
Boosted charge transport through Au-modified NiFe layered double hydroxide on silicon for efficient photoelectrochemical water oxidation†
Sungkyun
Choi
a,
Sol A
Lee
a,
Jin Wook
Yang
a,
Woonbae
Sohn
a,
Jaehyun
Kim
a,
Woo Seok
Cheon
a,
Jaemin
Park
b,
Jin Hyuk
Cho
c,
Chung Won
Lee
a,
Sang Eon
Jun
a,
Sung Hyuk
Park
a,
Jooho
Moon
 *b,
Soo Young
Kim
*c and
Ho Won
Jang
*ad
*b,
Soo Young
Kim
*c and
Ho Won
Jang
*ad
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr
bDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: jmoon@yonsei.ac.kr
cDepartment of Materials Science and Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: sooyoungkim@korea.ac.kr
dAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea
First published on 29th July 2023
Abstract
Designing an appropriate oxygen evolution reaction (OER) catalyst for photoelectrochemical (PEC) water splitting is an urgent issue for providing high-efficiency solar to hydrogen energy production. Transition metals have been central to OER catalyst research due to their plentifulness and specific electronic structure, but overcoming their water oxidation limits, including high overpotential and sluggish kinetics, remains challenging. The effective usage of noble metals for the OER, such as an intentional introduction of low-concentration noble metals into earth-abundant materials, can complement the limited reserve of noble metals and largely enhance the entire efficiency of solar water oxidation. Herein, we developed an OER photoelectrode of Au-incorporated NiFe layered double hydroxide (LDH) placed on a strong light absorber n-type silicon (Au-NiFe LDH/n-Si). With a minimal Au content of 2.7% in the catalyst structure, synergistic effects between noble metal Au and transition metal-based NiFe LDH notably accelerated the OER kinetics while stabilizing the Si-based photoanode structure in corrosive alkaline electrolyte. Optimally fabricated Au-NiFe LDH through a facile two-step electrodeposition process on n-Si exhibited a high saturated photocurrent density of ∼37 mA cm−2, and the saturated photocurrent density could be reached at an early underpotential point of 1.2 V vs. RHE. Moreover, it operated for ∼50 hours in pH 11.5 electrolyte, showing 5 times higher stability than NiFe LDH/n-Si under the same alkaline conditions. One step further, a 1/48 decrease in recombination kinetics could be achieved through doping Au atoms into NiFe LDH, revealing the efficacious defect site passivation effect with the minimum amount of noble metal usage.
Introduction
Production of future energy, which possesses competitiveness in yield, cost, and sustainability compared with fossil fuels, is an urgent issue to be addressed for the upcoming industrial era.1–5 Photoelectrochemical (PEC) water splitting, converting solar energy into storable hydrogen energy, can be a clean and effective pathway for new energy sources. However, the oxygen evolution reaction (OER), a process with high overpotential and sluggish kinetics, acts as a bottleneck in the overall water-splitting system.6,7 Hence, numerous catalytic materials such as noble metals, metal oxides, metal (oxy)hydroxides, layered double hydroxides (LDHs), metal phosphates, and metal phosphides are widely researched for OER catalysts.8–13 Nonetheless, noble metal oxides (RuO2 and IrO2) or noble metal catalysts (Pt, Pd, and Au) have been researched for their high intrinsic activity in water splitting, but scarcity, high cost, and composition instability under OER electrolyte conditions have obstructed their further catalytic applications.14–16 In addition, the usage of noble metals has been mainly focused on electrocatalysts, and still, applications of noble metals in photocatalytic water splitting, particularly in the OER process, have shown limited performances.17,18 A strategy of integrating these noble metals with earth-abundant transition metal-based catalysts along with an effective light-absorbing substrate could minimize the utilization of noble metals, stabilize the catalytic structure, and facilitate the solar water oxidation efficiency.19–21Transition metal-based LDHs contain several advantages such as high-water oxidation activities, material tunability, and earth abundance. Specifically, NiFe LDH has been reported as a prominent OER catalyst in alkaline media with surprisingly low OER overpotential.11,22–24 Accordingly, a photoanode consisting of NiFe LDH deposited on strong photo-absorber Si has revealed competent PEC water splitting performances.3,25,26 However, the as-deposited oxides or hydroxide films could contain natural defect sites of vacancies or various surface trap sites, increasing the recombination and trapping of charges and lowering the stability in strong alkaline electrolytes.27–32 Also, the open-channel architecture of LDH cannot effectively passivate Si from corrosive aqueous solutions.25,33 Therefore, an appropriate catalytic engineering strategy for existing LDHs with Si structures is necessary to overcome their overall PEC water oxidation limits.
Herein, a photoanode of NiFe LDH electrodeposited on n-type Si (n-Si) could substantially improve overall PEC characteristics with high durability through the introduction of Au atoms into the NiFe LDH structure. Au has emerged as the practical element for improving PEC water splitting properties of photoelectrodes for its virtues of facile light absorption, chemical stability, and electronic structure modification.34–37 An optimized amount of Au could be effectively incorporated into Ni and Fe vacancy sites through consecutive two-step electrodeposition processes. Only with a minimum Au precursor concentration, compared to the concentration of the NiFe precursor, can synergistic interactions between metallic elements derive efficient electronic and band structure modification in a way much more favorable to PEC water oxidation. On top of this, the addition of Au into NiFe LDH significantly reduces the existing defect densities through the defect passivation effect, resulting in a notable decrease in the charge recombination rate along with an increase in charge carrier lifetime.
An optimally fabricated Au-NiFe LDH OER catalyst electrodeposited on n-Si exhibited considerable enhancements in PEC water splitting properties compared with a pristine NiFe LDH/n-Si photoanode. The Au-NiFe LDH/n-Si photoanode exhibited a high saturated photocurrent density of ∼37 mA cm−2, and the saturated photocurrent density could be reached at an early underpotential point of 1.2 V vs. RHE, which is the highest PEC OER property among those of recently reported Si-based photoanodes without buried junctions, while 200 mV overpotential is needed for NiFe LDH/n-Si to reach saturated photocurrent density. In addition, Au-NiFe LDH/n-Si endured ∼50 hours in pH 11.5 electrolyte, with 5 times higher stability than NiFe LDH/n-Si under the same alkaline conditions. One step further, a 1/48 decrease in recombination kinetics could be achieved through doping Au atoms into NiFe LDH, revealing the efficacious defect site passivation effect with a minimal amount of noble metal usage. Our work distinctively demonstrated a facile synthetic strategy of noble metal and non-noble transition metal-based catalysts, combining their advantages for a highly efficient solar water oxidation photoelectrode, widening the application of noble metal utilization, which has been mainly focused on electrochemical energy conversion areas, along with breaking the PEC water splitting performance limits of Si-based photoanodes.
