mix (plant) | Proteome Discoverer | DDA (Orbitrap Velos) | Biological variability |
| Scaffold | Protein abundance estimates | |||
| MaxQuant | Protein fold change | |||
| Progenesis | ||||
In the present work, we analysed a real, clinically important dataset obtained from 23 human liver membrane samples. We used two software packages, MaxQuant and Progenesis, both commonly used for peptide/protein identification and quantification. MaxQuant36,37 uses its own search engine, Andromeda, for identification, which relies on a probability calculation for scoring a peptide-spectrum match.38 Quantification of proteins is based on maximum peptide ratio information from extracted peptide ion signal intensities. These are normalised to minimise the overall fold change of all peptides across all fractions.34 Progenesis uses Mascot for identification39 and quantifies proteins based on peptide ion peak intensity while allowing full operator control.34
The novelty of this study is that MaxQuant and Progenesis are evaluated for first time using healthy human liver samples from healthy volunteers and focusing on drug-metabolising enzymes and transporters. Human liver samples from healthy volunteers are very precious and very important as controls. Because of their rarity, several studies use ‘histologically normal’ livers from diseased patients as controls. However, our previous reports showed that livers from diseased subjects are different from healthy controls and therefore they are not ideal as controls.40,41 There is a particular ethical imperative therefore to generate as much information as possible from these very precious samples. This dataset was used to evaluate MaxQuant and Progenesis and to determine whether information could be maximised by the use of both software tools with a single dataset. We focused particularly on drug-metabolising enzymes and transporters because the liver is the primary site of drug metabolism in the body. Perturbations in the abundance of these proteins can therefore affect the toxicity and efficacy of drugs.
2. Materials and methods
2.1 Dataset
The dataset analysed in this study was previously generated by Couto et al.30 using 23 human liver microsomes (HLM) samples provided by Pfizer (Groton, CT, USA). Suppliers of these samples were Vitron (Tucson, AZ, USA) and BD Gentest (San Jose, CA, USA). Demographic details, sample preparation, LC-MS/MS analysis workflow and data analysis were reported previously.30 The primary goal for which these data were generated is to evaluate the expression of proteins responsible for the metabolism and transport of drugs and xenobiotic in human liver.302.2 Database fasta file
UniProtKB human proteome fasta file containing 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 599 entries (May 2017) was used for analysis by both Progenesis and MaxQuant.46
599 entries (May 2017) was used for analysis by both Progenesis and MaxQuant.46
2.3 Data processing
Data analysis was performed using MaxQuant 1.6.1.0 (Max Planck Institute of Biochemistry, Munich, Germany) and Progenesis QI 4.0 (Nonlinear Dynamics, Newcastle-upon-Tyne, UK). Replicates (two of each sample) were analysed in the same batch. Progenesis LC-MS takes raw data of the MS/MS scans and transforms them to peak lists. One sample was selected as a reference after checking the two-dimensional mapping (m/z versus retention time), and the retention times of the other samples within the batch were aligned. The 2D map uses as visual quality control and highlights any problems in a sample run. Default peak-picking settings were used and the resulting aggregate spectra were filtered to include +2, and +3 charge states only. These aligned spectra contain all peak information, allowing the detection of all ions. An “.mgf” file representing the aggregate spectra was exported and searched for peptide identification using in-house Mascot server (Matrix Science, London, UK) using human SwissProt and Tremble databases containing 75004 protein sequences. Search parameters used were: 5 ppm precursor mass tolerance, 0.5 Da fragment mass tolerance, cysteine carbamidomethylation was set as fixed modification, M oxidation, NQ deamidation, label 13C(6) (K), label 13C(6) were used as variable modifications. Trypsin/P was set as the proteolytic enzyme, and one missed cleavage was allowed (for more details on Progenesis and Mascot processing see ref. 30). The resulting “.xml” file was re-imported to assign peptides to features using the following thresholds: Mascot determined peptides with ion scores of 15 and above and only proteins with at least one unique peptide ranked as top candidate were considered and re-imported into Progenesis. Maximum number of hits was set to “AUTO” to ensure only statistically significant and high-quality identification is applied. Mascot scores corresponding to a false discovery rate (FDR) of <0.01 was set as a threshold for peptide identification. FDR of <0.01 was also used for protein-level identification. Quantitative analysis was carried out using the “Hi3” intensity-based method on Progenesis as previously described.5 The reference protein, in this case bovine serum albumin (BSA), was assigned at a known amount. Knowing the spiked amount of BSA and the accession number, abundance of all proteins in the sample was quantified from Progenesis output.
The parameters applied in MaxQuant were changed from default to match their counterparts in Progenesis and Mascot as presented in Table 2. Full details of all the parameter settings used for MaxQuant are listed in Table S1 (ESI†). No filters were applied for the scores in data processing and cut-off scores were applied manually after exporting the data. The ‘matching between runs’ feature was not enabled in MaxQuant.
| Parameter description | Parameter setting |
|---|---|
| Label free quantification | Yes |
| Multiplicity | 1 |
| Digestion enzyme | Trypsin/P |
| Variable modifications | Oxidation (M) & deamidation (NQ) |
| Fixed modifications | Carbamidomethyl (C) |
| Max number of modifications per peptide | 11 |
| Max charge | 7 |
| Main search peptide tolerance | 5 ppm |
| Min pep length | 7 |
| Min pep length for unspecific | 70 |
| Max peptide mass [Da] | 6000 Da |
| Peptides for quantification | Unique + razor |
| MS/MS match tolerance | 0.5 Da |
| False discovery rate (FDR) | 1% |
2.4 Comparison of peptides identified by MaxQuant and Progenesis
The comparison aimed to identify the differences between performance of the two software tools in terms of the number, nature and identity of identified peptides.2.5 Comparison at the protein level
All peptide sequences were matched against UniProt human proteome database, and accordingly each peptide was assigned to a certain human protein. The numbers of samples, in which a specific protein (CYP, UGT, or transporter) was identified based on unique peptides by each software, were counted and compared. In order to assign the detected peptides to appropriate human proteins, the following approach was applied:• All peptides were matched against the UniProt human proteome fasta file (May 2017).49 Proteins were prioritised according to the following criteria: (a) full length proteins were preferred over cDNA; (b) characterised sequences were prioritised over uncharacterised ones; and (c) longer sequences of the same proteins were preferred over shorter ones. The final order was arranged alphabetically.
• The remaining peptides that did not match any protein were deleted. Single peptides that appeared in two or fewer samples and did not appear in the UniProt fasta file were also deleted.
• A best-fit analysis was then run to minimise the number of accession codes that account for all the peptides.
For each sample, the number of proteins identified with at least one unique or razor peptide by each software package was determined. The number of CYP450s, UGTs, ABC and SLC transporters were calculated separately. Percentage identical proteins (PIPr) was calculated for all pairs of results, both inter- and intra-sample.
2.6 Quantification of DMEs and transporters
For the quantification of drug-metabolising enzymes and transporters, a widely used global proteomics approach – the total protein approach (TPA) was used,50 as previously described.51 This method does not require the use of standards for quantification and relies on MS signal intensity.2.7 Software availability and processing time
MaxQuant is as an open access cross-platform software available online from https://www.maxquant.org/, while Progenesis is a commercial software package provided by Waters Corporation (NYSE: WAT) and it requires a licence. The average time taken to process a sample was determined for both tools in hours and compared. MaxQuant processing time includes only one step from the raw data to the processed Excel sheets, while Progenesis requires an additional step to generate and export the “mgf” file, which represents the post-alignment aggregate spectrum, then this file is searched using Mascot.The raw files were processed by MaxQuant on personal computers that have the following specifications: Processor Intel® Core™ i7-6600U CPU@2.6 GHz; RAM 20 GB; 64-bit operating system; Windows 10. The computer used for Progenesis processing has the following specifications: Dell Precision T7600 Tower workstation; Processor 2x Intel Xeon-E5-2643 CPU@3.30 GHz; RAM 128 GB; 64-bit operating system; Windows 7.
