
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding and optimising the transfection of lipopolyplexes formulated in saline: the effects of peptide and serum†
Lili
Cui‡
 a,
Laila
Kudsiova§
a,
Frederick
Campbell¶
c,
David J.
Barlow
b,
Helen C.
Hailes
c,
Alethea B.
Tabor
*c and
M. Jayne
Lawrence
*ab
a,
Laila
Kudsiova§
a,
Frederick
Campbell¶
c,
David J.
Barlow
b,
Helen C.
Hailes
c,
Alethea B.
Tabor
*c and
M. Jayne
Lawrence
*ab
aInstitute of Pharmaceutical Science, King's College London, Franklin-Wilkins Building, 150 Stamford Street, Waterloo Campus, London SE1 9NH, UK
bDivision of Pharmacy & Optometry, School of Health Sciences, University of Manchester, Stopford Building, Oxford Road, Manchester, M13 9PT, UK. E-mail: jayne.lawrence@manchester.ac.uk
cDepartment of Chemistry, University College London, Christopher Ingold Laboratories, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: a.b.tabor@ucl.ac.uk
First published on 24th March 2023
Abstract
Lipopolyplexes (LPDs) are of considerable interest for use as gene delivery vehicles. Here LPDs have been prepared from cationic vesicles (composed of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of DOTMA with the neutral helper lipid, DOPE), singly branched cationic peptides and plasmid DNA. All peptides contained a linker sequence (cleaved by endosomal furin) attached to a targeting sequence selected to bind human airway epithelial cells and mediate gene delivery. The current study investigates the effects of novel Arg-containing cationic peptide sequences on the biophysical and transfection properties of LPDs. Mixed His/Arg cationic peptides were of particular interest, as these sequences have not been previously used in LPD formulations. Lengthening the number of cationic residues in a homopolymer from 6 to 12 in each branch reduced transfection using LPDs, most likely due to increased DNA compaction hindering the release of pDNA within the target cell. Furthermore, LPDs containing mixed Arg-containing peptides, particularly an alternating Arg/His sequence exhibited an increase in transfection, probably because of their optimal ability to complex and subsequently release pDNA. To confer stability in serum, LPDs were prepared in 0.12 M sodium chloride solution (as opposed to the more commonly used water) yielding multilamellar LPDs with very high levels of size reproducibility and DNA protection, especially when compared to the (unilamellar) LPDs formed in water. Significantly for the clinical applications of the LPDs, those prepared in the presence of sodium chloride retained high levels of transfection in the presence of media supplemented with fetal bovine serum. This work therefore represents a significant advance for the optimisation of LPD formulation for gene delivery, under physiologically relevant conditions, in vivo.
:1 molar ratio of DOTMA with the neutral helper lipid, DOPE), singly branched cationic peptides and plasmid DNA. All peptides contained a linker sequence (cleaved by endosomal furin) attached to a targeting sequence selected to bind human airway epithelial cells and mediate gene delivery. The current study investigates the effects of novel Arg-containing cationic peptide sequences on the biophysical and transfection properties of LPDs. Mixed His/Arg cationic peptides were of particular interest, as these sequences have not been previously used in LPD formulations. Lengthening the number of cationic residues in a homopolymer from 6 to 12 in each branch reduced transfection using LPDs, most likely due to increased DNA compaction hindering the release of pDNA within the target cell. Furthermore, LPDs containing mixed Arg-containing peptides, particularly an alternating Arg/His sequence exhibited an increase in transfection, probably because of their optimal ability to complex and subsequently release pDNA. To confer stability in serum, LPDs were prepared in 0.12 M sodium chloride solution (as opposed to the more commonly used water) yielding multilamellar LPDs with very high levels of size reproducibility and DNA protection, especially when compared to the (unilamellar) LPDs formed in water. Significantly for the clinical applications of the LPDs, those prepared in the presence of sodium chloride retained high levels of transfection in the presence of media supplemented with fetal bovine serum. This work therefore represents a significant advance for the optimisation of LPD formulation for gene delivery, under physiologically relevant conditions, in vivo.
Introduction
Gene therapy remains a promising strategy to cure human cancers, and genetic and orphan diseases, via regulating, repairing, or editing a genetic sequence and correcting the corresponding proteins.1,2 In the past two decades, numerous gene therapy clinical trials have been carried out, and over 20 gene therapy drugs licensed. Examples of licensed products include Strimvelis (for ADA-SCID), Kymriah/Yescarta (CAR-T therapies for leukaemias) and Luxturna (for Leber's congenital amaurosis which causes blindness).3 However, the majority of these systems rely on modified viruses to deliver the therapeutic genes. Such systems have several drawbacks including problems with immunogenicity, carcinogenesis and limited DNA packaging capacity.2 In comparison with viral vectors, non-viral vectors formulated from lipids, polymers/peptides and inorganic materials are promising alternatives due to their low immunogenicities, toxicities and high safety profiles.2,4–6 Formulations in which combinations of cationic lipids (L) and cationic polymers/peptides (P) are complexed with DNA (D), siRNA or mRNA (R) to give LPD or LPR lipopolyplexes, respectively, have received much attention in recent years,7 being reported to combine the advantages of lipoplexes and polyplexes. Indeed, lipopolyplexes have been shown to enhance the delivery of the nucleic acid cargo in vitro and in vivo, relative to peptide/nucleic acid (polyplex) or lipid/nucleic acid (lipoplex) formulations.7–16 For LPD formulations, the improved efficiency is believed to be related to the potentiation effect due to the polymers/peptides, such as improved DNA condensation due to the presence of extra charge, leading to smaller particle sizes and better protection of pDNA from enzyme degradation.8 Synthetic cationic polymers such as PEI often exhibit significant toxicity,17 limiting their use in vivo, whilst poly-L-Lys is biocompatible and nontoxic.18 Moreover, short cationic peptides, varying in their length, composition, charge, hydrophobicity and spacing can be readily synthesized, thereby allowing the effects of different sequences on the self-assembly, macromolecular organization and transfection efficiencies of the resulting lipopolyplexes to be understood and optimized.For all cationic peptide-containing lipopolyplex pDNA vectors, a balance must be struck between efficient condensation of the pDNA (resulting in small, compact particles with the pDNA protected from degradation) and intracellular release of the pDNA cargo.19 Linear sequences and dendrimers based on Lys have high cationic charge densities, and hence condense and protect pDNA efficiently. Lipopolyplexes (LPDs) incorporating Lys-based peptides are therefore frequently used for pDNA transfection.20–25 As LPDs are internalized into cells via endocytosis, efficient endosomal release is critical for efficient transfection,26 however a disadvantage of using Lys-based peptides is that the resulting LPDs do not trigger rupture and escape from the endosome. His-based peptides, in contrast can protonate within the acidic environment of the endosome, and swell and destabilize the endosomal membrane, a phenomenon known as the “proton sponge effect”.27 Some polyplexes (PD) and LPD based on His-rich peptides have been shown to be efficient gene delivery vectors.27 Unfortunately, in LPDs the reduced pDNA complexation abilities of His-rich peptides can lead to poor protection of the pDNA and low transfection efficiencies. To overcome these deficiencies, Mixson and co-workers have studied LPDs formulated using branched peptides with alternating His and Lys residues. Peptides with 4 branches with repeating His-Lys-Lys sequences, with either Lys or His-rich tails, gave particularly high transfection levels.28–31 Surprisingly, given the recent interest in Arg-based biomaterials for gene delivery,32 Arg-based peptides have hardly been used in LPD vectors to date. This is despite the potential of the guanidino side chain of Arg for bidentate interactions with the phosphate backbone of RNA,33 and the frequent occurrence of Arg-rich sequences in cell-penetrating peptides (CPP).34,35
Our group has developed LPDs for the targeted delivery of pDNA to different cell types.11,12,36–39 These contain peptide components with trifunctional sequences comprising: a cationic peptide domain to condense pDNA; a cell-targeting sequence that is displayed at the surface of the nanoparticle; and a linker sequence, RVRR, that is a substrate for the endosomal enzyme furin. The LPDs also include mixtures of cationic lipids which are co-formulated with the neutral lipid dioleoylphosphatidyl-ethanolamine (DOPE). We have studied the macromolecular organisation of the LPDs by a range of biophysical techniques, including dynamic light scattering (DLS), zeta potential, transmission electron microscopy (TEM), freeze-fracture electron microscopy (FFEM), cryoelectron microscopy (cryoEM) and small angle neutron scattering (SANS), and have shown that they consist of a tightly condensed pDNA/cationic peptide core, surrounded by a lipid bilayer through which the cell targeting peptide sequence partially protrudes. Recently, we have investigated the effects of branched and linear Lys, His or Arg sequences, as well as mixed His/Lys sequences, on the transfection efficiencies and macromolecular organisation of the LPDs formed.39 These showed that LPDs containing either branched Arg sequences or linear Lys sequences, gave the highest transfection efficiencies. LPD formulations incorporating either His-rich peptides or mixed His/Lys peptide sequences were very poor transfection agents by comparison.39 The His-rich peptides made only a small contribution to endosomal release, with the majority of the effect arising from the membrane destabilization properties of the helper lipid DOPE.40 In contrast, LPRs formulated from mixed His/Lys peptide sequences performed best as siRNA delivery agents, even in the presence of serum.41 Surprisingly, mixed His/Arg cationic peptide sequences have not been previously used in lipopolyplex formulations.
Non-viral vectors are frequently observed to have reduced transfection efficiency in the presence of serum in vitro and in vivo,42–47 and, as a consequence, therefore, most in vitro studies tend to be performed in serum-free media, such as OptiMEM. However, in order to successfully translate these gene transfer reagents to the clinic, it is necessary to understand the barriers to transfection in the presence of serum and to design vectors that can overcome them. The factors preventing successful transfection of polyplexes, lipoplexes and lipopolyplexes in serum are still imperfectly understood. They may include: the presence of enzymes such as DNase I that can degrade the cargo;47 interaction of serum components with the vectors;42 destabilization of the vectors leading to premature release of the pDNA;44,45 and complexation of negatively-charged serum components with positively-charged lipoplexes and lipopolyplexes.45 The adhesion of serum proteins to the surface of nanoparticles can also lead to aggregation and non-specific cell binding,45 although some studies suggest this may actually enhance uptake and transfection in vivo.48 Protein corona formation on the surface of nanoparticles can also alter the tissue distribution, depending on the protein composition and level of coating.49 Finally, nanoparticles can be eliminated from blood circulation by macrophages of the mononuclear phagocytic system, which is mediated by opsonin protein in serum. A commonly used strategy to prevent this unwanted elimination is to functionalise the nanoparticle surface using PEG.50
In this study, we have extended our previous work to investigate the effects of novel, mixed His/Arg cationic peptide sequences on the biophysical and transfection properties of LPDs. We have compared these with branched Lys, His or Arg sequences that are longer than those previously studied, to ascertain whether longer sequences confer greater stability and transfection efficiency on the lipopolyplexes. In parallel, we have carried out a detailed investigation of the effects of using different solvent systems and transfection media in the preparation of the complexes, to determine whether this could lead to LPDs with good transfection efficiencies in serum at physiologically relevant concentrations.
Materials and methods
Materials
Trimethyl [2,3-dioleyloxy-propyl] ammonium chloride (DOTMA) and dioleoylphosphotidylethanolamine (DOPE) were purchased from Tokyo Chemical Industry (Tokyo, Japan) and Avanti Polar Lipids (Alabama, USA) respectively. gWiz-luciferase plasmid (pDNA) purchased from Aldevron (Frago, USA) was used for all experiments with the exception of the small angle neutron scattering studies, where due to the larger amount of DNA required, calf thymus DNA (ctDNA), obtained from Sigma-Aldrich (Poole, UK) was used. ctDNA has been previously found to be a suitable substitute for such studies.39,41 Lipofectamine™ and Lipofectamine 2000 obtained from Invitrogen Life technologies (Paisley, UK) were used as positive controls. Luciferase assay kit and bicinchoninic acid (BCA) protein assay kit were purchased from Promega (Southampton, UK) and Thermo Scientific (Loughborough, UK). Reduced serum medium Opti-MEM®I, RPMI-1640 cell culture medium, PicoGreen reagent, and GelRed stain were purchased from Gibco life technologies, Life Technology (Paisley, UK), Sigma- Aldrich (Poole, UK), Invitrogen Life Technology (Paisley, UK) and Biotium (Heyward, USA), respectively. Deoxyribonuclease I (DNase 1) Type II from bovine pancreas, polyaspartic acid (pAsp) and all the other reagents were all from Sigma-Aldrich (Poole, UK).Peptide synthesis and purification
The synthesis, purification, and analytical data for the peptides are described in the ESI.†Preparation of the lipopolyplexes
LPDs consisting of lipid, peptide and DNA were prepared via a self-assembly process. In brief, a suspension of lipid vesicles (1 mg mL−1 of cationic lipid) was first prepared as previously described.51 The peptide solution (typically 50 μL) was then added to 50 μL of lipid vesicle suspension followed by the DNA solution (100 μL). The resulting mixture was then agitated using gentle pipette mixing and a minimum of 15 min allowed for complexation to occur before the LPDs were used. The order of mixing of the three components was kept the same, as changing the order has been previously shown to alter the structure of the resulting complex (see, for example, ref. 39). Specifically, if the lipid vesicles and DNA were mixed prior to the addition of peptide then lipoplexes (complexes of lipid and DNA) as opposed to lipopolyplexes are formed. Here, small angle neutron scattering and transfection studies were used to confirm the order of mixing was important using the Group 1 and Group 2 peptides ((H12BLY and HR)6BLY) (data not shown). The concentration of the various solutions/suspensions used to produce the LPDs was varied in order to prepare the required total concentration and lipid:peptide:DNA charge ratio, generally 0.5:6:1, unless otherwise stated – a few studies were also performed at a lipid:peptide:DNA charge ratio of 0.5:12:1. When investigating the effect of electrolyte on the formation of LPDs, the lipid vesicle suspension was prepared using either physiological saline (154 mM NaCl), 100, 50 or 10 mM NaCl in place of water, while stock peptide and DNA solutions were diluted with electrolyte solution such that the final concentration of NaCl present in the LPDs was either 120, 80, 40, or 4 mM, respectively. The order of mixing was the same as was used for lipopolyplexes prepared in water. Note that the order of mixing was kept the same as when preparing LPDs solely in water. LPDs prepared wholly in water are denoted as VwLPDw while those prepared in 0.12 M NaCl are described by VsLPDs. The effect of saline on the parent DOTMA:DOPE vesicles (Vw or Vs) was also investigated. If no subscript is used, then both the vesicles and the LPDs were prepared wholly in water.
Cell culture
A549 cells were cultured in RPMI-1640 media supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v of 100× strength nonessential amino acids, 1% v/v of 200 mM L-glutamine solution, and 1% of v/v penicillin/streptomycin solution (10000 IU mL−1/10 mg mL−1) at 37 °C in a 90% humidified atmosphere with 5% carbon dioxide. The cells were fed every other day and passaged every three to four days at around 70% confluency, using 0.25% trypsin-EDTA solution.