Experimental
Materials preparation
Nickel chloride hexahydrate (NiCl2·6H2O), potassium chloride (KCl), sodium chloride (NaCl), and 1 N sodium hydroxide standard solution (1 N NaOH) were purchased from Daejung Chemical. Iron chloride hexahydrate (FeCl2·6H2O) was procured from KANTO. Gold(III) chloride hydrate (HAuCl4·xH2O) and an indium gallium alloy (InGa alloy) were procured from Sigma Aldrich. Boric acid (H3BO3) was obtained from Junsei. An n-Si wafer (100) (1–10 Ω cm) was purchased from PlutoSemi.Electrode preparation for electrodeposition
The n-Si wafer (100) (1–10 Ω cm) was diced into 1.5 cm × 1.5 cm n-Si pieces. Diced pieces were cleaned progressively with acetone, isopropyl alcohol, and deionized (DI) water by ultrasonication. For forming ohmic contact, cleaned pieces were scratched on the backside, and the InGa alloy was applied. Subsequently, copper wires were attached to the InGa alloy by applying the silver paste. Then, leaving the center active area of 0.5 cm × 0.5 cm, Kapton tape was attached to the front side of the n-Si pieces to prohibit electrolyte contact. 1.5 cm × 2.5 cm size FTO glass substrates were cleaned in the same way as n-Si pieces, and the active area of 1.5 cm × 1.5 cm was adjusted by attaching Kapton tape on the front side of FTO. The silver paste was applied on the front upside of FTO for electronic contact during the electrodeposition process.Electrodeposition
Electrodeposition of NiFe LDH and Au-NiFe LDH catalysts was conducted in a standard three-electrode system with a prepared n-Si and FTO electrode as the working electrode, a Pt plate as the counter electrode, and an Ag/AgCl (saturated) electrode as the reference electrode. NiFe aqueous precursor solution was synthesized using 0.02 M of NiCl2·6H2O and 0.005 M of FeCl2·6H2O, and 0.015 M of KCl was added as the additive for constituting the nano-wall structure of LDH. Au aqueous precursor solution was prepared with varied HAuCl4·xH2O concentrations of 10 mg/1 L, 15 mg/1 L, 20 mg/1 L, 30 mg/1 L, 50 mg/1 L, 100 mg/1 L, and 200 mg/1 L with 0.05 M of NaCl as the additive for the facile transport of Au ions during electrodeposition. NiFe LDH electrodeposition was conducted by applying a constant current of −0.25 mA cm−2 to the n-Si and FTO electrodes in the prepared NiFe precursor solution. For the synthesis of Au-NiFe LDH, the as-synthesized NiFe LDH deposited substrates were electrodeposited in Au aqueous precursor solution through the CV method of applying voltage in the range of −0.5 ∼ 0 V vs. RHE. After the deposition process, substrates were rinsed with DI and dried under nitrogen gas.Electrode preparation for PEC measurements
For further PEC measurements after electrodeposition, electrode fabrication of catalyst deposited n-Si pieces has proceeded. Again, the backside of n-Si was scratched, and the InGa alloy was pasted to form an ohmic contact. Then, the Cu wire was attached to the InGa alloy by applying silver paste to both parts of the Cu wire and InGa alloy. For the encapsulation of Cu wire attached n-Si, the epoxy-based resin was covered on all parts of the silicon except the active area used for measurements. To curate resin, the samples were dried for over 12 h in the air.PEC measurements
PEC measurements (Ivium Technologies, Nstat) were carried out through a three-electrode system using Ag/AgCl (saturated) as the reference electrode and a Pt plate as the counter electrode in 1 M NaOH standard solution (pH 14) with a quartz cell to avoid UV absorption. For the light source, a XE arc lamp (Abet Technologies, LS150) was utilized, and its light intensity was adjusted to 1 sun (100 mW cm−2, AM 1.5G) through a reference photodiode. For J–V characteristics measurements, the anodic potential was swept with a scan rate of 20 mV s−1. Stability measurement was conducted by applying 1.5 V vs. RHE in buffered NaOH. Buffered NaOH electrolyte was synthesized by mixing 1 M NaOH and boric acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The IPCE was determined using the light source and a monochromator (MonoRa150) by applying 1.5 V vs. RHE, the voltage at which saturation current density is reached. The EIS was obtained in the frequency range of 250 kHz to 0.1 kHz by applying the potential near the onset potential of photoanodes (0.91 ± 0.1 V vs. RHE). EIS data were fitted to equivalent circuits using the Z plot 2.x software. The potential measured with the Ag/AgCl reference was calculated as the potential versus the reversible hydrogen electrode (RHE) by following the Nernst equation:
:1 molar ratio. The IPCE was determined using the light source and a monochromator (MonoRa150) by applying 1.5 V vs. RHE, the voltage at which saturation current density is reached. The EIS was obtained in the frequency range of 250 kHz to 0.1 kHz by applying the potential near the onset potential of photoanodes (0.91 ± 0.1 V vs. RHE). EIS data were fitted to equivalent circuits using the Z plot 2.x software. The potential measured with the Ag/AgCl reference was calculated as the potential versus the reversible hydrogen electrode (RHE) by following the Nernst equation:where ERHE is the potential versus RHE, EAg/AgCl is the experimentally measured potential versus Ag/AgCl, and
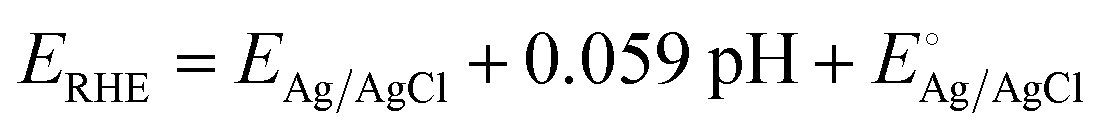
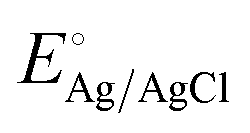 is 0.198 V at 25 °C.
is 0.198 V at 25 °C.
The Mott–Schottky analysis was conducted under light-off conditions with a frequency applied at 5 kHz. The flat band potential (Efb) was obtained by following the Mott–Schottky equation:
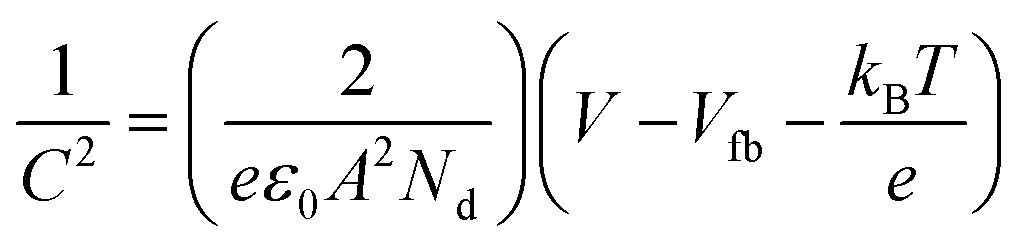
IMPS analysis was performed in the three-electrode configuration under white light illumination, along with a modulation intensity of 10% and frequency modulation range of 100 kHz to 0.1 Hz through a potentiostat (PP211, Zahner) and EC workstation (Zennium, Zahner). The ktrans and krec can be obtained from the following equations:
| ktrans + krec = 2πf |
| ktrans = transfer efficiency × (ktrans + krec) |
Characterization
The surface morphology of the synthesized catalysts was probed by FESEM (Merlin Compact, Jeiss). Crystalline structures and atomic distributions were obtained through TEM (JEM-2100F, JEOL) and Cs-corrected monochromated TEM/STEM (Themis Z, Thermo Fisher). Chemical valence states of the synthesized samples were analyzed through XPS (AXIS-His, Kratos). The oxygen gas production calculated through faradaic efficiency was measured by GC (FID-GC, PerkinElmer). XRD (X'Pert Pro, PANalytical) was carried out to identify the crystal phases of samples. For the verification of the chemical bonding of samples, Raman spectroscopy (Lab RAM HR, Horiba Jobin Yvon) was utilized. UV-vis transmittance (T) and reflectance (R) spectra were examined using a UV-vis spectrometer (Cary 5000, Agilent Technologies). The absorption (A) from UV-vis measurements could be calculated using the following equation:| A = 100 − T − R |
The absorption coefficient (α) was obtained using the following equation:
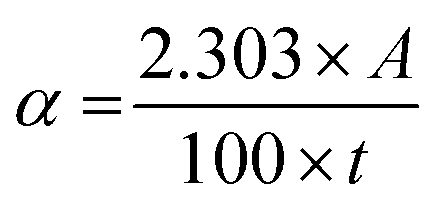
Indirect bandgap (Eg) calculation was fitted through Tauc plots using the formula:
| αhν = C(hν − Eg)1/2 |
In this study, all the spin-polarized density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP) with the projector augmented wave method for the core region and a plane-wave kinetic energy cut-off of 400 eV. The generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) for the exchange-correlation potentials was used. The optimized lattice constants of bulk FCC Au and NiFe LDH are a = 4.171 Å and a = 12.404 Å, b = 6.202 Å, c = 23.005 Å, α = 90°, β = 90°, and γ = 120°, respectively. One Ni or Fe atom was removed or replaced with an Au atom in order to simulate NiFe LDH with a Ni/Fe vacancy or Au single atom substituted model. A 2 × 1 × 1 gamma-centered Monkhorst–pack sampled k-point grid was employed to sample the reciprocal space for the slab models. Atoms in the systems are free to move in all directions until the convergence of energy and residual force on each atom is less than 1 × 10−4 eV and 0.05 eV Å−1, respectively.
Results and discussion
The schematic illustrations of the three-electrode system with a potentiostat and two-step sequential electrodeposition method for synthesizing Au-NiFe LDH on n-Si are shown in Fig. 1a. Recently, the sequential electrodeposition method has been effectively conducted for generating distinctive catalyst morphologies.4,38 Different types of electrodeposition modes were used to deposit Ni, Fe, and Au ions. As shown in Fig. 1b, a NiFe LDH film is deposited on n-Si by applying a constant current of −0.25 mA cm−2versus a reverse hydrogen electrode (RHE). The inset indicates a schematic of the nano-wall structure of NiFe LDH on n-Si. The introduction of Au precursors into the NiFe LDH structure was conducted through the cyclic voltammetry (CV) electrodeposition method, as shown in Fig. 1c. Linear sweep voltammetry (LSV) or CV methods can be useful in electrodeposition to deposit nanoparticles or single atom catalysts on specific substrates. The range of the CV potential, which can efficiently deposit Au on NiFe LDH for the highest water splitting LSV performance, could be verified through electrodepositions in different CV potential ranges for the reduction of Au ions (Fig. S1†). The Au-NiFe LDH photoanode in which Au is electrodeposited in the potential range of −0.5 V to 0 V versus RHE on NiFe LDH/n-Si exhibited the best PEC LSV performance, and Fig. 1c shows the obtained optimized CV deposition curve of Au on NiFe LDH/n-Si. The inset indicates a schematic of the Au-NiFe LDH/n-Si structure of Au doped into the NiFe LDH film. Field emission scanning electron microscopy (FESEM) was conducted to investigate the morphologies of catalysts. As shown in Fig. 1d, the wall-like structure of NiFe LDH was initially electrodeposited on the silicon surface. The adsorption of Au into the NiFe-based structure results in a distinctive change in morphology as shown in Fig. 1e. Nano-walls in LDH become denser and thicker by filling hollow spaces in nano-walls through the specific structure that originated from Au's incorporation into NiFe LDH. Insets show photographs of catalysts deposited with 0.5 × 0.5 cm2 on silicon substrates cut off into 1 × 1 cm2 area.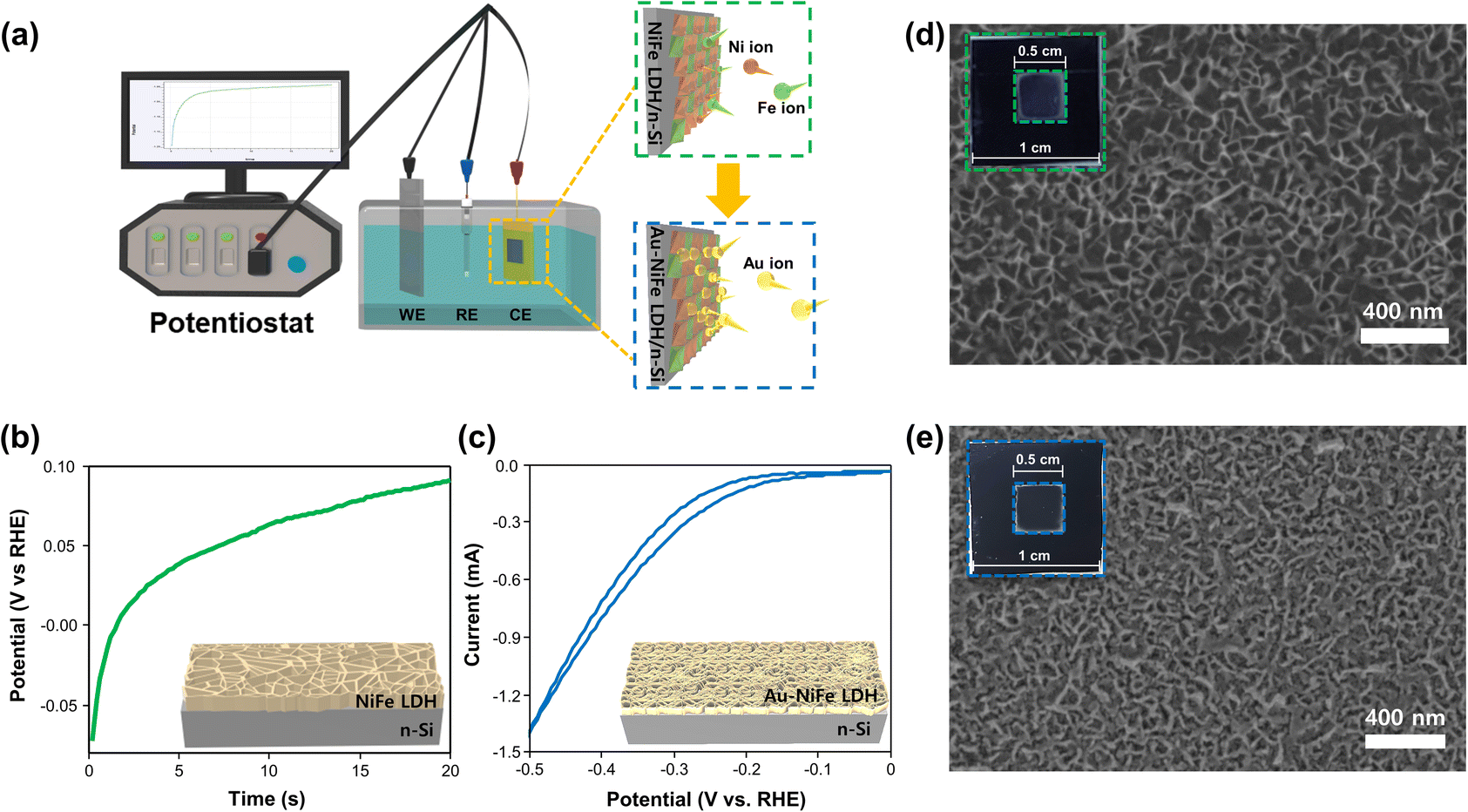 | ||
| Fig. 1 Schematic illustrations of the synthesis procedure. (a) Schematic of deposition of Au-NiFe LDH on n-Si by the sequential electrodeposition method. (b) Chronopotentiometry curve of electrodeposition of NiFe LDH on n-Si. (c) Cyclic voltammetry curve of electrodeposition of Au on NiFe LDH/n-Si. Insets show schematics of the NiFe LDH and Au-NiFe LDH on the silicon substrate. FESEM images of (d) NiFe LDH/n-Si and (e) Au-NiFe LDH/n-Si. Insets show photographs of each sample. | ||
For an in-depth study of the crystal structure and the element distributions, transmission electron microscopy (TEM) and aberration-corrected (AC) high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) were carried out. The TEM image of Au-NiFe LDH/n-Si (Fig. 2a) shows the Au-doped NiFe LDH structure of thin nanosheets with a height of 30 ∼ 40 nm on n-Si. A thin catalyst film below 50 nm can be advantageous for facile charge transport and light absorption for Si, facilitating PEC water oxidation.3 In the AC HAADF-STEM image and its corresponding energy dispersive spectroscopy (EDS) elemental mapping (Fig. 2b), uniformly doped Au into background NiFe LDH can be observed. The EDS spectrum reveals that the atomic ratio of Ni, Fe, and Au is 1:1.35:0.06 in Au-NiFe LDH (Fig. S2 and Table S1†), revealing the effective dispersion of Au atoms in the catalyst structure with a low atomic percentage of 2.76%. The STEM and TEM images of NiFe LDH and Au-NiFe LDH layers exfoliated from the substrate show the as-deposited catalyst's 3-dimensional (3-D) morphology. A layered structure composed of NiFe basal planes can be detected (Fig. S3a–c†), and this interconnected layer structure is maintained in Au-NiFe LDH with planes composed of Ni, Fe, and Au elements (Fig. S3d–f†). HAADF-STEM line-scan profiles of Ni, Fe, and Au across the basal plane of Au-NiFe LDH (Fig. 2c) reveal the consistency of Au doping along the NiFe LDH structure without specific aggregation. The atomic resolution AC HAADF-STEM image of Au-NiFe LDH (Fig. 2d) distinctively shows atomically dispersed Au atoms with a bright contrast in the NiFe LDH lattice of 0.25 nm interplanar distance which corresponds to the (012) plane of NiFe LDH.20 In order to verify the feasibility of atomic insertion of Au into the NiFe LDH structure as shown in TEM and STEM analyses, deeper atomic structure examination using density functional theory (DFT) calculations was carried out. We have calculated the total Gibbs free energy for the cases of Au fulfillments in Ni and Fe natural vacancy defects in the NiFe LDH basal plane. As shown in Table S2,† total Gibbs free energies of NiFe LDH when Ni vacancy sites are filled with Au atoms (AuNi = −915.05 eV) or Fe vacancy sites are filled with Au atoms (AuFe = −911.54 eV) were lower than the total Gibbs free energies of NiFe LDH which had left its Ni or Fe vacancy sites empty (VNi = −913.3 eV and VFe = −909.27 eV). Furthermore, the NiFe LDH structure schematic from DFT calculations in the case of Au occupation in Ni vacancy sites (Fig. 2e) reveals that the Gibbs free energy difference (ΔEf) by the addition of Au is −1.531 eV per site along with ΔEf = −2.031 eV per site for Au occupation in Fe vacancy sites (Fig. 2f), indicating the structural stabilization of the catalyst through Au introduction.
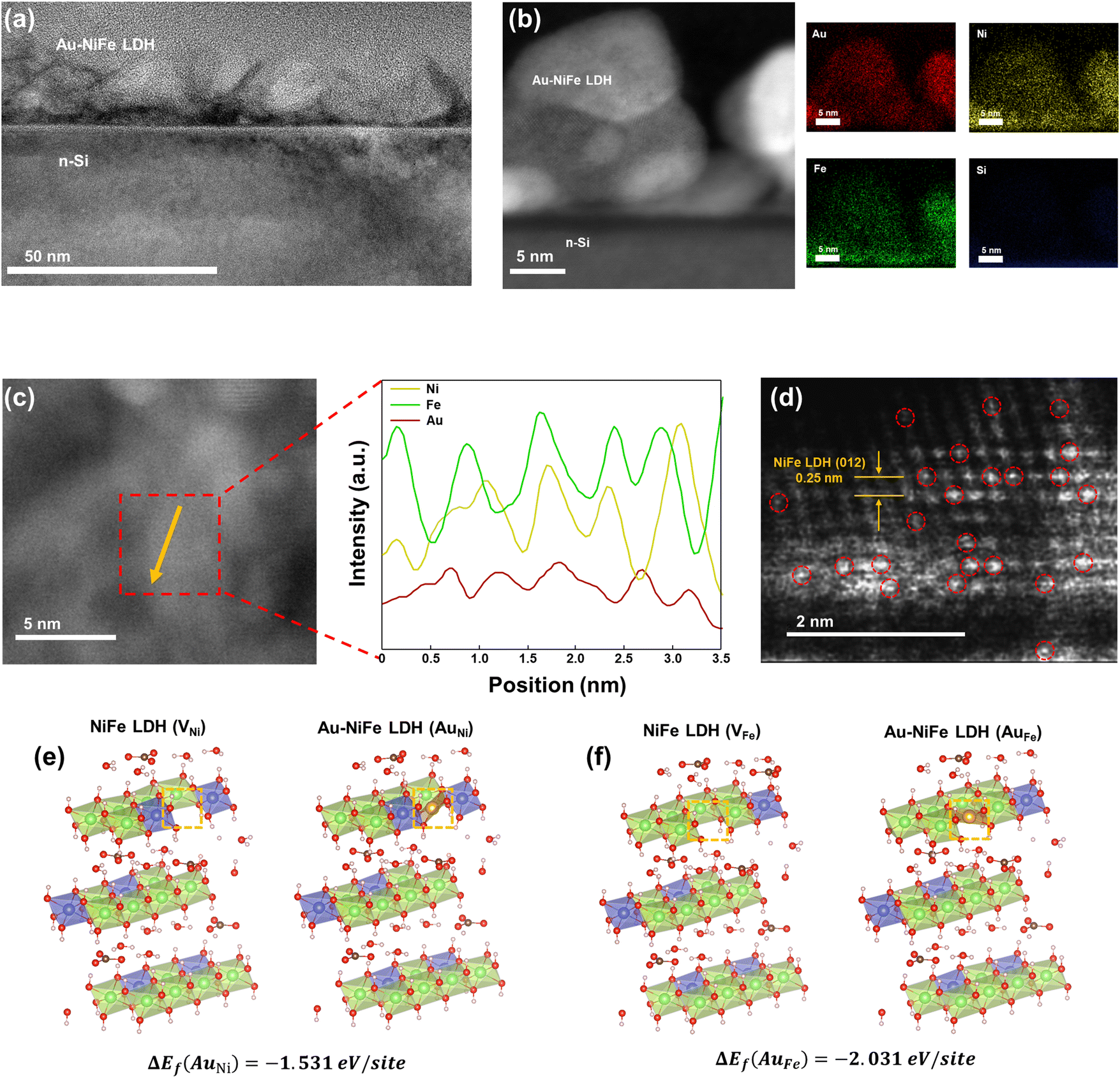 | ||
| Fig. 2 (a) TEM image of Au-NiFe LDH/n-Si. (b) HAADF-STEM image and the corresponding EDS maps of Au-NiFe LDH/n-Si. (c) HAADF-STEM image of Au-NiFe LDH with line-scan profiles of elements corresponding to the arrowed area. (d) Atomic resolution HAADF-STEM image of Au-NiFe LDH. Au atoms are represented by red circles. Schematics of the DFT calculation results of occupation of vacancy sites with Au atoms when vacancy sites are (e) Ni sites and (f) Fe sites in NiFe LDH. | ||
X-ray photoelectron spectroscopy (XPS) measurements were carried out to examine the chemical valence state of the as-deposited catalysts on the Si substrate. For the comparison of Au valence status with the existence of NiFe LDH, the Au film was directly electrodeposited on Si through the same Au precursor solution and electrodeposition mode as it was performed on NiFe LDH. Fig. S4 and S5† show Au 4f XPS spectra of Au film/n-Si and Au-NiFe LDH/n-Si. Peaks at 87.9 eV and 84.3 eV correspond to Au 4f5/2 and 4f7/2 of metallic Au0 species (Fig. S5a†).39 While only two distinct Au metallic peaks appear in the Au film, Au 4f XPS spectra in Au-NiFe LDH (Fig. S5b†) exhibit deconvolution into Au0 (87.5 eV at Au 4f5/2 and 83.8 eV at Au 4f7/2) and Auδ+ of 0 < δ < 3 (88 eV at Au 4f5/2 and 84.3 eV at Au 4f7/2) species due to the electronic interaction between Au and NiFe.40 In addition, the XPS peak shift of Ni and Fe elements could be detected as a positive 0.3 eV peak shift of Ni and a negative 0.6 eV peak shift of Fe in Au-NiFe LDH compared with NiFe LDH, as shown in Fig. S6.† The deconvolution of component peaks in Au and peak shifts in Ni and Fe would come from the bonding interaction and charge flow between Au and NiFe elements, leading to the restructuring of the surface chemical state.41,42
PEC water oxidation activities of demonstrated photoanodes were investigated, as shown in Fig. 3. Current–potential (J–V) characteristics of Au-NiFe LDH/n-Si photoanodes were optimized with the concentration of Au precursor solution, which is electrodeposited on NiFe LDH/n-Si (Fig. S7†). The photoanode synthesized with a concentration of 50 mg L−1 Au precursor solution of HAuCl4·3H2O powder dissolved in NaCl electrolyte, which is a comparably far lower concentration than that of NiFe precursor solution (5744 mg L−1), showed the highest water oxidation performance. Excess amounts of Au addition prohibited silicon's light absorption, leading to the deterioration of PEC performances. The optimized Au-NiFe LDH/n-Si photoanode exhibited superior LSV characteristics of ∼37 mA cm−2 saturated photocurrent density, and the saturated photocurrent density could be reached at an early underpotential point of 1.2 V vs. RHE. The improvement of PEC J–V properties by introducing Au atoms into NiFe LDH can be observed in Fig. 3a. 230 mV potential could be decreased in Au-NiFe LDH/n-Si compared with pristine NiFe LDH/n-Si to reach saturation photocurrent density along with an overall cathodic shift. Exhibiting high saturated photocurrent density in early potential regions indicates that the photoanode has fast charge transfer ability and a low recombination rate, leading to a rapid approach to its maximum water oxidation properties. A comparison of photocurrent density at 1.23 V vs. RHE and the potential to reach saturated photocurrent density with those of previously reported n-Si-based photoanodes without buried junction structures is shown in Fig. 3b.4,9,43–53 Au-NiFe LDH/n-Si demonstrated in this work required the minimum potential to reach saturated photocurrent density, exhibiting the highest photocurrent density at a water oxidation potential of 1.23 V vs. RHE, representing superior charge transport properties and water oxidation performance. Stability tests were carried out in pH 11.5 buffered NaOH electrolyte by applying a constant voltage of 1.5 V vs. RHE (Fig. 3c). Notably, Au-NiFe LDH/n-Si steadily retained its initial photocurrent for over 50 hours, while NiFe LDH/n-Si revealed photocurrent decay before 10 hours of the stability test. The durability of the Au-NiFe LDH photoanode could be identified by comparing J–V characteristics before and after the stability test (Fig. S8†). Even though a small anodic shift occurred, photocurrent density was nearly maintained after the stability test, inferring the adequate durability of the photoanode under alkaline conditions. The chemical environment and crystal structure changes after the stability test in Au-NiFe LDH have been unraveled through XPS and TEM analysis. XPS wide curves in Fig. S9a and b† show that the main element peaks of Ni 2p, Fe 2p, and Au 4f are distinctively represented, and no big difference in total curve fitting occurred after the stability test. A detailed valence state change of Au is shown in Fig. S9c and d.† A negative peak shift of 0.5 eV and an increase in Auδ+ content after the stability test imply an increase in oxidized Au states after the water oxidation reaction.6,40 TEM investigation revealed that the catalyst morphology of Au-NiFe LDH had been nearly retained even after the long-term OER under electrolyte conditions (Fig. S9e and f†).
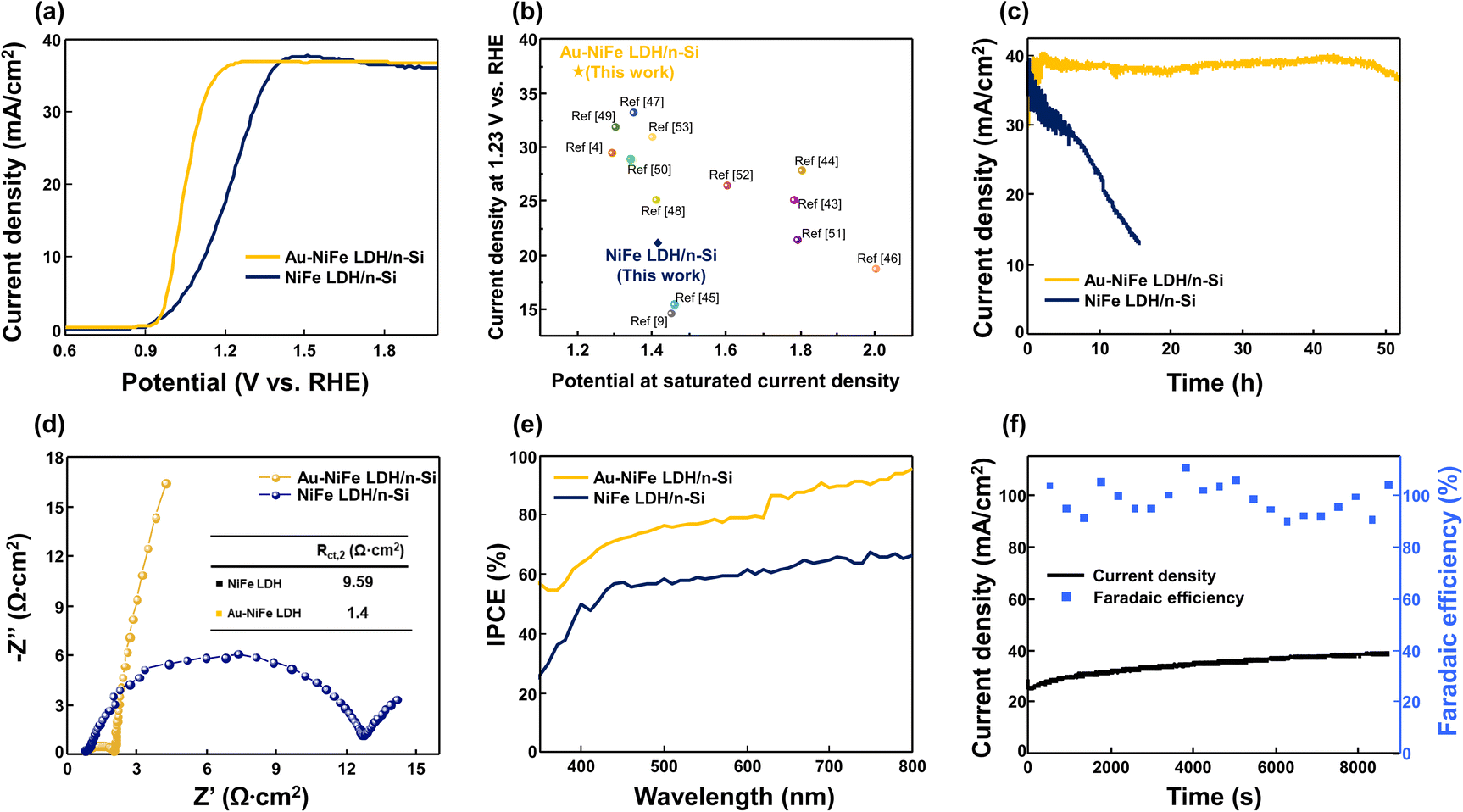 | ||
| Fig. 3 PEC performances of Au-NiFe LDH/n-Si and NiFe LDH/n-Si photoanodes under AM 1.5G sunlight (100 mW cm−2). (a) J–V curves of Au and NiFe LDH based silicon photoanodes in 1 M NaOH aqueous solution. (b) Comparison of photocurrent density at water oxidation potential (1.23 V vs. RHE) and potential to reach saturated photocurrent density with those of previously reported n-Si based photoanodes. (c) Stability tests of photoanodes in buffered NaOH aqueous solution. (d) EIS spectra and (e) IPCE curves of Au-NiFe LDH/n-Si and NiFe LDH/n-Si photoanodes in 1 M NaOH aqueous solution. (f) Faradaic efficiency and the corresponding chronoamperometric curve of the Au-NiFe LDH/n-Si photoanode in 1 M NaOH electrolyte. | ||
In order to examine the effect of annealing on each photoanode, heat treatment at 300 °C was conducted in an N2 atmosphere for 1 hour. As shown in SEM images of annealed samples (Fig. S10a and b†), morphologies of annealed NiFe LDH (a-NiFe LDH) and annealed Au-NiFe LDH (a-Au-NiFe LDH) were transformed into more dense and aggregated structures. While J–V characteristics were drastically diminished in NiFe LDH through the annealing process, a-Au-NiFe LDH nearly retained its water oxidation activities with a slight increase in overpotential (100 mV to reach saturation photocurrent density) and gradual decrease of photocurrent density in the stability test in comparison with the Au-NiFe LDH photoanode (Fig. S10c and d†). Electrochemical impedance spectroscopy (EIS) was implemented to probe charge kinetics during the OER process with an external bias near onset potential (Fig. 3d). An equivalent circuit consisting of charge transfer resistance (Rct) and capacitance (C) was utilized to match the measured EIS data (Fig. S11, Tables S3, and S4†) with real values. Au-NiFe LDH/n-Si showed a remarkable decrease in the Rct2 value of 1.4 Ω cm2 compared to NiFe LDH of 9.59 Ω cm2 as can be seen in the semicircle arc size and inset in Fig. 3d, demonstrating the enhancement of the charge transfer rate between the silicon substrate and deposited catalysts. Au-NiFe LDH/n-Si revealed the cathodic shift of flat-band potential compared with NiFe LDH/n-Si, as shown in Fig. S12a,† indicating the improvement of charge transport through the increased surface band bending.3 High light sensitivity of the Au-NiFe LDH/n-Si photoanode could be detected through the chronoamperometric measurements at 1.5 V vs. RHE under light chopping on/off conditions (Fig. S12b†), also inferring the low charge recombination with neglectable leakage current in the photoanode.4 The above PEC results indicate that the synergistic effects exerted by Au and NiFe LDH could critically contribute to the enhancement of charge transfer ability and the stability of photoanodes in alkaline electrolytes.
To inspect the overall water splitting efficiency of photoanodes, incident photon-to-current efficiency (IPCE) was obtained at the voltage where saturation current density was reached (Fig. 3e). IPCE of Au-NiFe LDH/n-Si exceeds all the measured wavelengths of NiFe LDH/n-Si, reaching nearly 95% of superior efficiency at a wavelength near 800 nm, while NiFe LDH/n-Si showed ∼60% efficiency in the same wavelength range. For the investigation of oxygen production in the optimized Au-NiFe LDH/n-Si photoanode, faradaic efficiency was calculated through gas chromatography (GC), as shown in Fig. 3f. For the measured time of 9000 s, Au-NiFe LDH/n-Si retained constant photocurrent density, outperforming 100% faradaic efficiency. Few points higher than 100% are due to the surface block from O2 bubbles during the OER.
Insightful analysis has been subsequently carried out to elucidate the considerable expedition of PEC water oxidation properties through the incorporation of Au into an as-deposited NiFe LDH structure. Photoluminescence (PL) spectroscopy was employed to explore the charge flow behaviors of catalysts. In steady-state PL spectra (Fig. 4a), Au-NiFe LDH revealed largely increased PL intensity than NiFe LDH in around 500–600 nm range, revealing a drastic increase in the photo-generated charge flow rate by incorporating Au into NiFe LDH, which narrows down the electron–hole recombination gap and passivates defect sites in pristine NiFe LDH.54,55 Time-resolved photoluminescence (TRPL) was implemented to investigate charge transport dynamics. PL lifetime was investigated as presented in the Fig. 4b inset. Au-NiFe LDH exhibited a ∼17% increase in charge carrier lifetime compared to NiFe LDH, indicating a reduced recombination rate of charge carriers with increased charge separation efficiency.56,57 Raman spectroscopy was employed to identify the chemical bonding of elements in the demonstrated catalytic materials, as shown in Fig. 4c. Strong Si substrate peaks at around 513–526 cm−1 could be detected in both NiFe LDH/n-Si and Au-NiFe LDH/n-Si photoanodes. A distinct red shift in the Fe3+/Ni2+–O–Ni2+ metal–oxygen bond peak has occurred in Au-NiFe LDH/n-Si (464.6 cm−1) compared to NiFe LDH/n-Si (434.6 cm−1), implying the decrease of bond energy and the increase of bond length through the effective insertion of Au atoms into Ni or Fe sites. Intriguingly, the cation vacancy peak at around ∼600 cm−1 in NiFe LDH disappeared in Au-NiFe LDH, revealing the successful vacancy defect passivation role of Au in the Au-NiFe LDH catalyst.42,58,59 Significantly reduced defect sites could promote the stabilization of the catalyst layer, which is consistent with the DFT calculation analysis of doping Au into NiFe LDH (Fig. 2e and f), resulting in a substantial improvement in stability (Fig. 3c).
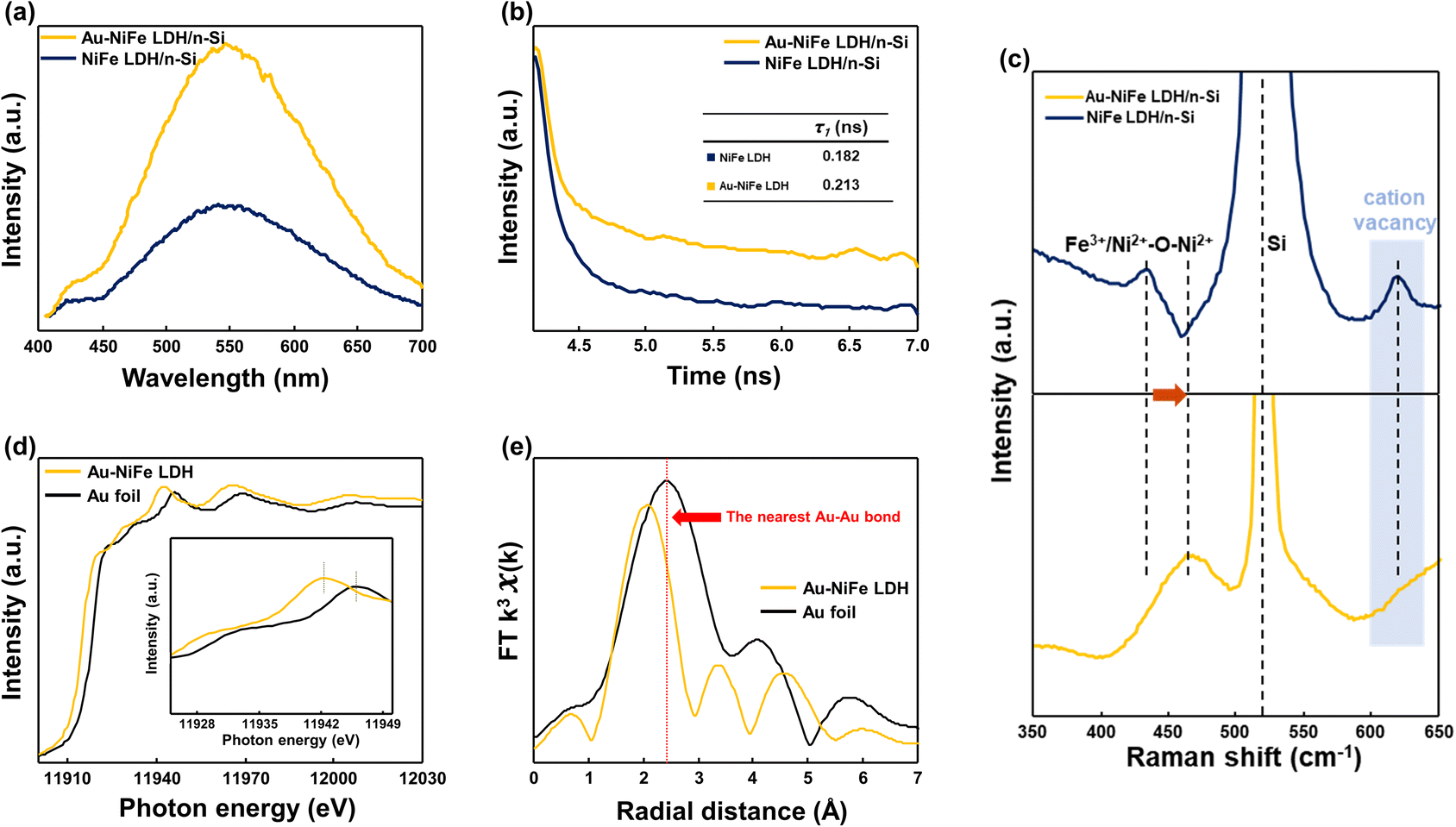 | ||
| Fig. 4 (a) Steady-state photoluminescence (PL) spectra, (b) time-resolved photoluminescence (TRPL) spectra, and (c) Raman spectra of Au-NiFe LDH/n-Si and NiFe LDH/n-Si photoanodes. (d) XANES and (e) EXAFS spectra in the Au L3-edge for Au-NiFe LDH and Au foil. Inset in (d) shows an expanded pre-edge region. | ||
X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) were carried out to probe chemical configuration and electronic structures of the Au element in our electrodeposited Au-NiFe LDH and Au foil for comparison. The absorption intensity in the Au L3-edge region of 11925–11945 eV is related to the electron transition from 2p2/3 to unoccupied 5d states (Fig. 4d). As shown in the Fig. 4d inset of the extended pre-edge region, Au-NiFe LDH shows a larger absorption intensity than Au foil, indicating that the depletion of the Au d-band is larger in Au-NiFe LDH than in Au foil. The deviation of white line peaks between Au-NiFe LDH and Au foil also implies a dissimilar gold atomic electronic structure due to the electronic interaction of Au with neighboring Ni or Fe elements in Au-NiFe LDH.14,60 The coordination environment of Au in NiFe LDH was further investigated through EXAFS analysis (Fig. 4e). The nearest Au–Au bond peak position of Au-NiFe LDH shows a meaningful negative shift compared with Au foil, originating from the geometrical bonding interaction between Au–Ni and Au–Fe.61 Through the EXAFS fitting results (Fig. S13 and Table S5†), the derived Au–Au radial distance of Au-NiFe LDH (2.63 ± 0.3 Å) is lower than that of Au foil (2.87 Å), inferring the scattered distribution and the decreased thickness of Au in the NiFe LDH structure instead of existing in a bulk state of Au.62
Enhancements in charge transport ability and photoactivity in Au-NiFe LDH were systematically verified through band structures. In the first step, the optical bandgaps (Eg) of NiFe LDH and Au-NiFe LDH were obtained through UV-vis spectroscopy measurements. Then, using the UV-vis transmittance and reflectance spectra, the absorption spectra were acquired, which could be utilized for calculating absorption coefficients (α) (Fig. S14a and b†). Lastly, the Eg of NiFe LDH (2.25 eV) and Au-NiFe LDH (2.2 eV) were measured through Tauc plots (Fig. S14c and d†).63,64 Ultraviolet photoelectron spectroscopy (UPS) was conducted to determine materials' band edge positions and work functions. In the high binding energy region of the UPS, secondary electron emission (SEE) spectra can be observed (Fig. 5a).65 Through calculating the energy difference between SEE spectra cut-off energy and He(I) photon source energy (21.22 eV), the work functions of NiFe LDH (3.96 eV) and Au-NiFe LDH (4.1 eV) could be obtained. Fig. 5b shows the low binding energy region of the UPS, and the energy difference between the Fermi level and valence band (VB) maximum (EF − EVBM) can be measured through these VB spectra. The first-derivative of the VB spectra is used to determine EF − EVBM of NiFe LDH (0.67 eV) and Au-NiFe LDH (0.43 eV). The resulting photoanode band diagrams of NiFe LDH/n-Si and Au-NiFe LDH/n-Si are shown in Fig. 5c and d. Intriguingly, compared with NiFe LDH/n-Si, Au-NiFe LDH showed a type II heterojunction with larger band bending, which has more positive catalyst VB energy than silicon VB energy. Type II heterojunction construction in the Au-NiFe LDH/n-Si photoanode system stems from the low value of EF − EVBM in Au-NiFe LDH (0.43 eV), exhibiting p-type semiconducting catalyst characteristics. Through constructing a distinct type II heterojunction in the Au-NiFe LDH/n-Si photoanode, the hole charge carrier can effectively transfer between silicon of the light absorbing substrate and Au-NiFe LDH of the water splitting catalyst, expediting PEC water oxidation through a facile charge travel pathway.
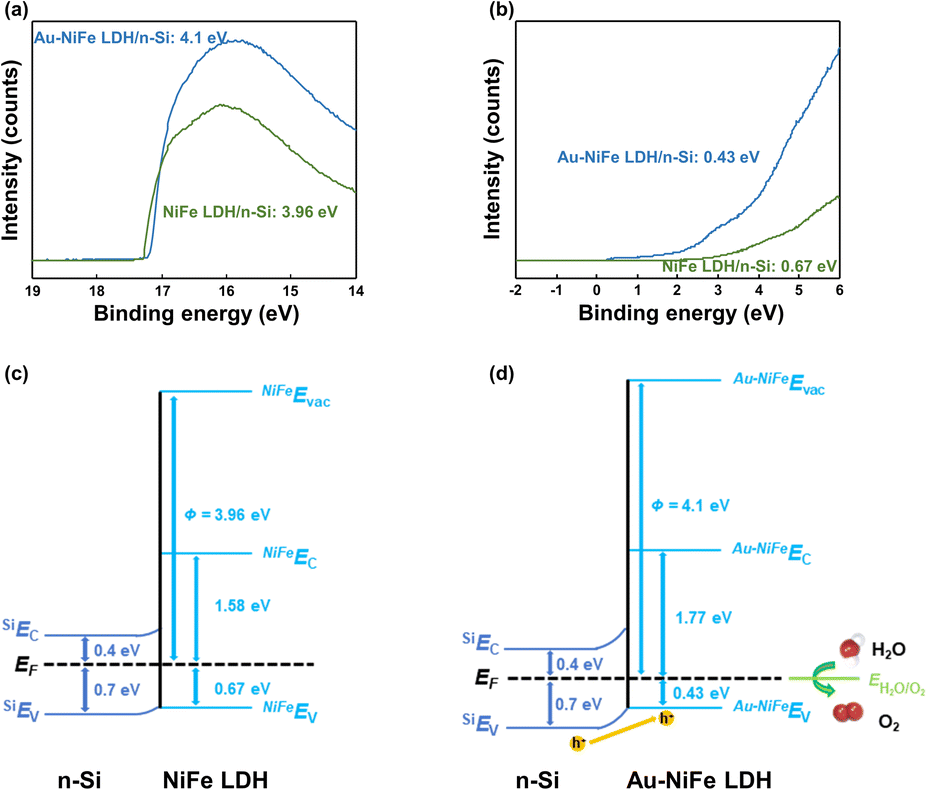 | ||
| Fig. 5 (a) The secondary electron emission spectra of Au-NiFe LDH/n-Si and NiFe LDH/n-Si. (b) Valence band spectra (EF − EVBM) of Au-NiFe LDH/n-Si and NiFe LDH/n-Si. Energy band diagram schematics of the (c) NiFe LDH/n-Si and (d) Au-NiFe LDH/n-Si. | ||
In PEC water splitting photoanodes, hole charge accumulation at the catalyst-electrolyte interface and charge lifetime are important factors. Specifically, charge kinetics can be hampered when the surface recombination rate constant (krec) surpasses the charge transfer rate constant (ktrans). The intensity-modulated photocurrent spectroscopy (IMPS) method investigates the rate constants of charge transfer and recombination in PEC devices by analyzing photocurrent in accordance with sinusoidal perturbations in the DC illumination. In the semicircle plot of the normalized IMPS graph, the apex of the semicircle refers to ktrans + krec and the ratio between normalized real photocurrent intercepts at the low frequency and high frequency indicates the charge transfer efficiency of ktrans/(ktrans + krec). Through the above values obtained from the semicircle apex and the normalized real photocurrent intercepts, ktrans and krec can be calculated, respectively.4,57Fig. 6a and b show the IMPS results of NiFe LDH/n-Si and Au-NiFe LDH/n-Si photoanodes at various water oxidation potentials. The calculated ktrans values of NiFe LDH/n-Si and Au-NiFe LDH/n-Si are similar, which are 0.735 s−1 and 0.214 s−1, respectively, in the onset potential region of 0.9 V vs. RHE. On the other hand, the obtained krec of NiFe LDH is 40756 s−1 and krec of Au-NiFe LDH is 846 s−1, which show a substantial difference in comparison with the photoanodes' ktrans values. Schematics of NiFe LDH-based Au-modified catalysts are presented in Fig. 6c and d. The as-prepared electrodeposited NiFe LDH possesses a couple of natural defective sites such as oxygen, Ni, and Fe vacancies. These defective sites in the catalyst induce the charge recombination state (Ф) and hinder hole charge transport for water oxidation by the trapping and recombination of charges (Fig. 6c).6,66 By modifying the existing NiFe LDH structure through the adequate addition of Au by the sequential electrodeposition process, Au atoms can effectively passivate the surface charge trap sites in NiFe LDH. Surface defect passivation by the introduction of Au atoms prevents charge recombination, facilitating facile charge transport into the catalyst/electrolyte interface (Fig. 6d).67 Furthermore, the electronic environment of NiFe LDH changes due to the atomic interaction between Au and NiFe LDH, accelerating water oxidation activities with remarkable enhancement in stability under strongly alkaline conditions.
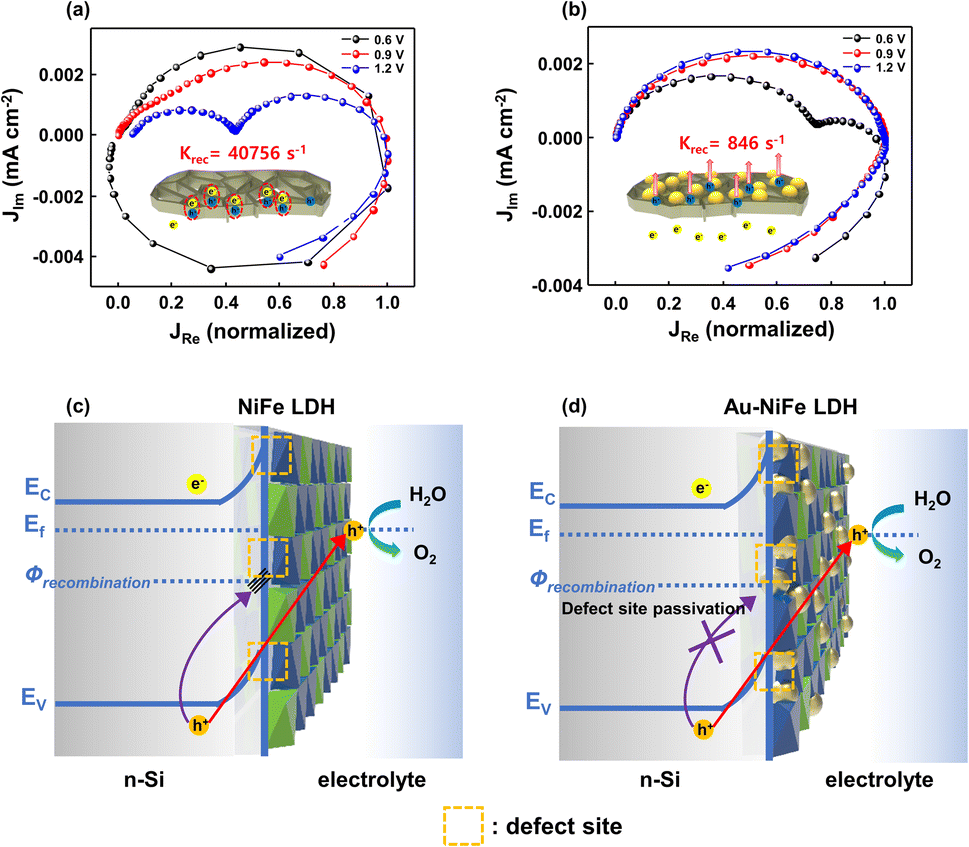 | ||
| Fig. 6 IMPS Nyquist plot of (a) NiFe LDH/n-Si and (b) Au-NiFe LDH/n-Si. Insets show schematics of charge recombination and transfer with charge recombination rates. Schematics of charge transfer & recombination processes with band structures during the OER in (c) NiFe LDH/n-Si and (d) Au-NiFe LDH/n-Si. | ||
Conclusions
As a strategy for efficient utilization of noble metals for PEC water oxidation, we successfully synthesized the catalyst structure of Au incorporated into NiFe LDH (Au-NiFe LDH) by adjusting an adequate amount of Au with a facile two-step electrodeposition process. As a result, Au could be effectively substituted for Ni or Fe vacancy sites, energetically stabilizing the entire catalyst structure. Synergistic effects exerted between Au and NiFe LDH catalysts could drastically enhance solar water splitting performances in the photoanode, exhibiting a photocurrent density of ∼37 mA cm−2 at 1.2 V vs. RHE, which has not been reported for Si-based photoanodes without buried junctions, retaining more than 50 hours under the alkaline conditions of pH 11.5 with overall improvements in PEC characteristics. These catalytic developments by the introduction of Au arise from two major factors: (1) electronic modification of the band structure in a more advantageous way for the transport of hole charges and (2) passivation of existing defect sites in NiFe LDH, largely lowering the recombination rates during water oxidation. Through UV-vis and UPS analysis, Au-NiFe LDH was revealed as a p-type semiconducting catalyst, altering the n-Si-based photoanode as a distinctive type II junction, which could expedite the hole transport. Steady-state PL, TRPL, and IMPS investigations could verify the significantly reduced charge recombination rate in Au-NiFe LDH, which is 1/48 of pristine NiFe LDH with an increased charge lifetime. In addition, the XANES and EXAFS results reveal that Au could be effectively merged into the NiFe structure with specific chemical coordination. All of the beneficial effects for PEC water oxidation by doping a small portion of Au into NiFe LDH could stem from substantially enhanced charge transport capability, which also contributes to the stability of the entire photoanode structure. We believe that our study of synthesizing outperforming OER catalysts composed of transition metals and noble metals through a facile electrodeposition process would pave the way for designing high-performance silicon-based photoanodes while providing efficacious application of noble metals by maximizing their synergistic effects between heterogeneous elements.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Research Foundation of Korea (NRF) funded by the Korea government Ministry of Science and ICT (MSIT) (2021M3H4A1A0305740311, 2021M3D1A2039641). Ho Won Jang acknowledges the KRISS (Korea Research Institute of Standards and Science) MPI Lab. Program.Notes and references
- J. Ran, H. Zhang, S. Fu, M. Jaroniec, J. Shan, B. Xia, Y. Qu, J. Qu, S. Chen, L. Song, J. M. Cairney, L. Jing and S.-Z. Qiao, Nat. Commun., 2022, 13, 4600 CrossRef CAS PubMed.
- B. Xia, Y. Yang, Y. Zhang, Y. Xia, M. Jaroniec, J. Yu, J. Ran and S.-Z. Qiao, Chem. Eng. J., 2022, 431, 133944 CrossRef CAS.
- S. Choi, S. A. Lee, H. Yang, T. H. Lee, C. Kim, C. W. Lee, H. Shin and H. W. Jang, ACS Appl. Energy Mater., 2020, 3, 12298–12307 CrossRef CAS.
- S. A. Lee, I. J. Park, J. W. Yang, J. Park, T. H. Lee, C. Kim, J. Moon, J. Y. Kim and H. W. Jang, Cell Rep. Phys. Sci., 2020, 1, 100219 CrossRef CAS.
- M. G. Lee, J. W. Yang, H. Park, C. W. Moon, D. M. Andoshe, J. Park, C.-K. Moon, T. H. Lee, K. S. Choi, W. S. Cheon, J.-J. Kim and H. W. Jang, Nano-Micro Lett., 2022, 14, 1–15 CrossRef PubMed.
- S. A. Lee, T. H. Lee, C. Kim, M.-J. Choi, H. Park, S. Choi, J. Lee, J. Oh, S. Y. Kim and H. W. Jang, ACS Catal., 2019, 10, 420–429 CrossRef.
- Y. Liu, D. Zhou, T. Deng, G. He, A. Chen, X. Sun, Y. Yang and P. Miao, ChemSusChem, 2021, 14, 5359–5383 CrossRef CAS PubMed.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- S. A. Lee, T. H. Lee, C. Kim, M. G. Lee, M.-J. Choi, H. Park, S. Choi, J. Oh and H. W. Jang, ACS Catal., 2018, 8, 7261–7269 CrossRef CAS.
- H. Park, I. J. Park, M. G. Lee, K. C. Kwon, S.-P. Hong, D. H. Kim, S. A. Lee, T. H. Lee, C. Kim, C. W. Moon, D.-Y. Son, G. H. Jung, H. S. Yang, J. R. Lee, J. Lee, N.-G. Park, S. Y. Kim, J. Y. Kim and H. W. Jang, ACS Appl. Mater. Interfaces, 2019, 11, 33835–33843 CrossRef CAS PubMed.
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC.
- H. Zhao and Z.-Y. Yuan, ChemCatChem, 2020, 12, 3797–3810 CrossRef CAS.
- K. Liu, C. Zhang, Y. Sun, G. Zhang, X. Shen, F. Zou, H. Zhang, Z. Wu, E. C. Wegener, C. J. Taubert, J. T. Miller, Z. Peng and Y. Zhu, ACS Nano, 2018, 12, 158–167 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- P.-C. Chen, M. Li, J. Jin, S. Yu, S. Chen, C. Chen, M. Salmeron and P. Yang, ACS Mater. Lett., 2021, 3, 1440–1447 CrossRef CAS.
- J. He, P. Liu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 6835–6871 RSC.
- Q. Zhang and J. Guan, Sol. RRL, 2020, 4, 2000283 CrossRef CAS.
- Y. Yang, W.-J. Wang, Y.-B. Yang, P.-F. Guo, B. Zhu, K. Wang, W.-T. Wang, Z.-H. He and Z.-T. Liu, J. Electrochem. Soc., 2022, 169, 024503 CrossRef CAS.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- Q.-Q. Chen, C.-C. Hou, C.-J. Wang, X. Yang, R. Shi and Y. Chen, Chem. Commun., 2018, 54, 6400–6403 RSC.
- H. Boumeriame, E. S. Da Silva, A. S. Cherevan, T. Chafik, J. L. Faria and D. Eder, J. Energy Chem., 2022, 64, 406–431 CrossRef CAS.
- B. W. Xue, C. H. Zhang, Y. Z. Wang, W. W. Xie, N.-W. Li and L. Yu, Nanoscale Adv., 2020, 2, 5555–5566 RSC.
- R. Yang, Y. Zhou, Y. Xing, D. Li, D. Jiang, M. Chen, W. Shi and S. Yuan, Appl. Catal., B, 2019, 253, 131–139 CrossRef CAS.
- B. Guo, A. Batool, G. Xie, R. Boddula, L. Tian, S. U. Jan and J. R. Gong, Nano Lett., 2018, 18, 1516–1521 CrossRef CAS PubMed.
- X. Yu, P. Yang, S. Chen, M. Zhang and G. Shi, Adv. Energy Mater., 2017, 7, 1601805 CrossRef.
- J. Strand, M. Kaviani, D. Gao, A.-M. El-Sayed, V. V. Afanas'ev and A. L. Shluger, J. Phys.: Condens. Matter, 2018, 30, 233001 CrossRef PubMed.
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 2373 CrossRef PubMed.
- F. Le Formal, N. Tétreault, M. Cornuz, T. Moehl, M. Grätzel and K. Sivula, Chem. Sci., 2011, 2, 737–743 RSC.
- Y. Lee, J. H. Choi, H. J. Jeon, K. M. Choi, J. W. Lee and J. K. Kang, Energy Environ. Sci., 2011, 4, 914–920 RSC.
- M. Li, T. Liu, Y. Yang, W. Qiu, C. Liang, Y. Tong and Y. Li, ACS Energy Lett., 2019, 4, 1983–1990 CrossRef CAS.
- X. Ning, P. Du, Z. Han, J. Chen and X. Lu, Angew. Chem., 2021, 60, 3504–3509 CrossRef CAS PubMed.
- M. Gong and H. Dai, Nano Res., 2014, 8, 23–39 CrossRef.
- B. T. Diroll, S. Jeong and X. Ye, Small Sci., 2022, 2, 2100103 CrossRef CAS.
- J. Lee, M. H. Naveen, J. Park, K. Pyo, H. Kim, D. Lee and J. H. Bang, ACS Energy Lett., 2021, 6, 2305–2312 CrossRef CAS.
- C. W. Moon, M.-J. Choi, J. K. Hyun and H. W. Jang, Nanoscale Adv., 2021, 3, 5981–6006 RSC.
- A. Abdullah, A. Waseem, I. V. Bagal, M. A. Johar, M. A. Kulkarni, J. K. Lee and S.-W. Ryu, ACS Appl. Energy Mater., 2021, 4, 13759–13765 CrossRef CAS.
- X. Li, Y. Wang, J. Wang, Y. Da, J. Zhang, L. Li, C. Zhong, Y. Deng, X. Han and W. Hu, Adv. Mater., 2020, 32, 2003414 CrossRef CAS PubMed.
- C.-H. Kuo, W. Li, L. Pahalagedara, A. M. El-Sawy, D. Kriz, N. Genz, C. Guild, T. Ressler, S. L. Suib and J. He, Angew. Chem., Int. Ed., 2015, 54, 2345–2350 CrossRef CAS PubMed.
- X. Duan, X. Tian, J. Ke, Y. Yin, J. Zheng, J. Chen, Z. Cao, Z. Xie and Y. Yuan, Chem. Sci., 2016, 7, 3181–3187 RSC.
- A. Y. Klyushin, T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2014, 16, 7881–7886 RSC.
- X.-P. Li, W.-K. Han, K. Xiao, T. Ouyang, N. Li, F. Peng and Z.-Q. Liu, Catal. Sci. Technol., 2020, 10, 4184–4190 RSC.
- Q. Cai, W. Hong, C. Jian and W. Liu, Nanoscale, 2020, 12, 7550–7556 RSC.
- Z. Luo, B. Liu, H. Li, X. Chang, W. Zhu, T. Wang and J. Gong, Small Methods, 2019, 3, 1900212 CrossRef CAS.
- F. Li, Y. Li, Q. Zhuo, D. Zhou, Y. Zhao, Z. Zhao, X. Wu, Y. Shan and L. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 11479–11488 CrossRef CAS PubMed.
- W. Hong, Q. Cai, R. Ban, X. He, C. Jian, J. Li, J. Li and W. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6262–6268 CrossRef CAS PubMed.
- S. A. Lee, J. W. Yang, T. H. Lee, I. J. Park, C. Kim, S. H. Hong, H. Lee, S. Choi, J. Moon, S. Y. Kim, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2022, 317, 121765 CrossRef CAS.
- C. Li, M. Huang, Y. Zhong, L. Zhang, Y. Xiao and H. Zhu, Chem. Mater., 2019, 31, 171–178 CrossRef CAS.
- D. Liu, T. Jiang, D. Liu, W. Zhang, H. Qin, S. Yan and Z. Zou, ChemSusChem, 2020, 13, 6037–6044 CrossRef CAS PubMed.
- Z. Liu, C. Li, Y. Xiao, F. Wang, Q. Yu, M. B. Faheem, T. Zhou and Y. Li, J. Phys. Chem. C, 2020, 124, 2844–2850 CrossRef CAS.
- Q. Cai, W. Hong, C. Jian, J. Li and W. Liu, ACS Catal., 2017, 7, 3277–3283 CrossRef CAS.
- Q. Cai, W. Hong, C. Jian, J. Li and W. Liu, ACS Catal., 2018, 8, 9238–9244 CrossRef CAS.
- C. Zhao, B. Guo, G. Xie, C. Li, W. Xie, Y. Dai, J. Gong and J. R. Gong, ACS Appl. Energy Mater., 2020, 3, 8216–8223 CrossRef CAS.
- S. Majumder, B. Satpati, S. Kumar and S. Banerjee, ACS Appl. Nano Mater., 2018, 1, 3945–3955 CrossRef CAS.
- W. Su, L. Jin, X. Qu, D. Huo and L. Yang, Phys. Chem. Chem. Phys., 2016, 18, 14001–14006 RSC.
- J. Liu, M. Shahid, Y.-S. Ko, E. Kim, T. K. Ahn, J. H. Park and Y.-U. Kwon, Phys. Chem. Chem. Phys., 2013, 15, 9775–9782 RSC.
- W. Yang, S. Lee, H.-C. Kwon, J. Tan, H. Lee, J. Park, Y. Oh, H. Choi and J. Moon, ACS Nano, 2018, 12, 11088–11097 CrossRef CAS PubMed.
- B. Sriram, J. N. Baby, S.-F. Wang, R. R. Mathiarasu, M. Govindasamy and M. George, ACS Sustainable Chem. Eng., 2020, 8, 17772–17782 CrossRef CAS.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. S. Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Angew. Chem., Int. Ed., 2021, 60, 24612 CrossRef CAS PubMed.
- G. Chen, K. A. Kuttiyiel, D. Su, M. Li, C.-H. Wang, D. Buceta, C. Du, Y. Gao, G. Yin, K. Sasaki, M. B. Vukmirovic and R. R. Adzic, Chem. Mater., 2016, 28, 5274–5281 CrossRef CAS.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS PubMed.
- L. Wang, Y. Zhu, J.-Q. Wang, F. Liu, J. Huang, X. Meng, J.-M. Basset, Y. Han and F.-S. Xiao, Nat. Commun., 2015, 6, 6957 CrossRef CAS PubMed.
- C. Maheu, L. Cardenas, E. Puzenat, P. Afanasiev and C. Geantet, Phys. Chem. Chem. Phys., 2018, 20, 25629–25637 RSC.
- J. W. Yang, I. J. Park, S. A. Lee, M. G. Lee, T. H. Lee, H. Park, C. Kim, J. Park, J. Moon, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2021, 293, 120217 CrossRef CAS.
- K. C. Kwon, S. Choi, K. Hong, C. W. Moon, Y.-S. Shim, D. H. Kim, T. Kim, W. Sohn, J.-M. Jeon, C.-H. Lee, K. T. Nam, S. Han, S. Y. Kim and H. W. Jang, Energy Environ. Sci., 2016, 9, 2240–2248 RSC.
- L. Xi and K. M. Lange, Catalysts, 2018, 8, 497 CrossRef.
- J. Xiao, l. Fan, Z. Huang, J. Zhong, F. Zhao, K. Xu, S.-F. Zhou and G. Zhan, Chin. J. Catal., 2020, 41, 1761–1771 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta03075j |
| This journal is © The Royal Society of Chemistry 2023 |