3. Results
3.1 Comparison of peptide scores between Progenesis and MaxQuant
The scores of peptides identified by the two software packages were plotted against one another, as shown in Fig. 1(A) for sample HLM76 as an representative example. Linear regression gave rise to a best fit equation in the form y = mx, with R2 values (typically around 0.23–0.43) reflecting considerable scatter; peptide scores were far from consistent between the two software packages. Equations linking scores for all samples are shown in Table S2 (ESI†). Peptides with scores below 40 in MaxQuant were disregarded and the corresponding cut-off scores in Progenesis were calculated according to these equations. For sample HLM76, the Progenesis cut-off score was 13.4. The average for all samples was 14.03, so a cut-off score of 14 for Progenesis could be used as an ad hoc equivalent to MaxQuant 40. Fig. 1(B) depicts all the trend lines for all the samples. The red line represents the trend line for collated data from all samples. Eqn (1) represents the fit of data from all the samples, allowing a slope of 0.35 to be used in the general case.| Progenesis score = 0.3508 × MaxQuant score | (1) |
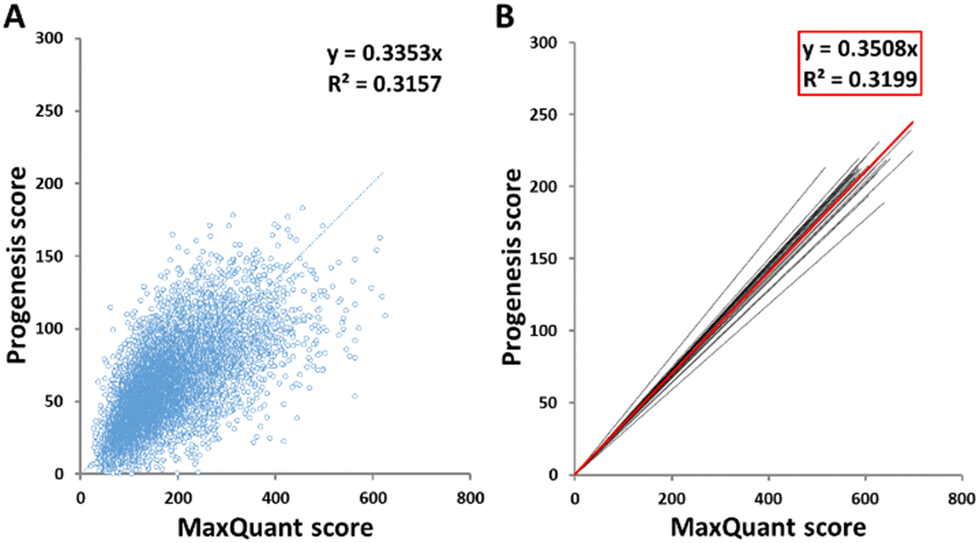 | ||
| Fig. 1 Linear regression of MaxQuant and Progenesis peptide scores. A representative linear regression analysis for one sample, HLM76, is shown (A), with the trend lines for the linear regression equations for each sample shown in black and for the collated data from all samples shown in red (B). | ||
3.2 Total number of peptides and modified peptides
Prior to filtering, the total number of peptides identified by the two packages averaged 20736 for Progenesis and 17963 for MaxQuant (Table S4, ESI†). Filtering the data led to identification of 14870 (range 11490–16126) by Progenesis, compared with 17534 (range 15991–20129) by MaxQuant. The default parameters in MaxQuant have a cut-off for modified peptides of 40, and these are generally the peptides with the lowest scores. Modified peptides in this study represent peptides with asparagine/glutamine deamidation and/or methionine oxidation. Table 3 summarises the numbers of peptides detected by the two software packages after data filtering. There was from 52–72% overlap (65% on average) between the peptides detected by the two packages; 10% of the peptides identified by Progenesis were modified but only 6% of those identified by MaxQuant.
| Sample | MaxQuant total peptides | Progenesis total peptides | MaxQuant only peptides | Progenesis only peptides | Overlap | MaxQuant modified | Progenesis modified | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | Percent | Number | Percent | Number | Percent | Number | Percent | Number | Percent | |||
| HLM01 | 15991 |
11490 |
3280 | 18% | 2193 | 12% | 12711 |
70% | 673 | 4% | 1337 | 9% |
| HLM02 | 20129 |
14838 |
7313 | 33% | 2022 | 9% | 12816 |
58% | 1182 | 6% | 1397 | 9% |
| HLM06 | 18343 |
14437 |
6092 | 30% | 2186 | 11% | 12251 |
60% | 892 | 5% | 1405 | 10% |
| HLM08 | 17563 |
15876 |
3906 | 20% | 2219 | 11% | 13657 |
69% | 789 | 4% | 1267 | 8% |
| HLM11 | 17509 |
15947 |
3931 | 20% | 2369 | 12% | 13578 |
68% | 833 | 5% | 1507 | 9% |
| HLM25 | 18421 |
15483 |
5407 | 26% | 2469 | 12% | 13014 |
62% | 961 | 5% | 1553 | 10% |
| HLM38 | 17480 |
16126 |
3735 | 19% | 2381 | 12% | 13745 |
69% | 861 | 5% | 1461 | 9% |
| HLM41 | 17335 |
14589 |
5076 | 26% | 2330 | 12% | 12259 |
62% | 1338 | 8% | 1828 | 13% |
| HLM48 | 17270 |
15113 |
4265 | 22% | 2108 | 11% | 13005 |
67% | 910 | 5% | 1343 | 9% |
| HLM71 | 16012 |
15644 |
2746 | 15% | 2378 | 13% | 13266 |
72% | 786 | 5% | 1522 | 10% |
| HLM72 | 16883 |
15406 |
3637 | 19% | 2160 | 11% | 13246 |
70% | 829 | 5% | 1425 | 9% |
| HLM73 | 16727 |
15225 |
3707 | 20% | 2205 | 12% | 13020 |
69% | 2921 | 17% | 3137 | 21% |
| HLM74 | 17828 |
14352 |
5691 | 28% | 2215 | 11% | 12137 |
61% | 827 | 5% | 1391 | 10% |
| HLM75 | 16447 |
15058 |
3537 | 19% | 2148 | 12% | 12910 |
69% | 752 | 5% | 1388 | 9% |
| HLM76 | 18744 |
14454 |
6447 | 31% | 2157 | 10% | 12297 |
59% | 1006 | 5% | 1431 | 10% |
| HLM77 | 16802 |
14847 |
3872 | 21% | 1917 | 10% | 12930 |
69% | 907 | 5% | 1285 | 9% |
| HLM78 | 18361 |
14256 |
6294 | 31% | 2189 | 11% | 12067 |
59% | 987 | 5% | 1457 | 10% |
| HLM80 | 16918 |
15075 |
4037 | 21% | 2194 | 12% | 12881 |
67% | 758 | 4% | 1365 | 9% |
| HLM89 | 17379 |
15941 |
3827 | 19% | 2389 | 12% | 13552 |
69% | 1492 | 9% | 2004 | 13% |
| HLM90 | 17071 |
12205 |
4009 | 20% | 2731 | 14% | 13062 |
66% | 931 | 5% | 1140 | 9% |
| HLM91 | 16812 |
15111 |
4045 | 21% | 2344 | 12% | 12767 |
67% | 790 | 5% | 1399 | 9% |
| HLM100 | 17722 |
15666 |
4334 | 22% | 2278 | 11% | 13388 |
67% | 854 | 5% | 1388 | 9% |
| HLM117 | 19535 |
14884 |
6998 | 32% | 2347 | 11% | 12537 |
57% | 921 | 5% | 1517 | 10% |
| Mean |
17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 534 534
|
14870
|
4617 | 23% | 2258 | 11% |
12917
|
65% | 1009 | 6% | 1519 | 10% |
| SD | 1027 | 1102 | 1273 | 5% | 165 | 1% | 495 | 5% | 458 | 3% | 394 | 3% |
| CV | 6% | 7% | 28% | 23% | 7% | 8% | 4% | 7% | 45% | 47% | 26% | 26% |
Sample HLM73 (and to a lesser extent HLM41) is an interesting case, with much higher levels of modification than the norm, identified by both software packages. It is not clear whether the high level of modification is the result of technical differences in handling the samples, or biological differences (for example in response to ageing).