Transfection of the lipopolyplexes
After passaging, A549 cells in RPMI-1640 supplemented media were plated in 96 transparent-well plates at a seeding density of 1.2 × 104 cells per well and cultured for 24 h. On the day of the experiment, the media was removed from the A549 cells and replaced with either 50 μL of OptiMEM or RPMI 1640 media containing 10% FBS, followed by the addition of an equal volume of LPD dispersion. All LPD suspensions were added at a DNA concentration of 0.25 μg per 100 μL per well. Unless otherwise stated, the LPDs were incubated for 4 h after which time the LPD containing media was removed from the cells and replaced with 100 μL of RPMI-1640 supplemented media and maintained at 37 °C in a 90% humidified atmosphere with 5% carbon dioxide for a further 44 h, described as an incubation time of 4 + 44 h. The A549 cells were then washed with 50 μL PBS and lysed with 50 μL of lysis buffer (200 nM Tris-HCl, pH 7.8, 2 mM EDTA and 0.05% v/v Triton X-100) at ambient temperature for 1 h. The lysed A549 cells were then frozen at −80 °C for 30 min before defrosting and agitating using an orbital incubator at 37 °C for 1 h. Luciferase activity of the lysed cells was measured using a luciferase assay kit in accordance with the manufacturer's protocol. In brief, 30 μL aliquot of cell lysate was added to a white 96-well plate and the luminescence determined using a FLUOstar Omega luminometer (BMG LABTECH GmbH, Ortenberg, Germany) which automatically delivered 100 μL of reconstituted luciferase assay reagent into each well. Luciferase activity was expressed as relative light units (RLU) per mg of protein (RLU per mg protein). For protein determination of the transfected and lysed cells a BCA protein assay (Thermo Scientific, UK) was used. In brief, 20 μL of cell lysate was added into a transparent 96-well plate, 200 μL of protein assay reagent added and the plate incubated with slight shaking at 37 °C for 30 min before its absorbance was measured at 562 nm using a SpectraMax 190 plate reader (Molecular Device, USA). Lipofectamine 2000 was used throughout as a positive control. All determinations within an experiment were performed in triplicate allowing the mean and SD to be calculated. The transfections were repeated on at least two separate occasions and frequently three times. Where data were obtained in triplicate, the statistical significance of differences was assessed by means of a students’ t-test, accepting a significance level of <0.05. In all cases the same trends in the data were seen, although the absolute level of transfection may have varied slightly between experiments.Agarose gel retardation assay
The complexation, release and protection of pDNA from DNase I degradation afforded by the LPD complexes prepared in water or different NaCl concentrations were determined using agarose gel electrophoresis. Here LPDs were prepared at a final pDNA concentration of 0.25 μg per 10 μL per well at a 0.5:6:1 LPD charge ratio. To determine the extent of complexation, a 10 μL aliquot of LPD suspension was used. For assessment of pDNA release, the LPD suspensions were treated with 0.625 μL of a 10 mg mL−1 solution of polyaspartic acid (pAsp). To establish the level of pDNA protection, a 10 μL aliquot of LPD suspension was treated with 0.5 μL of 500U mL−1 DNase I, followed by 0.5 μL of MgCl2 (0.1 M) to activate enzymes. After 10 min incubation at 37 °C, 1.25 μL of EDTA (0.5 M) was added to deactivate the enzymatic reaction and the resulting mixture incubated at room temperature for 10 min to ensure complete deactivation. 0.625 μL of 10 mg mL−1 pAsp solution was then added to the above mixture to release any DNA remaining associated with the LPDs for detection on the gel. Free pDNA and pDNA treated with DNase I were used at DNA concentration of 0.25 μg per 10 μL per well as controls. 2 μL of loading buffer containing 40% w/v sucrose and 0.25% w/v bromophenol blue was added to each of the samples before the samples were loaded into wells in a 0.8% w/v agarose gel containing GelRed® in Tris-acetate-EDTA (TAE) buffer (pH 8.0) at 80 mV for 40 min (Fisher Brand, Model HU12 electrophoreses chamber, Loughborough, UK). The resulting gel was visualized under UV light using an Alphalmage EP MultiImage Light Cabinet (Randpark, South Africa). All experiments were performed in triplicate.
PicoGreen fluorescence assay
To determine the extent of pDNA complexation, the binding of PicoGreen to the LPDs was measured. A 50 μL aliquot of PicoGreen reagent (1:150 v:v PicoGreen in 3× Tris-EDTA buffer) was added to 100 μL of LPD suspension in a black 96-well plate containing either 0.1 or 0.2 μg of pDNA per 100 μL per well in saline or water respectively. These DNA concentrations were chosen to ensure a linear relationship between fluorescence and concentration in the different solvents. The plates were incubated at ambient for 5 min and fluorescence measured with excitation and emission wavelengths of 485/520 nm, with a gain of 1000 using a FLUOstar Omega fluorimeter (BMG LABTECH GmbH, Ortenberg, Germany) equipped with a plate reader. All fluorescence readings were normalized against the response from a free (naked) pDNA control, and were expressed as a percentage change (in relative fluorescence units, RFU). All experiments were performed in triplicate and are presented as the mean ± the standard deviation.
Particle size and zeta-potential measurement of the LPD complexes
The apparent hydrodynamic size and zeta potential of the LPDs were measured using a Malvern Zetasizer Nano ZS, (Worcestershire, UK). All LPDs were measured in triplicate at a scattering angle of 173° at 25 ± 0.1 °C. For both measurement of the apparent hydrodynamic size and zeta potential, the concentration of LPDs was sufficiently low to ensure the absence of any inter-particle interactions, namely 0.01 mg mL−1 of cationic lipid for the lipid vesicles and 0.5 μg mL−1 of DNA for the LPDs. At least 3, but usually 6 repeat measurements were performed on each sample, with each composition being tested on at least 2 but typically 3 separate occasions.Small angle neutron scattering of the LPD complexes
The internal structure of the parent vesicles and LPDs was established using small angle neutron scattering (SANS) experiments performed on freshly prepared samples using the instruments LoQ and SANS2D at the ISIS pulsed neutron source (Rutherford-Appleton Laboratories, Didcot, UK). All samples were measured in disk-shaped, fused silica banjo cells of 2 mm path length. The scattering intensity, I(Q), of the samples as a function of the scattering vector, Q = (4π/λ)sin(θ/2) where sin(θ/2) is the scattering angle, was determined by normalizing the scattering to the appropriate sample transmission after subtraction of the scattering for the relevant solvent. All fitting procedures included flat background corrections to account for any mismatch in the incoherent and inelastic scattering between the sample and the D2O solvent. The levels of the fitted backgrounds were checked to ensure that they were of a physically reasonable magnitude.The SANS profiles were model fitted using the SASVIEW package52 assuming the presence of isolated infinite planar (lamellar) sheets (i.e. unilamellar vesicles) called the ‘sheet model’, and where necessary, the presence of one-dimensional paracrystals (stacks) to account for any multilamellar vesicles present, i.e. the mixed ‘sheet and stack model’.53,54 The lamellar stack model uses a total of eight parameters, namely the scale factor for the lamellar stack, sample background, the thickness of the bilayers (L) in the stack, the number of bilayers per stack, the d-spacing of the bilayers in the stack and the polydispersity on this spacing and the scattering length densities of the solvent and the layer. The total variability on each of the fitted d-spacings was taken as the convolution of the statistical uncertainty in the spacing, and the associated polydispersity index converted to a standard deviation.
Results
Design of branched peptides
The use of trifunctional cationic/linker/targeting peptides to formulate LPDs has previously demonstrated that complexes prepared with singly branched cationic sequences were effective transfection agents, particularly for Arg-containing peptides.39 Further branching of the cationic peptide region had a detrimental effect. In contrast, the number of cationic residues is important, with branched peptides containing 12 Lys residues giving more effective transfection reagents than the equivalent peptides containing 8 Lys residues. Here, a series of singly branched, B, cationic peptides (Fig. 1, Group 1) with longer branched cationic His, Arg and Lys sequences were synthesised. All peptides contained a linker sequence, L, which has been hypothesised to be cleaved by furin within the endosome12 after the LPD complex is internalised.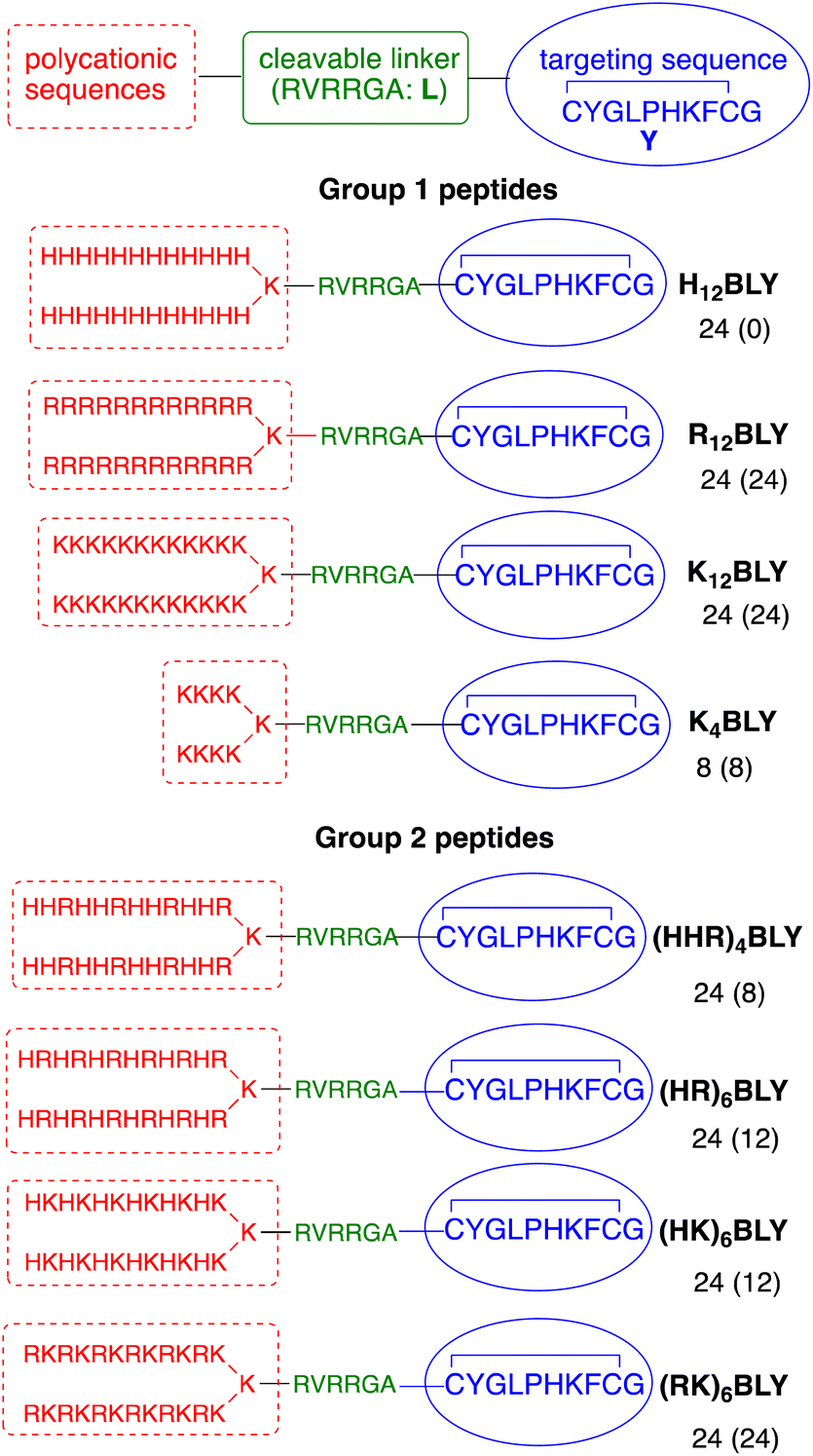 | ||
| Fig. 1 Sequences of the singly branched cationic peptides used in this study. The number of cationic residues (and the number of those which are fully protonated at pH 7) in each sequence are indicated. | ||
This linker was attached to a targeting sequence, Y, which was previously selected by phage display to bind human airway epithelial cells and mediate gene delivery.55 The sequences contained 24 cationic residues in total (H12BLY, R12BLY, K12BLY) and were compared with the previously studied shorter K4BLY peptide.39,56 In previous studies39 we have shown that LPD lipopolyplexes incorporating mixed His/Lys sequences gave generally poor transfection. In contrast, PDs formulated with branched sequences based on His-His-Lys repeats, such as (HHK)4BLY, have shown good transfection efficiencies.29,39,56
In view of recent reports that Arg-based peptides and dendrimers show potential for gene delivery,33–35,41 we wished to explore the use of branched peptides containing mixed His/Arg and Lys/Arg sequences (Fig. 1, Group 2). (HHR)4BLY was therefore synthesised as an Arg-containing analogue of (HHK)4BLY. The other three peptides, (HR)6BLY, (HK)6BLY and (RK)6BLY, were designed to compare the effects of incorporating Arg residues into singly branched, alternating cationic sequences.
Do the sequences of the cationic peptides and the presence of electrolyte affect the in vitro transfection of the lipopolyplexes?
In previous work,39 optimal transfection of LPDs was observed with lipid:peptide:pDNA charge ratios in the range of 0.5:6:1 and 0.5:12:1, and with incubation times of 24 h + 24 h or 4 h + 44 h. Here, we carried out preliminary studies with both of the Group 1 and Group 2 peptides and demonstrated that the lipid:peptide:pDNA ratio of 0.5:6:1 and 4 h + 44 h incubation time gave the highest level of transfection efficiency (ESI, Fig. S1†). These ratios and incubation times were therefore used throughout the rest of this study, unless otherwise stated.
In the initial in vitro experiments, both the vesicles used to prepare the LPDs and the LPDs themselves were prepared fully in water (VwLPDw) and then diluted 4-fold in OptiMEM (VwLPDw/OptiMEM) before testing their transfection ability (Fig. 2a). Under these conditions, LPDs formulated using the longer chain cationic Group 1 peptides (H12BLY, R12BLY, K12BLY) gave significantly poorer transfection levels than was obtained with LPDs prepared using the shorter cationic chain K4BLY peptide, and also significantly lower transfections levels (with p generally < 0.01) than was achieved with the LPDs made using the Group 2 peptides.
