
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards new coordination modes of 1,2,3-triazolylidene: controlled by the nature of the 1st metalation in a heteroditopic bis-NHC ligand†
Praseetha Mathoor
Illam
,
Chandra Shekhar
Tiwari
and
Arnab
Rit
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: arnabrit@iitm.ac.in
First published on 21st October 2022
Abstract
An unusual effect of the nature of the first metal coordination of a heteroditopic N-heterocyclic carbene ligand (L2) towards the coordination behavior of 1,2,3-tzNHC is explored. The first metal coordination at the ImNHC site (complexes 3 and 4) was noted to substantially influence the electronics of the 1,2,3-triazolium moiety leading to an unprecedented chemistry of this MIC donor. Along this line, the RhIII/IrIII-orthometalation in complexes 4 makes the triazolium C4–H more downfield shifted than C5–H, whereas a reverse trend, although to a lesser extent, is observed in the case of the non-chelated PdII-coordination. This difference in behavior assisted us to achieve the selective activation of triazole C4/C5 positions, not observed before, as supported by the isolation of the homo- and hetero-bimetallic complexes, 5, 6 and 7–9via C5- and C4-metalation, respectively. Furthermore, the %Vbur calculations eliminate any considerable steric influence and the DFT studies strongly support the selectivity observed during bimetalation.
Introduction
N-heterocyclic carbenes (NHCs), with their characteristic strong σ-electron donating nature1 coupled with the tuneable stereoelectronic properties,2 have proven to be a versatile class of ligands to anchor one, two or more metals for diversified applications.3 Along with the very common Arduengo type imidazolylidenes, triazole derived 1,2,4-triazolylidenes have also been widely utilized for various applications.4 After Bertrand's early discovery of 1,2,4-triazol-3,5-diylidene supported organometallic polymer,5 the Peris group successfully utilized this bis-NHC system to access diverse homo- as well as heterometallic complexes.3c,6 Later in 2008, the group of Albrecht introduced 1,2,3-triazolylidenes into the NHC family as an abnormal congener (mesoionic carbenes, MICs) of normal NHCs.7 They exhibit superior donor properties, due to less heteroatom stabilization, which influence the reactivity of their transition metal complexes.8 Furthermore, easy accessibility of the precursor triazolium salts via a simple [3 + 2] cycloaddition reaction of azides and alkynes (‘click reaction’)8a,9 have made 1,2,3-triazolylidenes attractive in organometallic chemistry.10 Among them, the 1,2,3-triazol-5-ylidenes derived from the corresponding C4-protected triazolium salts are well explored,10c,11 whereas the related MICs, generated from the analogous C4- and C5-unsubstituted triazolium salts, are rarely studied (Fig. 1).12,13 This might be due to the availability of two backbone carbons (C4 and C5), which could, in principle, be deprotonated to yield either C4- or C5-ylidenes restricting the selective metalation.12 This was indeed supported by the formation of a mixture of C4- and C5-coordinated products during the metalation of unsubstituted 1,2,3-triazolylidenes.12 Moreover, the reported complexes of unsubstituted MICs are only of the cyclometalated type and they possess the metal center at the triazolylidene C5-position (Fig. 1B).12,13 The additional stability offered by chelation could possibly be the driving force for such metalation at the C5-position.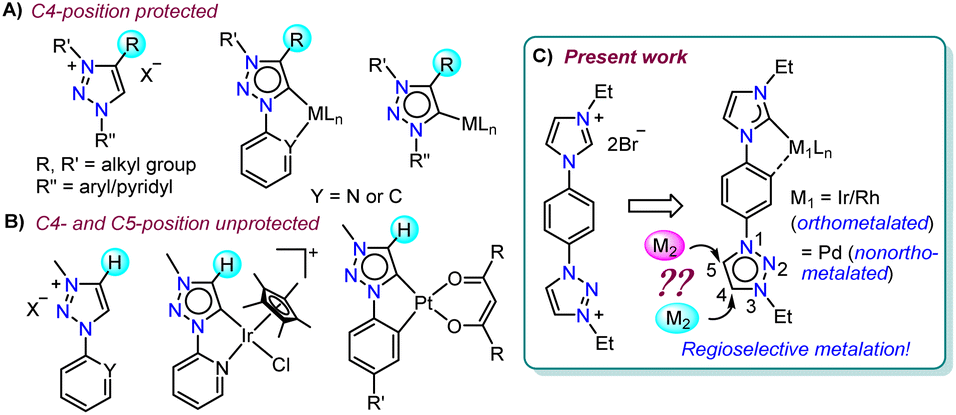 | ||
| Fig. 1 Comparison of previous reports on 1,2,3-triazolylidene complexes with the present work. | ||
Based on these literature reports, we engaged ourselves in developing strategies for selective activation of the backbone protons (C4– and C5–H) of the unsubstituted 1,2,3-triazolium salt as this will open up a new family of metal-NHC complexes for various applications including catalysis (activities of these regioisomeric metal complexes are expected to be different). We hypothesized that either some electronic or steric modulation could help in achieving this. In this line, along with our interest14a towards hetero-bimetallic complexes,14a,b we have now designed an unsymmetrical bis-azolium salt [L2-H2]Br2 which possesses a triazolium group substituted with an N-phenyl-para-imidazolium moiety. The first metalation occurs at the imidazolium end as its C2–H acidity is more compared to that of the triazolium backbone protons and thus, provides us the unique opportunity to control the subsequent metalation either at the C4- or C5-position of the triazolium group. Accordingly, mono-metalation of [L2-H2]Br2 with PdII- and IrIII/RhIII precursors yielded non-orthometalated (3) and orthometalated (4a and b) analogues, respectively which were detected to impart an unprecedented influence on the triazolium C4/C5–H chemical shifts and thus, essentially on their reactivities. This effect was utilized for the synthesis of their bimetallic counterparts (5, 6 from 3 and 7–9 from 4) via selective activation of either the C4– or C5–H which was unequivocally supported by detailed NMR analyses along with the X-ray crystallographic studies.