3.3 Correlation between intensities in MaxQuant and Progenesis
Good correlations were observed between intensities of peptide signals reported by MaxQuant and Progenesis, with each sample giving a relationship in the form y = mx, with average R2 of 0.75 (Fig. 2(B)). Although each sample had an independent linear regression equation (Fig. 2(A)), intensities reported by MaxQuant were always higher than corresponding intensities reported by Progenesis. Individual regression equations for intensities for each individual sample are shown in Table S3 (ESI†). The average trend line for all data was described by the equation below:| Progenesis intensity = 0.0149 × MaxQuant intensity | (2) |
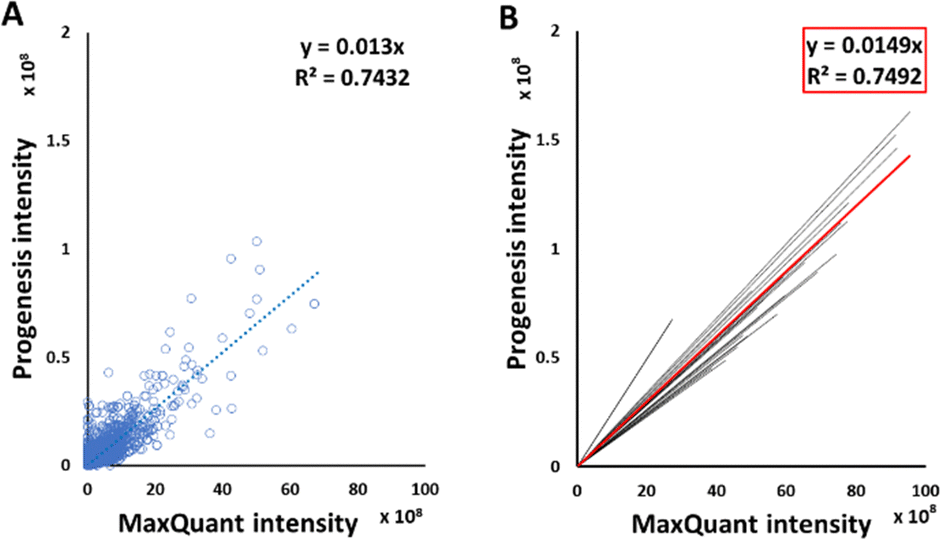 | ||
| Fig. 2 Linear regression of MaxQuant and Progenesis peptide signal intensities. A representative linear regression analysis for one sample, HLM76, is shown (A). The trend lines for the linear regression equations for each sample are shown in black and for the collated data from all samples shown in red (B). | ||
3.4 Peptide characteristics
Several characteristics of the peptides detected by a single tool were now investigated, as these were thought to be indicative of any possible bias by the software algorithm. These are illustrated in Fig. 3, using sample HLM76 as an example. Firstly, any bias towards long or short peptides was probed. The median and mode lengths of MaxQuant specific peptides (n = 6447, non-Gaussian distribution) were 13 and 11, whereas median and mode lengths of Progenesis-specific peptides (n = 2157, non-Gaussian distribution) were 13 and 7, showing that Progenesis favoured relatively shorter peptides. The scores of software-specific peptides were treated similarly. The ranges of median and mode scores of the MaxQuant specific peptides for all the samples were 74.6 to 199.7 and 46.5 to 367.2, whereas ranges of median and mode scores of the Progenesis-specific peptides were 23.9 to 53.1 and 13.6 to 88.6, which, when adjusted to be equivalent to the MaxQuant values (using equations in Table S2, ESI†) were 57.9 to 164.7 and 40.4 to 257.2 (Table S7, ESI†). Thus, MaxQuant detects a higher number of software-specific peptides with relatively greater confidence than Progenesis. The same trends were observed across all samples.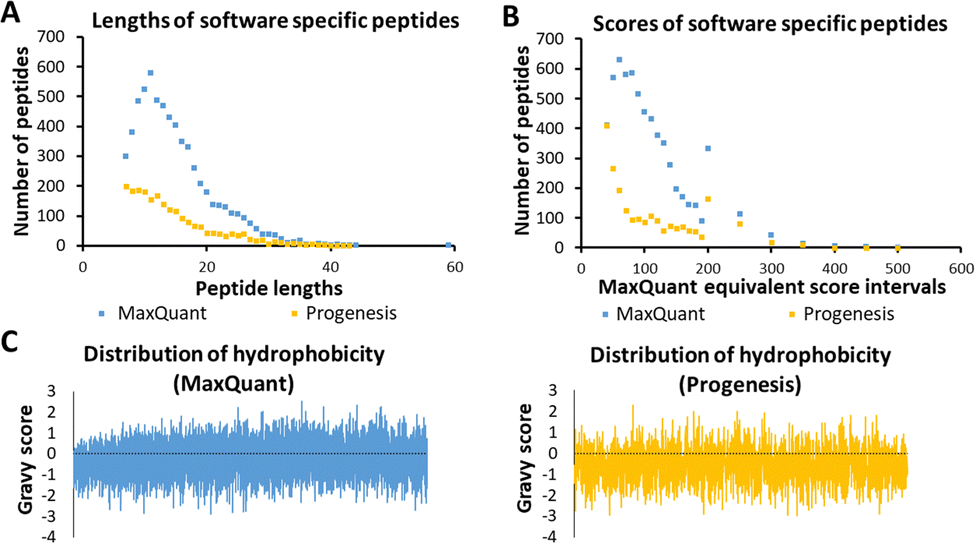 | ||
| Fig. 3 Characteristics of sample HLM76 peptides identified by the two software packages in terms of length (A), score (B) and hydrophobicity (C). In panel (C), each peptide is represented by a line starting from 0 on the y axis and ending either in the positive or the negative side of the y axis, depending on the actual value of hydrophobicity (GRAVY score). The mean Gravy score for Progenesis-specific peptides is more negative. | ||
Fig. 3(C) shows GRAVY scores for MaxQuant-specific and Progenesis-specific peptides; the more negative the value, the more hydrophilic the peptide. The median and mode GRAVY scores of the MaxQuant specific peptides in all samples ranged from −0.35 to 0.09 and from −0.7 to 0.4, respectively, whereas median and mode GRAVY scores of the Progenesis-specific peptides were ranging from −0.53 to −0.43 and from −0.9 to 0.1 (Table S8, ESI†). Therefore, the peptides identified by Progenesis (Fig. 3(C)) had more negative GRAVY scores, indicating higher hydrophilicity than those identified solely by MaxQuant (Fig. 3(C)).
Table S5 (ESI†) provides an example of statistical analysis in relation to the peptide length, GRAVY score (hydrophobicity), isoelectric point (PI), and molecular weight of peptides from sample HLM76. Comparison of these characteristics showed that Progenesis-specific peptides were generally shorter, more hydrophilic, and more basic, with lower mass.
3.5 Multivariate analysis of peptide and protein data
PIP (percentage identical peptides) and PIPr (percentage identical proteins) were calculated between samples for each software package as previously described,48 and the results were analysed by principal components analysis (PCA). PCA results are represented in Fig. 4. PCA on PIP and PIPr values returned two distinct clusters for each package. Clustering of PIP and PIPr data generated with Progenesis and MaxQuant were quite similar and the % variance explained by each dimension were almost identical. The clusters contained the same patient samples and the difference between PIP and PIPr (regardless of the software) was the outlier with PIP (sample HLM73) and PIPr (sample HLM2). Sample HLM73 is different at the peptide level possibly due to extensive modification as shown in Table 3. Importantly, PCA provides more information in relation to explained variance when technical and biological factors are tractable.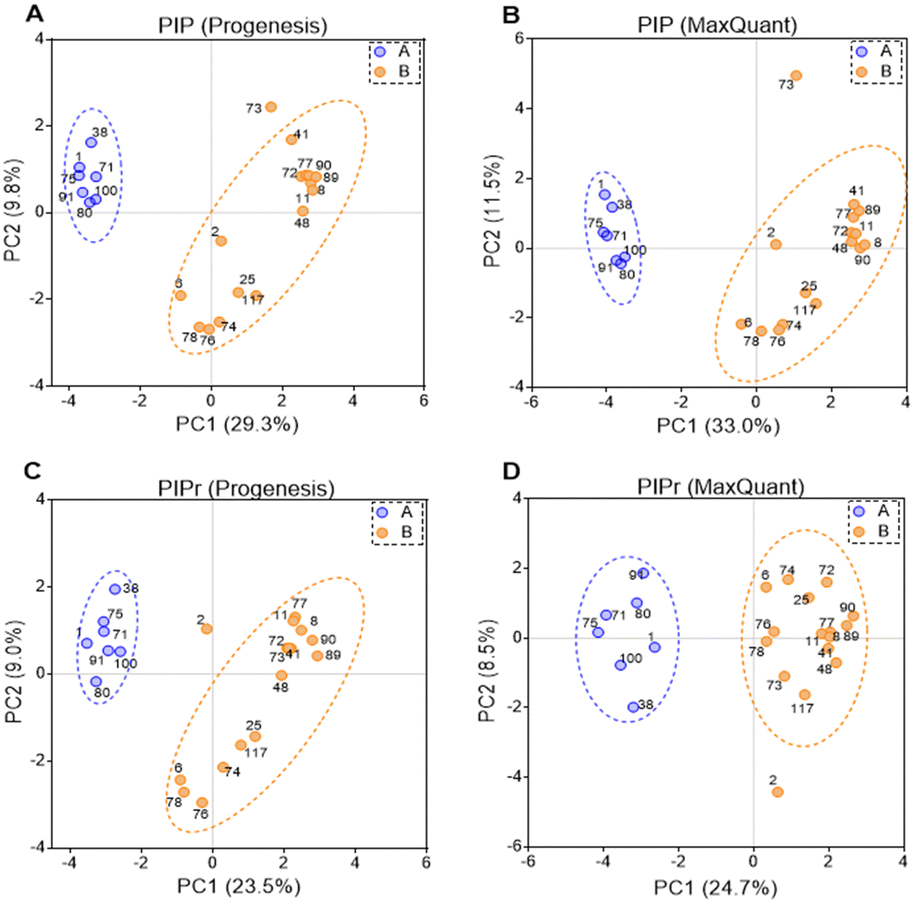 | ||
| Fig. 4 Principal components analysis (PCA) for 23 human liver samples based on percentage identical peptides (PIP) identified by Progenesis (A) and MaxQuant (B), and percentage identical proteins (PIPr) identified by Progenesis (C) and MaxQuant (D). | ||
3.6 Drug-metabolising enzymes and transporters
