
Hydrogen bonding-promoted tunable approach for access to aza-bicyclo-[3.3.0]octanes and cyclopenta[b] pyrroles†
Sourav
Pramanik
a,
Subhadeep
Hazra‡
a,
Ayan
Chatterjee‡
a and
Jaideep
Saha§
 *b
*b
aDivision of Molecular Synthesis and Drug Discovery, Centre of Biomedical Research (CBMR), Lucknow 226014, India
bDepartment of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Mohali 160062, India. E-mail: jdsaha2000@gmail.com; jsaha@niper.ac.in
First published on 4th April 2024
Abstract
A unified strategy is disclosed that builds on successfully engaging the aniline nitrogen of 1,3-amphoteric γ-aminocyclopentenone for a tandem annulation with electron-poor alkynes, solely assisted by the H-bonding network of HFIP. This metal-free mild strategy provides access to medicinally relevant aza-bicyclo-octanes en route to another important scaffold: cyclopenta[b]pyrrole.
Pyrrole1 and its saturated congeners such as dihydropyrrole and pyrrolidine2 have found widespread applications in medicinal chemistry and drug discovery. Several synthetic strategies are currently available for pyrrole functionalization, which can anchor distinct sets of functional groups, site-selectively, into the ring. Besides, carbo- or heterocycle-annulated (fused) pyrroles have also emerged as useful synthetic targets owing to their various pharmacophoric attributes.3 Cyclopenta[b]pyrrole represents one such motif in which a carbocyclic ring is juxtaposed to the C2/C3-position of a pyrrole. A selective set of compounds bearing this unit is depicted in Fig. 1a (top panel).4 Aza-bicyclo-[3.3.0]octanes, on the other hand, represent the partial or fully saturated version of the former scaffold. This motif is also prevalent in various natural products and drug molecules (Fig. 1a; bottom panel).5
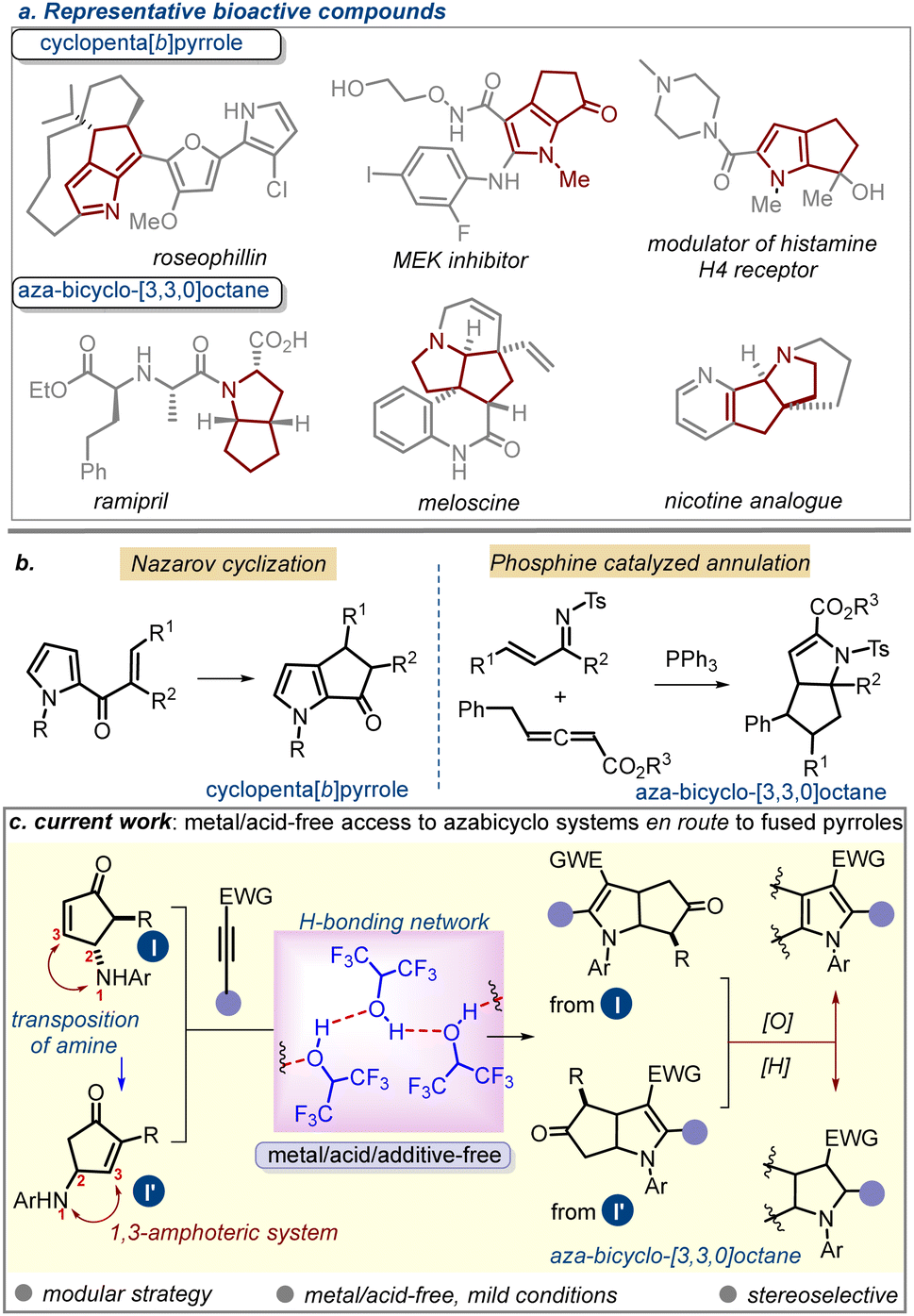 | ||
| Fig. 1 (a–b). Overview of the importance of fused pyrroles and aza-bicyclo-[3,3,0]octanes. (c) Strategy for their current development. | ||
Surprisingly, fewer strategies are available for accessing these compound classes,6–8 which in turn restricts the opportunity to find new drug-like chemical spaces around such scaffolds. To date, Nazarov cyclization remains the major means to prepare cyclopenta[b]pyrrole derivatives.6 Some reports have appeared sporadically that include Au(I)- or Pt-catalyzed cascade cyclization-rearrangement of strained rings,7a,b Brønsted acid-mediated pyrrole annulation with α-oxoketene