Results and discussion
Previous reports on unsubstituted 1,2,3-triazolium salts with IrIII- and PtII-centers12,13 suggest that metalation occurs at the C5-position of the triazole ring. However, it should be noted that these complexes are of the cyclometalated type which might have some influence on the metalation behavior. To understand this in detail, at the outset, we intended to study the chemistry of simple C4/C5-backbone unsubstituted mesoionic carbene precursors such as the click derived [L1-H]Br with PdII and AuI centers (Scheme 1a) as they do not generally prefer the chelate complex formation via orthometalation. After synthesis, the NMR analyses of [L1-H]Br reveal that the C4– and C5–H resonances have very close chemical shift values (δ = 10.05 and 10.16 ppm, respectively), which might pose difficulties to its selective deprotonation cum metalation.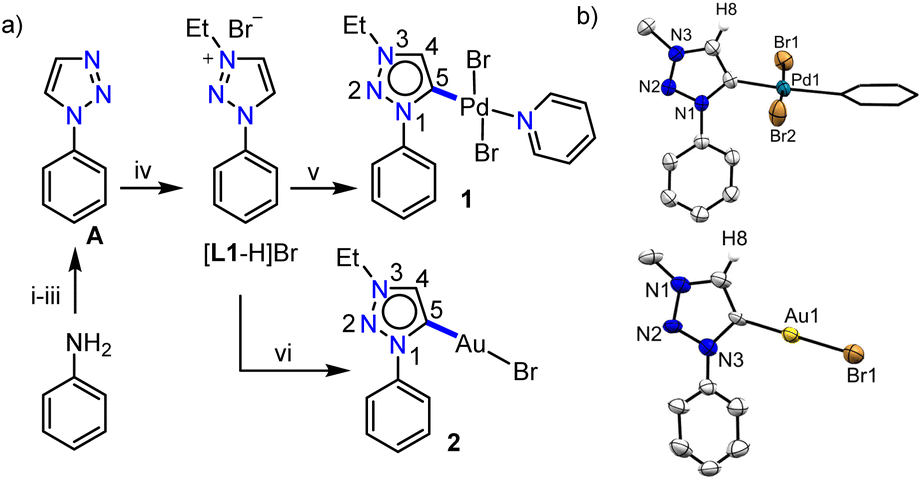 | ||
| Scheme 1 (a) Synthesis of [L1-H]Br and its palladium (1) and gold (2) complexes: (i) NaNO2, HCl/H2O (10% solution), 0 °C, 1 h; (ii) NaN3, 0 °C-RT, 12 h; (iii) vinyl acetate, reflux, 24 h; (iv) EtBr, CH3CN, reflux, 24 h; (v) [Pd(CH3CN)2Cl2], Cs2CO3, KBr, CH3CN/pyridine, 70 °C, 24 h; (vi) [Au(SMe2)Cl], Cs2CO3, CH3CN, 70 °C, 24 h; (b) molecular structures of 1 and 2 with ellipsoids at a 50% probability level. Hydrogen atoms except H8 and Me moieties of the N–Et groups are omitted for clarity. Pyridine is shown in capped stick. | ||
Nevertheless, [L1-H]Br was first reacted with [Pd(CH3CN)2Cl2] under suitable conditions (Scheme 1a) and the 1H NMR spectrum with only one triazolylidene backbone proton along with a PdII-MIC 13C{1H} signal at δ = 139.5 ppm suggest the formation of a monopalladium complex. The X-ray crystallographic analysis disclosed the structure of complex 1 (Scheme 1b) in which the non-cyclometalated PdII-center is attached at the C5-position of MIC ligand L1. To ascertain whether this is the preferred coordination mode of the ligand under consideration (L1) or not, we then attempted the synthesis of an analogous AuI-complex (Scheme 1a). We first proceeded with the most common transmetalation strategy via AgI–NHC complex formation using Ag2O. To our surprise, formation of a mixture of C4- and C5-ylidene coordinated AuI-complexes15 was observed even at room temperature, which could be due to the formation of both C4– and C5–ylidene coordinated AgI-complexes having similar stability and/or comparable carbene transfer efficiency.12 This observation clearly suggests that both the backbone protons (C4/C5–H) are susceptible towards deprotonation. However, a C5–ylidene coordinated AuI-complex 2 was exclusively obtained in good yield (80%) via a Cs2CO3 assisted metalation strategy and the multinuclear NMR data along with the 1H–1H NOESY spectrum (Fig. S10†) confirmed the coordination of AuI at the C5-position of L1. This conclusion was established by the molecular structure determination via X-ray crystallography (Scheme 1b).