We now focused on membrane proteins of particular interest in drug metabolism and disposition: CYPs, UGTs, ABC and SLC transporters. For each of these proteins, the number of samples in which the protein could be detected (with unique peptides) by each software package is shown in Table S6 (ESI†). Fig. 5–7 shown the results for (CYP and UGT) enzymes, ABC transporters and SLCs, respectively. The more abundant proteins (for example, CYP3A4, UGT1A1, ABCD3, SLC3A1) were found in all 23 samples, regardless of the software. More interesting in the context of this paper are examples such as CYP1A1, CYP2F1, UGT1A7, ABCA2, and many SLC transporters which, in many samples, achieve a positive identification using one software package only (Fig. 5–7). In these cases, the use of two software packages permits additional identification and higher coverage of important proteins relative to the use of a single package. The most important cases are summarised in Table 4.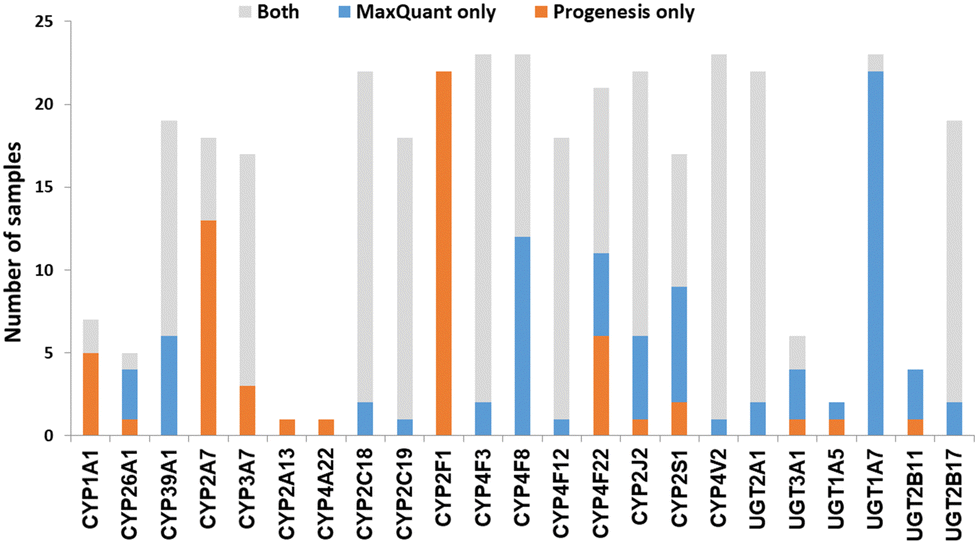 | ||
| Fig. 5 The number of samples in which CYPs and UGTs identified by each software tool. Other CYPs and UGTs that have been identified by both software (overlap) in all samples are not included. | ||
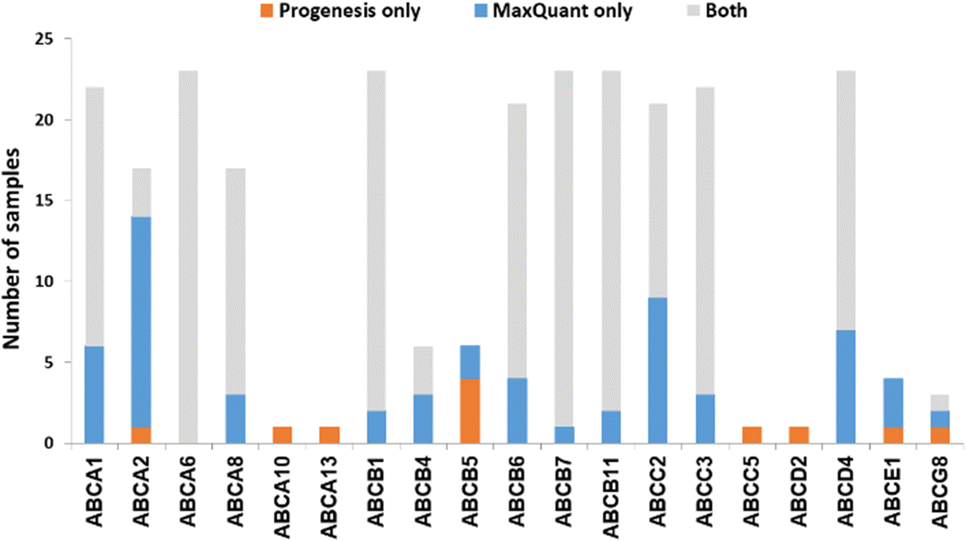 | ||
| Fig. 6 The number of samples in which ABC transporters were identified by each software tool. Other ABC transporters that have been identified by both software (overlap) in all samples are not included. | ||
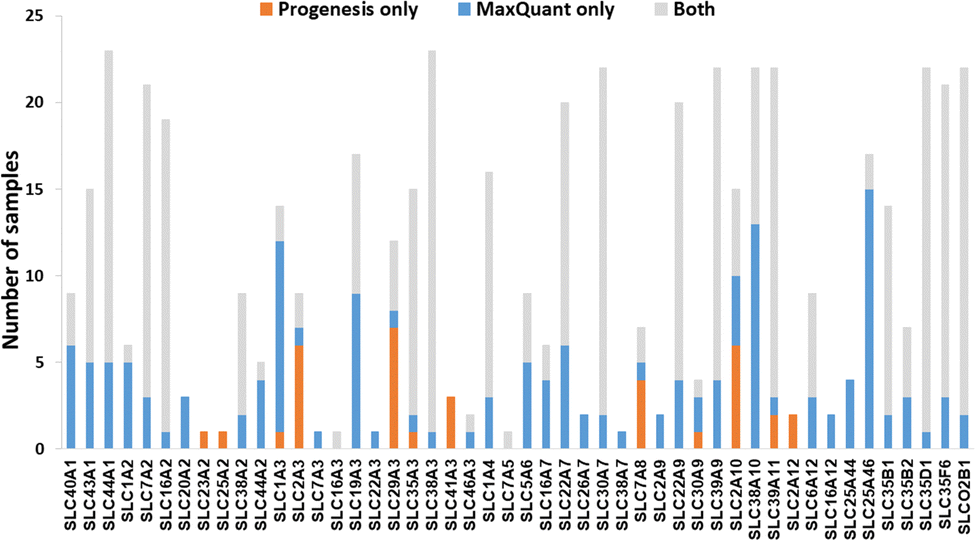 | ||
| Fig. 7 The number of samples in which solute carriers (SLCs) were identified by each software tool. Other SLC transporters that have been identified by both software (overlap) in all samples are not included. | ||
| Protein | Samples with reliable detection by: | Comments | |
|---|---|---|---|
| Pro-genesis | Max-Quant | ||
| CYP1A1 | 7 | 2 | Involved in steroid hormone biosynthesis,52 fatty acid,53 and retinol metabolism.54 |
| CYP39A1 | 13 | 19 | Involved in cholesterol degradation and bile acid biosynthesis.55 |
| CYP2A7 | 18 | 5 | |
| CYP2F1 | 22 | 0 | Possibly involved in the metabolism of naphthalene.56 |
| CYP4F8 | 11 | 23 | Involved in fatty acid metabolism.57 |
| CYP4F22 | 16 | 15 | Autosomal recessive loss of function mutations associated with congenital ichthyosiform erythroderma.58,59 |
| CYP2J2 | 17 | 21 | Involved in arachidonate metabolism60 |
| CYP2S1 | 10 | 15 | Involved in fatty acid metabolism.61 |
| ABCA1 (ABC-1) | 16 | 22 | Involved in the transport of cholesterol and high-density lipoproteins.62 Mutations lead to Tangier disease.63 |
| ABCA2 (ABC2) | 4 | 16 | Associated with drug resistance in cancer cells, and one SNP of ABCA2 is linked to early onset of Alzheimer's disease.64 |
| ABCB5 (ABCB5 P-gp) | 4 | 2 | Associated with drug resistance in colorectal cancer and melanoma.65,66 |
| ABCC2 (MRP2) | 12 | 22 | Mutations are associated with Dubin–Johnson syndrome.67 |
| ABCD4 (PMP70) | 16 | 23 | Involved in vitamin B12 transport.68 |
| SLC2A1 (GLUT-1) | 10 | 5 | Involved in glucose transport and when mutated, associated with GLUT1 deficiency syndrome.69 |
| SLC29A1 (ENT1) | 12 | 17 | Mutations are associated with inherited H syndrome, pigmented hypertrichosis with insulin-dependent diabetes, and Faisalabad histiocytosis.70 |
| SLC29A3 (ENT3) | 11 | 5 | Mutations associated with disorders, such as H syndrome, pigmented hypertrichotic dermatosis with insulin-dependent diabetes syndrome, and histiocytosis with massive lymphadenopathy.71 |
| SLC22A7 (OAT2) | 14 | 20 | Acts as sodium-independent organic anion/dimethyldicarboxylate exchanger.72 |
Additionally, all the identified CYPs, UGTs, ABC and SLC transporters were quantified using TPA. Fig. 8 illustrates the correlation of the abundance of these proteins between MaxQuant and Progenesis. The more abundant proteins, CYPs, UGTs and SLC transporters show good correlation, clustering around lines of y = x as expected. The ABC transporters, with the exceptions of ABCD3 and MRP3 (not shown on the graph), are, however, of very low abundance, close to the limit of detection and are poorly enriched in microsomes compared with endoplasmic reticulum proteins, such as CYPs and UGTs. Fig. 8(C) and Table S9 (ESI†) now show much more scatter from y = x. This is not surprising. In general, the biases that lead to Progenesis favouring short, basic, hydrophilic peptides and MaxQuant favouring longer, hydrophobic, more acidic peptides cancel extremely well for abundant proteins with many detectable peptides, leading to consistent quantification, despite the differences in detected peptides. For low abundance proteins, such as ABC transporters, many peptides fall below the scoring threshold for at least one of the packages, leading to bigger discrepancies. The precision of quantification is poor, and it is not possible to judge which package is better for any particular protein. The advantage of analysing data with both packages is that it allows us to confirm the presence of more proteins than we could detect with a single package. However, quantification of low abundance proteins is perilous, and several criteria must be taken in consideration, including the number of peptides corresponding to that protein identified by each software, the uniqueness of the peptides, their quality (modifications, missed cleavages), the number of samples where the protein was identified, and, where possible, quantification with different methods (TPA, HiN, QconCATs, iBAQ, etc.).