dithioacetals,7c cascade reaction of 2-furylcarbinol,7d cyclization of isocyanide,7e or the use of a tailored aza-Piancatelli/rearrangement sequence.7f Conversely, only a handful of reports on the access to the aza-bicyclo-[3.3.0]octane core are available.8 Notable examples include the Cope ring rearrangement of bicyclo[3.2.1]octene system8a and a phosphine-catalyzed sequential [2+3] and [3+2] annulation process.8b
The preceding discussion indicates the importance of developing a broader platform for accessing diversely functionalized aza-bicyclo-[3.3.0]octane derivatives or annulated pyrroles. To achieve this goal, we contemplated a strategy for building the aza-bicyclo-[3.3.0]octane framework by deploying a 1,3-amphoteric system, namely γ-aminocyclopentenone (ACP). The ready accessibility of this moiety with a large variation of substitution patterns was expected to be an advantage for accessing elaborate assemblies of aza-bicyclo-[3.3.0]octanes upon the successful realization of the desired annulation with various dipolarophiles. γ-Amino cyclopentenones (ACPs) are valuable building blocks bearing multiple reactive centres.9 Recently, we showed their unique uses in accessing polycyclic indole derivatives and other natural-product-like structures through Lewis acid-mediated tandem processes.10 The presence of a nucleophilic amino group at the γ-position and a β-electrophilic carbon centre qualifies this system as a 1,3-amphoteric molecule.11 This feature can be harnessed to construct important classes of bicyclic systems through annulation. In this vein, we questioned whether ACPs could be used for developing a formal [3+2]-cycloaddition with electron-poor alkynes, specifically through the intervening alkyne-hydroamination12 and a C–C bond-forming step. While intermolecular alkyne hydroamination is typically performed in the presence of a transition-metal catalyst and/or at higher temperature,13 we aimed for a metal/acid-free mild process. Herein, we showcase the successful realization of the strategy in HFIP, which involves highly regio- and stereoselective annulation through tandem C–N and C–C bond formation facilitated through the H-bonding network of HFIP.14
To make the strategy more versatile, we developed a sequential oxidative strategy (step-wise or one-pot telescopic) that could transform the former scaffolds into cyclopenta[b]pyrrole derivatives. This process was also realized successfully.