After studying the coordination behavior of the simple MIC ligand L1, we started investigating the possibilities of selective triazolium backbone activation. In this direction, as per our postulated electronic modulation, we designed an unsymmetrical bis-azolium salt [L2-H2]Br2, containing an imidazolium group along with a 1,2,3-triazolium moiety, the precursor for a biscarbene ligand. [L2-H2]Br2 was synthesized as an air stable white powder in excellent yield (94%) following the multistep procedure as detailed in Scheme 2. The 1H NMR analysis reveals the most downfield shifted imidazolium N–CH–N proton resonance at δ = 10.20 ppm, whereas the two triazolium backbone N–CH–C protons were observed at δ = 9.79 (C5–H) and 9.37 (C4–H) ppm (confirmed by 2D NMR spectroscopy).
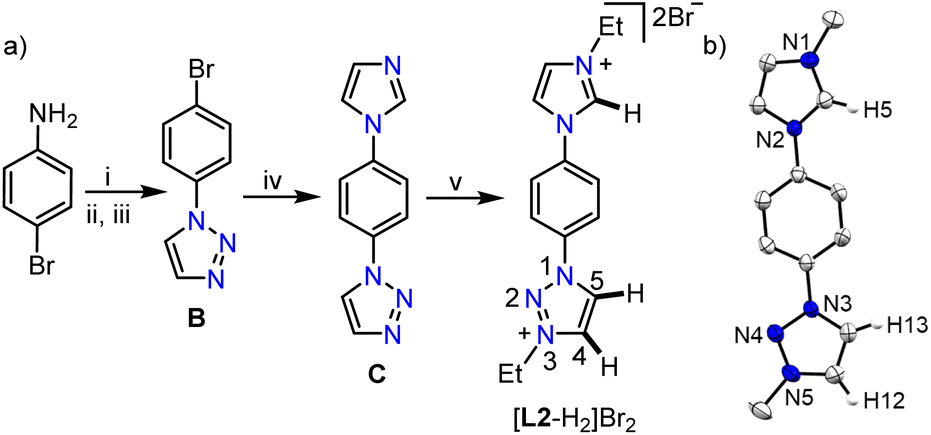 | ||
| Scheme 2 (a) Synthesis of bisazolium salt, [L2-H2]Br2: (i) NaNO2, HCl/H2O (10% solution), 0 °C, 1 h; (ii) NaN3, 0 °C–RT, 12 h; (iii) vinyl acetate, reflux, 24 h; (iv) imidazole, K2CO3, CuO, DMSO, 150 °C, 48 h; (v) EtBr, DMF, reflux, 24 h. (b) Molecular structure of [L2-H2]Br2 with ellipsoids at a 50% probability level. Hydrogen atoms except H5, H12, and H13, counterions, solvent of crystallization and Me moieties of the N–Et groups are omitted for clarity. | ||
It is worth mentioning that the difference in chemical shifts of the triazolium backbone protons became more prominent after the installation of the imidazolium moiety in [L2-H2]Br2 as compared to that in [L1-H]Br. This indicates some electronic influence of the installed imidazolium moiety on the triazolium unit, which would probably be beneficial for the selective activation of its backbone protons. Moreover, the relatively higher acidity of the imidazolium N–CH–N proton than that of the triazolium ones implies that the first metalation should happen preferably at the imidazolylidene site, which would provide us with a unique opportunity to fine tune the electronics of the triazolium moiety via first metalation. Similar behavior was observed previously with a related unsymmetrical bis-azolium salt and was attributed to the difference in acidities16a of the azolium moieties.16b Finally, the structure of [L2-H2]Br2 was confirmed by X-ray crystallographic analysis (Scheme 2b).