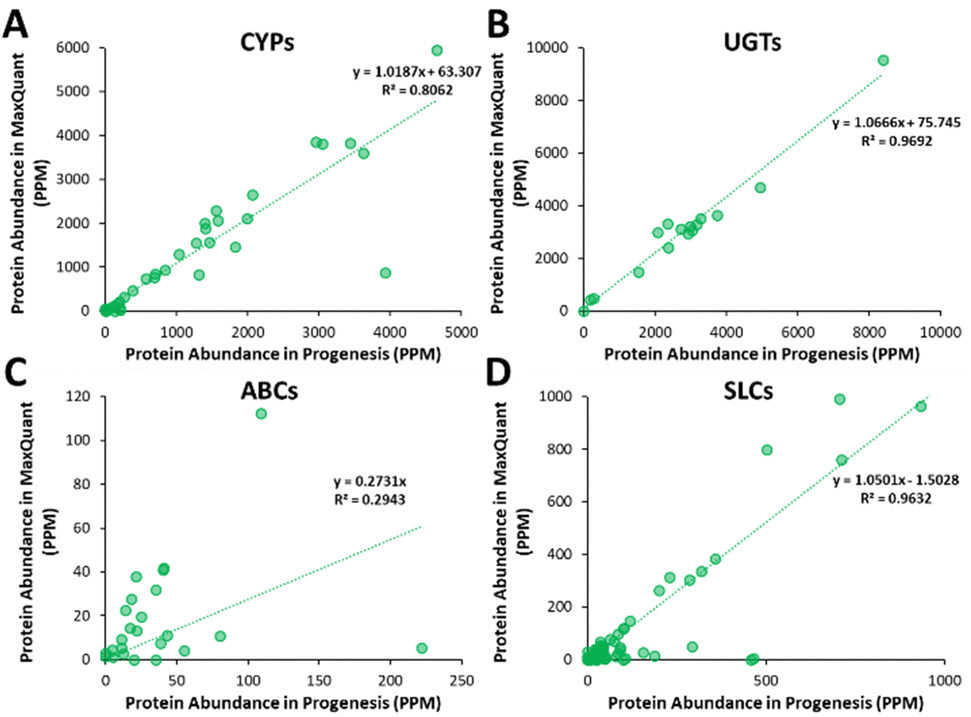 | ||
| Fig. 8 Correlation of the abundance of CYPs (A), UGTs (B), ABC (C) and (SLC) transporters between MaxQuant and Progenesis. The abundance was measured using the total protein approach, and expressed as parts per million (PPM). | ||
3.7 Processing time
Although computer specifications were superior with Progenesis, it took approximately 3 hours to process 2 raw files (2 replicates of the same sample with average size 1.5 GB) with Progenesis and 2–4 hours by MaxQuant. Notably, Progenesis requires an additional step to run a search on Mascot in order to generate the final output for identification and quantification of the protein targets, which might take 1 extra hour. Both software processing procedures are time consuming but being an open access tool, MaxQuant can be used on personal computers while commercially available tools (e.g. Progenesis) are expensive and are normally operated on dedicated PCs.4. Discussion
Mass spectrometry-based global proteomics is a powerful tool, allowing thousands of proteins to be identified and quantified simultaneously, with very high sensitivity and selectivity. Many commentators have noted, however, that such sensitivity and selectivity come at a price – the lack of independent verification. Sample preparation and sampling by the mass spectrometer can lead to reasonably well-understood differences between the results reported by different laboratories, even when many replicates are run and/or many fractionation steps performed. It remains, however, somewhat disturbing that different processing software, even when (as here) well-respected packages are used, can yield different results using the same input.There have been a relatively small number of studies devoted to understanding the role of the processing package in interpreting global proteomic data and many of these focus on quite simple model systems, such as yeast and plants.73,74 The real importance of differences in processing will only be apparent when different packages are used to process clinical samples, especially precious human samples where sample availability is limited and where the proteins under study are of low abundance, membrane bound, or show high homology and therefore yield few unique peptides.
Duplicate MS output files, generated from duplicate tryptic digests of 23 human liver samples were processed by two different software packages, Progenesis and MaxQuant. Peptide score correlation obtained for each sample by the two software tools was performed and an average trend line was created to establish a score cut-off equivalent to a MaxQuant score of 40. A comparison between the remaining sets of peptides was performed. The overlap between the peptides detected by the two packages ranged from 52–72% (mean 65%) with the total number of peptides identified by MaxQuant typically 18% higher. Progenesis, on average detected more modified peptides (10% compared to 6% for MaxQuant). A comparison of the characteristics of the software-specific peptides showed that, in general, Progenesis identified shorter peptides than MaxQuant, and they tended to be more basic and more hydrophilic.
We used consistent parameters for both software tools (mass tolerance, enzyme specificity, missed cleavages and modifications) and both search engines use a peptide score to match the experimental MS/MS data with a theoretical spectrum. The scoring of the peptide-spectrum match (PSM) by both tools is based on a probability calculation. The more recently developed Andromeda (MaxQuant) tool bases the scores on a binomial distribution probability, taking into account peptide fragments, neutral losses (water, ammonia) and diagnostic peaks.38,75 Mascot (Progenesis) scoring uses peptide fragments for spectral correlation with a probabilistic modelling approach and applies an ion score cut-off to filter the PSMs.76 Although the scoring systems seem very similar, the processes necessary for assigning a PSM can yield different outcomes because the algorithms used for peak picking and subsequent peptide sequencing differ between search engines.77 False positive PSMs present a challenge, as the false peptide/protein identification interferes with the interpretation of the data. Therefore, ways to measure and control the number of false identifications are required. These measures discriminate correct PSMs from false identifications and ultimately allow controlling the false discovery rate (FDR).78
The scoring algorithms aim to describe the match quality, for instance, the number of shared fragment ions between a spectrum and a candidate peptide sequence39 or similarity in general. In the case of Mascot/Andromeda the number of shared fragment ions is converted into a probabilistic match score using the negative logarithm of the determined probability that the computed PSM is an incorrect assignment.38 This generates a measure of match quality with high scores representing more likely hits and a high proportion of matching fragment ions. An expectation value is calculated for all sequence candidates based on the score distribution. Low quality peaks can either be used for scoring or filtered out by the search engine, leading to differences in the quality of the PSMs. Matches of medium to high quality spectra tend to be scored robustly by the two software, leading to the observed significant overlap.