During our initial forays to develop annulation between the model substrate 1a (ACP) and diethyl acetylene dicarboxylate (2a), we considered nucleophilic catalysis, first to transform the electron-poor alkyne into an activated zwitterionic intermediate, and then initiated the desired annulation with ACP. Accordingly, we screened phosphines15 and tertiary amines in various solvents. This approach, however, turned out to be completely futile and did not yield any productive reaction at room temperature or under heating in various solvents (entries 1–3; Table 1). Intriguingly, performing the reaction in neat 1,1,1,3,3,3-hexafluoroisopropanol (HFIP; 0.1 M) at room temperature afforded the desired compound 3 in a 94% yield (entry 4). Trifluoroethane (TFE; 0.1 M) was not efficient in the same scale as HFIP (entry 5), whereas no reaction proceeded in iPrOH (entry 6). Also, a higher concentration of HFIP or elevated reaction temperature proved detrimental to the reaction (entries 7–8).
|
|
|||
|---|---|---|---|
| Entry | Catalysts/additives | Solvent | Yieldb [%] |
| a Reaction conditions: compound 1 (1.0 equiv.), diethyl acetylene dicarboxylate (2; 2.0 equiv.), solvent (0.1–0.5 M), 12 h. b Isolated yields. c THF or CH2Cl2 or CH3CN at rt or 50 °C. d Reaction performed at 50 °C. e Conc.[0.5 M]. | |||
| 1 | PPh3 or PBu3 | 0 | |
| 2 | DBU | 0 | |
| 3 | — | 0 | |
| 4 | — | HFIP | 94 |
| 5 | — | TFE | 64 |
| 6 | — | iPrOH | 0 |
| 7d | — | HFIP | 86 |
| 8e | — | HFIP | 73 |

Given the operational simplicity and uniqueness of the process, featuring metal-free, hydrogen-bonding-activated tandem C–N/C–C bond formation, we examined the scope (Scheme 1). Substituent variation was evaluated for both partners, i.e. γ-amino cyclopentenone and electron-deficient alkyne. First, we investigated the electronic effect of aniline nitrogen in the reaction. A weakly donating or withdrawing group on aniline had almost no influence on the efficiency (4–6), whereas para-NO2 (strongly electron-withdrawing) completely shuttered the process (not shown). Substituents on aryl residues at the α,α′-position of the cyclopentenone ring had only a subtle influence (7–9). A heterocyclic motif at the α-position was tolerated (10). Entries 9–10 were obtained as a mixture of diastereomers, which was rooted in the use of the starting material as a mixture of diastereomers. A pure diastereomer of ACP (1i/1j) led to stereoselective transformations (11–12). ACP having an alkyl substituent at the α,α′-position and dimethyl acetylene dicarboxylate as a partner resulted in successful reactions (13–14).
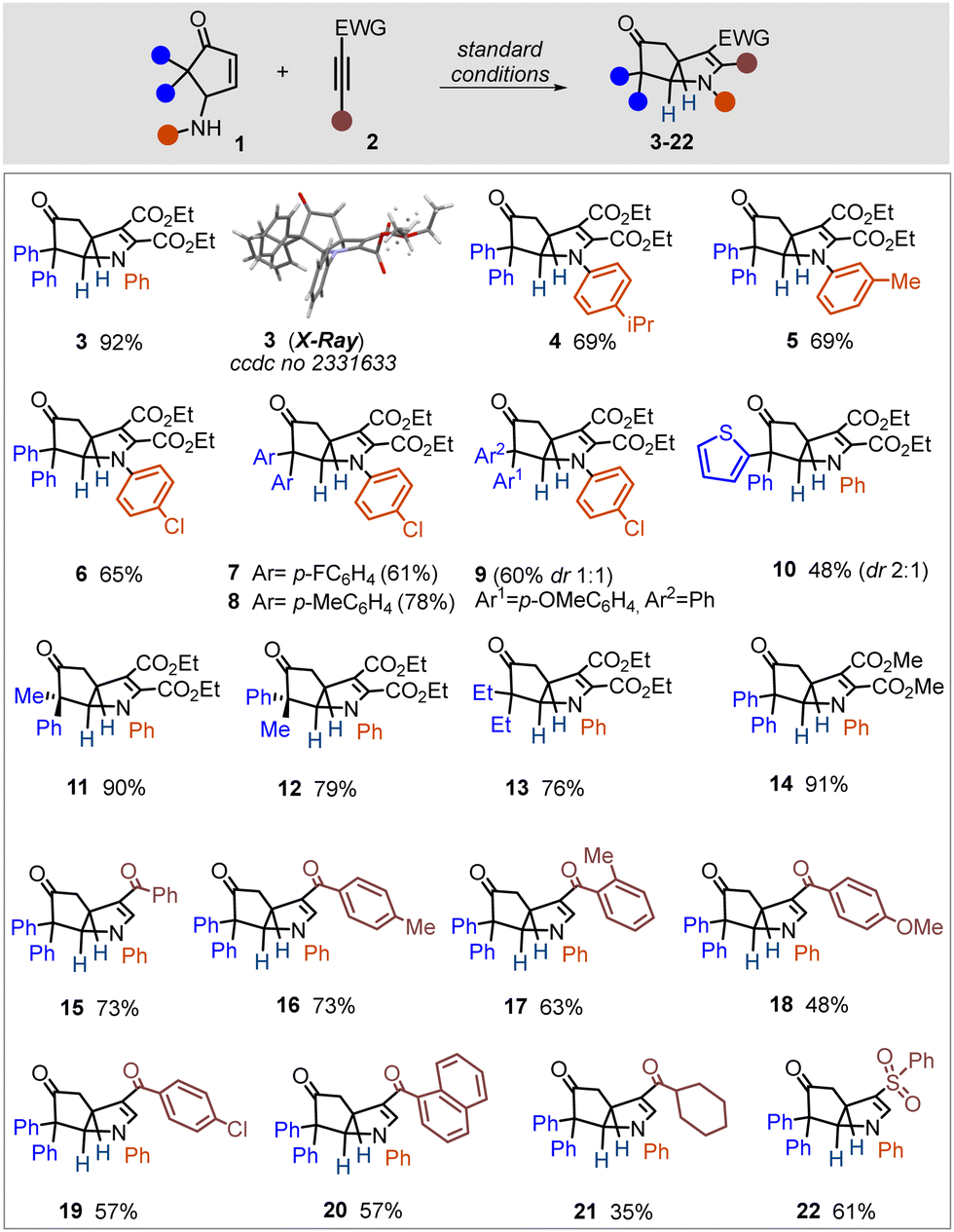 | ||
| Scheme 1 Scope of the reactions for diaryl-substituted ACPs with electron-poor alkynes. | ||
Next, we investigated various mono-substituted electron-poor alkynes in the reaction. Those bearing an aryl ketone residue readily participated in the reaction and delivered aza-bicyclo-[3.3.0]octane derivatives in moderate to good yields (15–20). Regioselectivity for the annulation was in accordance with the matching polarity of the two reactants, and the transformation was found to proceed with excellent diastereoselectivity. Alkyl ketone and aryl sulfone residues could be accommodated on the alkynes (21–22), albeit the former was less efficient. A terminal phenyl group on an electron-poor alkyne was found to impede the reaction (not shown). Importantly, we could not trace any acyclic product (i.e., only the hydroamination product of the alkyne) in the reaction mixture, which in turn suggested an excellent cooperativity between the two steps (hydroamination/C–C bond formation), which could potentially be aided through the HFIP hydrogen-bonding cluster.
To broaden the scope, next we examined various α-mono-aryl-substituted γ-aminocyclopentenones (Scheme 2). Such compounds are usually more reactive and unstable under acidic conditions compared to α,α-disubstituted analogues. Pleasingly, these substrates proved equally competent and afforded compounds 23–27 in good yields. In the same vein, we explored the isomeric γ-ACPs (1′). These substrates (with varying substitution pattern) also could be readily transformed into the desired aza-bicyclo-[3.3.0]octane derivatives (28–34). Of note, in compounds 28–34, the N-substituent and α-aryl group on the cyclopentanone ring were in an anti-relationship.
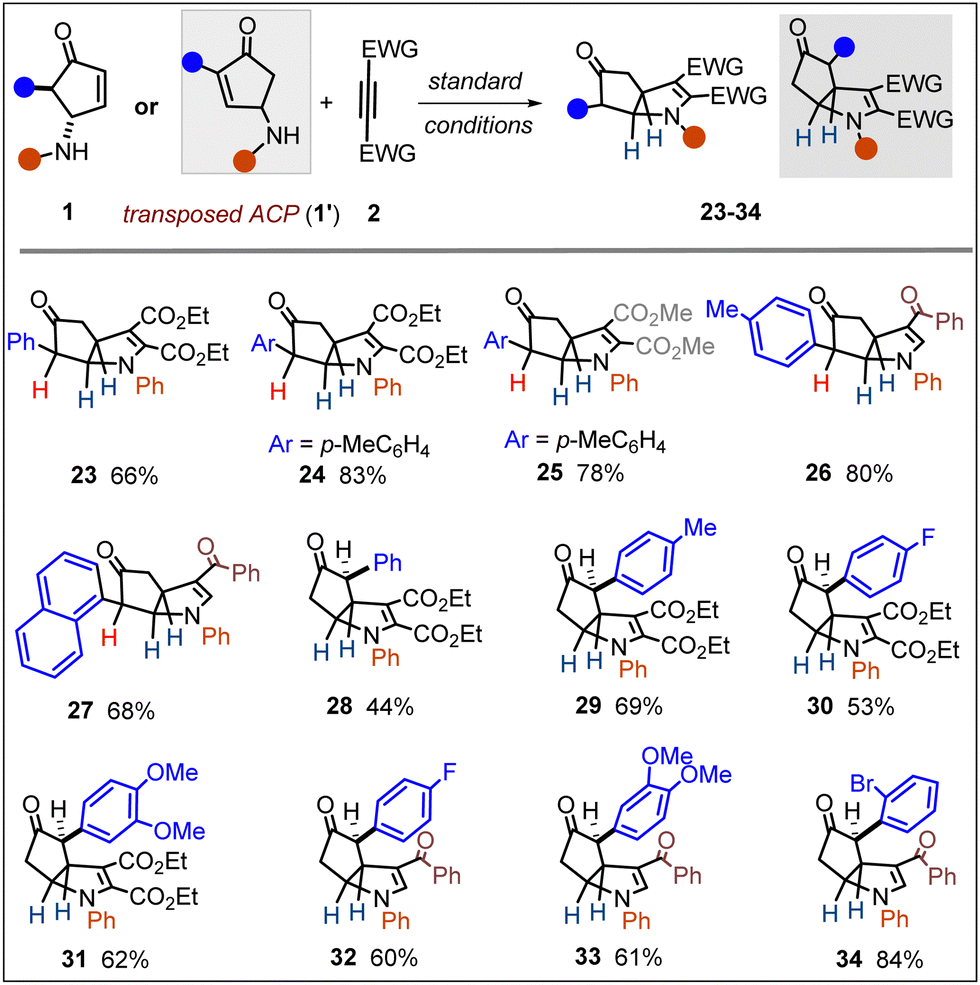 | ||
| Scheme 2 Annulation scope for α-mono-aryl-substituted ACP and structural isomers. | ||
It is also important to note that the mono-substituted γ-ACPs rendered remarkable diastereoselectivity in affording 23–34, with the formation of three new contiguous stereocenters.16 After establishing a reliable process for the stereoselective access to aza-bicyclo-[3.3.0]octanes, we first attempted a one-pot strategy to transform them into another medicinally attractive motif: cyclopenta[b]pyrrole. Oxidation of the aniline nitrogen into an iminium followed by a deprotonation/aromatization sequence was expected to deliver a pyrrole unit. A few trials revealed DDQ as the most effective oxidant in the process, which could transform 1a to 35 (43%) through a one-pot telescoped process in HFIP. However, we found that this yield could be significantly improved (80%) in a step-wise reaction by subjecting the aza-bicyclo octane 3 (vide Scheme 1) with DDQ in toluene (35 (80%)b; Scheme 3).
 | ||
Scheme 3 (a) Conversion of aza-bicyclo-[3,3,0]octanes to fused pyrroles or saturated systems. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) one-pot process: compound 1/1′ (1.0 equiv.), alkyne (1.2 equiv.), HFIP, rt, then DDQ (1.5 equiv.); bstep-wise process from aza-bicyclo-octanes: 11/13–14/20/24/29 (1.0 equiv.), DDQ (1.5 equiv.), toluene (0.1 M), rt; yields for step-2 are provided. one-pot process: compound 1/1′ (1.0 equiv.), alkyne (1.2 equiv.), HFIP, rt, then DDQ (1.5 equiv.); bstep-wise process from aza-bicyclo-octanes: 11/13–14/20/24/29 (1.0 equiv.), DDQ (1.5 equiv.), toluene (0.1 M), rt; yields for step-2 are provided. | ||
Applying the latter conditions, different fused-pyrrole derivatives bearing diaryl, dialkyl, or monaryl substitution at the α-position on cyclopentenone unit (35–37/39–40) and ketoaryl residues on the heterocyclic ring (38) were obtained in synthetically useful yields.
Next, we attempted to access fully saturated derivatives of the aza-bicyclo-[3.3.0]octanes prepared herein through the hydrogenation of the olefinic residue present in the dihydropyrrole unit. While the catalytic hydrogenation (Pd–C/H2) was unsuccessful, the use of pinacol borane (H-BPin) turned out to be the most effective reducing agent for the desired outcome.17 Olefinic reduction (possibly via intermediate 43) is showcased with two examples (15/20 as the starting), which gave good yields and high diastereoselectivity (41–42; Scheme 3b).
Of note, while an array of HFIP-assisted chemical transformations have been reported in the literature,14 the current transformation involving γ-amincyclopentenone as a 1,3-amphoteric system and electron-poor alkyne, however, underscores a distinct type of reaction manifold being facilitated through the H-bonding network of HFIP. Some other features of the current reaction also merit note. The poor nucleophilicity of aniline nitrogen generally hinders its addition onto carbon-based π-systems and this is often circumvented under high pressure/temperature.18 This current development, however, needs no such requirement. Furthermore, the addition of aniline to the electron-poor alkyne solely occurs through the N-centre in the present reaction and does not involve the para-position of the phenyl ring.19 When an equivalent yet intermolecular reaction mimicking the current transformation was conducted (Scheme 4a), C–C bond formation did not proceed (MVK; 42 or cyclopentenone; 43). This suggested that C–C bond formation from the intervening enamine was much faster in the intramolecular setting, which was in agreement with the untraceable acyclic enamine intermediate in the reaction mixture. For an unsymmetrical alkyne, a stronger electron-withdrawing group sets the regioselectivity of the annulation (Scheme 4b; 46). Based on the experimental observations, a mechanistic proposal is laid out in Scheme 4c.
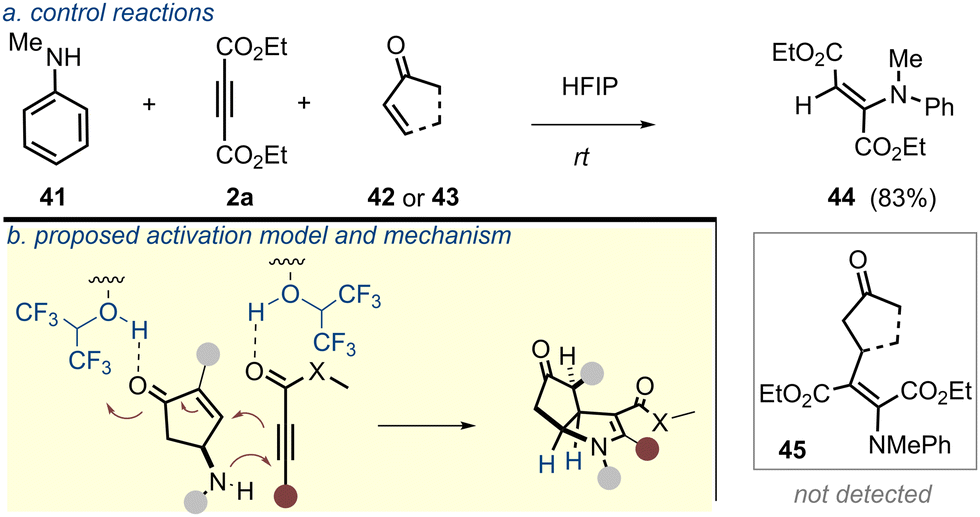 | ||
| Scheme 4 Control experiments and the mechanism. | ||
In summary, an operationally simple, metal-free, modular, and stereoselective approach was developed for the assembly of aza-bicyclo-[3.3.0]octane and cyclopenta[b]pyrrole derivatives. The process operates through a unique HFIP hydrogen-bonding network and capitalizes on the tandem alkyne hydroamination of alkyne and Csp2–Csp3 bond formation, constituting an overall [3+2]-annulation between various isomeric γ-amino cyclopentenones and electron- deficient alkynes. The current method, in addition to complementing existing processes for preparing similar scaffolds, also highlights an instance of HFIP-promoted new chemical transformation.
Funding from SERB (CRG/2020/000381) and CSIR-EMR-II (02(0395)/21/EMR-II) to JS is acknowledged. SP and SH thank CSIR for fellowship.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- (a) S. Thirumalairajan, B. M. Pearce and A. Thompson, Chem. Commun., 2010, 46, 1797–1812 RSC; (b) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801–1839 RSC; (c) S. S. Gholap, Eur. J. Med. Chem., 2016, 110, 13–31 CrossRef CAS PubMed.
- (a) D. Wang, Y. Fan, P. Yu and L. Désaubry, Chem. Commun., 2020, 56, 5584–5592 RSC; (b) N. A. Frolov and A. N. Vereshchagin, Int. J. Mol. Sci., 2023, 24, 2937 CrossRef CAS PubMed.
- C. Pramanik, P. Barik, S. A. Ali, D. S. Nayak, M. Ikbal, A. Mandal, R. Jana, S. Giri and S. Samanta, New J. Chem., 2023, 47, 6476–6527 RSC.
- For selected examples, see: (a) D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116, 7818–7853 CrossRef CAS PubMed; (b) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef PubMed; (c) M. L. R. Heffernan, Q. K. Fang, R. J. Foglesong, S. C. Hopkins, C. O. Ogbu, M. Soukri and K. L. Spear, PCT Int. Appl., 2011, WO2011011330 A2 Search PubMed; (d) M. E. Adams, M. B. Wallace, T. Kanouni, N. Scorah, S. M. O’Connell, H. Miyake, L. Shi, P. Halkowycz, L. Zhang and Q. Dong, Bioorg. Med. Chem. Lett., 2012, 22, 2411–2414 CrossRef CAS PubMed; (e) X.-T. Xu, X.-Q. Mou, Q.-M. Xi, W.-T. Liu, W.-F. Liu, Z.-J. Sheng, X. Zheng, K. Zhang, Z.-Y. Du, S.-Q. Zhao and S.-H. Wang, Bioorg. Med. Chem. Lett., 2016, 26, 5334–5339 CrossRef CAS PubMed; (f) Y. Tachibana, M. Miyagawa, T. Masuda and T. Hasegawa, PCT Int. Appl., 2011, WO2011013752 A1 Search PubMed.
- (a) T. Nagata, M. Nakagawa and A. Nishida, J. Am. Chem. Soc., 2003, 125, 7484–7485 CrossRef CAS PubMed; (b) I. S. Young and M. A. Kerr, J. Am. Chem. Soc., 2007, 129, 1465–1469 CrossRef CAS PubMed; (c) D. B. Martin and C. D. Vanderwal, Angew. Chem., Int. Ed., 2010, 49, 2830–2832 CrossRef CAS PubMed.
- For selected references, see: (a) C. Song, D. W. Knight and M. A. Whatton, Org. Lett., 2006, 8, 163–166 CrossRef CAS PubMed; (b) M. Fujiwara, M. Kawatsura, S. Hayase, M. Nanjo and T. Itoh, Adv. Synth. Catal., 2009, 351, 123–128 CrossRef CAS; (c) J. A. Malona, J. M. Colbourne and A. J. Frontier, Org. Lett., 2006, 8, 5661–5664 CrossRef CAS PubMed; (d) C. Huang, R. Ding, C. Song, J. Lu, L. Liu, X. Han, J. Wu, H. Hou and Y. Fan, Chem. – Eur. J., 2014, 20, 16156–16163 CrossRef CAS PubMed.
- (a) X.-Q. Mou, Z.-L. Xu, S.-H. Wang, D.-Y. Zhu, J. Wang, W. Bao, S.-J. Zhou, C. Yang and D. Zhang, Chem. Commun., 2015, 51, 12064–12067 RSC; (b) M. Yoshida, Y. Maeyama, M. A.-Amin and K. Shishido, J. Org. Chem., 2011, 76, 5813–5820 CrossRef CAS PubMed; (c) X. Yang, K. Wu, P. Wu, J. Chen, C. Sun and Z. Yu, Chem. – Eur. J., 2015, 21, 9323–9327 CrossRef CAS PubMed; (d) C. Wang, C. Dong, L. Kong, Y. Li and Y. Li, Chem. Commun., 2014, 50, 2164–2166 RSC; (e) L. Zhang, X. Xu, W. Xia and Q. Liu, Adv. Syn. Catal., 2011, 353, 2619–2623 CrossRef CAS; (f) L. Marin, V. Gandon, E. Schulz and D. Leboeuf, Adv. Syn. Catal., 2017, 359, 1157–1163 CrossRef CAS.
- (a) E. Semenova, O. Lahtigui, I. Ghiviriga and A. J. Grenning, Org. Lett., 2021, 23, 2263–2267 CrossRef CAS PubMed; (b) E. Li, P. Jia, L. Liang and Y. Huang, ACS Catal., 2014, 4, 600–603 CrossRef CAS.
- G. K. Veits, D. R. Wenz and J. R. d.Alaniz, Angew. Chem., Int. Ed., 2010, 49, 9484–9487 CrossRef CAS PubMed.
- (a) C. Jagadeesh, B. Mondal, S. Pramanik, D. Das and J. Saha, Angew. Chem., Int. Ed., 2021, 60, 8808–8812 CrossRef CAS PubMed; (b) B. Mondal, C. Jagadeesh, D. Das and J. Saha, Chem. Commun., 2022, 58, 2504–2507 RSC.
- (a) Z. He, A. Zajdlik and A. K. Yudin, Acc. Chem. Res, 2014, 47, 1029 CrossRef CAS PubMed also see:; (b) X.-Q. Zhang, T. Xu, C. Zhang, C. Wang and Y. Wang, Org. Chem. Front., 2023, 10, 680 RSC.
- (a) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef CAS PubMed; (b) X. Y. Chen, L. Zhang, Y. Tang, S. Yuan, B. Zhu, G. Chen and X. Cheng, Synlett, 2020, 878–882 CrossRef CAS.
- (a) X. Li, M. Chen, X. Xie, N. Sun, S. Li and Y. Liu, Org. Lett., 2015, 17, 2984–2987 CrossRef CAS PubMed; (b) Z. Tashrifi, M. M. Khanaposhtani, M. Biglar, B. Larijani and M. Mahdavi, Asian J. Org. Chem., 2020, 9, 969–991 CrossRef CAS; (c) X. Y. Chen, L. Zhang, Y. Tang, S. Yuan, B. Zhu, G. Chen and X. Cheng, Synlett, 2020, 878–882 CrossRef CAS.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aube, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- K. Kishi, S. Takizawa and H. Sasai, ACS Catal., 2018, 8, 5228 CrossRef CAS.
- Relative orientation was determined from 3J coupling constant.
- R. Fu, Y. Liu, T. Wu, X. Zhang, Y. Zhu, J. Luo, Z. Zhang and Y. Jiang, Chem. Commun., 2022, 58, 3525 RSC.
- (a) A. Fedotova, B. Crousse, I. Chataigner, J. Maddaluno, A. Y. Rulev and J. Legros, J. Org. Chem., 2015, 80, 10375 CrossRef CAS PubMed; (b) J. Aleksic, M. Stojanovic, J. Boskovic and M. Baranac-Stojanovic., Org. Biomol. Chem., 2023, 21, 1187 RSC.
- I. Colomer, ACS Catal., 2020, 10, 6023–6029 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2331633. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01065e |
| ‡ Equal contribution. |
| § Ex-faculty member of CBMR Lucknow. |
| This journal is © The Royal Society of Chemistry 2024 |