With the well characterized bis-azolium salt [L2-H2]Br2 in hand, we proceeded to study its metalation behavior. Initially [L2-H2]Br2 was treated with [Pd(CH3CN)2Cl2] in the presence of Cs2CO3 under the conditions shown in Scheme 3a. The NMR analyses confirmed the formation of complex 3via coordination of a PdII-center to the imidazolylidene moiety, suggested by the absence of the most downfield shifted proton in the 1H NMR spectrum as well as the PdII-bound ImNHC 13C{1H} NMR signal at δ = 150.2 ppm.17c,d The chemical shift values of the backbone protons (δ = 9.92 (C4–H) and 10.24 (C5–H) ppm) were assigned from the NOESY spectrum (Fig. S15†). Finally, the X-ray crystallographic analysis confirmed the coordination of PdII to the imidazolylidene donor (Scheme 3b). Before proceeding towards the synthesis of bimetallic complexes from 3, we also synthesized the IrIII-complex of [L2-H2]Br2, 4a/a′ in good yields of 78–80%. The absence of the imidazolium proton of [L2-H2]Br2 and the observed integration of 3 instead of 4 aryl protons in the 1H NMR spectrum along with the 13C{1H} NMR signal for the IrIII-ImNHC at δ = 165.8 ppm17a,b suggest the attachment of IrIII to the imidazolylidene donor and the phenyl ring in an orthometalated fashion in complex 4a. This is further substantiated by the diastereotopic nature of the imidazolium N–CH2 protons (two multiplets of one proton intensity at δ = 4.24 and 4.40 ppm). The X-ray crystallographic analysis of a single crystal of 4a′ establishes the structure of the monoiridium complex (Scheme 3b) as concluded from NMR analyses. Interestingly, the 2D-NOESY (Fig. 2) spectrum of 4a reveals that the triazolium C4–H is significantly downfield shifted (δ = 9.86 ppm) compared to C5–H (δ = 9.05 ppm).
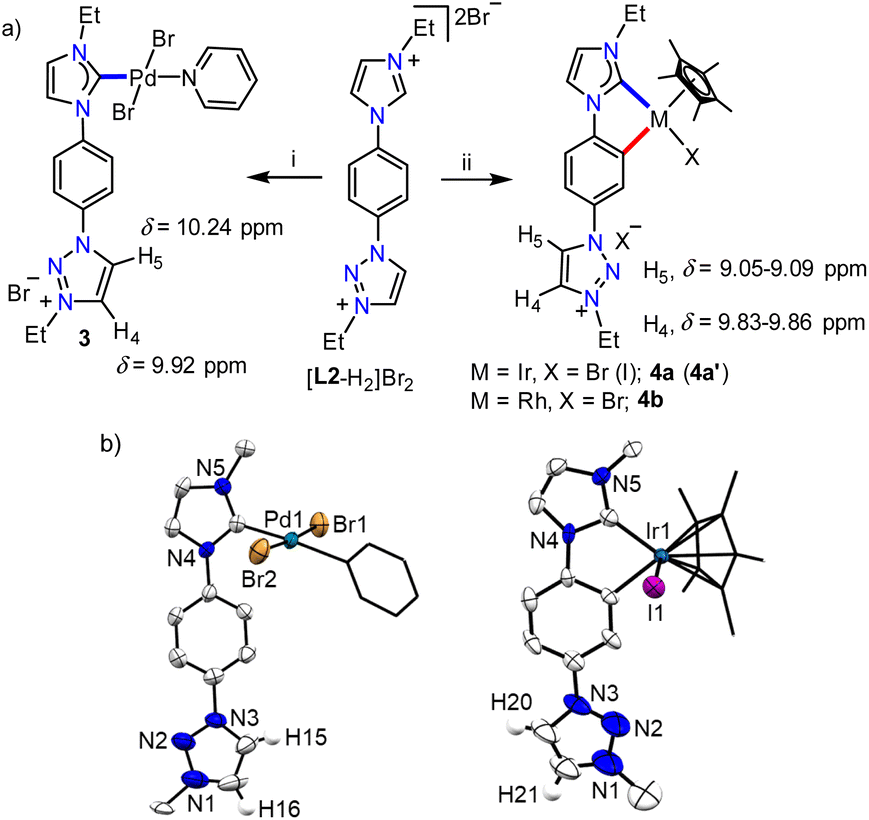 | ||
| Scheme 3 (a) Synthesis of palladium (3), iridium (4a and a′), and rhodium (4b) complexes: (i) [Pd(CH3CN)2Cl2], Cs2CO3, KBr, CH3CN/pyridine, RT, 12 h. (ii) [M(Cp*)Cl2]2 (M = Ir/Rh), NaOAc, K2CO3/Cs2CO3, KBr (for 4a and b) or KI (for 4a′), CH3CN, 75 °C, 24 h. (b) Molecular structures of 3 and 4a′ with ellipsoids at 50% probability level. Hydrogen atoms except H15/H16 in 3 and H20/H21 in 4a′, counterions, and the Me groups of N–Et moieties are omitted for clarity. Pyridine and Cp* moieties are shown in capped stick. | ||
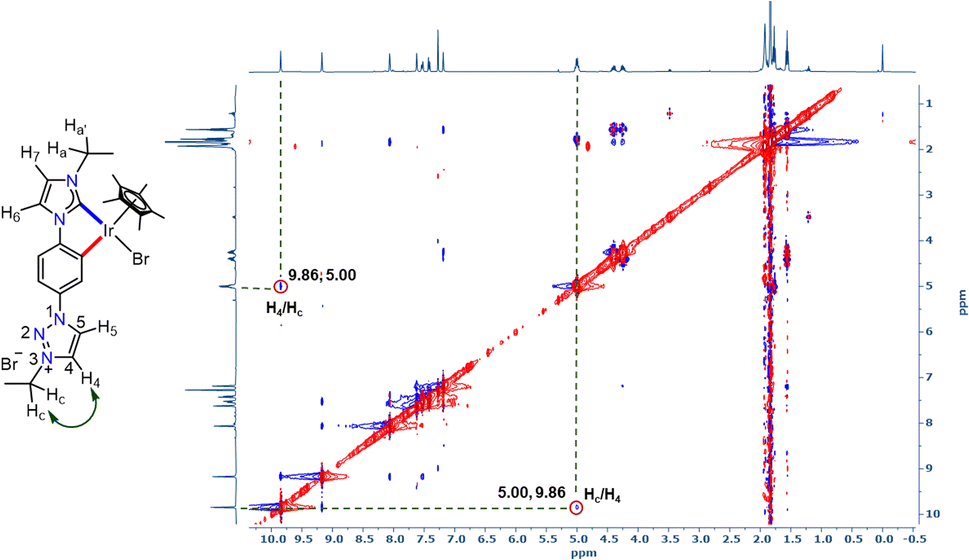 | ||
| Fig. 2 2D-NOESY NMR spectrum of 4a showing the interaction of triazolium C4–H with the N–CH2 protons of the ethyl group. | ||
In order to confirm this switching of triazolium backbone proton chemical shifts upon orthometalation, we also synthesized the analogous RhIII-complex, 4b (Scheme 3). The NMR analysis confirmed that the RhIII-center also coordinates to the ImNHC in a similar way to IrIII and importantly, has a comparable influence on the triazolium proton chemical shifts (δ = 9.83 and 9.09 ppm for C4–H and C5–H, respectively), in sharp contrast to that observed in the case of the non-orthometalated PdII-complex 3. This may be attributed to the planar orthometalated RhIII/IrIII-center, which possibly attracts some electron density from the triazolium ring making the C4–H more acidic than C5–H.
All the above findings from the 1st metalation of ligand [L2-H2]Br2 ascertain that the nature of metal coordination offers substantial electronic influence on the triazolium moiety and thus, governs the chemical shifts, which would essentially control the activity of its backbone protons. It is in line with previous observations by several research groups that the coordination mode of a metal centre and its orientation after metalation strongly influence the site-selective C–H activation due to some electronic effect in the resulting system.18
With this definite idea about the electronic effect of the first metal coordination, we focused on the synthesis of the corresponding bimetallic complexes from 3 and 4. First, the monopalladium complex, 3 was reacted with [Pd(CH3CN)2Cl2] (Scheme 4a) and the product was isolated in good yield (85%). The NMR spectroscopic analyses provided primary evidence for the homobimetallic complex (5) formation. First of all, the 13C{1H} resonances at δ = 150.6 and 140.4 ppm, concluded to be PdII-ImNHC and PdII-MIC, respectively from the HMBC spectrum (Fig. S31†), confirm the attachment of two PdII-centers to ligand L2, which was also supported by the ESI-mass analysis. Furthermore, a resonance at δ = 7.74 ppm in the 1H NMR spectrum was assigned to the triazole backbone C4–H based on the NOESY spectrum (Fig. S32†), indicating the PdII-coordination at the triazolylidene C5-position. Finally, the single crystal X-ray crystallographic analysis confirmed the coordination of the second PdII-center to the C5-position of 1,2,3-tzNHC (Scheme 4b), which was observed to have a more downfield shifted proton in 3 as per the detailed NMR analyses. To validate this further, we also synthesized another heterobimetallic (PdII–AuI) complex 6 from 3 (Scheme 4) and the 1H NMR spectrum of the obtained complex suggested the formation of the expected complex. Further support for the formation of the heterobimetallic complex, 6 was obtained from the ESI-MS analysis, exhibiting the most intense peak at m/z 729.8502 for [M–Cl–py]+ (calcd. m/z = 729.8540) with the isotopic patterns matching perfectly. Coordination of AuI to the 1,2,3-tzNHC C5-position was finally established via single crystal X-ray crystallographic analysis of 6. All the above results reinforce that the more downfield shifted triazolium proton is activated during the second metalation of a PdII–NHC complex (3) of [L2-H2]Br2.
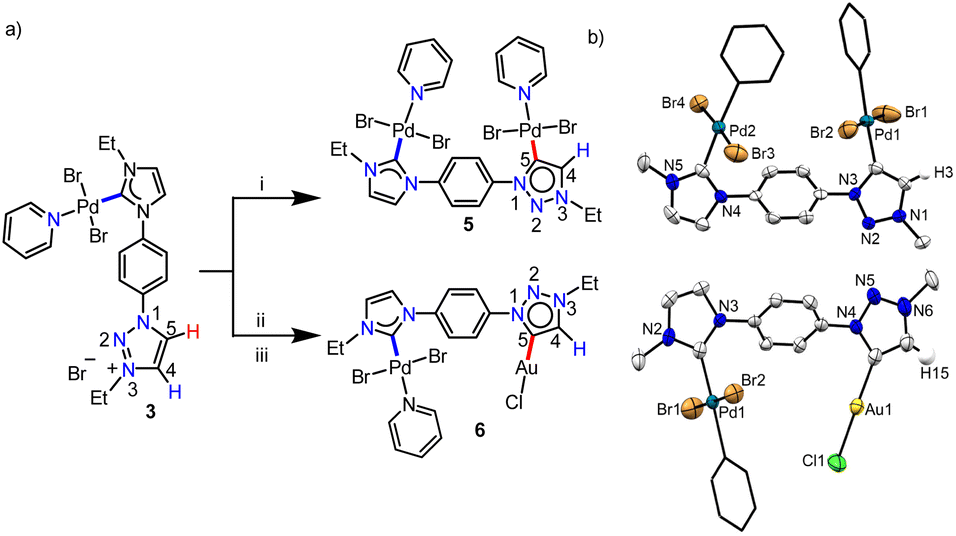 | ||
| Scheme 4 (a) Synthesis of the bimetallic complexes 5 and 6 from 3: (i) [Pd(CH3CN)2Cl2], Cs2CO3, KBr, CH3CN/pyridine, 60 °C, 24 h; (ii) Ag2O, DCM, RT, 12 h; (iii) [Au(SMe2)Cl], RT, 12 h. (b) Molecular structures of 5 and 6 with ellipsoids at a 50% probability level. Hydrogen atoms except H3 in 5, H15 in 6, and the Me groups of N–Et moieties are omitted for clarity. Pyridine groups are shown in capped stick. | ||
Keeping this in mind, we proceeded with the second metalation of the orthometalated IrIII-complex 4a with the anticipated activation of the triazolium C4–H. In this direction, complex 4a was reacted with [Pd(CH3CN)2Cl2] using Cs2CO3 as the base in the CH3CN/pyridine solvent mixture and the desired IrIII–PdII bimetallic complex 7 was obtained in 64% yield (Scheme 5a). The 1H NMR spectrum of 7 unveils that one of the triazolium protons of the precursor complex 4a is missing as expected and the 2D NMR data confirmed the PdII-coordination to the C4- instead of the C5-position. This establishes the deprotonation of carbon having a more downfield shifted proton in 4a during sequential metalation and notably, the C5–H resonance is upfield shifted to δ = 7.93 ppm in 7 from 9.05 ppm in 4a. Furthermore, the attachment of PdII to the mesoionic carbene was supported by the 13C{1H} NMR carbene signal at δ = 136.3 ppm, which was upfield shifted compared to the corresponding PdII-bound C5-MIC signal at 140.4 ppm in 5. Eventually, the X-ray crystallographic studies authenticate the attachment of PdII to the triazole C4 position (Scheme 4b).
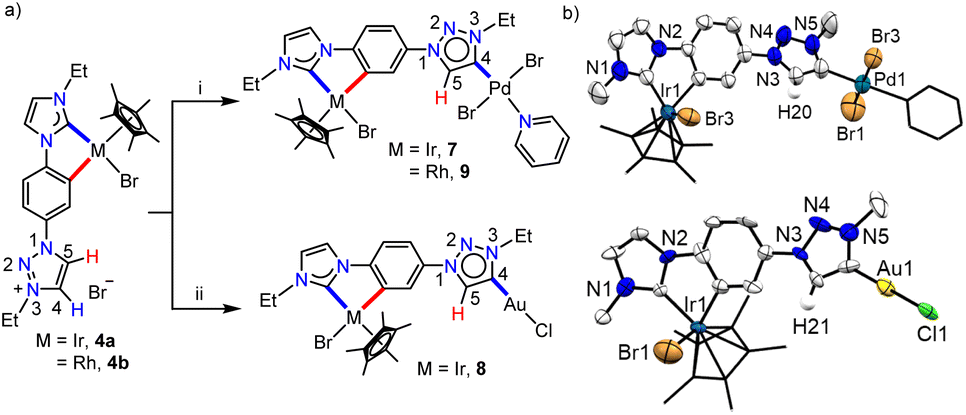 | ||
| Scheme 5 (a) Synthesis of the heterobimetallic complexes 7–9: (i) [Pd(CH3CN)2Cl2], Cs2CO3, KBr, CH3CN/pyridine, 70 °C, 24 h. (ii) Ag2O, DCM, RT, 12 h and then [Au(SMe2)Cl], RT, 12 h. (b) Molecular structures of 7 and 8 with ellipsoids at a 50% probability level. Hydrogen atoms except H20 in 7 and H21 in 8, and the Me moieties of N–Et groups are omitted for clarity. Pyridine and Cp* moieties are shown in capped stick. | ||
In order to affirm the activation of C4- rather than the C5-position of the triazolium moiety during the second metalation of the monoiridium complex 4a, we further synthesized the related AuI complex, 8 in 61% yield, following the transmetalation procedure (Scheme 5a). The HMBC spectrum, displaying a correlation of the AuI-MIC 13C{1H} resonance with the Tz–N–CH2 resonance (Fig. S41†), provides strong evidence for AuI-coordination at the triazolylidene C4 position which was ultimately established by the X-ray diffraction analysis (Scheme 5b). Furthermore, to prove the generality of this finding, we also utilized the mono-RhIII-complex, 4b for the synthesis of a RhIII–PdII bimetallic complex 9 following a similar procedure used for the synthesis of complex 7 (Scheme 5). Multinuclear NMR spectroscopic along with mass spectrometric data analyses reveal the formation of the expected heterobimetallic complex, 9. Moreover, the HMBC NMR spectrum (Fig. S45†) confirms the coordination of the PdII-center to the triazole C4 position by exhibiting a correlation between the PdII bound tzNHC 13C{1H} signal at δ = 136.5 ppm and the triazole Tz–N–CH2 protons at δ = 5.09 ppm, as observed in the case of the complex 7. All of the above results establish that the orthometalated coordination of the IrIII/RhIII-center has a distinct electronic impact on the triazolium moiety in 4a and b which plays a crucial role in achieving the selective activation of the triazole backbone C4–H proton during the second metalation. It is worth mentioning that the complexes 7–9 represent the first ever isolated metal complexes of a C5-unprotected 1,2,3-triazol-4-ylidene donor.