For the purpose of comparison in this study, the score cut-off values were normalised based on a predefined cut-off score of 40 for MaxQuant. An equivalent value was determined for Mascot (ranging from 11.9 to 16.5). This finding is in agreement with the literature, which reported that MaxQuant score is about three times Mascot score.38 The cut-off values of ≥40 for MaxQuant and ≥20 for Mascot were reported to offer a high identification probability in proteomics.74,79 Higher score was associated with unmodified peptides, with a clear indication of higher confidence in unmodified peptide identification across the 23 analysed samples; the average proportion of unmodified peptides associated with scores ≥40 for MaxQuant and Mascot was 94% and 90%, respectively. This is in line with a previous assessment reporting 89.1% unmodified peptides (in mouse dendritic cells).38
Searching software algorithms and comparing the data based on the algorithm of the compared software tools is generally beyond the scope of this paper. Our aim is not to find the element of the algorithm that may lead to differences in the identification of peptides and quantification of proteins between the two software tools. Instead, we aim to identify the differences between performance of the two software tools in terms of the number, nature and identity of identified peptides, and quantity of clinically important proteins. This has been achieved by keeping the setup parameters consistent between the two software tools.
At the protein level, our comparison focused on hepatic drug-metabolising enzymes and transporters involved in drug metabolism and disposition. There is considerable inter-individual variability in the expression of these proteins, and this results in different efficacy and toxicity of drugs among different patients.80 The distribution and abundances of these proteins can be used for the prediction of the pharmacokinetics of drugs in pharmacologically based pharmacokinetics models. More specifically, they can be used as scaling factors for the in vitro to in vivo extrapolation of drug clearance.23 Most hepatic drug-metabolising enzymes identified herein are of high abundances. This is because the samples are enriched microsomal fractions which are the main fractions harbouring these proteins within the hepatocyte. Identification of proteins of interest require additional rigour to establish confidence in their identification using unique peptides for this specific protein as explained in the Methods (Section 2.5).
In most of the samples, unique peptides corresponding to CYP and UGT proteins were detected by both software tools; in general, Progenesis and MaxQuant identified similar numbers of CYP and UGT peptides (Chi-squared test, p > 0.05). There were some discrepancies, however, with the most interesting cases being CYP1A1, 2A7, 2F1, 4F8 and UGT1A7 (Table 4 and Fig. 5). These are important for the metabolism of steroids, pneumotoxicants, naphthalene, fatty acids, and many other endogenous and xenobiotic substances (Table 4).81
Transporters are generally expressed at very low levels and in the plasma membrane, rather than endoplasmic reticulum, so they are not well enriched in microsomal preparations. We have previously demonstrated that microsomes are a crude membrane fraction that comprises membranes from various intracellular compartments as well as the plasma membrane.30,82 Endoplasmic reticulum is highly enriched in microsomes while plasma membrane tends to be less enriched; enrichment factors are normally less than 2 fold for plasma membrane, whereas reticular proteins have higher enrichment (>5 fold)83 This is mainly because of different levels of loss of membrane protein; in-house data showed 50–80% recovery of reticular protein compared to 30–60% recovery of cell membrane protein.84 Microsomal crude membrane extracts are not perfect, but they are the best available enriched membrane preparation. Extracting purer fractions such as plasma membrane fractions is fraught with unmitigated levels of protein loss. Like UGTs, transporters are membrane embedded, and, like UGTs, they tended to be more readily detected by MaxQuant. However, count differences (Chi-squared statistics) showed non-significant differences. Table 4 and Fig. 5–7 show that in some cases, MaxQuant identifies more unique peptides for CYPs, UGTs and transporters, whereas in other cases the opposite trend is observed. Table 4 also illustrates how the peptides detected only by Progenesis (for example, GNGIAFSSGDRWK and KSPAFMPFSAGR from CYP2F1) tend to be hydrophilic and basic whereas those detected only in MaxQuant (for example, TLDFIDVLLLSEDKNGK and SVINTSDAITDK from CYP4F8) tend to be slightly longer, less hydrophilic and weak acids, in line with the characteristics preferred by MaxQuant compared to Progenesis. The ABC transporters’ dataset illustrates that any search conditions will inevitably lead to some loss of genuine peptides together with the noise. When this dataset was subjected to MaxQuant processing with deamidation not permitted, most of the peptides detected here only with Progenesis appeared.
The quantification of DMEs and transporters with both software tools indicated that there is a reassuring consistency in the quantification of CYPs, UGTs and SLC transporters between MaxQuant and Progenesis. However, this is not observed in the case of the low abundance ABC transporters. This finding indicates that in the case of low abundance proteins, it may be very useful to use both software tool in a complementary way to increase the information extracted from the data. This will allow for more proteins of low abundance to be quantified, at least approximately.
The PCA analysis of the data shown in Fig. 4 is gratifying. The two software packages are in broad agreement, especially with respect to inter-individual variability. For example, both packages agree that sample 75 is similar to 71, and 77 is similar to 89. Where they disagree, we have developed some understanding of the reasons. It is therefore possible carefully to augment the data obtained using a Progenesis single package30 with the additional data obtained here using MaxQuant.
5. Conclusion
When two software packages (in this case MaxQuant and Progenesis) are used to analyse the same proteomic LC-MS/MS dataset, different results are obtained with on average 65% identical peptides. MaxQuant favours hydrophobic, more acidic peptides while Progenesis favours hydrophilic, basic peptides, including those with post-translational modification. Both software tools favour short peptides, with MaxQuant favouring slightly longer peptides. The overlap gives a set of very robust identifications, and these are sufficient for many purposes where abundant proteins from reproducible samples are being detected. The present samples, however, are precious, from human donors and the proteins under study are of low abundance. In this case, the additional effort of extracting information readily verifiable with only one of the software packages is worthwhile.Data availability
The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD020910 (Adult Liver Set 1). All data generated or analysed during this study are included in this published article and its ESI.†Author contributions
Study conception, design, and coordination: JB, AR-H, BA, ZA. Sample analysis: NC, ZA. Experimental design and implementation: AMV, EE, SA, BA, ZA, NC. Data analysis, writing the manuscript: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Centre for Applied Pharmacokinetic Research (CAPKR), University of Manchester. The authors would like to thank the Biological Mass Spectrometry Core Facility (BioMS), University of Manchester and the ChELSI Institute, University of Sheffield, for access to LC-MS/MS instrumentation, and Pfizer (Groton, CT) for supplying the liver microsomal samples. Financial support from the Egyptian government (The Egyptian Missions Sector), the Saudi Ministry of Education, KSU, and Merck KGaA to EE, SA, and AMV, respectively, is acknowledged.References
- T. Yan, L. Lu, C. Xie, J. Chen, X. Peng, L. Zhu, Y. Wang, Q. Li, J. Shi, F. Zhou, M. Hu and Z. Liu, Mol. Cancer Ther., 2015, 14, 2874–2886 CrossRef CAS.
- T. Yan, S. Gao, X. Peng, J. Shi, C. Xie, Q. Li, L. Lu, Y. Wang, F. Zhou, Z. Liu and M. Hu, Pharm. Res., 2015, 32, 1141–1157 CrossRef CAS PubMed.
- S. Billington, A. S. Ray, L. Salphati, G. Xiao, X. Chu, W. G. Humphreys, M. Liao, C. A. Lee, A. Mathias, C. E. C. A. Hop, C. Rowbottom, R. Evers, Y. Lai, E. J. Kelly, B. Prasad and J. D. Unadkat, Drug Metab. Dispos., 2018, 46, 189–196 CrossRef CAS.
- B. Prasad, D. K. Bhatt, K. Johnson, R. Chapa, X. Chu, L. Salphati, G. Xiao, C. Lee, C. E. C. A. Hop, A. Mathias, Y. Lai, M. Liao, W. G. Humphreys, S. C. Kumer and J. D. Unadkat, Drug Metab. Dispos., 2018, 46, 943–952 CrossRef CAS.
- Z. M. Al-Majdoub, H. Al Feteisi, B. Achour, S. Warwood, S. Neuhoff, A. Rostami-Hodjegan and J. Barber, Mol. Pharmaceutics, 2019, 16, 1220–1233 CrossRef CAS PubMed.
- C. Wegler, F. Z. Gaugaz, T. B. Andersson, J. R. Wiśniewski, D. Busch, C. Gröer, S. Oswald, A. Norén, F. Weiss, H. S. Hammer, T. O. Joos, O. Poetz, B. Achour, A. Rostami-Hodjegan, E. van de Steeg, H. M. Wortelboer and P. Artursson, Mol. Pharmaceutics, 2017, 14, 3142–3151 CrossRef CAS.
- T. Välikangas, T. Suomi and L. L. Elo, Briefings Bioinf., 2018, 19, 1344–1355 Search PubMed.
- H. Al Feteisi, B. Achour, A. Rostami-hodjegan and J. Barber, Expert Opin. Drug Metab. Toxicol., 2015, 11, 1357–1369 CrossRef CAS PubMed.
- R. Aebersold and M. Mann, Nature, 2016, 537, 347–355 CrossRef CAS PubMed.
- S. Pan, R. Aebersold, R. Chen, J. Rush, D. R. Goodlett, M. W. McIntosh, J. Zhang and T. A. Brentnall, J. Proteome Res., 2009, 8, 787–797 CrossRef CAS.