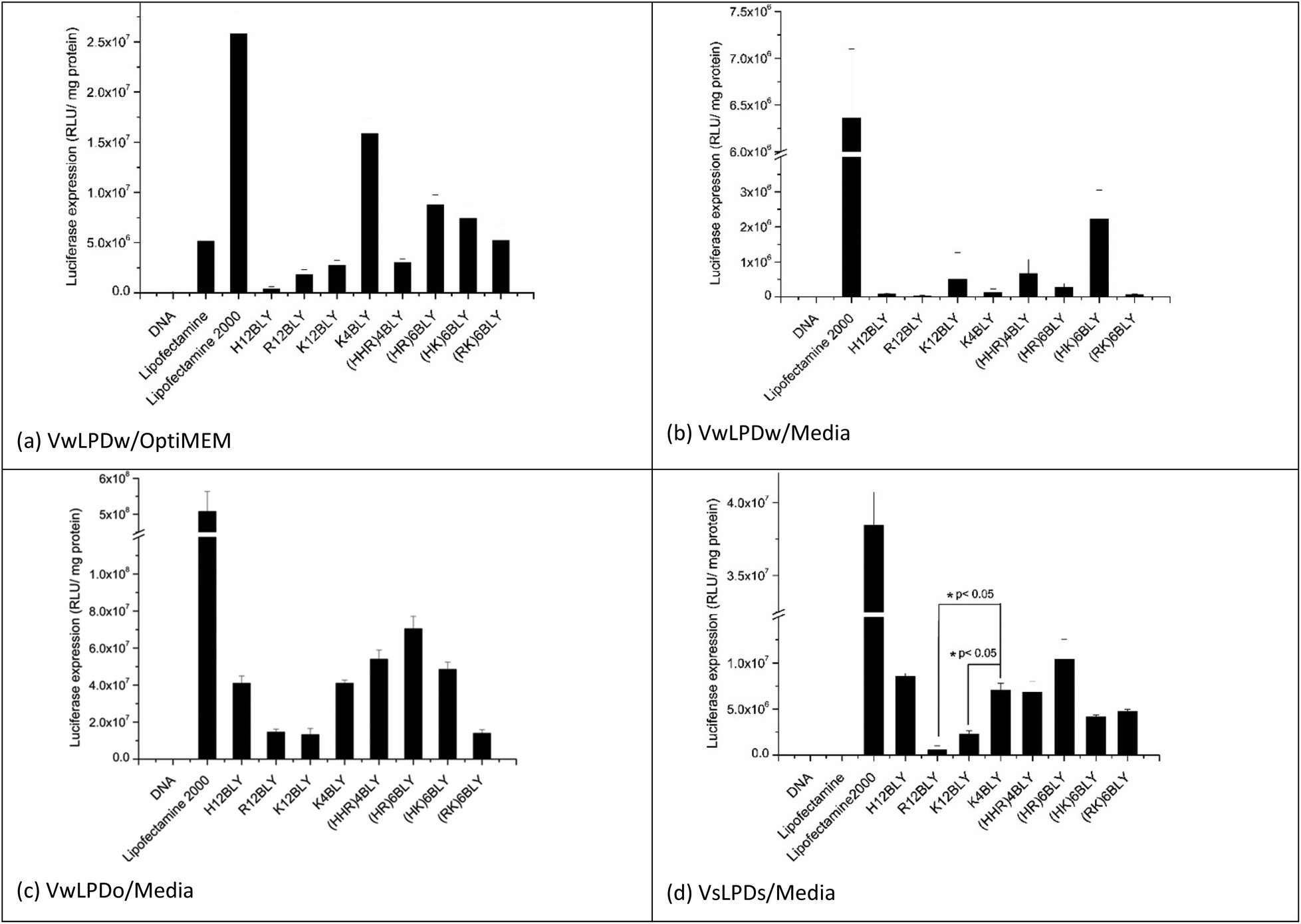 | ||
| Fig. 2 Transfection efficiency of LPDs containing various peptides and made at a lipid:peptide:DNA charge ratio of 0.5:6:1 in A549 cells in the presence of OptiMEM (a) and RPMI-1640 medium containing 10% FBS (b-d). Vw and Vs indicate that lipid vesicles used to make LPD were prepared in water and 0.15 M NaCl solution, respectively. LPDw, LPDo and LPDs mean that LPD complexes were prepared in water, OptiMEM and NaCl solution at a final concentration of 120 mM, respectively. The incubation time of LPD with A549 cells was 4 h. Cationic vesicles used to prepare the LPDs were composed of DOTMA:DOPE at 1:1 molar ratio. Data are mean values ± standard deviation of three replicates. | ||
In order to prepare LPDs suitable for parenteral administration to humans, it is necessary to make the LPD suspension isotonic with the body fluids. This is usually achieved by the addition, to the suspending media, of electrolyte or sugars. Interestingly, the dilution of LPDs prepared wholly in water (VwLPDw) with OptiMEM provides for good levels of in vitro transfection, although unfortunately this method cannot be translated to the clinic. In contrast, replacing OptiMEM with serum-containing RPMI-1640 media (supplemented with 10%v/v of fetal bovine serum (FBS)) greatly reduced transfection to such a level that little or none was observed when the LPDs were prepared in water (VwLPDw) (Fig. 2b), a result which is in line with previous reports.43 Surprisingly, however, when the LPDs were prepared partially in OptiMEM (VwLPDo), the subsequent level of transfection achieved in the presence of serum-containing RPMI-1640 media was high (Fig. 2c) suggesting that the presence of OptiMEM was particularly beneficial to the transfection.
As OptiMEM is a buffered modification of Eagle's minimum essential medium, we reasoned that it might be possible to use another modification of the Eagles medium in place of OptiMEM, which should lead to LPDs retaining a high level of transfection when subsequently diluted in serum-containing RPMI-1640 media. The electrolyte selected for the present study was NaCl as it is widely used in a range of clinical applications and is used to produce isotonic solutions of medicinal preparations. Consequently, vesicles and their corresponding LPDs, were prepared at a final NaCl concentration of 120 mM NaCl (VsLPDs) and diluted using RPMI-1640 medium containing 10% FBS. Significantly, when their transfection ability was measured after dilution in serum-containing media, a good level of transfection was observed (Fig. 2d) confirming the importance of the presence of electrolyte on the transfection ability of the LPDs.
When examining the level of transfection achieved when the VwLPDo and VsLPDs LPDs were diluted in RPMI-1640 medium containing 10% FBS (Fig. 2c and d), it was seen that the mixed cationic sequence peptides of Group 2 tended to give a higher level of transfection, although good levels of transfection were also observed for H12BLY and K4BLY. Comparison of the transfection data obtained for the LPDs prepared using K4BLY in 120 mM NaCl and diluted in media, show a significantly lower level of transfection for the corresponding LPDs made with K12BLY and R12BLY (p < 0.005), but no statistically significant difference for H12BLY and the Group 2 peptides.
In all these experiments, commercially available Lipofectamine 2000 was used as a positive control. Although this reagent gives very high levels of transfection, it is also known to be very toxic.57 Indeed, analysis of the protein content of the wells after the treatment of A549 cells with Lipofectamine 2000 indicated the toxicity of this reagent, whereas the VwLPDo and VsLPDs LPD formulations prepared here exhibited little toxicity towards the A549 cells (ESI Fig. S2†).
To further investigate the influence of the presence of NaCl on the transfection efficiency of LPDs in the presence of serum-containing medium, LPDs containing the Group 2 peptide (HHR)4BLY, were prepared in a range of differing final concentrations of NaCl. LPDs containing (HHR)4BLY were selected for further study because, although LPDs prepared using (HR)6BLY gave higher levels of transfection, the (HHR)4BLY containing LPDs gave consistently good levels of transfection. To study the effect of electrolyte concentration on transfection, cationic vesicles were first prepared either in water or using varying concentrations of NaCl, and then mixed with peptide and DNA solutions to formulate the LPDs (either VwLPDs or VsLPDs) to different final strength NaCl solutions of either 8, 40, 80 and 120 mM NaCl.
The resultant transfection results are shown in Fig. 3a. As can be seen there was a concentration-dependent effect of NaCl on the transfection efficiency of the (HHR)4BLY-containing LPD complexes, with the highest level of transfection being observed in LPDs prepared using cationic vesicles prepared in 120 mM saline and using the highest concentration of NaCl, namely 120 mM, which was encouraging as this concentration of NaCl is the most relevant to the potential in vivo use of the LPDs. It is noteworthy that the highest level of transfection was seen when both the cationic vesicles and the final LPDs were prepared in NaCl solution; when only the LPDs were prepared in NaCl, with the cationic vesicles being prepared in water, transfection was noticeably lower. A similar trend in transfection was observed with Lipofectamine 2000 in that higher levels of transfection were observed with complexes prepared using solutions of increasing NaCl concentration.
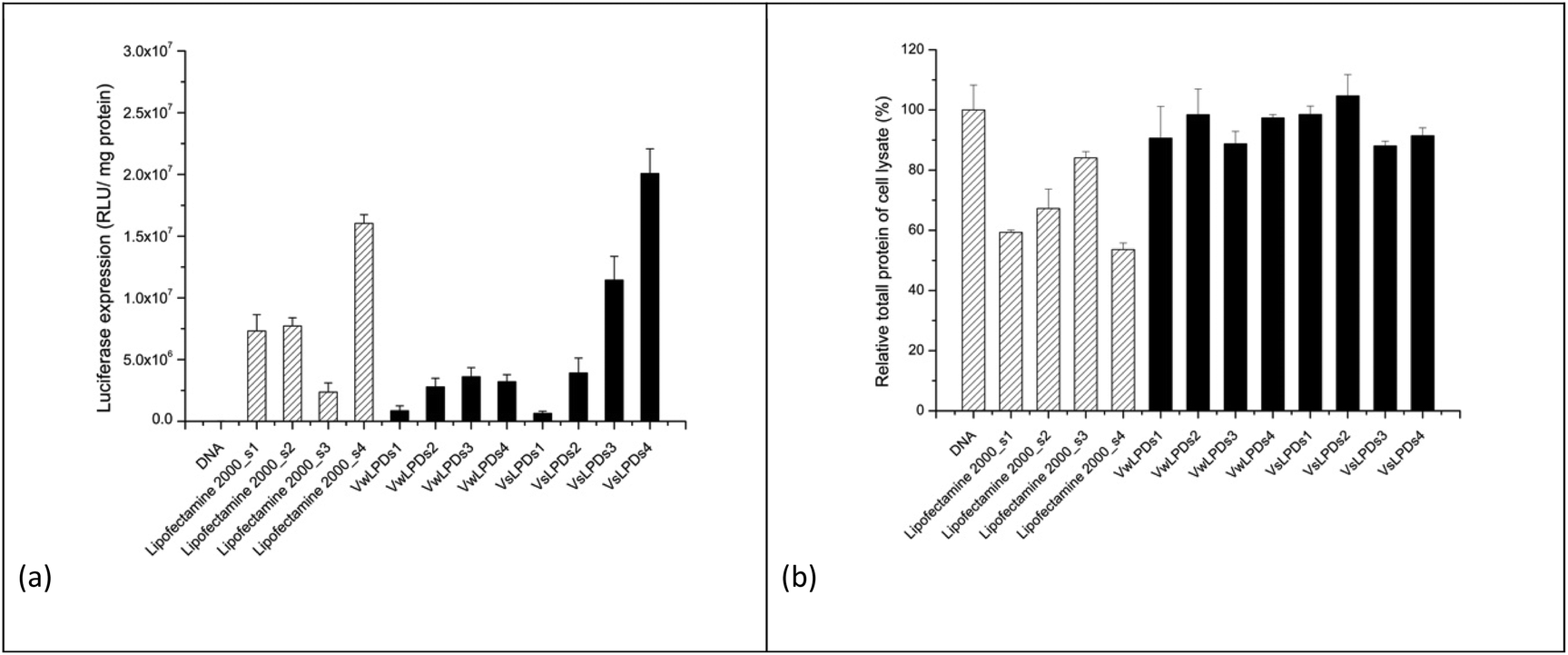 | ||
| Fig. 3 Luciferase transfection efficiency of LPD-(HHR)4BLY on A549 cells using a 24 + 24 h incubation. LPD-(HHR)4BLY were prepared partly (VwLPDs) or fully (VsLPDs) in various NaCl solutions (s1 = 8 mM; s2 = 40 mM; s3 = 80 mM; s4 = 120 mM) and transfected in 75% v/v RPMI-1640 media containing 10% v/v FBS. (a) The level of luciferase transfection and (b) the level of protein assay. LPDs were prepared at a L:P:D charge ratio of 0.5:6:1. Cationic vesicles were composed of DOTMA/DOPE lipids at 1:1 molar ratio. Data are mean values ± standard deviation of three replicates. | ||
Fig. 3b shows the total protein content of the lysate of the A549 cells after transfection using the various LPD, and Lipofectamine 2000, formulations as a proxy of toxicity. As previously reported, all the Lipofectamine 2000 group displayed to varying extents a negative (or toxic) effect on cell growth, independent of NaCl concentration. Importantly, however, no detrimental effect of VwLPDs and VsLPDs LPD formulations on the growth of A549 cells was observed, as the protein content in such cases was comparable to that of the free or naked pDNA control. In order to understand the reasons for the high transfection efficiencies of LPD formulated in saline solutions, we investigated and compared the physicochemical properties of LPD complexes prepared in water and in varying concentrations of NaCl. We were particularly interested to study the degree of protection of the pDNA afforded by the various cationic peptide sequences in NaCl solutions, and the effects that formulation in NaCl solutions had on the structural properties of the LPD complexes.
Is the condensation of DNA in the lipopolyplexes influenced by cationic peptide sequence and electrolyte?
To examine and compare the effect of the nature of the peptide and the presence of saline on the interaction of DNA in the complex, gel retardation and PicoGreen® fluorescence assays were performed. Agarose gel electrophoresis was used to investigate the condensation, release and protection of pDNA in LPD complexes prepared fully in water (VwLPDw) and fully in 120 mM NaCl (VsLPDs) solution. In Fig. 4, Lane A shows the condensation/complexation of the pDNA in the presence of both lipid vesicles and peptide, while in Lanes B and C the release of pDNA (displacement by the anionic polymer of pAsp) and the protection of pDNA from DNase I degradation afforded by the LPDs complexes, respectively are seen. As expected, free (uncomplexed) pDNA exhibited two bands in Lane A, an intense lower band and a faint upper band, attributable to the pDNA's supercoiled and open circular forms, respectively. When DNase I was added to the free pDNA, Lane C exhibited no band comparable to that seen with intact pDNA, only low molecular weight fragments were seen as a diffuse band towards the bottom of the gel, suggesting the complete degradation of pDNA by DNase I. In the case of the LPD complexes, DNA appeared to be fully condensed, regardless of which peptides were used to prepare the complexes and whether the LPDs were prepared all in water or all in the presence of 120 mM NaCl, as shown in Lane A. It is worth commenting here that while the assumption in these studies is that if the pDNA is associated with/contained in the lipopolyplex it will be retained in the well due to its positive charge. However, if the pDNA is complexed solely with peptide (i.e. forms a polyplex as opposed to a lipopolyplex) and is not incorporated in the lipopolyplex, it is also likely to stay in the well and not migrate down the gel due to its positive charge. The results of the release study, seen in Lane B, show that pDNA can be released from all the LPD complexes whether formulated using Group 1 or Group 2 peptides. Significantly, compared with the VwLPDw complexes, the VsLPDs complexes appeared to display a more complete release of pDNA, due to a release profile more comparable to that of free pDNA in Lane A. Furthermore, the results in Lane C indicated a significant difference between the extent of protection afforded by the VwLPDw and VsLPDs. In the case of the VwLPDw complexes, only limited protection was observed, while VsLPDs exhibited the greatest level of protection as evidenced by the presence of bands identical to that of free pDNA in Lane A, with the exception of the LPDs prepared using H12BLY where no DNA release was observed.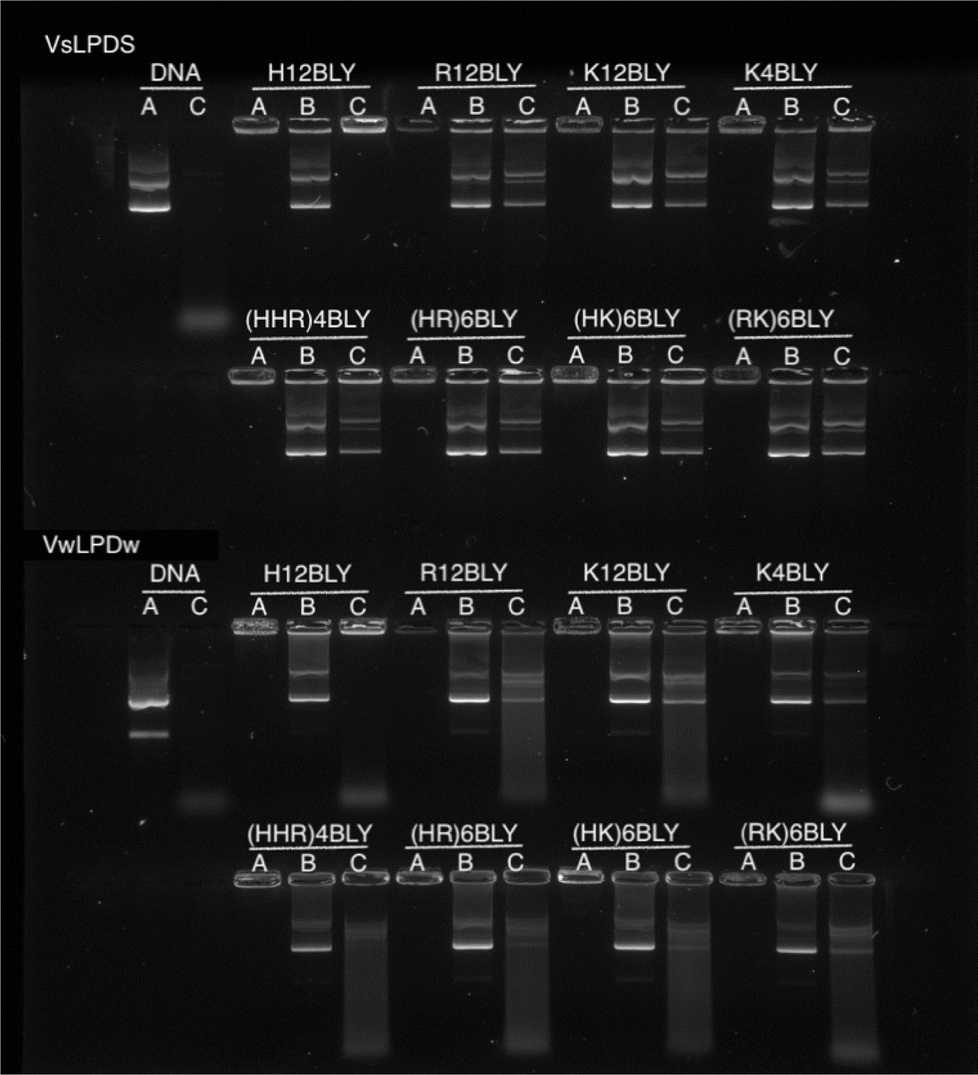 | ||
| Fig. 4 Agarose gel electrophoresis of VwLPDw and VsLPDs (0.25 μg pDNA/10 μL per well). Lane A: DNA or LPD. Lane B: LPD treated with pAsp. Lane C: LPD treated with DNAse I at 37 ± 0.1 °C followed by pAsp. In all cases, LPDs were prepared at a lipid:peptide:DNA charge ratio of 0.5:6:1. Cationic vesicles used to prepare the LPDs were composed of DOTMA:DOPE at 1:1 molar ratio. | ||
To study further the effect of NaCl content on the pDNA protection afforded by the LPDs, LPDs (both VwLPDs and VsLPDs) were prepared using varying NaCl concentrations. Two peptides, namely K12BLY and (HHR)4BLY were selected as representative of Group 1 and Group 2 peptides, respectively. The results are shown in Fig. 5. Here, controls of pDNA dissolved either in water or varying concentrations of NaCl solution were used. As can be seen, pDNA behaved in a similar manner regardless of NaCl content. When DNase I was added to the pDNA (Lane C) no band comparable to intact pDNA was seen; only a diffuse band of low molecular weight fragments towards the bottom of the gel were observed, suggesting the degradation of pDNA by DNase I. It should be noted, however, that the intensity of the low molecular degradation band appeared to increase slightly with increasing saline concentration from 8–120 mM NaCl. When the LPDs were examined, DNA was fully condensed (Lane A) and can be fully released (Lane B) irrespective of the peptide and the NaCl concentration used to prepare LPD complexes and where the LPDs were partially or fully prepared using NaCl solution. It was clear, however, from the results in Lane C that the protection afforded to the pDNA by the complexes increased as the concentration of NaCl increased. Interestingly, there was no significant difference in the pDNA protection afforded by VsLPDs and VwLPDs.
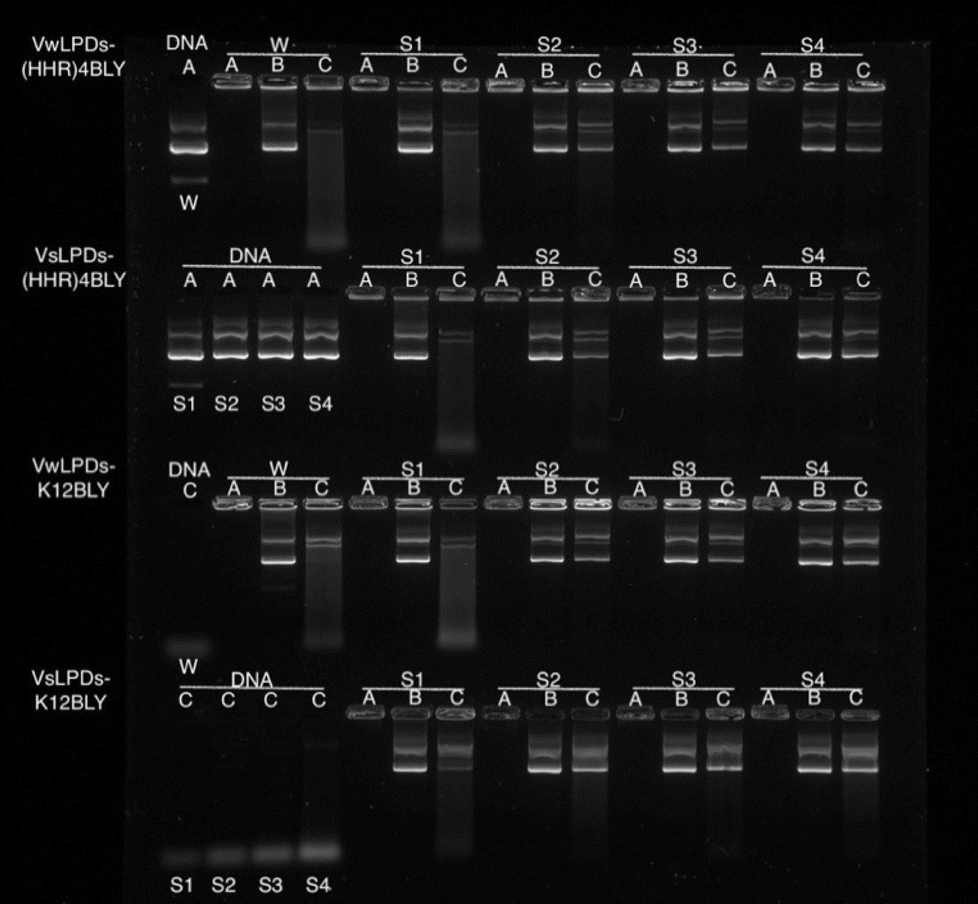 | ||
| Fig. 5 Agarose gel electrophoresis of VwLPDw and VsLPDs (0.25 μg pDNA/10 μL per well). Lane A: DNA or LPD. Lane B: LPD treated with pAsp. Lane C: LPD treated with DNAse I at 37 ± 0.1 °C followed by pAsp. W = water; S1 = 8 mM NaCl; S2 = 40 mM NaCl; S3 = 80 mM NaCl; S4 = 120 mM NaCl. In all cases, LPDs were prepared at a lipid:peptide:DNA charge ratio of 0.5:6:1. Cationic vesicles used to prepare the LPDs were composed of DOTMA:DOPE at 1:1 molar ratio. | ||
To determine the extent of pDNA condensation in the LPDs and to compare the amount of pDNA encapsulated in complexes formulated totally in water (VwLPDw) with those formulated totally in the presence of NaCl (VsLPDs), fluorescence studies using PicoGreen® were carried out. PicoGreen® is used to detect the level of free ‘unentrapped’ DNA in the VsLPDs suspensions. Since PicoGreen® only fluoresces when bound to pDNA, the lower the sample fluorescence compared to free pDNA control the better the DNA condensation efficiency. LPD complexes were formulated at 0.5:1:1 to 0.5:18:1 lipid:peptide:pDNA charge ratios whereby the lipid:pDNA ratio was kept constant but the peptide ratio in the LPD complex increased.
In order to ensure a linear relationship was obtained between the DNA concentration and fluorescence upon interaction with PicoGreen®, different starting pDNA concentrations were used, namely 0.2 and 0.1 μg per 100 μL per well for samples prepared in water and 120 mM NaCl, respectively (ESI, Fig. S3†). In this manner, any possible effects of NaCl on the binding of PicoGreen® to dsDNA, which has been previously reported at higher NaCl concentrations, should be eliminated.58
The results obtained for the VwLPDw and VsLPDs LPD complexes are shown in Fig. 6a and b, respectively. In both figures, the upper graphs show the results obtained using the Group 1 peptides while the lower graphs detail those for Group 2 peptides. Considering the LPDs prepared fully in water (VwLPDw) using Group 1 peptides (Fig. 6a), with the exception of H12BLY at a L:P:D charge ratio of 0.5:1:1, less than 25% fluorescence compared to that produced by free DNA control remained; in the case of H12BLY it was about 45%. Considering the corresponding LPDs prepared using the Group 2 peptide-based LPD complexes, about 40% fluorescence was measured at an L:P:D charge ratio of 0.5:1:1, suggesting that VwLPDw LPDs containing the Group 1 peptides have a greater capacity to condense the pDNA than the corresponding Group 2 complexes. Regardless of whether a Group 1 or Group 2 peptide was used to prepare the complex, upon increasing the peptide ratio up to 0.5:6:1, the condensation of pDNA by the LPD complexes increased slightly, but thereafter no further increase in condensation capacity was seen, with the exception of the (HHR)4BLY containing complex. When considering the LPD complexes prepared totally in 120 mM NaCl solution (VsLPDs) (Fig. 6b), irrespective of the Group of peptides used, an apparently lower ability to condense pDNA was observed. For example, for the Group 1 peptides, the VsLPDs LPDs prepared using H12BLY and K4BLY, at an L:P:D charge ratio of 0.5:6:1 showed ∼50 and ∼70% fluorescence, respectively. With the exception of (RK)6BLY, the VsLPDs LPDs using Group 2 peptides appeared to condense pDNA even less well.
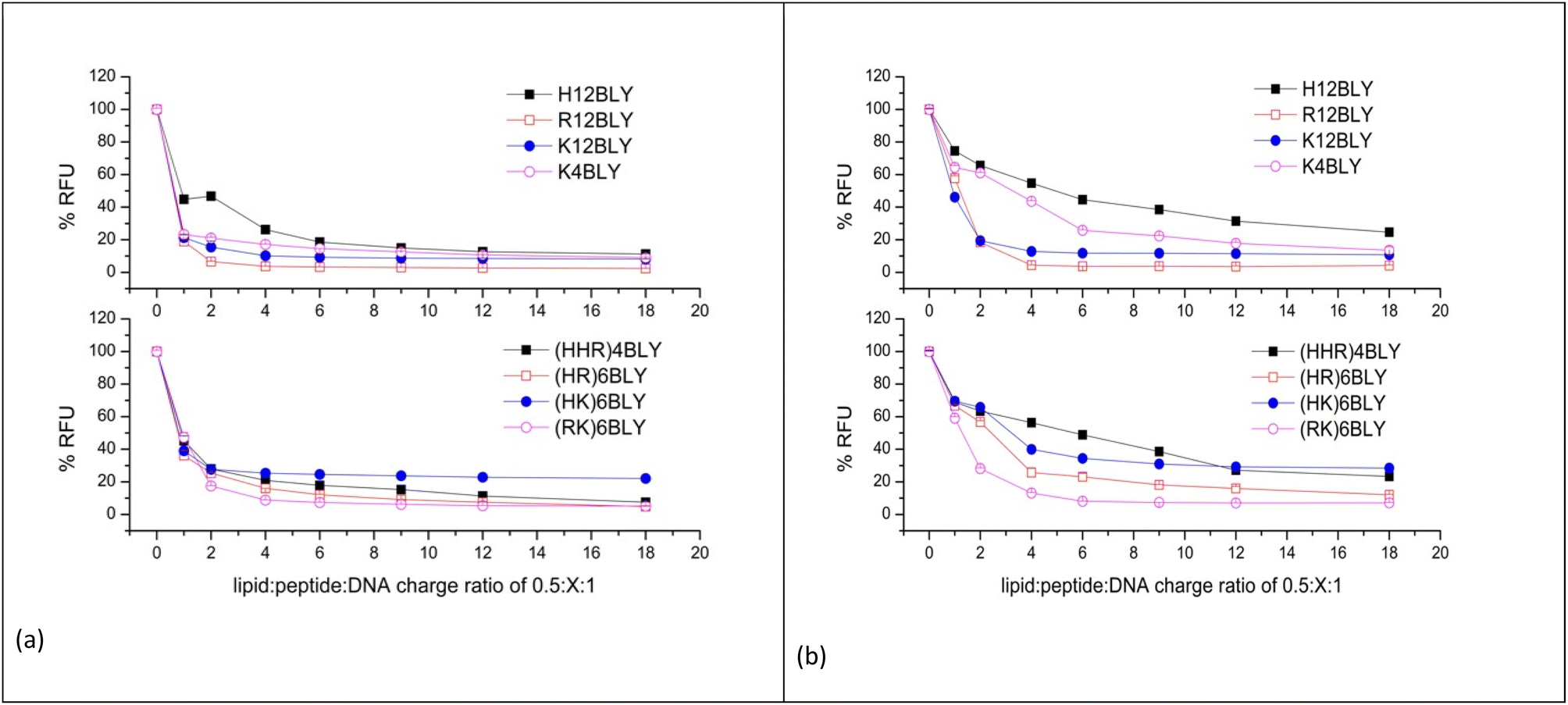 | ||
| Fig. 6 PicoGreen fluorescence assay of DNA uncondensed in LPD complexes containing increasing concentrations of peptide. The LPD complexes were prepared in water (VwLPDw) at 0.2 μg of pDNA per 100 μL per well (a) or 120 mM NaCl solution (VsLPDs) at 0.1 μg of pDNA per 100 μL per well (b). The lipid:DNA charge ratio in the LPD complexes was kept constant at 0.5:1. Measurements were carried out at 25 ± 0.1 °C. Data are mean values ± standard deviation of three replicates. | ||
The reduced ability of VsLPDs to condense pDNA compared to the LPDs prepared completely in water as determined by the PicoGreen® fluorescence assay was at variance with the results obtained using agarose gel electrophoresis. Part of this apparent discrepancy may be because the presence of NaCl is expected to weaken the electrostatic interactions between DNA and the peptide resulting in the PicoGreen® being able to detect both free DNA and DNA weakly complexed with peptide in the presence of NaCl.
Is lipopolyplex size and structure influenced by the cationic peptide sequence and electrolyte?
The apparent hydrodynamic size and ζ-potential of the LPDs at a charge ratio of 0.5:6:1 in water (VwLPDw) and in the presence of 120 mM NaCl (VsLPDs) were determined by dynamic light scattering (DLS) and electrophoretic light scattering techniques, respectively (Fig. 7). As seen, the apparent hydrodynamic size and the ζ-potential of the parent cationic lipid vesicles prepared in water were about 65 nm and 52 mV, respectively. In contrast, the complexes made from these vesicles, VwLPDw, displayed larger apparent hydrodynamic sizes in the range 90–170 nm and ζ-potential of 25–45 mV.
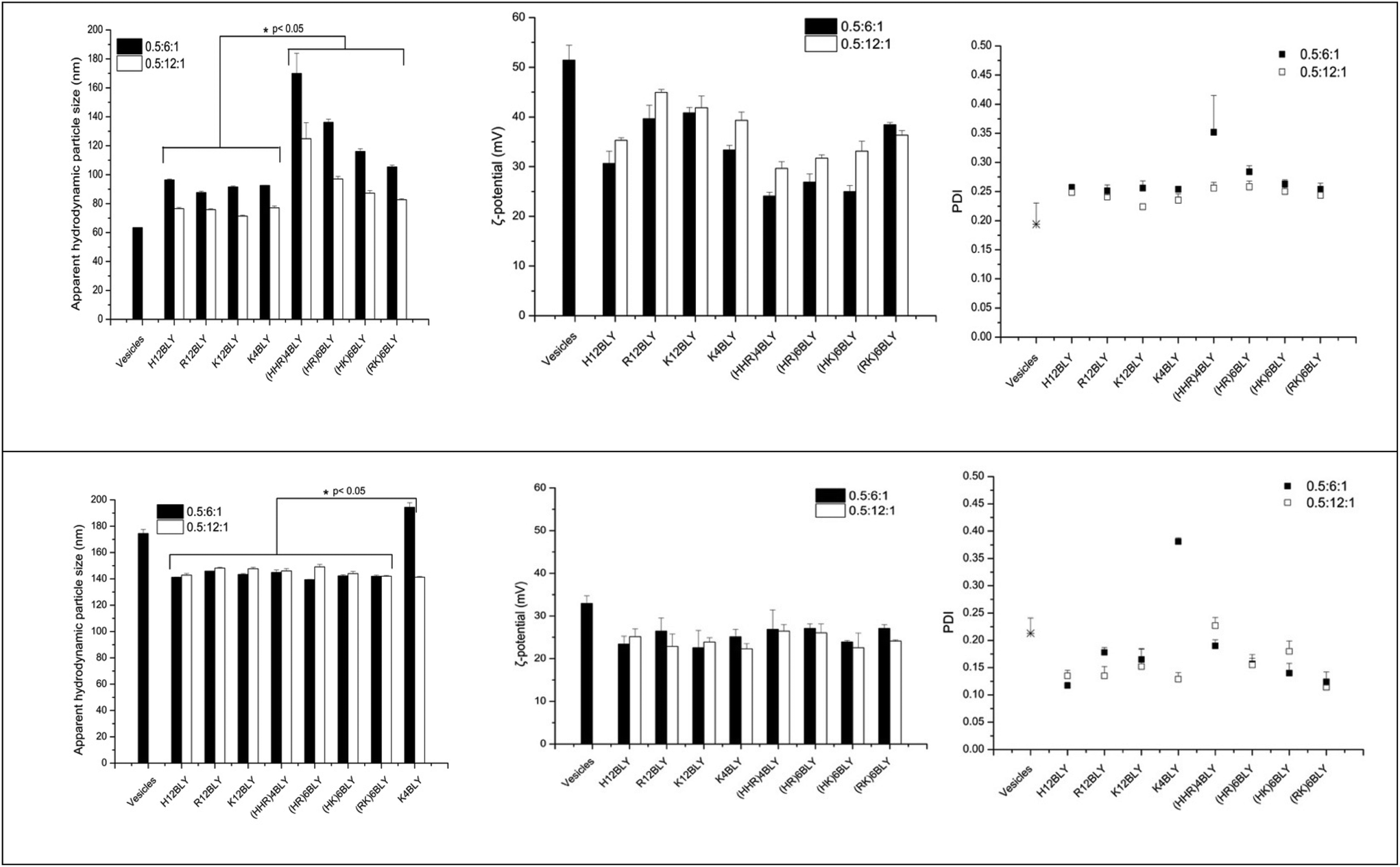 | ||
| Fig. 7 Apparent hydrodynamic particle size, polydispersity index (PDI) and ζ-potential of LPDs made in water (VwLPDw; upper panels) and 120 mM NaCl solution (VsLPDs; lower panels). In all samples, the lipid:peptide:DNA charge ratio was kept at 0.5:6:1 and 0.5:12:1, respectively (final DNA concentration of 0.01 mg mL−1). Cationic vesicles were composed of DOTMA/DOPE at 1:1 molar ratio. Measurements were performed at 25 ± 0.1 °C (n = 3). Data are mean values ± standard deviation of three replicates within the same experiment. The experiments were repeated three times. | ||
The apparent hydrodynamic size of LPDs prepared using Group 2 peptides were generally larger than those containing Group 1 peptides, while the ζ-potentials were lower at around 25–30 mV as compared to 30–45 mV. The lowest ζ-potential for Group 1 peptides was seen with H12BLY, while the highest ζ-potential for Group 2 peptides is exhibited by (RK)6BLY. These observations correlate with the number of His residues each of the cationic peptides possess.
The LPDs prepared in water (VwLPDw) at a lipid:DNA:charge ratio of 0.5:6:1 using a Group 1 peptides have a significantly smaller mean size compared with those containing Group 2 peptides (viz., 92 nm vs. 132 nm, p < 0.05) and the same holds true for the LPDs prepared at a lipid:DNA:charge ratio of 0.5:12:1 (viz., 75 nm vs. 98 nm). There are no significant differences in the ζ-potentials found for the LPDs prepared in water and containing Group 1 versus Group 2 peptides.
In comparison, the apparent hydrodynamic particle size of the parent cationic vesicles prepared in 120 mM NaCl were much larger at about 175 nm than when these vesicles are prepared in water, while their ζ-potential was much lower at 25 mV. However, the LPD formulations, VsLPDs prepared from these vesicles in 120 mM NaCl were, regardless of which cationic peptide used, smaller at 140–145 nm, although they exhibited a similar charge ∼ 25 mV (Fig. 7). LPDs prepared in 120 mM saline (VsLPDs) at both lipid:DNA:charge ratios of 0.6:6:1 and 0.5:12:1 showed no statistically significant difference in size nor in polydispersity.
The LPDs prepared with K4BLY at a lipid:DNA charge ratio of 0.5:6:1 are significantly larger than those prepared with any of the other peptides (viz. 194 nm vs. a mean of 143 ± 2 nm, p < 0.001).
Of particular note, is the more uniform apparent hydrodynamic particle size of the VsLPDs LPDs. Consideration of the polydispersity index (PDI) also shows that, with the exception of VsLPDs LPD containing K4BLY, values of ∼0.15 or less were obtained indicating a narrow size distribution of LPDs. By contrast, VwLPDw LPDs exhibited higher PDI values than the VsLPDs LPDs, suggesting that these VwLPDw LPD preparations were more heterogenous in size. The larger size of vesicles prepared in the presence of the electrolyte, NaCl is most likely due to the presence of multilayers in the vesicles due to the ability of NaCl to partly screen charge. Similarly, the lower ζ-potential observed for the vesicles and LPDs prepared in the presence of NaCl is also likely to be a consequence of the charge effect of the NaCl.
Consideration of the ζ-potential of the formulations would suggest that the more highly positively charged vesicles and VwLPDw LPDs would be more stable than the corresponding preparation made in saline. We therefore studied LPD stability with respect to apparent hydrodynamic size and PDI of the various types of LPD formulations (Fig. 8, Fig. S4†).
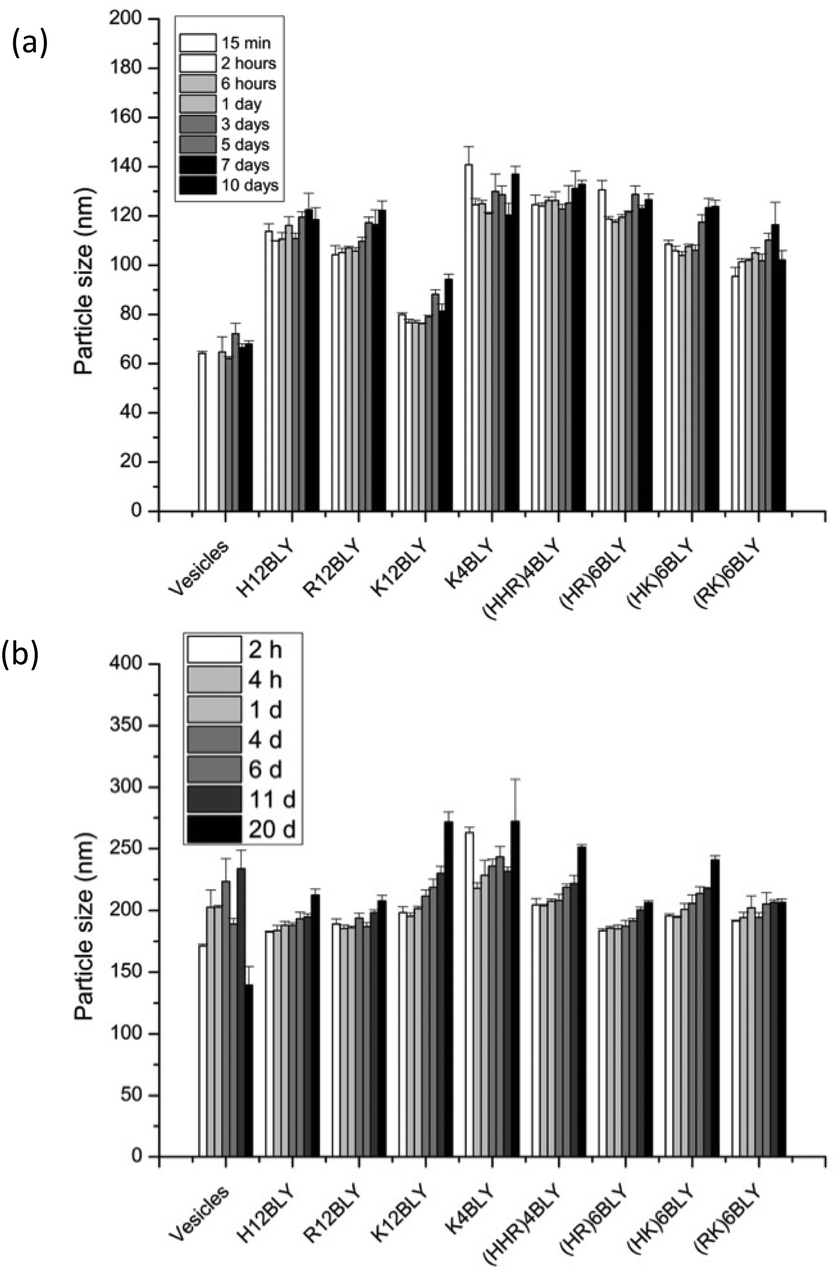 | ||
| Fig. 8 Mean hydrodynamic particle size stability of LPDs made in water (VwLPDw: (a)) and in saline (VsLPDs: (b)). All LPDs were prepared at lipid:peptide:DNA charge ratio of 0.5:6:1 (0.01 mg mL−1 of pDNA). Cationic vesicles were composed of DOTMA/DOPE at 1:1 molar ratio. Data are mean values ± standard deviation of three replicates at 25 ± 0.1 °C. The experiment was repeated once with a similar trend of results being obtained. | ||
This showed that while the VwLPDw LPD formulations were stable with respect to apparent hydrodynamic size over a period of 10 days, (Fig. 8a), the PDI of the preparations started to increase after about 5–7 days (Fig. S4a†) suggesting the presence of some larger particles. Longer periods were not tested. In contrast, the VsLPDs LPDs retained their apparent hydrodynamic size (Fig. 8b) and PDI value (Fig. S4b†) for 10 days and thereafter there was some increase in particle size. The very slightly greater size stability of the VsLPDs complexes may be a consequence of their relatively more homogeneous size reducing Ostwald ripening compared to the VwLPDw LPDs.59
To further study the effect of NaCl content on the apparent hydrodynamic particle size and PDI of LPDs (both VwLPDs and VsLPDs) were prepared using varying NaCl concentrations, namely 0.008, 0.04, 0.08 and 0.12 M NaCl. For this study, we prepared VwLPDs and VsLPDs formulations containing the Group 2 cationic peptide, (HHR)4BLY. The results of particle size and PDI measurements are shown in Fig. 9. The vesicles prepared in saline (Vs) exhibited consistently larger apparent hydrodynamic sizes than those prepared in water (Vw). Similarly, the apparent hydrodynamic size of the LPD complexes prepared fully in the various strength NaCl solutions (VsLPDs), were larger than those prepared using LPDs made fully or partially in water (VwLPDw/VwLPDs).
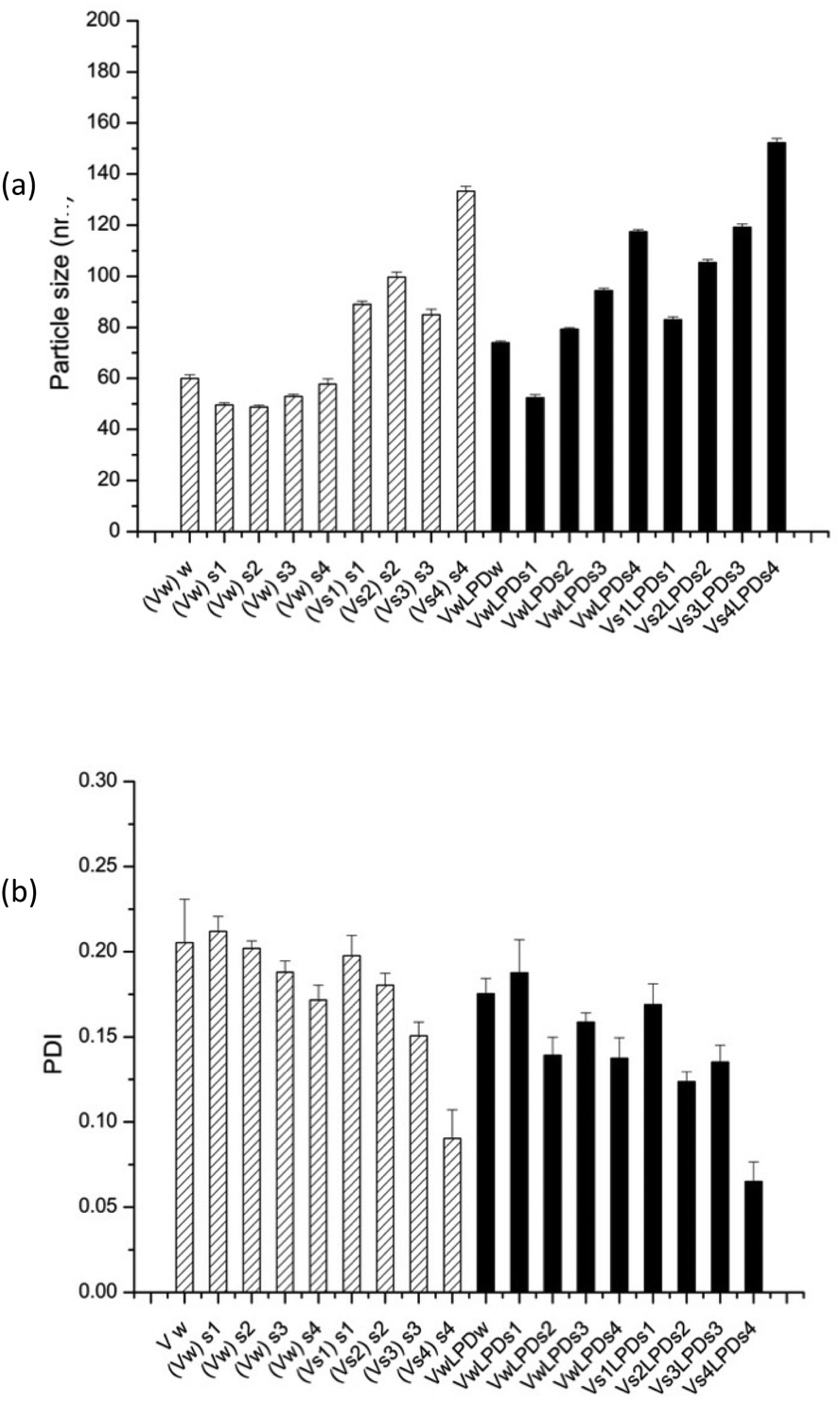 | ||
| Fig. 9 Apparent hydrodynamic particle size (a) and PDI (b) of (HHR)4BLY-based LPD prepared in various NaCl solutions (S1 = 8; S2 = 40; S3 = 80; S4 = 120 mM NaCl). Vw and Vs indicate that lipid vesicles used to make LPD were prepared in water and NaCl solution, respectively. (Vw)s means vesicles prepared in water and diluted in NaCl solution, while (Vs)s means vesicles prepared and diluted in NaCl solutions. LPDs mean that LPD complexes were prepared in NaCl solution. In all samples, lipid:peptide:DNA charge ratio was kept at 0.5:6:1 (0.01 mg mL−1 pDNA). Cationic vesicles were composed of DOTMA/DOPE at 1:1 molar ratio. Measurements were performed at 25 ± 0.1 °C. Data are mean values ± standard deviation of three replicates. The experiment was repeated once with a similar trend of results being obtained. | ||
Furthermore, for both VsLPDs and VwLPDs formulations, particle size significantly increased in a NaCl concentration-dependent manner, accompanied by decreasing PDI values. Thus, increasing concentrations of NaCl during both the preparation of the initial vesicles and the formulation of the LPDs results in complexes that are larger, but more uniform in size, have less surface charge and give higher levels of transfection when diluted in serum-containing media.
To understand the effects that assembly of the LPD complexes in NaCl solution have on the structure and macromolecular organization of these nanoparticles, and to elucidate how this translates into enhanced transfection in the presence of serum, we performed a series of small angle neutron scattering (SANS) experiments using ctDNA. We have investigated LPDs made in water (VwLPDw) and compared these with LPDs prepared in water containing varying concentrations of NaCl (VsLPDs) and containing the peptide, (HHR)4BLY, chosen as representative of the peptides used in the present study. Note that in the present study we have not examined the structure of the complexes formed by either lipid vesicles or peptide with DNA, however previous studies39,41,51,57,60 have shown these compositions in water form lipoplexes and polyplexes, respectively. It would be anticipated that, even in the presence of NaCl, lipoplexes and polyplexes would still be formed, albeit with larger d-spacings in the case of the lipoplexes and as a looser, fluffier complex in the case of the polyplexes.
The structures of the parent DOTMA:DOPE vesicles prepared in either D2O or various concentrations of NaCl were also investigated. Fig. 10 shows the SANS data (plotted as neutron scattering intensity (I) as a function of momentum transfer Q, plotted against Q) along with the best fit to the data for the DOTMA:DOPE vesicles while Table 1 shows the parameters used to obtain the best fit. The results obtained for the vesicles prepared in water agree well with results previously reported by our group.39,41,51,57,60 It is clear both from the nature of the scattering curve and the parameters used to obtain the best fit, that the presence of salt has a significant effect on the structure of the vesicles which become increasingly multilamellar as the salt concentration is increased. Significantly, while it was possible to fit vesicles prepared in D2O assuming the presence of unilamellar vesicles only (the ‘sheet’ model), it was necessary to invoke a ‘mixed sheet and stack’ model to account for the increasing number of multilamellar vesicles with increasing NaCl concentration. A few points are clear from Table 1. Firstly, there is little change in the thickness of the vesicle's bilayer as the concentration of NaCl increases (38.4 ± 1 Å in the absence of NaCl (Vw) to 36.6 ± 1 Å in the presence of 120 mM NaCl (Vs)). The d-spacing of the vesicle bilayers in the presence of NaCl is about 130 Å although there is quite a large variability on this value. It is also clear from the analysis that the proportion of multilamellar vesicles increased with NaCl concentration, in that when 40 mM of NaCl was present only 3 vol% of the vesicles contained more than 3 bilayers, and when 120 mM NaCl is present, this rose to 30 vol%. It should be noted that the vesicles were prepared and measured on several occasions with a very similar trend of results being observed.
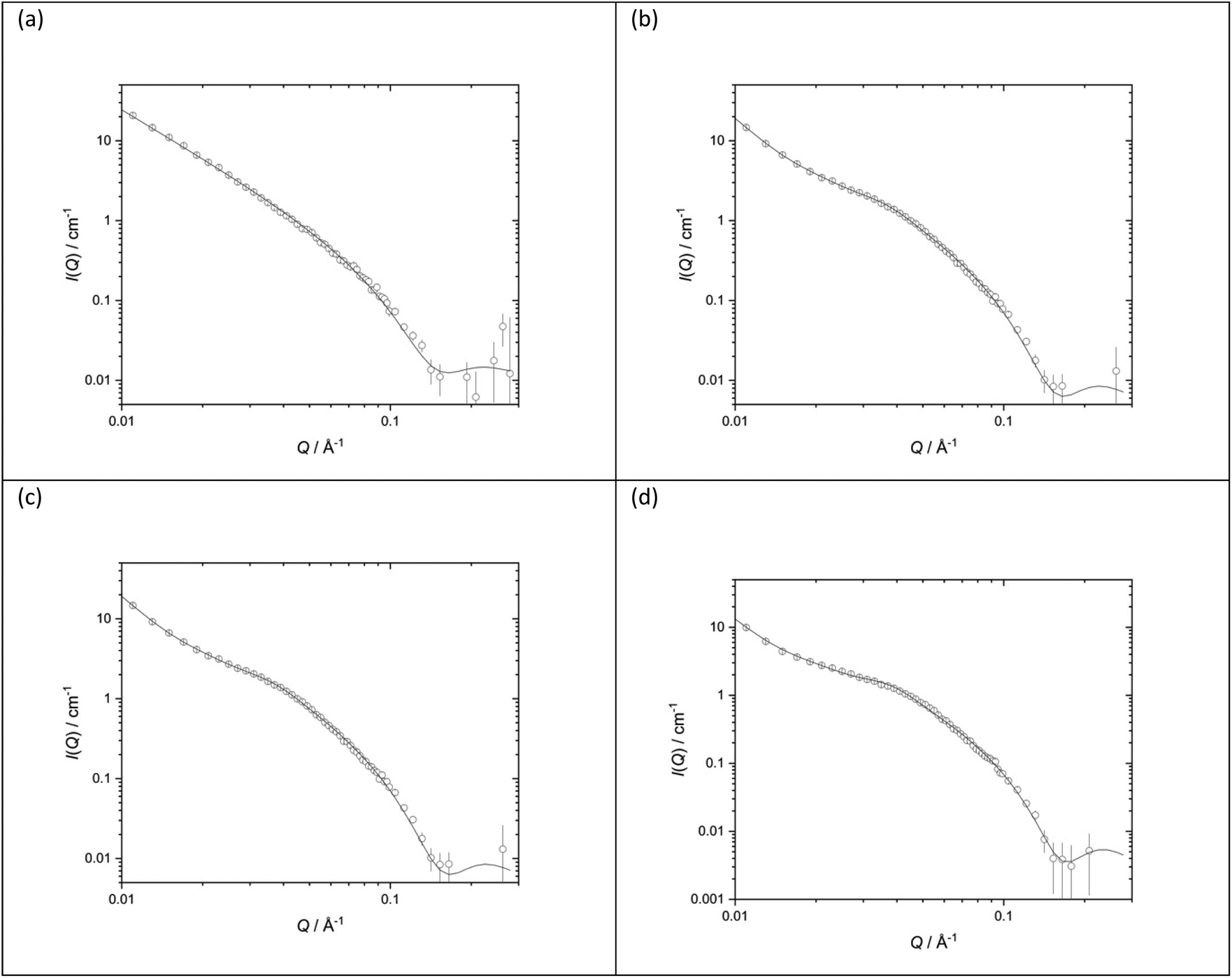 | ||
| Fig. 10 Small angle neutron scattering (SANS) data (dots) and best fit (solid line) for vesicles prepared using DOTMA:DOPE at 1:1 molar ratio (1.0 mg mL−1 DOTMA) in various concentrations of aqueous NaCl solution (a) D2O (b) 40 mM (c) 80 mM (d) 120 mM. SANS was measured at 25 ± 0.1 °C. | ||
:DOPE at 1:1 molar ratio (1.0 mg mL−1 DOTMA) in various concentrations of aqueous NaCl solution (a) D2O (b) 50 mM (c) 100 mM (d) 150 mM. SANS was measured at 25 ± 0.1 °C. Figures in brackets indicate the standard errors on the fitted parameter values
| Vesicles | D2O | 40 mM NaCl | 80 mM NaCl | 120 mM NaCl |
|---|---|---|---|---|
| d-Spacing (Å) | — | 134.3 (8) | 129 (58) | 132 (53) |
| Thickness (Å) | 38.4 (1) | 38.6 (1) | 37.7 (1) | 36.6 (1) |
| Unilamellar | — | 29% | 34% | 24% |
| Bi/trilamellar | — | 68% | 57% | 46% |
| >Trilamellar | — | 3% | 9% | 30% |
Fig. 11 shows the SANS data (plotted as neutron scattering intensity (I) as a function of momentum transfer Q, was plotted against Q), along with the best fit obtained to the data, of the LPDs prepared from the parent DOTMA:DOPE vesicles in D2O or various concentrations of NaCl. Table 2 shows the parameters used to obtain the best fit to the SANS data. In previous experiments39,41,60 we used the fit obtained for the parent vesicles to determine if there was any significant change to the thickness of the bilayer when used to prepare the LPDs. However, in the present study, because of the complexity of the model and the presence of multilamellar vesicles, such an approach was not taken and so all the LPDs were fitted with the ‘mixed sheet and stack model’. Table 2 indicates that the thickness of the DOTMA:DOPE bilayers in the LPDs prepared in water was slightly thicker than in the parent vesicles (42.4 ± 1 Å as opposed to 38.4 ± 1 Å), and increasing NaCl concentration in the LPD preparations lead to no change in bilayer thickness (42.4 ± 1 Å in the absence of NaCl to 40.4 ± 1 Å in the presence of 120 mM NaCl). Again, as with the vesicles, the polydispersity on the d-spacing is high, while the mean d-spacing has slightly decreased to ∼85 Å, most probably due to the NaCl screening the charge on the lipid bilayer. In line with the vesicles, the amount of multilamellar LPDs increased with NaCl concentration. From these observations it is likely that the peptide/ctDNA complex is predominately located in the core of the LPDs prepared, with only a small amount of complex being present in the water layers trapped between the cationic lipid bilayers in those LPDs made fully in the presence of NaCl (VsLPDs), possibly accounting for the high polydispersity observed in the d-spacing.
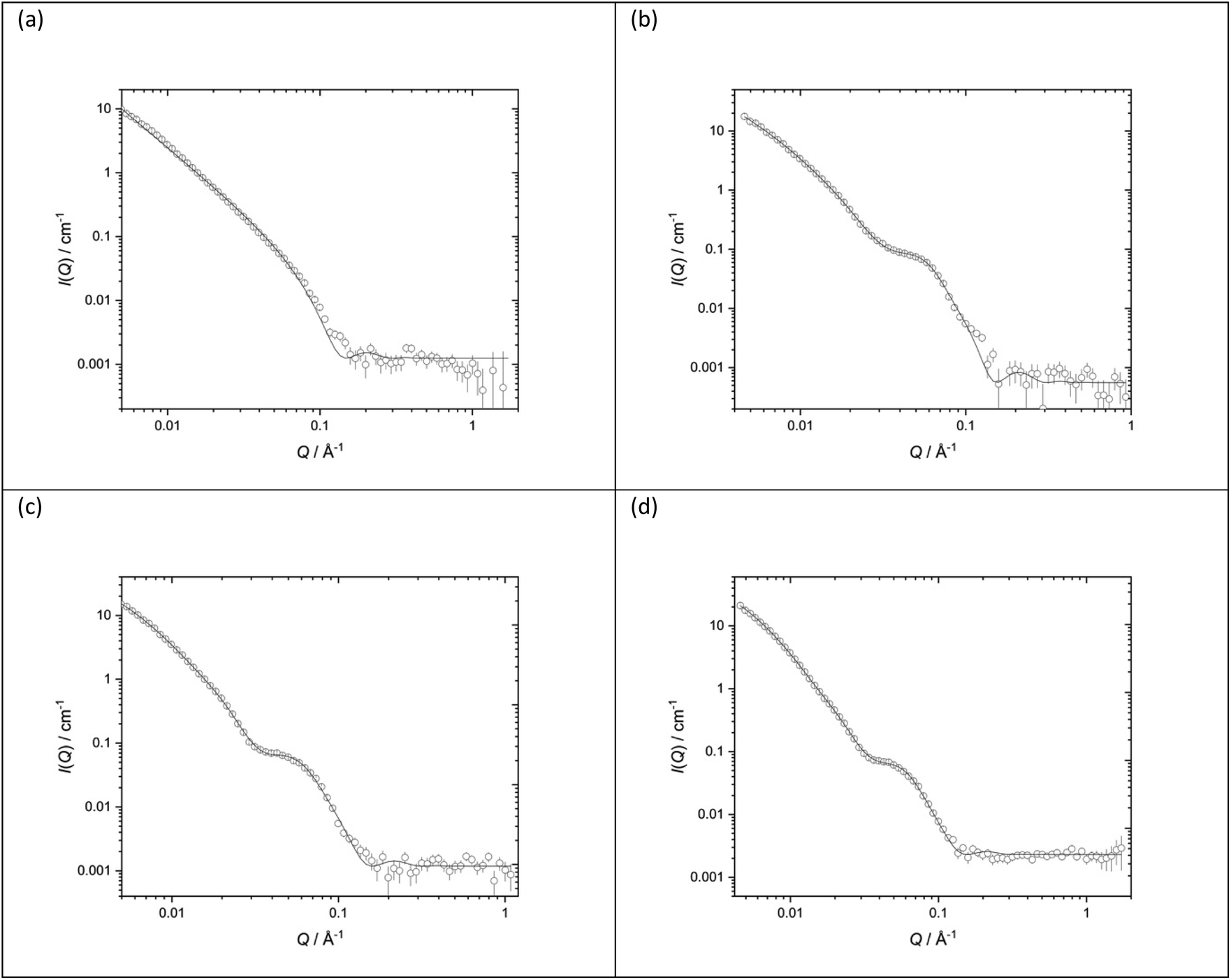 | ||
| Fig. 11 Small angle neutron scattering (SANS) data (dots) and best fit (solid line) for LPDs containing the peptide (HHR)4BLY at a lipid:peptide:ctDNA charge ratio 0.5:6:1 and made from vesicles prepared using DOTMA:DOPE at 1:1 molar ratio (1.0 mg mL−1 DOTMA) in various concentrations of aqueous NaCl solution (a) D2O (b) 40 mM (c) 80 mM (d) 120 mM. SANS was measured at 25 ± 0.1 °C. | ||
:peptide:ctDNA charge ratio 0.5:6:1 and made from vesicles made using DOTMA:DOPE at 1:1 molar ratio (1.0 mg mL−1 DOTMA) in various concentrations of aqueous NaCl solution (a) D2O (b) 40 mM (c) 80 mM (d) 120 mM. SANS was measured at 25 ± 0.1 °C. Figures in brackets indicate the standard errors on the fitted parameter values
| LPDs | D2O | 40 mM NaCl | 80 mM NaCl | 120 mM NaCl |
|---|---|---|---|---|
| d-Spacing (Å) | 97 (25) | 85 (30) | 83 (31) | |
| Thickness (Å) | 42.4 (1) | 40.9 (1) | 39.5 (1) | 40.4 (1) |
| Unilamellar | 30% | 0% | 0% | |
| Bi/trilamellar | 66% | 84% | 65% | |
| >Trilamellar | 4% | 16% | 35% |
To ensure that the results obtained for the LPDs containing (HHR)4BLY were representative of LPDs containing other peptides, LPDs prepared in the absence and presence of NaCl and containing peptides from either Group 1 (R12BLY, K12BLY) or Group 2, ((HK)6BLY) were examined at two L:P:D mixing ratios (Table 3). Significantly, the bilayer thickness obtained for this batch of LPDs prepared in water and in 120 mM NaCl were very comparable to those detailed in Table 2, which were prepared and measured on a different occasion. Moreover, there is no significant difference in the bilayer thickness of the LPD complexes containing the various peptides studied, thereby supporting the hypothesis that the nature of the peptide and the presence of the ctDNA does not influence the structure of the bilayers (with these molecules residing predominately in the aqueous compartments of the particles). Although not shown in Table 3, the volume percentage of the various types of multilamellar vesicles remains largely unchanged with peptide type and L:P:D mixing ratio. It is noticeable, however, that while the d-spacings obtained for the LPDs reported in Table 3 were comparable to those obtained for the vesicles detailed in Table 1, they were larger than those seen for the LPDs noted in Table 2. The reason for this difference is not clear but possibly indicates that more of the complex is present in the water layer between the bilayers in the batch of LPDs reported in Table 2, although it should be noted that there is a large polydispersity on all of the d-spacings.
:DOPE at 1:1 molar ratio (1.0 mg mL−1 DOTMA) and containing the peptide (HHR)4BLY in various concentrations of aqueous NaCl solution (a) D2O (b) 40 mM (c) 80 mM (d) 120 mM and ctDNA. SANS was measured at 25 ± 0.1 °C. Figures in brackets indicate the standard errors on the fitted parameter values
| LPDs | LPDs (D2O) | LPDs (D2O) | LPDs (120 mM NaCl) | LPDs (120 mM NaCl) |
|---|---|---|---|---|
| L:P:D ratio |
0.5:6:1 |
0.5:12:1 |
0.5:6:1 |
0.5:12:1 |
| (HHR)4BLY | ||||
| Thickness (Å) | 42.4 (1) | 46 (2) | 41 (5) | 34 (4) |
| d-Spacing (Å) | — | — | 144 (78) | 156 (81) |
| (HK)6BLY | ||||
| Thickness (Å) | 46.0 (2) | 45.2 (2) | 38 (3) | 39 (6) |
| d-Spacing (Å) | — | — | 153 (70) | 146 (81) |
| R12BLY | ||||
| Thickness (Å) | 42.7 (2) | 41.5 (2) | 37 (5) | 35 (5) |
| d-Spacing (Å) | — | — | 144 (78) | 143 (79) |
| K12BLY | ||||
| Thickness (Å) | 41.3 (2) | 42 (2) | ND | ND |
| d-Spacing (Å) | — | — | ||
| (HHR)4BLY | ||||
| Thickness (Å) | 42.4 (1) | 46 (2) | 41 (5) | 34 (4) |
| d-Spacing (Å) | — | — | 144 (78) | 156 (81) |
Discussion
In order to design gene delivery vectors with high transfection efficiencies in vivo, many competing factors have to be taken into account. High levels of protection of the DNA cargo are desirable, particularly when administering the vectors in the presence of serum. Conversely, once the vector has been internalised within the target cell, efficient release from the endosome and trafficking to the nucleus become of paramount importance. Here, we have investigated two different approaches to fine-tune the balance between these competing requirements, and to optimise the formulation of ternary lipid–peptide–DNA LPD for administration in the presence of serum. The effects of different singly branched cationic peptide sequences were investigated, containing Lys, His and/or Arg residues, combined with peptide sequences previously shown to target human airway epithelial cells and to be cleavable within the endosome.Formulating these complexes in the presence of NaCl solutions gave LPDs with greatly enhanced transfection efficiencies in the presence of serum, and a detailed study of the effects of electrolyte on the formulation and structure was performed.
Initially we studied the properties of a series of peptides with long homogeneous cationic residue sequences (H12BLY, R12BLY, K12BLY) (Group 1 peptides), along with a second series of novel peptides with alternating His/Arg, Lys/Arg and His/Lys sequences (Group 2 peptides: (HHR)4BLY, (HR)6BLY, (HK)6BLY and (RK)6BLY). Although in previous work,39 branched Arg and linear Lys-containing cationic sequences significantly outperformed mixed His/Lys and homogeneous His sequences, here the Group 2 peptides were more effective transfection agents. It has previously been suggested that His-rich peptide sequences promote endosomal escape of polyplexes, and subsequent decomplexation of the pDNA cargo, through the “proton sponge” effect.28–31 However, these LPD complexes incorporated high levels of DOPE, which can induce endosomal membrane destabilisation61 and our previous studies have shown that this is a more significant factor than the presence of His residues in the cationic peptide sequence.
The superior performance of the Group 2 peptides can be attributed to other factors. Firstly, the Group 1 peptides in this study are longer with more cationic residues than in our previous work. The increased DNA compaction of the R12BLY and K12BLY sequences, reflected in their greater capacity to complex pDNA in both water and saline (Fig. 6) is likely to be offset by poorer eventual release of the pDNA cargo. Secondly, most of the Group 2 sequences have Arg residues, alternating with His or Lys residues, cationic motifs not previously reported for lipopolyplex delivery. For transfection to be successful in gene delivery vectors, the pDNA must overcome the barrier to translocation to the nucleus through the nuclear pore complex. In nature, this process is mediated by recognition of the karyopherin family of nuclear protein transporters by nuclear localisation sequences (NLS)62 found in viral targeting proteins63 and histones.64 These NLS are characterised by cationic motifs rich in Arg and Lys residues, with bipartite ((K/R)–(K/R)–X10–12–(K/R)3/5) and monopartite (K–(K/R)–X–(K/R)) motifs being particularly well characterised.63 Indeed, the NLS from the SV40 large tumour antigen has been used to enhance uptake of pDNA into the nucleus in a variety of gene delivery systems65 including LPDs.66 It is therefore also possible that the Arg-rich repeating motifs in the Group 2 peptides serve an additional function as an unidentified NLS, enhancing transfection further, particularly as we have shown that the cationic peptide moiety of similar LPD formulations remains complexed to the pDNA and is trafficked to the nucleus.12 Finally, the presence of alternating Arg residues in the cationic sequences of the Group 2 peptides may also lead to improved pDNA condensation and release properties of the LPD complexes, compared to the LPD complexes formulated from alternating His-Lys cationic peptides previously studied. Theoretical, NMR and molecular dynamics studies have established that the interaction between Arg side chains and nucleic acids is highly rigid compared to that between Lys side chains and nucleic acids.67 We have previously demonstrated that homo-Arg and homo-Lys cationic peptides have different modes of DNA condensation in LPD complexes.39 Comparative studies on peptide-pDNA polyplexes have also shown that polyplexes formulated from homo-Arg sequences are more likely than those formulated from homo-Lys sequences to form multimolecular condensed nanostructures that completely release DNA.68 Also, that the distribution of His residues in polyplexes formulated from mixed His-Arg cationic peptide sequences can fine-tune the balance between condensation and pDNA release.69,70 The probable effect of the Arg side chain in modulating the pKa of the His side chains must also be considered.71
Importantly, this study has demonstrated the importance of formulating lipopolyplexes in the presence of NaCl. Independent of the peptide sequence, the VsLPDs have considerably enhanced transfection in serum, and this increased at higher concentrations of NaCl (Fig. 2). We have shown that VsLPDs tend to form larger particles that have lower ζ-potential and are more homogeneous in size (Fig. 7). This slightly larger particle size may explain, at least in part, some of the improvement in transfection efficiency; large particles are known to sediment more rapidly, resulting in a greater number being in contact with the cells, thus increasing transfection. Our SANS experiments suggest a probable explanation for this increase in size in the presence of NaCl, namely the VwLPDw are unilamellar whereas the VsLPDs are multilamellar. Previous SANS studies, performed by our group, in which we determined the structure of a variety of LPDs prepared from vesicles, cationic peptide, polylysine or cationic peptide (polylysine) conjugated to a targeting lipid and either DNA or siRNA, and prepared in water, indicated that complexes comprising cationic peptide and nucleic acid were contained in the core of the lipopolyplex which was surrounded by a single bilayer – the number of lipopolyplexes containing more than one bilayer was extremely small. Indeed, the present study was the first time the presence of (a significant amount) of multilamellar structures has been reported.39,41,51,57,60
To our knowledge, this is the first time that the effect on structure of formulating lipopolyplexes in the presence of NaCl has been reported. However, our results are consistent with previous work investigating the interactions of electrolytes with surfactant-DNA and polycation-DNA, and with lipoplex (DNA–lipid) formulations72,73 while a range of theoretical and experimental studies have shown the effect of NaCl on DNA complexation. SAXS measurements74 and computational simulations75 have indicated that complexes of DNA with either polyLys or polyArg are tightly packed at low salt concentrations, transitioning to loosely packed and then dissociated structures at increasing salt concentrations. In the case of lipoplexes, salt induces partial dissociation between lipid and DNA, an effect which can be modulated by DOPE. At lower concentrations of NaCl, compact lipoplexes are observed which still show enhanced transfection in serum and DNA release.76 Higher concentrations of NaCl, similar to those used in the present study (0.1 M–0.2 M), yielded larger lipoplexes with reduced surface charge and better serum transfection,43,77,78 which is consistent with our observations in the present study, that VsLPDs complexes have lower ζ-potential than VwLPDw (Fig. 7). Finally, increasing NaCl concentration has previously been shown to lead to an increase in pDNA encapsulation during lipoplex formulation.79,80
For the lipopolyplexes prepared in the present study to be effective transfection agents in serum, they must also form stable nanocomplexes that protect the pDNA from enzymatic degradation. Our agarose gel electrophoresis results (Fig. 4 and 5) indicate that DNA complexation is complete in all VsLPDs formulations (Lane A), and that these are very effective in protecting pDNA from DNase I degradation (Lane C), in contrast the VwLPDw preparations did not afford protection from DNase I (Lane C). Furthermore, increasing the concentration of NaCl enhances the protection, independent of the peptide sequence used for the formulations. However, the PicoGreen® fluorescence assays (Fig. 6) are in apparent variance with respect to the extent of DNA entrapment observed with the agarose gel electrophoresis (Lane A). Specifically, significant PicoGreen fluorescence was observed in the VsLPDs suspensions, even those prepared at peptide:DNA ratios higher than 6:1, with this effect being particularly pronounced for some Group 1 (K4BLY, H12BLY) and Group 2 ((HHR)4BLY, (HK)6BLY) peptides. This is not the first time that apparent discrepancies between agarose gel electrophoresis and PicoGreen® assays have been reported.25
In the present study, the apparent differences observed between the two assays are the result of the method of preparation of the lipoplexes which means that the majority, if not all, of unentrapped DNA in the LPD suspension is likely to be complexed with cationic peptide (present in at least a 6:1 charge ratio), rather than (free) DNA. The implications of the presence of peptide complexed DNA is significant because instead of having free (negatively charged) DNA in the well of the agarose gel, there instead will be positively charged complex which will not travel down the gel once the voltage is applied but rather remain stationary in the well. As a consequence, it will appear that there is no free/unentrapped DNA in the LPD preparation in Lane A. It is worth commenting that, by comparison, all the DNA controls in the agarose gel electrophoresis experiments contained only DNA.
In contrast, the PicoGreen® assay detects the level of unentrapped DNA in the VsLPDs suspensions. In this assay, the PicoGreen® is added in solution to the suspension where it interacts with any free DNA. Significantly these PicoGreen® results indicate a lower level of DNA entrapped in the VsLPDs than in the VwLPDsw.81 This result accords well with both the weakened electrostatic interactions between DNA and the peptide resulting in the PicoGreen® detecting both free DNA and DNA weakly complexed with peptide in the presence of NaCl and the multilamellar nature of the lipopolyplexes formed in the presence of NaCl as it is widely recorded that, for the same amount of lipid, multilamellar structures entrap less material in their aqueous compartments than their unilamellar counterparts.82
The higher protection afforded to the DNA entrapped in VsLPDs compared to VwLPDw (as seen in gel electrophoresis experiments) may be due to the inhibition of DNase I at high NaCl concentrations83 or the modulation of PicoGreen fluoresence in the presence of NaCl.58 It is far more likely however that the absence of any DNA release/degradation with the VsLPDs lies in the fact that the majority of the entrapped DNA/peptide complex (or polyplex) is protected by its incorporation in the central core of the lipopolyplex. Indeed, our SANS data indicate that while some of the DNA/peptide complex may reside in the water layers between the lamellae, most of the DNA/peptide complex is likely to be located in the core of the lipopolyplex where it is largely shielded from the milieu. These results suggest that while less of the DNA may be entrapped in the VsLPDs formulations, it is better protected and as a consequence more efficiently delivered to the nucleus where it can exert its activity.
Furthermore, it is possible that the presence of Na+ and Cl− ions in the LPD formulation could reduce the electrostatic interaction between the DNA and the peptide in the complex.84 Since DNA dissociation upon cellular internalisation is highly important for efficient transfection, the enhancement in transfection seen in the presence of NaCl could be attributed to this more loosely bound DNA structure.
Conclusions
The present study has a number of important implications for the development of improved lipopolyplexes for gene delivery: firstly, the design of the peptides. The results from the current study, in combination with those from an earlier study of ours39 have established that the length of the cationic sequence is important in determining the level of transfection obtained. Here, we observe that lengthening the number of cationic residues in the homopolymer from H/R/K6BLY to H/R/K12BLY results in a decrease in transfection. This observation is most likely a consequence of the increased DNA compaction seen with the longer cationic sequences, which is offset by poorer eventual release of the pDNA cargo within the target cell. Furthermore, the nature of the alternating cationic peptide sequence is important in determining the level of transfection obtained, specifically if arginine is present in an alternative residue pattern, wherein there is an increase in transfection over the mixed peptides prepared in its absence, e.g. LPDs containing (HR)6BLY exhibit greater transfection than those containing (HK)6BLY. The importance of the eventual release of the DNA from the peptide, is demonstrated by the greater transfection achieved when the second peptide is histidine rather than lysine. Significantly, despite their potential as complexing agents, these cationic arginine-containing motifs have not been previously reported for lipopolyplex delivery.Secondly, making lipopolyplexes in the presence of sodium chloride yields preparations that possess a number of very important attributes that make them very exciting for use as gene delivery vehicles. Significantly, VsLPDs are multilamellar in nature compared to the LPDs prepared only in water (VwLPDw) which are unilamellar, meaning that they have only one bilayer surrounding a core containing complexed DNA and peptide. The resulting attributes in these multilamellar LPDs include the high levels of size reproducibility and protection afforded to entrapped DNA. Indeed, the protection afforded the entrapped DNA more than outweighs the fact that the VsLPDs entrap slightly lower amounts of DNA/peptide complex than the VLPDs. It also means that it is possible to entrap ‘loosely’ bound DNA/peptide complex in the VsLPDs allowing for efficient dissociation of the complex and enhanced transfection once internalised in the cell. Finally, if the LPDs are to be exploited clinically they need to be stable in serum. Reassuringly, the good levels of transfection obtained in RPMI-1640 medium containing 10% FBS with the VsLPDs demonstrate that there is a considerable advantage gained by formulating LPDs in the presence of sodium chloride.
One significant advantage of the LPDs used in the present study is the presence of a cationic lipid with a permanent charge which means that the stability of the resulting gene delivery vehicle is unaffected by the buffer used in its preparation, or by a change in pH, yielding LPDs. This behaviour contrasts strongly with the behaviour of the lipid nanoparticles (LNPs) widely used for the delivery of the COVID vaccine, where the properties of the ionisable cationic lipid used in their formation are critically dependent upon pH and buffer salts. The results of the present study suggest that a profitable way forward to reduce the toxicity of the permanent cationic lipids used in the preparation of LPDs would be to design a cationic lipid that can be readily degraded in the cell once it has released its nucleic acid payload.
Author contributions
The peptide synthesis was carried out by L.C. and F.C. LPD formulation and characterisation, transfection, gel retardation and PicoGreen assays were carried out by L.C. and L.K. SANS measurements were carried out by L.C. and M.J.L., and the data was analysed by D.J.B. and M.J.L. The project conceptualisation was by all authors and the project was supervised by M.J.L., A.B.T., H.C.H. and L.K. The original draft of the paper was written by ABT, and the manuscript has been reviewed and edited by all contributing authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
King's College London is thanked for the award of a KORS-China studentship to L.C. The EPSRC is thanked for the award of a Nanotechnology Grand Challenge Grant (EP/G061521/1) to L.K. and F.C. Experiments at the ISIS Neutron and Muon Source were supported by beamtime allocation from STFC, and the SANS data are available at https://doi.org/10.5286/ISIS.E.RB1210318 and https://doi.org/10.5286/ISIS.E.RB1220379. This work benefited from the use of the SasView application, originally developed under National Science Foundation Award DMR-0520547. SasView also contains a code developed with funding from the European Union's Horizon 2020 Research and Innovation Program under the SINE2020 project, Grant No. 654000. The expert help of Dr Ann Terry (ISIS, The Rutherford Appleton Laboratories) is gratefully acknowledged for SANS measurements and data analysis. A549 cells (adenocarcinomic human alveolar basel epithelial) were obtained from ATCC (Manassas, USA).References
- L. Naldini, Nature, 2015, 526, 351–360 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. G. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- C.-C. Ma, Z.-L. Wang, T. Xu, Z.-Y. He and Y.-Q. Wei, Biotechnol. Adv., 2020, 40, 107502 CAS.
- O. S. Thomas and W. Weber, Front. Bioeng. Biotechnol., 2019, 7, 415 CrossRef PubMed.
- A. Wahane, A. Wagmode, A. Kapphahn, K. Dhuri, A. Gupta and R. Bahal, Molecules, 2020, 25, 2866 CrossRef CAS PubMed.
- J. D. Torres-Vanegas, J. C. Cruz and L. H. Reyes, Pharmaceutics, 2021, 13, 428 CrossRef CAS PubMed.
- M. Rezaee, R. K. Oskuee, H. Nassirli and B. Malaekeh-Nikouei, J. Controlled Release, 2016, 236, 1–14 CrossRef CAS PubMed.
- X. Gao and L. Huang, Biochemistry, 1996, 35, 1027–1036 CrossRef CAS PubMed.
- S. L. Hart, C. V. Arancibia-Carcamo, M. A. Wolfert, C. Mailhos, N. J. O'Reilly, R. R. Ali, C. Coutelle, A. J. T. George, R. P. Harbottle, A. M. Knight, D. F. P. Larkin, R. J. Levinsky, L. W. Seymour, A. J. Thrasher and C. Kinnon, Hum. Gene Ther., 1998, 9, 575–585 CrossRef CAS PubMed.
- S. Li, M. A. Rizzo, S. Bhattacharya and L. Huang, Gene Ther., 1998, 5, 930–937 CrossRef CAS PubMed.
- A. D. Tagalakis, R. J. McAnulty, J. Devaney, S. E. Bottoms, J. B. Wong, M. Elbs, M. J. Writer, H. C. Hailes, A. B. Tabor, C. O'Callaghan, A. Jaffe and S. L. Hart, Mol. Ther., 2008, 16, 907–915 CrossRef CAS PubMed.
- M. F. M. Mustapa, S. M. Grosse, L. Kudsiova, M. Elbs, E. A. Raiber, J. B. Wong, A. P. R. Brain, H. E. J. Armer, A. Warley, M. Keppler, T. Ng, M. J. Lawrence, S. L. Hart, H. C. Hailes and A. B. Tabor, Bioconjugate Chem., 2009, 20, 518–532 CAS.
- S. D. Li, S. Chono and L. Huang, Mol. Ther., 2008, 16, 163–169 CrossRef CAS PubMed.
- H. Song, G. Wang, B. He, L. Li, C. Li, Y. Lai, X. Xu and Z. Gu, Int. J. Nanomed., 2012, 7, 4637–4648 CAS.
- K. D. Murray, C. J. Etheridge, S. I. Shah, D. A. Matthews, W. Russell, H. M. D. Gurling and A. D. Miller, Gene Ther., 2001, 8, 453–460 CrossRef CAS PubMed.
- J. W. Wiseman, E. S. Scott, P. A. Shaw and W. H. Colledge, J. Gene Med., 2005, 7, 759–770 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122–142 CrossRef CAS PubMed.
- S. Manouchehri, P. Zarrintaj, M. R. Saeb and J. D. Ramsey, Mol. Pharmaceutics, 2021, 18, 3652–3670 CrossRef CAS PubMed.
- S. Berger, A. K. Levačić, E. Hörterer, U. Wilk, T. Benli-Hoppe, Y. Wang, Ö. Öztürk, J. Luo and E. Wagner, Biomacromolecules, 2021, 22, 1282–1296 CrossRef CAS PubMed.
- A. Akinc and R. Langer, Biotechnol. Bioeng., 2002, 78, 503–508 CrossRef CAS PubMed.
- M. E. Martin and K. G. Rice, AAPS J., 2007, 9, E18–E29 CrossRef CAS PubMed.
- M. S. Wadhwa, W. T. Collard, R. C. Adami, D. L. McKenzie and K. G. Rice, Bioconjugate Chem., 1997, 8, 81–88 CrossRef CAS PubMed.
- R. C. Adami, W. T. Collard, S. A. Gupta, K. Y. Kwok, J. Bonadio and K. G. Rice, J. Pharm. Sci., 1998, 87, 678–683 CrossRef CAS PubMed.
- A. Kwok, G. A. Eggimann, J.-L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- A. Kwok, D. McCarthy, S. L. Hart and A. D. Tagalakis, Chem. Biol. Drug Des., 2016, 87, 747–763 CrossRef CAS PubMed.
- A. Ahmad, J. M. Khan and S. Haque, Biochimie, 2019, 160, 61–75 CrossRef CAS PubMed.
- P. Midoux, C. Pichon, J. J. Yaouanc and P. A. Jaffres, Br. J. Pharmacol., 2009, 157, 166–178 CrossRef CAS PubMed.
- Q.-R. Chen, L. Zhang, S. A. Stass and A. J. Mixson, Nucleic Acids Res., 2001, 29, 1334–1340 CrossRef CAS PubMed.
- Q.-R. Chen, L. Zhang, P. W. Luther and A. J. Mixson, Nucleic Acids Res., 2002, 30, 1338–1345 CrossRef CAS PubMed.
- Q. X. Leng and A. J. Mixson, Nucleic Acids Res., 2005, 33, e40 CrossRef PubMed.
- Q. X. Leng, P. Scaria, O. B. Ioffe, M. Woodle and A. J. Mixson, J. Gene Med., 2006, 8, 1407–1415 CrossRef CAS PubMed.
- Y. Zhou, S. Han, Z. Liang, M. Zhao, G. Liu and J. Wu, J. Mater. Chem. B, 2020, 8, 5564–5577 RSC.
- B. J. Calnan, B. Tidor, S. Biancalana, D. Hudson and A. D. Frankel, Science, 1991, 252, 1167–1171 CrossRef CAS PubMed.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS PubMed.
- N. Sakai, T. Takeuchi, S. Futaki and S. Matile, ChemBioChem, 2005, 6, 114–122 CrossRef CAS PubMed.
- M. F. Mustapa, P. C. Bell, C. A. Hurley, A. Nicol, E. Guénin, S. Sarkar, M. J. Writer, S. E. Barker, J. B. Wong, M. A. Pilkington-Miksa, B. Papahadjopoulos-Sternberg, P. A. Shamlou, H. C. Hailes, S. L. Hart, D. Zicha and A. B. Tabor, Biochemistry, 2007, 46, 12930–12944 CrossRef CAS PubMed.
- S. M. Grosse, A. D. Tagalakis, M. F. Mustapa, M. Elbs, Q. H. Meng, A. Mohammadi, A. B. Tabor, H. C. Hailes and S. L. Hart, FASEB J., 2010, 24, 2301–2313 CrossRef CAS PubMed.
- R. Bofinger, M. Zaw-Thin, N. J. Mitchell, P. S. Patrick, C. Stowe, A. Gomez-Ramirez, H. C. Hailes, T. L. Kalber and A. B. Tabor, J. Pept. Sci., 2018, 24, e3131 CrossRef PubMed.
- K. Welser, F. Campbell, L. Kudsiova, A. Mohammadi, N. Dawson, S. L. Hart, D. J. Barlow, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Pharmaceutics, 2013, 10, 127–141 CrossRef CAS PubMed.
- A. D. Miller, Angew. Chem., Int. Ed., 1998, 37, 1769–1785 CAS.
- L. Kudsiova, K. Welser, F. Campbell, A. Mohammadi, N. Dawson, L. Cui, H. C. Hailes, M. J. Lawrence and A. B. Tabor, Mol. Biosyst., 2016, 12, 934–951 RSC.
- V. Escriou, C. Ciolina, F. Lacroix, G. Byk, D. Scherman and P. Wils, Biochim. Biophys. Acta, Biomembr., 1998, 1368, 276–288 CrossRef CAS PubMed.
- J. Turek, C. Dubertret, G. Jaslin, K. Antonakis, D. Scherman and B. Pitard, J. Gene Med., 2000, 2, 32–40 CrossRef CAS PubMed.
- S. Li, W.-C. Tseng, D. B. Stolz, S.-P. Wu, S. C. Watkins and L. Huang, Gene Ther., 1999, 6, 585–594 CrossRef CAS PubMed.
- O. Zelphati, L. S. Uyechi, L. G. Barron and F. C. Szoka, Biochim. Biophys. Acta, Lipids Lipid Metab., 1998, 1390, 119–133 CrossRef CAS PubMed.
- L. García, M. Buñuales, N. Düzgünes and C. Tros de Ilarduya, Eur. J. Pharm. Biopharm., 2007, 67, 58–66 CrossRef PubMed.
- X. Gao and L. Huang, Biochemistry, 1996, 35, 1027–1036 CrossRef CAS PubMed.
- G. Caracciolo, L. Callipo, S. C. De Sanctis, C. Cavaliere, D. Pozzi and A. Lagana, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 536–543 CrossRef CAS PubMed.
- D. Chen, N. Parayath, S. Ganesh, W. Wang and M. Amiji, Nanoscale, 2019, 11, 18806–18824 RSC.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS PubMed.
- L. Kudsiova, A. Mohammadi, M. F. M. Mustapa, F. Campbell, K. Welser, D. Vlaho, H. Story, D. J. Barlow, A. B. Tabor, H. C. Hailes and M. J. Lawrence, Biomater. Sci., 2019, 7, 149–158 RSC.
- R. D. Harvey, R. K. Heenan, D. J. Barlow and M. J. Lawrence, Chem. Phys. Lipids, 2005, 133, 27–36 CrossRef CAS PubMed.
- D. Ahmadi, N. Mahmoudi, P. Li, K. Ma, J. Doutch, F. Foglia, R. K. Heenan, D. J. Barlow and M. J. Lawrence, Sci. Rep., 2020, 10, 4082 CrossRef CAS PubMed.
- D. Ahmadi, D. J. Barlow, M. J. Lawrence, N. Mahmoudi, L. Peixun and J. Tellam, Simple Creams, Complex Structures, in Nanoparticle and Molecular Assemblies, American Chemical Society, Washington, DC, USA, 2020, ch. 6 Search PubMed.
- M. J. Writer, B. Marshall, M. A. Pilkington-Miksa, S. E. Barker, M. Jacobsen, A. Kritz, P. C. Bell, D. H. Lester, A. B. Tabor, H. C. Hailes, N. Klein and S. L. Hart, J. Drug Targeting, 2004, 12, 185–193 CrossRef CAS PubMed.
- In our previous papers ref. 39 and 41 we carried out a detailed investigation of the effects on transfection/knockdown of peptides with differing degrees of branching (linear peptides B0; singly branched peptides, B1; doubly branched peptides, B2) and on endosomally cleavable (L1) or non-cleavable (L2) sequences. In the present work we have just studied singly branched peptides with cleavable sequences, and we have therefore simplified the nomenclature. Thus, peptide (HHK)4B1-L1-[Y] is referred to as (HHK)4BLY in the present paper.
- A. Mohammadi, L. Kudsiova, M. F. M. Mustapa, F. Campbell, D. Vlaho, K. Welser, H. Story, A. D. Tagalakis, S. L. Hart, D. J. Barlow, A. B. Tabor, M. J. Lawrence and H. C. Hailes, Org. Biomol. Chem., 2019, 17, 945–957 RSC.
- A. I. Dragan, J. R. Casas-Finet, E. S. Bishop, R. J. Strouse, M. A. Schenerman and C. D. Geddes, Biophys. J., 2010, 99, 3010–3019 CrossRef CAS PubMed.
- M. Kahlweit, Adv. Colloid Interface Sci., 1975, 5, 1–35 CrossRef CAS.
- L. Kudsiova, B. Fridrich, J. Ho, M. F. M. Mustapa, F. Campbell, K. Welser, M. Keppler, T. Ng, D. J. Barlow, A. B. Tabor, H. C. Hailes and M. J. Lawrence, Mol. Pharmaceutics, 2011, 8, 1831–1847 CrossRef CAS PubMed.
- B. Ma, S. Zhang, H. Jiang, B. Zhao and H. Lv, J. Controlled Release, 2007, 123, 184–194 CrossRef CAS PubMed.
- C. Dingwall and R. A. Laskey, Trends Biochem. Sci., 1991, 16, 478–481 CrossRef CAS PubMed.
- M. Soniat and Y. M. Chook, Biochem. J., 2015, 468, 353–362 CrossRef CAS PubMed.
- N. E. Bernardes and Y. M. Chook, Biochem. Soc. Trans., 2020, 48, 2753–2767 CrossRef CAS PubMed.
- M. A. Zanta, P. Belguise-Valladier and J.-P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 91–96 CrossRef CAS PubMed.
- J. W. Wiseman, E. S. Scott, P. A. Shaw and W. H. Colledge, J. Gene Med., 2005, 7, 759–770 CrossRef CAS PubMed.
- B. Yu, B. M. Pettitt and J. Iwahara, Acc. Chem. Res., 2020, 53, 1802–1810 CrossRef CAS PubMed.
- A. Mann, G. Thakur, V. Shukla, A. K. Singh, R. Khanduri, R. Naik, Y. Jiang, N. Kalra, B. S. Dwarakanath, U. Langel and M. Ganguli, Mol. Pharmaceutics, 2011, 8, 1729–1741 CrossRef CAS PubMed.
- A. Mann, V. Shukla, R. Khanduri, S. Dabral, H. Singh and M. Ganguli, Mol. Pharmaceutics, 2014, 11, 683–696 CrossRef CAS PubMed.
- E. M. McErlean, M. Ziminska, C. M. McCrudden, J. W. McBride, S. P. Loughran, G. Cole, E. J. Mulholland, V. Kett, N. E. Buckley, T. Robson, N. J. Dunne and H. O. McCarthy, J. Controlled Release, 2021, 330, 1288–1299 CrossRef CAS PubMed.
- R. Nasanit, P. Iqbal, M. Soliman, N. Spencer, S. Allen, M. C. Davies, S. S. Briggs, L. W. Seymour, J. A. Preece and C. Alexander, Mol. BioSyst., 2008, 4, 741–745 RSC.
- Y. H. Kim, K. Lee and S. Li, Chem. Mater., 2021, 33, 7923–7943 CrossRef CAS.
- C. Leal, E. Moniri, E. Pegado and H. Wennerstrom, J. Phys. Chem. B, 2007, 111, 5999–6005 CrossRef CAS PubMed.
- J. Rouchey, R. R. Netz and J. O. Rädler, Eur. Phys. J. E: Soft Matter Biol. Phys., 2005, 16, 17–28 CrossRef PubMed.
- H. S. Antila and M. Sammalkorpi, J. Phys. Chem. B, 2014, 118, 3226–3234 CrossRef CAS PubMed.
- S. Kawakami, Y. Ito, S. Fumoto, F. Yamashita and M. Hashida, J. Gene Med., 2005, 7, 1526–1533 CrossRef CAS PubMed.
- S. Evan-Chen, R. Cohen and Y. Barenholz, Chem. Phys. Lipids, 2012, 165, 414–423 CrossRef PubMed.
- Y. Hattori, W.-X. Ding and Y. Maitani, J. Controlled Release, 2007, 120, 122–130 CrossRef CAS PubMed.
- L. B. Jeffs, L. R. Palmer, E. G. Ambegia, C. Giesbrecht, S. Ewanick and I. MacLachlan, Pharm. Res., 2005, 22, 362–372 CrossRef CAS PubMed.
- Y. Sakurai, T. Matsuda, T. Hada and H. Harashima, Biol. Pharm. Bull., 2015, 38, 1185–1191 CrossRef CAS PubMed.
- The high level of DNA entrapment observed in the VwLPDw is the reason why no discrepancy between the results of the agarose gel electrophoresis and PicoGreen® assays have been previously observed.
- W. R. Perkins, S. R. Minchey, P. L. Ahl and A. S. Janoff, Chem. Phys. Lipids, 1993, 64, 197–217 CrossRef CAS PubMed.
- C. Q. Pan and R. A. Lazarus, J. Biol. Chem., 1998, 273, 11701–11708 CrossRef CAS PubMed.
- Y. Wang, R. Y. K. Chang, W. J. Britton and H.-K. Chan, Adv. Drug Delivery Rev., 2022, 180, 114066 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods for synthesis and purification of peptides; characterisation data for all new peptides; transfection, protein assay and biophysical data Fig. S1–S4. See DOI: https://doi.org/10.1039/d2bm01905a |
| ‡ Present address: Advanced Drug Delivery, Pharmaceutical Sciences, BioPharmaceuticals R&D, AstraZeneca, Cambridge CB21 6GH, UK. E-mail: lilicuinike@gmail.com. |
| § Present address: University of Birmingham Dubai, Dubai International Academic City, PO Box 341799, E-mail: L.kudsiova@bham.ac.uk. |
| ¶ Present address: NanoVation Therapeutics UK Ltd, Rutherford Appleton Laboratory, Harwell Oxford, Didcot OX11 0FA, UK. |
| This journal is © The Royal Society of Chemistry 2023 |