More insight into the contrasting influence of the 1st PdII- and IrIII-metal centres on the second metalation was obtained from the %Vbur analysis and DFT calculations. The %Vbur calculated at the triazole C5 position of the monometallic complexes, 3 and 4a′ was found to be essentially similar (50.1 vs. 50.4, calculated with a sphere radius of 3.5 Å, see the ESI†). This observation clearly suggests that the selectivity observed during the formation of complexes 5/6 from 3 and 7/8/9 from 4 is primarily controlled by electronic factors. Furthermore, the difference of DFT calculated ground state energy between the representative bimetallic systems 5 with its C4-analogue, 5′ and 7 with its C5-analouge, 7′ was noted to be significant (0.7 kcal mol−1 and 1.4 kcal mol−1, respectively, see the ESI†) and is in favour of the experimentally observed regioisomers. Thus, DFT calculations also endorse the isolation of C5- and C4-bound bimetallic systems (5 and 7, respectively) based on the nature of the 1st metal coordination to L2.
Conclusions
In conclusion, we have uncovered an unprecedented electronic influence of the 1st metal coordination on altering the reactivity/metalation behavior of the C4/C5-unprotected 1,2,3-triazolium moiety of a bis-azolium salt, [L2-H2]Br2. Importantly, the impact of monometalation in a non-chelated (complex 3) or chelated (complexes 4a and b) fashion on the triazole backbone (C4/C5) protons could be ascertained easily by 2D NMR analysis. Crucially, these changes in the electronic nature of triazole protons assisted us to access the first ever selective metalation at either the C4- or C5-position of 1,2,3-triazolylidene as undoubtedly supported by the synthesis of bimetallic complexes 5–9. Furthermore, the %Vbur calculations suggest that the observed selectivity is primarily controlled by the electronic nature of the first metal coordination. Moreover, the DFT studies of selected complexes (5 and 7) strongly support the exclusive formation of a particular regioisomer as observed experimentally. The tandem catalytic activity studies of the synthesized complexes are underway in our laboratory.Data availability
Experimental data including experimental procedures and characterization data (NMR, ESI-MS, and single crystal X-ray); the NMR and ESI-MS spectra of the new compounds; detailed computational studies are available in the ESI.†Author contributions
A. R. conceived and designed the project. P. M. I. and C. S. T. performed the experiments. P. M. I. and A. R. wrote the manuscript and all authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge SERB, India (Grant No. CRG/2020/000780) and IITM (IRDA) for financial support and the department of chemistry/SAIF, IIT Madras for the instrumental facility. P. M. I and C. S. T thank IIT Madras for a research fellowship. We also thank Dr P. K. S. Antharjanam and Mr Shashi Kumar for X-ray structure analyses and theoretical calculations, respectively.Notes and references
- For selected reviews, see: (a) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed; (b) S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef CAS; (c) D. L. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 CAS; (d) M. C. Jahnke and F. E. Hahn, Introduction to N-Heterocyclic Carbenes: Synthesis and Stereoelectronic Parameters, in N-Heterocyclic Carbenes from Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, Cambridge, U.K, 2011, vol. 6, pp. 1–41 Search PubMed.
- For selected references, see: (a) R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed; (c) H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed; (d) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- (a) L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed; (b) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (c) J. A. Mata, F. E. Hahn and E. Peris, , Chem. Sci., 2014, 5, 1723–1732 RSC; (d) S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390 CrossRef CAS PubMed; (e) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (f) K. J. Evans and S. M. Mansell, Chem.–Eur. J., 2020, 26, 5927–5941 CrossRef CAS PubMed; (g) N. Sinha and F. E. Hahn, Acc. Chem. Res., 2017, 50, 2167–2184 CrossRef CAS; (h) R. C. Nishad and A. Rit, Chem.–Eur. J., 2021, 27, 594–599 CrossRef CAS.
- For selected references, see: (a) D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J.-P. Melder, K. Ebel and S. Brode, Angew. Chem., Int. Ed. Engl., 1995, 34, 1021–1023 CrossRef CAS; (b) D. Enders and K. Breuer, J. Prakt. Chem., 1997, 339, 397–399 CrossRef CAS; (c) M. T. S. Metz, I. Münster, G. Wagenblast and T. Strassner, Organometallics, 2013, 32, 6257–6264 CrossRef; (d) V. K. Singh, S. N. R. Donthireddy, P. M. Illam and A. Rit, Dalton Trans., 2020, 49, 11958–11970 RSC; (e) P. M. Illam and A. Rit, Catal. Sci. Technol., 2022, 12, 67–74 RSC.
- O. Guerret, S. Solé, H. Gornitzka, M. Teichert, G. Trinquier and G. Bertrand, J. Am. Chem. Soc., 1997, 119, 6668–6669 CrossRef CAS.
- For selected references, see: (a) E. Mas-Marzá, J. A. Mata and E. Peris, Angew. Chem., Int. Ed., 2007, 46, 3729–3731 CrossRef; (b) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531–14537 CrossRef CAS PubMed; (c) S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 CrossRef PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- For selected references (a) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed; (b) D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Z. Anorg. Allg. Chem., 2017, 643, 554–584 CrossRef CAS; (c) P. M. Illam, S. N. R. Donthireddy, S. Chakrabartty and A. Rit, Organometallics, 2019, 38, 2610–2623 CrossRef CAS.
- (a) J. C. Sheehan and C. A. Robinson, J. Am. Chem. Soc., 1951, 73, 1207–1210 CrossRef CAS; (b) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef; (c) S. N. R. Donthireddy, P. M. Illam and A. Rit, Inorg. Chem., 2020, 59, 1835–1847 CrossRef CAS PubMed.
- (a) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC; (b) P. I. P. Elliott, Organomet. Chem., 2014, 39, 1–25 CAS; (c) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (d) R. Maity and B. Sarkar, JACS Au, 2022, 2, 22–57 CrossRef CAS PubMed.
- For selected references (a) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Organometallics, 2011, 30, 6017–6021 CrossRef CAS PubMed; (b) R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem.–Eur. J., 2014, 20, 9952–9961 CrossRef CAS PubMed; (c) L.-A. Schaper, L. Graser, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, B. Bechlars, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 52, 6142–6152 CAS; (d) E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 6215–6221 CrossRef CAS; (e) A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588–2598 CrossRef CAS; (f) R. Maity, A. Mekic, M. van der Meer, A. Verma and B. Sarkar, Chem. Commun., 2015, 51, 15106–15109 RSC.
- A. Petronilho, J. A. Woods, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Chem.–Eur. J., 2014, 20, 15775–15784 CrossRef CAS PubMed.
- (a) J. Soellner, M. Tenne, G. Wagenblast and T. Strassner, Chem.–Eur. J., 2016, 22, 9914–9918 CrossRef CAS PubMed; (b) J. Soellner and T. Strassner, Chem.–Eur. J., 2018, 24, 5584–5590 CrossRef CAS PubMed; (c) J. Soellner and T. Strassner, ChemPhotoChem, 2019, 3, 1–6 CrossRef.
- (a) R. C. Nishad, S. Kumar and A. Rit, Organometallics, 2021, 40, 915–926 CrossRef CAS; (b) H. Jin, T. T. Y. Tan and F. E. Hahn, Angew. Chem., Int. Ed., 2015, 54, 13811–13815 CrossRef CAS PubMed.
- The formation of a mixture of C4/C5-bound AuI-MIC complexes (in a 0.45:0.55 ratio), obtained by transmetallation of the in situ generated Ag-carbene complex of L1, was confirmed from the multinuclear (Fig. S46 and S47†) and 2D (HMBC) NMR data (Fig. S48†).
- (a) E. M. Higgins, J. A. Sherwood, A. G. Lindsay, J. Armstrong, R. S. Massey, R. W. Alder and A.-M. C. O'Donoghue, Chem. Commun., 2011, 47, 1559–1561 RSC; (b) M. T. Zamora, M. J. Ferguson, R. McDonald and M. Cowie, Organometallics, 2012, 31, 5463–5477 CrossRef CAS.
- (a) S. Semwal, I. Mukkatt, R. Thenarukandiyil and J. Choudhury, Chem.–Eur. J., 2017, 23, 13051–13057 CrossRef CAS PubMed; (b) S. Semwal, D. Ghorai and J. Choudhury, Organometallics, 2014, 33, 7118–7124 CrossRef CAS; (c) T. Mondal, S. Yadav and J. Choudhury, J. Organomet. Chem., 2021, 953, 122047–122056 CrossRef; (d) C. J. Adams, M. Lusi, E. M. Mutambi and A. G. Orpen, Chem. Commun., 2015, 51, 9632–9635 RSC.
- For selected references, see: (a) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (b) L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045–6048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and NMR and mass spectra of the synthesized compounds. CCDC 2176097–2176105. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05024b |
| This journal is © The Royal Society of Chemistry 2022 |