- H. Kawakami, S. Ohtsuki, J. Kamiie, T. Suzuki, T. Abe and T. Terasaki, J. Pharm. Sci., 2011, 100, 341–352 CrossRef CAS PubMed.
- S. Ohtsuki, O. Schaefer, H. Kawakami, T. Inoue, S. Liehner, A. Saito, N. Ishiguro, W. Kishimoto, E. Ludwig-Schwellinger, T. Ebner and T. Terasaki, Drug Metab. Dispos., 2012, 40, 83–92 CrossRef CAS PubMed.
- B. Achour, M. R. Russell, J. Barber and A. Rostami-Hodjegan, Drug Metab. Dispos., 2014, 42, 500–510 CrossRef PubMed.
- J. Li, L. Zhou, H. Wang, H. Yan, N. Li, R. Zhai, F. Jiao, F. Hao, Z. Jin, F. Tian, B. Peng, Y. Zhang and X. Qian, Analyst, 2015, 140, 1281–2190 RSC.
- H. Wang, H. Zhang, J. Li, J. Wei, R. Zhai, B. Peng, H. Qiao, Y. Zhang and X. Qian, Anal. Methods, 2015, 7, 5934–5941 RSC.
- A. Vildhede, J. R. Wiśniewski, A. Norén, M. Karlgren and P. Artursson, J. Proteome Res., 2015, 14, 3305–3314 CrossRef CAS PubMed.
- H.-F. Zhang, H.-H. Wang, N. Gao, J.-Y. Wei, X. Tian, Y. Zhao, Y. Fang, J. Zhou, Q. Wen, J. Gao, Y.-J. Zhang, X.-H. Qian and H.-L. Qiao, J. Pharmacol. Exp. Ther., 2016, 358, 83–93 CrossRef.
- N. Couto, Z. M. Al-Majdoub, S. Gibson, P. J. Davies, B. Achour, M. D. Harwood, G. Carlson, J. Barber, A. Rostami-Hodjegan and G. Warhurst, Drug Metab. Dispos., 2020, 48, 245–254 CrossRef CAS.
- B. Achour, A. Dantonio, M. Niosi, J. J. Novak, J. K. Fallon, J. Barber, P. C. Smith, A. Rostami-Hodjegan and T. C. Goosen, Drug Metab. Dispos., 2017, 45, 1102–1112 CrossRef CAS PubMed.
- L. C. Wienkers and T. G. Heath, Nat. Rev. Drug Discovery, 2005, 4, 825–833 CrossRef CAS PubMed.
- M. D. Harwood, B. Achour, S. Neuhoff, M. R. Russell, G. Carlson and G. Warhurst, Drug Metab. Dispos., 2016, 44, 297–307 CrossRef CAS PubMed.
- B. Achour, Z. M. Al-Majdoub, A. Rostami-Hodjegan and J. Barber, Annu. Rev. Anal. Chem., 2020, 13, 223–247 CrossRef PubMed.
- A. Rostami-Hodjegan, Clin. Pharmacol. Ther., 2012, 92, 50–61 CrossRef CAS PubMed.
- M. Jamei, Curr. Pharmacol. Rep., 2016, 2, 161–169 CrossRef CAS PubMed.
- S. Sharma, D. Suresh Ahire and B. Prasad, J. Clin. Pharmacol., 2020, 60, S17–S35 CAS.
- B. Prasad, B. Achour, P. Artursson, C. E. C. A. Hop, Y. Lai, P. C. Smith, J. Barber, J. R. Wisniewski, D. Spellman, Y. Uchida, M. A. Zientek, J. D. Unadkat and A. Rostami-Hodjegan, Curr. Pharmacol. Rep., 2019, 106, 525–543 Search PubMed.
- M. Howard, B. Achour, Z. Al-Majdoub, A. Rostami-Hodjegan and J. Barber, Proteomics, 2018, 1800200 CrossRef.
- D. K. Bhatt and B. Prasad, Curr. Pharmacol. Rep., 2018, 103, 619–630 CAS.
- L. C. Gillet, A. Leitner and R. Aebersold, Annu. Rev. Anal. Chem., 2016, 9, 449–472 CrossRef.
- N. Couto, Z. M. Al-Majdoub, B. Achour, P. C. Wright, A. Rostami-Hodjegan and J. Barber, Mol. Pharmaceutics, 2019, 16, 632–647 CrossRef CAS.
- B. Prasad, R. Evers, A. Gupta, C. E. C. A. Hop, L. Salphati, S. Shukla, S. V. Ambudkar and J. D. Unadkat, Drug Metab. Dispos., 2013, 42, 78–88 CrossRef PubMed.
- N. Jehmlich, K. H. D. Dinh, M. Gesell-Salazar, E. Hammer, L. Steil, V. M. Dhople, C. Schurmann, B. Holtfreter, T. Kocher and U. Völker, J. Periodontal Res., 2013, 48, 392–403 CrossRef CAS PubMed.
- E. Leoni, M. Bremang, V. Mitra, I. Zubiri, S. Jung, C.-H. Lu, R. Adiutori, V. Lombardi, C. Russell, S. Koncarevic, M. Ward, I. Pike and A. Malaspina, Sci. Rep., 2019, 9, 4478 CrossRef PubMed.
- M. R. Al Shweiki, S. Mönchgesang, P. Majovsky, D. Thieme, D. Trutschel and W. Hoehenwarter, J. Proteome Res., 2017, 16, 1410–1424 CrossRef CAS.
- A. Chawade, M. Sandin, J. Teleman, J. Malmström and F. Levander, J. Proteome Res., 2015, 14, 676–687 CrossRef CAS.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Mol. Cell. Proteomics, 2014, 13, 2513–2526 CrossRef CAS.
- J. Cox and M. Mann, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef CAS.
- J. Cox, N. Neuhauser, A. Michalski, R. A. Scheltema, J. V. Olsen and M. Mann, J. Proteome Res., 2011, 1794–1805 CrossRef CAS.
- D. N. Perkins, D. J. C. Pappin, D. M. Creasy and J. S. Cottrell, Electrophoresis, 1999, 20, 3551–3567 CrossRef CAS PubMed.
- A. M. Vasilogianni, B. Achour, D. Scotcher, S. A. Peters, Z. M. Al-Majdoub, J. Barber and A. Rostami-Hodjegan, Drug Metab. Dispos., 2021, 49, 563–571 CrossRef CAS.
- A. M. Vasilogianni, Z. M. Al-Majdoub, B. Achour, S. Annie Peters, J. Barber and A. Rostami-Hodjegan, Clin. Pharmacol. Ther., 2022, 112, 699–710 CrossRef CAS PubMed.
- J. Merl, M. Ueffing, S. M. Hauck and C. von Toerne, Proteomics, 2012, 12, 1902–1911 CrossRef CAS.
- A. Chawade, M. Sandin, J. Teleman, J. Malmström and F. Levander, J. Proteome Res., 2015, 14, 676–687 CrossRef CAS PubMed.
- T. Välikangas, T. Suomi and L. L. Elo, Briefings Bioinf., 2017, 19, 1344–1355 Search PubMed.
- M. R. Al Shweiki, S. Mönchgesang, P. Majovsky, D. Thieme, D. Trutschel and W. Hoehenwarter, J. Proteome Res., 2017, 16, 1410–1424 CrossRef CAS PubMed.
- A. Bateman, M. J. Martin, C. O’Donovan, M. Magrane, E. Alpi, R. Antunes, B. Bely, M. Bingley, C. Bonilla, R. Britto, B. Bursteinas, H. Bye-AJee, A. Cowley, A. Da Silva, M. De Giorgi, T. Dogan, F. Fazzini, L. G. Castro, L. Figueira, P. Garmiri, G. Georghiou, D. Gonzalez, E. Hatton-Ellis, W. Li, W. Liu, R. Lopez, J. Luo, Y. Lussi, A. MacDougall, A. Nightingale, B. Palka, K. Pichler, D. Poggioli, S. Pundir, L. Pureza, G. Qi, S. Rosanoff, R. Saidi, T. Sawford, A. Shypitsyna, E. Speretta, E. Turner, N. Tyagi, V. Volynkin, T. Wardell, K. Warner, X. Watkins, R. Zaru, H. Zellner, I. Xenarios, L. Bougueleret, A. Bridge, S. Poux, N. Redaschi, L. Aimo, G. ArgoudPuy, A. Auchincloss, K. Axelsen, P. Bansal, D. Baratin, M. C. Blatter, B. Boeckmann, J. Bolleman, E. Boutet, L. Breuza, C. Casal-Casas, E. De Castro, E. Coudert, B. Cuche, M. Doche, D. Dornevil, S. Duvaud, A. Estreicher, L. Famiglietti, M. Feuermann, E. Gasteiger, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz-Gumowski, U. Hinz, C. Hulo, F. Jungo, G. Keller, V. Lara, P. Lemercier, D. Lieberherr, T. Lombardot, X. Martin, P. Masson, A. Morgat, T. Neto, N. Nouspikel, S. Paesano, I. Pedruzzi, S. Pilbout, M. Pozzato, M. Pruess, C. Rivoire, B. Roechert, M. Schneider, C. Sigrist, K. Sonesson, S. Staehli, A. Stutz, S. Sundaram, M. Tognolli, L. Verbregue, A. L. Veuthey, C. H. Wu, C. N. Arighi, L. Arminski, C. Chen, Y. Chen, J. S. Garavelli, H. Huang, K. Laiho, P. McGarvey, D. A. Natale, K. Ross, C. R. Vinayaka, Q. Wang, Y. Wang, L. S. Yeh and J. Zhang, Nucleic Acids Res., 2017, 45, D158–D169 CrossRef CAS.
- S. Ellis and J. Barber, Pract. Res. High. Educ., 2016, 10, 121–129 Search PubMed.
- H. Al Feteisi, Z. M. Al-Majdoub, B. Achour, N. Couto, A. Rostami-Hodjegan and J. Barber, J. Neurochem., 2018, 146, 670–685 CrossRef CAS PubMed.
- UniProt Consortium, Nucleic Acids Res., 2019, 47, D506–D515 CrossRef.
- J. R. Wiśniewski and D. Rakus, J. Proteomics, 2014, 109, 322–331 CrossRef.
- A. M. Vasilogianni, E. El-Khateeb, Z. M. Al-Majdoub, S. Alrubia, A. Rostami-Hodjegan, J. Barber and B. Achour, J. Proteomics, 2022, 263, 104601 CrossRef CAS PubMed.
- D. Schwarz, P. Kisselev, S. S. Ericksen, G. D. Szklarz, A. Chernogolov, H. Honeck, W.-H. Schunck and I. Roots, Biochem. Pharmacol., 2004, 67, 1445–1457 CrossRef CAS PubMed.
- D. Schwarz, P. Kisselev, S. S. Ericksen, G. D. Szklarz, A. Chernogolov, H. Honeck, W.-H. Schunck and I. Roots, Biochem. Pharmacol., 2004, 67, 1445–1457 CrossRef CAS PubMed.
- H. Chen, W. N. Howald and M. R. Juchau, Drug Metab. Dispos., 2000, 28, 315–322 CAS.
- A. R. Stiles, J. Kozlitina, B. M. Thompson, J. G. McDonald, K. S. King and D. W. Russell, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4006–E4014 CrossRef CAS PubMed.
- L. Li, S. Carratt, M. Hartog, N. Kovalchuk, K. Jia, Y. Wang, Q.-Y. Zhang, P. Edwards, L. Van Winkle and X. Ding, Environ. Health Perspect., 2017, 125, 067004 CrossRef PubMed.
- J. Bylund, M. Hidestrand, M. Ingelman-Sundberg and E. H. Oliw, J. Biol. Chem., 2000, 275, 21844–21849 CrossRef CAS PubMed.
- K. Sugiura, T. Takeichi, K. Tanahashi, Y. Ito, T. Kosho, K. Saida, H. Uhara, R. Okuyama and M. Akiyama, J. Dermatol. Sci., 2013, 72, 193–195 CrossRef CAS PubMed.
- K. Sugiura and M. Akiyama, J. Dermatol. Sci., 2015, 79, 4–9 CrossRef CAS PubMed.
- D. Lucas, S. Goulitquer, J. Marienhagen, M. Fer, Y. Dreano, U. Schwaneberg, Y. Amet and L. Corcos, J. Lipid Res., 2010, 51, 1125–1133 CrossRef CAS PubMed.
- P. Bui, S. Imaizumi, S. R. Beedanagari, S. T. Reddy and O. Hankinson, Drug Metab. Dispos., 2011, 39, 180–190 CrossRef CAS PubMed.
- F. Quazi and R. S. Molday, J. Biol. Chem., 2013, 288, 34414–34426 CrossRef CAS PubMed.
- S. Rust, M. Rosier, H. Funke, J. Real, Z. Amoura, J.-C. Piette, J.-F. Deleuze, H. B. Brewer, N. Duverger, P. Denèfle and G. Assmann, Nat. Genet., 1999, 22, 352–355 CrossRef CAS PubMed.
- J. T. Mack, C. B. Brown and K. D. Tew, Expert Opin. Ther. Targets, 2008, 12, 491–504 CrossRef CAS PubMed.
- B. J. Wilson, T. Schatton, Q. Zhan, M. Gasser, J. Ma, K. R. Saab, R. Schanche, A.-M. Waaga-Gasser, J. S. Gold, Q. Huang, G. F. Murphy, M. H. Frank and N. Y. Frank, Cancer Res., 2011, 71, 5307–5316 CrossRef CAS PubMed.
- B. J. Wilson, K. R. Saab, J. Ma, T. Schatton, P. Putz, Q. Zhan, G. F. Murphy, M. Gasser, A. M. Waaga-Gasser, N. Y. Frank and M. H. Frank, Cancer Res., 2014, 74, 4196–4207 CrossRef CAS PubMed.
- J. D. Schuetz, P. W. Swaan and D. J. Tweedie, Drug Metab. Dispos., 2014, 42, 541–545 CrossRef PubMed.
- J. C. Deme, M. A. Hancock, X. Xia, C. A. Shintre, M. Plesa, J. C. Kim, E. P. Carpenter, D. S. Rosenblatt and J. W. Coulton, Mol. Membr. Biol., 2014, 31, 250–261 CrossRef CAS PubMed.
- E. E. Lee, J. Ma, A. Sacharidou, W. Mi, V. K. Salato, N. Nguyen, Y. Jiang, J. M. Pascual, P. E. North, P. W. Shaul, M. Mettlen and R. C. Wang, Mol. Cell, 2015, 58, 845–853 CrossRef CAS.
- A. Bolze, A. Abhyankar, A. V. Grant, B. Patel, R. Yadav, M. Byun, D. Caillez, J.-F. Emile, M. Pastor-Anglada, L. Abel, A. Puel, R. Govindarajan, L. de Pontual and J.-L. Casanova, PLoS One, 2012, 7, e29708 CrossRef CAS PubMed.
- N. Kang, A. H. Jun, Y. D. Bhutia, N. Kannan, J. D. Unadkat and R. Govindarajan, J. Biol. Chem., 2010, 285, 28343–28352 CrossRef CAS PubMed.
- Y. Kobayashi, N. Ohshiro, R. Sakai, M. Ohbayashi, N. Kohyama and T. Yamamoto, J. Pharm. Pharmacol., 2005, 57, 573–578 CrossRef CAS PubMed.
- A. I. Nesvizhskii, J. Proteomics, 2010, 73, 2092–2123 CrossRef CAS PubMed.
- S. T. Tsai, C. C. Tsou, W. Y. Mao, W. C. Chang, H. Y. Han, W. L. Hsu, C. L. Li, C. N. Shen and C. H. Chen, Proteome Sci., 2012, 10, 69 CrossRef CAS PubMed.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS PubMed.
- C. Tu, J. Li, S. Shen, Q. Sheng, Y. Shyr and J. Qu, PLoS One, 2016, 11, e0160160 CrossRef PubMed.
- J. A. Paulo, Webmedcentral, 2013, 4, WMCPLS0052 Search PubMed.
- A. I. Nesvizhskii, J. Proteomics, 2010, 73, 2092–2123 CrossRef CAS PubMed.
- K. Dudekula and T. Le, Data Brief, 2016, 8, 494–500 CrossRef PubMed.
- R. M. Turner, B. K. Park and M. Pirmohamed, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2015, 7, 221–241 Search PubMed.
- G. Tournel, C. Cauffiez, I. Billaut-Laden, D. Allorge, D. Chevalier, F. Bonnifet, E. Mensier, J.-J. Lafitte, M. Lhermitte, F. Broly and J.-M. Lo-Guidice, Mutat. Res., 2007, 617, 79–89 CrossRef CAS PubMed.
- B. Achour, H. Al Feteisi, F. Lanucara, A. Rostami-Hodjegan and J. Barber, Drug Metab. Dispos., 2017, 45, 666–675 CrossRef CAS PubMed.
- F. Weiß, H. S. Hammer, K. Klein, H. Planatscher, U. M. Zanger, A. Norén, C. Wegler, P. Artursson, T. O. Joos and O. Poetz, Drug Metab. Dispos., 2018, 46, 387–396 CrossRef PubMed.
- B. Achour, Z. M. Al-Majdoub, A. Grybos-Gajniak, K. Lea, P. Kilford, M. Zhang, D. Knight, J. Barber, J. Schageman and A. Rostami-Hodjegan, Clin. Pharmacol. Ther., 2021, 109, 222–232 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mo00144j |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |