
Direct intercalation of MoS2 and WS2 thin films by vacuum filtration†
Ding-Yuan
Kuo
 and
Brandi M.
Cossairt
*
and
Brandi M.
Cossairt
*
Department of Chemistry, University of Washington, Seattle, WA 98195, USA. E-mail: cossairt@uw.edu
First published on 15th September 2021
Abstract
In the development of next-generation electronics and energy devices, intercalation compounds of transition metal dichalcogenides (TMDCs) are gaining attention for their unique properties that result from synergistic interactions between guest species and host materials. Nowadays, intercalation compounds of MoS2 and WS2 are commonly prepared by a two-step process: (1) exfoliation to form single-layer and/or few-layer nanosheets and (2) restacking the nanosheets with the guest species by vigorously mixing the exfoliated suspension with the solution of guest species. While a wide variety of intercalation compounds have been synthesized using this approach, the intercalation process is often time-consuming, and the product slurry limits material quality and impedes characterization and applications. Herein, we report a versatile method for preparing intercalated TMDCs in a thin-film morphology. Using this approach, we successfully prepared a range of existing intercalation compounds of MoS2 and WS2 (e.g., ferrocene and amine intercalated MoS2 and WS2). Additionally, by leveraging the versatility of this intercalation method, we intercalated phenazine and benzoquinone into MoS2 and WS2 for the first time.
New conceptsWe demonstrate for the first time a versatile new approach for preparing thin-film intercalation compounds of transition metal dichalcogenides (TMDCs). Because of their transformative potential in a wide variety of catalytic, electronic, sensing, and energy storage applications, intercalation compounds of TMDCs have been explored, often in the form of a slurry. This limits the quality and processability of the materials. These synthetic procedures also lack generality and often need to be modified and re-optimized for different classes of compounds. Here we present an easy route to intercalate molecules into TMDCs in a thin-film morphology. We use vacuum filtration to accelerate the intercalation of guest species into MoS2 and WS2 films restacked from exfoliated nanosheets. This work demonstrates the intercalation of electron–proton transfer mediators into the MoS2 and WS2 for the first time. Our approach also accommodates a great variety of intercalants from organometallics to alkylamines. This method is easily generalizable to other layered materials in a thin-film morphology, paving the way for their expanded investigation and application in next-generation technologies. |
Introduction
Intercalation compounds of transition metal dichalcogenides (TMDCs) offer a wide variety of physicochemical properties through the combination of guest species and intrinsic traits of the host TMDCs.1–4 The building blocks of TMDCs are layers represented as TX2 where T is the transition metal atom and X is a chalcogen atom. These layers are loosely bound to each other via van der Waals interactions. This unique layered structure enables intercalation with a great variety of guest species into the van der Waals gap between these layers.4,5 Among all the TMDCs, intercalation compounds of group VI TMDCs (e.g., MoS2 and WS2) have attracted attention because of their great potential in applications of energy storage, electronics, and catalysis.6–8 The expansion of the interlayer distance due to intercalation has been applied to reduce the energy barrier for alkali ion batteries.9 Ye et al. have also demonstrated superconductivity of intercalated MoS2.10 Moreover, intercalation compounds of MoS2 and WS2 have shown enhanced activity for the hydrogen evolution reaction by modifying the electronic structure of the catalysts.11,12Group VI TMDCs are, however, one of the most challenging host materials among the TMDCs for intercalation. Intercalation generally involves a charge transfer process from the guest species (electron donors) to the layered hosts (electron acceptors). Because of their low electron affinity, MoS2 and WS2 are much weaker electron acceptors.13 Only strong electron donors (e.g., n-butyllithium) can be intercalated into MoS2 and WS2 directly; in contrast, much weaker electron donors can be intercalated into group IV and V TMDCs. For example, direct intercalation of ammonia into TiS214 and intercalation of pyridine into TaS2 are facile.15
To expand the variety of guest species in group VI TMDCs, several strategies have been explored. Electrochemical intercalation has enabled a great variety of intercalation compounds of MoS2 with positively charged intercalants from small alkali cations16–18 to alkylammonium cations.19,20 Instead of accepting electrons from electron donors, this approach injects electrons into the conduction band of MoS2 by applying a reducing potential. The Coulombic interaction between the negatively charged MoS2 layers and the cations further drives the intercalation process.21 The disadvantage of this approach is the limitation of guest species to cations. Alternatively, restacking the exfoliated MoS2 and WS2 layers in a solution with guest species is another method to prepare intercalation compounds of MoS2 and WS2. Guest species such as cations,22,23 polymers,24,25 and clusters26 have been successfully intercalated into MoS2 and WS2 using this method. This strategy has also been applied to intercalating organometallic compounds (e.g., metallocenes) and organic molecules that are not electron donors (e.g., naphthalene) into MoS2.27,28 The intercalation occurs after vigorously mixing an aqueous suspension of single layer MoS2 and (usually) an immiscible organic solvent that contains the guest species.
While there are many different categories of guest species that have been intercalated into MoS2 and WS2, the experimental procedure needs to be designed separately in each case and some of the syntheses require several days.1,29 Moreover, the products are often generated in the form of a slurry, which may limit the processability and quality of the intercalated materials. For example, sonication is commonly required to form a suspension for deposition of a high-quality film for electrochemical characterization. However, these intercalation compounds may de-intercalate during sonication30,31 which complicates the characterization of the intercalation compounds.
Herein, we developed a versatile method for intercalating different categories of guest species into restacked MoS2 and WS2 thin films. Pioneering work by Talyzin et al. has shown that solvents can intercalate into restacked graphene oxide membranes.32 Similarly, Ries et al. have demonstrated that the functionalization of the MoS2 can be achieved in the form of restacked MoS2via vacuum filtration.33 These studies suggest that small molecules can easily diffuse into the space in between the sheets of layered materials. Inspired by their work, we used vacuum filtration to accelerate the diffusion of guest species to encapsulate them in the van der Waalss gap. We applied this approach to three different categories of guest species including organometallic compounds, n-alkylamines, and electron–proton transfer mediators in both MoS2 and WS2. Our approach enables the facile synthesis of a great variety of intercalation compounds of MoS2 and WS2 in a thin-film morphology, which opens new doors to developing electrocatalysts and energy storage materials.
Results and discussion
Fig. 1a shows an optical image of a restacked MoS2 film obtained by vacuum filtration. The thickness of the film can be controlled by the volume of exfoliated MoS2 suspension used in the filtration.34 From our scanning electron microscopy characterization, the thickness of a representative MoS2 film is ∼1 μm (Fig. 1b). As shown by previous studies,35 the phase of chemically exfoliated MoS2 is predominantly 1T phase (∼70%) agreeing with our Raman and UV-Vis spectroscopic analysis (Fig. S1 and S2, ESI†). Fig. 1c shows that this thin film can be transferred to other surfaces such as conductive substrates (e.g., gold) for further applications.34 The WS2 films prepared via vacuum filtration (Fig. S3 and S4, ESI†) demonstrate similar features, including having controllable thickness and being easily transferrable. | ||
| Fig. 1 (a) Optical image of a representative restacked MoS2 film on a nitrocellulose membrane. (b) SEM cross sectional image of a representative restacked MoS2 film. (c) Optical image of an electrode prepared by transferring a restacked MoS2 film onto a gold substrate. The edges of the electrode were sealed using silver paint. | ||
The first class of guest species we explored were metallocenes, which have been intercalated into TMDCs previously owing to their interesting spin and electronic properties for superconductivity and catalysis.1,11 We intercalated ferrocene (Cp2Fe), 1,1′-dimethylferrocene ((MeCp)2Fe), and decamethylferrocene ((Me5Cp)2Fe) into MoS2 and WS2. X-ray diffraction (XRD) clearly demonstrates the expected lattice expansion arising from intercalation (Fig. 2). The XRD results show that the distance between layers in our restacked MoS2 and WS2 are ∼6.2 Å which agree with the bulk materials.36,37 In the XRD patterns of intercalation compounds the peaks corresponding to the restacked MoS2 and WS2 disappear, indicating no residual restacked MoS2 and WS2. We found that the distances between host sheets in the Cp2Fe intercalated MoS2 and WS2 are both 11.8 Å consistent with previous literature.22,27 The expansion (∼5.6 Å) is close to the van der Waals diameter of a cyclopentadienyl (Cp) ring, suggesting that the ring is perpendicular to the host layers.38 Similar expansion (∼5.6 Å) was observed in the (MeCp)2Fe intercalated MoS2 and WS2. We hypothesize that the Cp ring is still perpendicular to the host sheets, but the methyl group in MeCp is pointing away from the host sheets. In contrast, the distance between the host sheets is ∼13.4 Å in (Me5Cp)2Fe intercalated MoS2 and WS2. The expansion (∼7.2 Å) is close to the size of Me5Cp, consistent with the ring laying perpendicular to the host sheets. Among the restacked and intercalated films, we only observed diffraction peaks from the {001} plane, indicating a preferred orientation along the z-axis in the thin film samples. The distances between the host sheets in the intercalation compounds are summarized in Table S1 (ESI†).
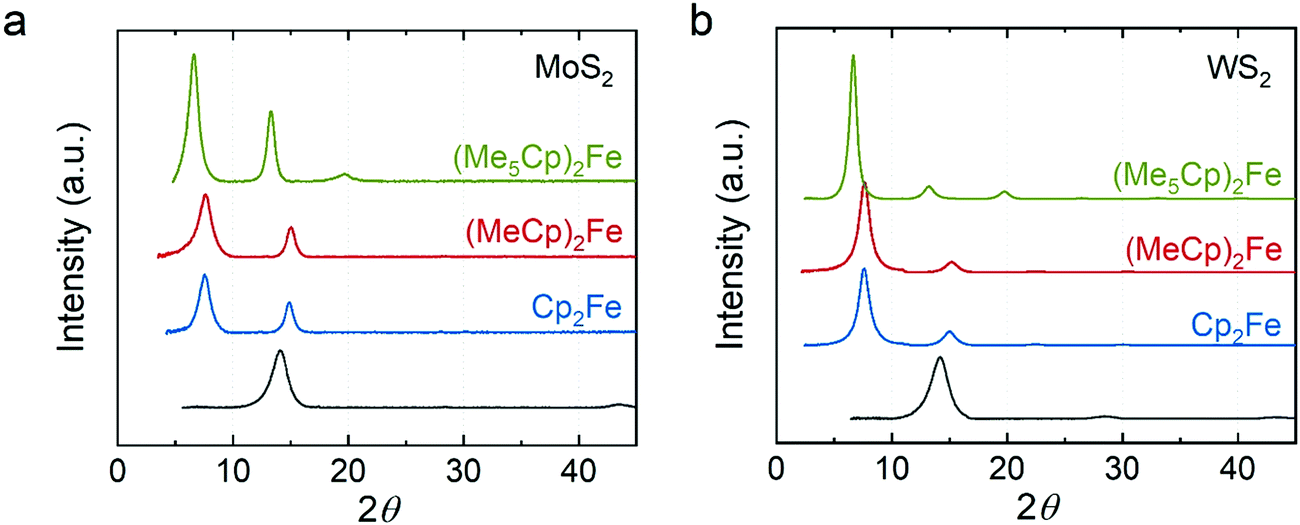 | ||
| Fig. 2 XRD of metallocene intercalated (a) MoS2 and (b) WS2. Green: decamethylferrocene-intercalated; red: 1,1′-dimethylferrocene-intercalated; blue: ferrocene-intercalated; dark gray: restacked. | ||
We quantified the compositions of the metallocene intercalated MoS2 and WS2 using inductively coupled plasma optical emission spectroscopy (ICP-OES, Table 1). We found that the S/Mo ratios are ∼2 consistent with the stoichiometry of the bulk MoS2. The Fe/Mo ratios are 0.118, 0.093 and 0.135 in Cp2Fe, (MeCp)2Fe, and (Me5Cp)2Fe intercalated MoS2, respectively. The Fe ratio in our compounds is higher than the ferrocene intercalated MoS2 restacked at the interface of two immiscible solutions (Fe/Mo ∼ 0.05).27 However, our ICP-OES results show that the S/W ratios in the WS2 intercalation compounds are only ∼1.8. To understand this discrepancy, we examined the S/W ratio in the bulk WS2 and found it is also ∼1.8. We hypothesized that the ratio of S/W is lower than the theoretical stoichiometry because of the loss of S during the intense digestion of WS2 which involves HF and HNO3. A previous study39 suggested the formation of volatile S compounds during sample digestion, resulting in an underestimation of S concentration deduced by ICP-OES analysis. We assumed no formation of volatile Fe and W compounds during the digestion and found that the Fe/W ratios are 0.058, 0.072, and 0.092 in three different intercalated WS2 samples.
| Sample | Mo/W | S | Fe |
|---|---|---|---|
| Cp2Fe–MoS2 | 1 | 1.99 | 0.118 |
| (MeCp)2Fe–MoS2 | 1 | 2.02 | 0.093 |
| (Me5Cp)2Fe–MoS2 | 1 | 2.00 | 0.135 |
| Cp2Fe–WS2 | 1 | 1.83 | 0.058 |
| (MeCp)2Fe–WS2 | 1 | 1.82 | 0.072 |
| (Me5Cp)2Fe–WS2 | 1 | 1.79 | 0.092 |
Our results show that the ratio of (Me5Cp)2Fe in the intercalation compounds is the highest of the metallocene intercalants; however, no clear trend was observed in the ratio of Cp2Fe and (MeCp)2Fe. It has been suggested that a more reducing guest species tends to form a more stable intercalation compound.40 Since the (Me5Cp)2Fe is the most reducing guest species ((Me5Cp)2Fe > (MeCp)2Fe > Cp2Fe),41 we expect that (Me5Cp)2Fe would form the most stable intercalation compound and therefore be present in the highest ratio. Besides their reducing power, the size of the molecule also affects the ratio of the guest species in the intercalation compounds by determining the maximum packing density. Due to the orientation of the guest species in the intercalation compounds, the maximum packing density of Cp2Fe is higher than (MeCp)2Fe. Taken together, the ratio of Cp2Fe and (MeCp)2Fe may be a delicate balance influenced by reducing power and the size of intercalant.
In addition, we demonstrated that our approach is applicable to intercalation of n-alkylamines into MoS2 and WS2. Alkylamines, which interact with the host materials through their N lone pair, have been successfully intercalated into many layered oxides42–44 and TMDCs.2,30,31 Yet ammonium cations have been commonly observed as the end product in intercalation compounds of MoS2 and WS2 arising from cation exchange reactions in the syntheses.30,45 One striking property of these intercalation compounds is that the lattice expansion can be tuned by the chain length of the alkylamine. We chose three different chain lengths of alkylamine – octylamine (OA), dodecylamine (DDA), and hexadecylamine (HDA) – to reveal the chain length dependency in our intercalation compounds.
Fig. 3 shows the XRD data for the alkylamine intercalated MoS2 and WS2. We found that the distance between the TMDC sheets increases with the chain length of alkylamine, where OA–MoS2 is ∼10.1 Å, DDA–MoS2 is 14.1 to 14.5 Å, and HDA–MoS2 is ∼32.4 Å. We observed a similar trend in WS2 intercalation compounds, where OA–WS2 is ∼10.1 Å, DDA–WS2 is ∼14.3 Å, and HDA–MoS2 is ∼32.4 Å. Our results concur with reported n-alkylamine intercalated MoS2 and WS2 synthesized through a direct reaction of lithium intercalated TMDCs with an aqueous solution of alkylamine/ammonia.35 However, unlike these compounds, which show a diffraction peak at 2θ ∼ 32°, our compounds only showed diffraction peaks from the {001} plane indicating, again, a preferred orientation along the z-axis.
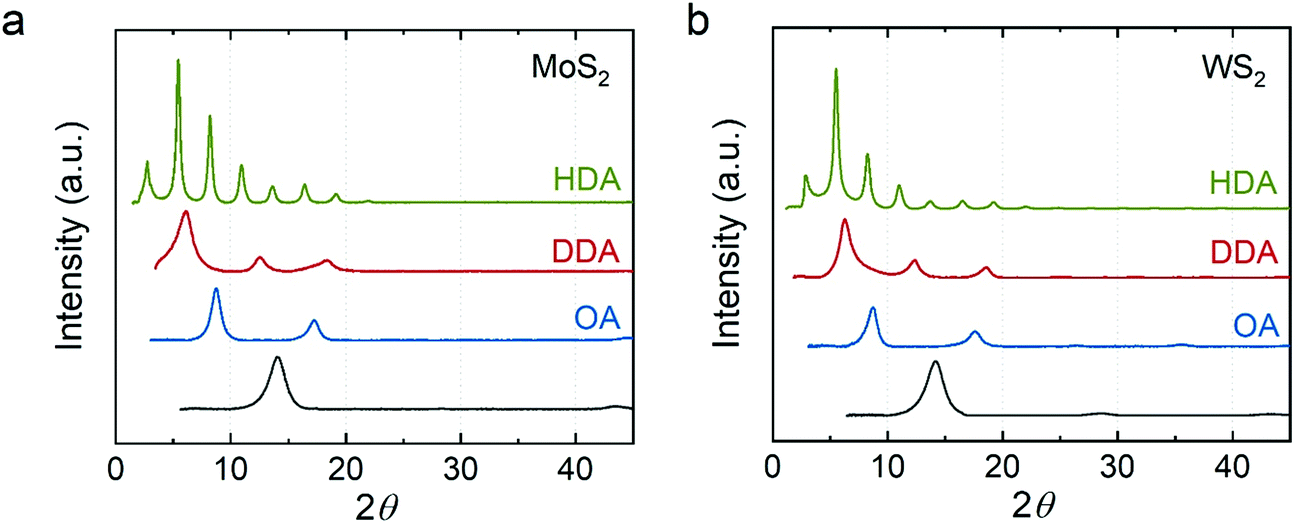 | ||
| Fig. 3 XRD of the n-alkylamine intercalated (a) MoS2 and (b) WS2. Green: hexadecylamine-intercalated; red: dodecylamine-intercalated; blue: octylamine-intercalated; dark gray: restacked. | ||
The non-linear increase of the distance between basal planes is attributed to the orientation of the alkylamine. We hypothesize that a short chain alkylamine (e.g., OA) prefers lying down in the van der Waals gap of the TMDCs. As we showed, the expansion in the OA–MoS2 and OA–WS2 are ∼3.9 Å. This value is much shorter than the chain length of OA but close to the van der Waals diameter of the alkyl chain (∼4 Å). This observation suggests that the alkyl chain lies parallel to the host sheets. A similar phenomenon has been reported in alkylammonium intercalated MoS245 and alkylamine intercalated TaS22 where the lattice expansion in short chain amine (n < 5) intercalation compounds is ∼4 Å. Interestingly, we found that the expansion in DDA–MoS2 and DDA–WS2 is ∼8.1 Å, which is close to twice the size of the alkyl chain. We proposed that two layers of DDA sandwiched between the host sheets. However, previous studies have suggested that the alkylamine may form a tilted bilayer structure at an angle from 56° to 68°.45,46 If we assume a tilted monolayer of DDA in between the host sheets, the tilt angle would be 25.8°, much smaller than in the previously reported structures.
In contrast, the distance between host sheets expands to >32 Å in the HDA intercalated compounds. This expansion is close to the length of the alkyl chain implying a monolayer of HDA perpendicular to the host sheets. We attributed this structural difference to longer alkyl chains having stronger van der Waals interactions among themselves. This interaction may assist the assembly of the amine and stabilize the perpendicular orientation. Considering that the guest species need to diffuse into the film, we note that our materials could be one of several possible kinetically stable phases. Mixtures of intercalation compounds have been found in the alkylamine intercalation TaS2.2
We used CHN combustion elemental analysis to quantify the stoichiometry of OA, DDA, and HDA in both MoS2 and WS2 intercalation compounds (Table 2). We observed that the stoichiometry of HDA is higher than that of OA and DDA in both MoS2 and WS2 likely because of the orientation of the alkylamines. Compared with the parallel orientation, the perpendicular orientation occupies less projected area on the sheets leading to a higher packing density. We note that the mass percentage of carbon from CHN analysis is higher (∼2 to 3%) than the calculated values from the proposed formula in MoS2 intercalation compounds, which we attributed to residual carbon compounds adsorbed on the MoS2. The physical adsorption of solvent has been commonly found on high-surface-area materials.47,48 We examined the composition of restacked MoS2 and WS2 without alkylamine and found that the carbon mass percentage is 2.3% in MoS2 and only ∼ 0.25% in WS2.
| Sample | Mass percentage | Approximate formula | ||
|---|---|---|---|---|
| C | H | N | ||
| OA–MoS2 | 9.918 | 1.719 | 1.074 | OA0.136MoS2 |
| (7.356) | (1.466) | (1.072) | ||
| DDA–MoS2 | 14.414 | 2.086 | 1.093 | DDA0.145MoS2 |
| (11.178) | (2.111) | (1.086) | ||
| HDA–MoS2 | 29.020 | 4.781 | 1.869 | HDA0.319MoS2 |
| (25.855) | (4.746) | (1.884) | ||
| OA–WS2 | 7.463 | 1.404 | 0.989 | OA0.203WS2 |
| (7.113) | (1.418) | (1.037) | ||
| DDA–WS2 | 9.705 | 1.651 | 0.975 | DDA0.194WS2 |
| (9.848) | (1.859) | (0.957) | ||
| HDA–WS2 | 17.155 | 2.849 | 1.254 | HDA0.283WS2 |
| (17.194) | (3.156) | (1.253) | ||
Finally, our approach enables synthesis of intercalation compounds beyond those that have been synthesized previously. We demonstrated that electron–proton transfer mediators (e.g., phenazine and p-benzoquinone) can be intercalated into MoS2 and WS2. Fig. 4 shows the XRD of p-benzoquinone and phenazine intercalated MoS2 and WS2. The expansion from the intercalation is ∼3.4 Å close to the van der Waals diameter of a carbon atom (3.4 Å). This observation suggests that the aromatic rings of the intercalants lie parallel to the host sheets. We used the size of p-benzoquinone (8.3 × 6.6 Å2) and phenazine (11.3 × 7.2 Å2) to estimate their monolayer maximum packing density. In intercalation compounds of MoS2, the maximum ratio of p-benzoquinone is ∼0.213 whereas the maximum ratio of phenazine is ∼0.121. They both show slightly higher packing density in the intercalation compounds of WS2 (∼0.223 for p-benzoquinone and ∼0.127 for phenazine) due to the larger lattice constant of WS2.
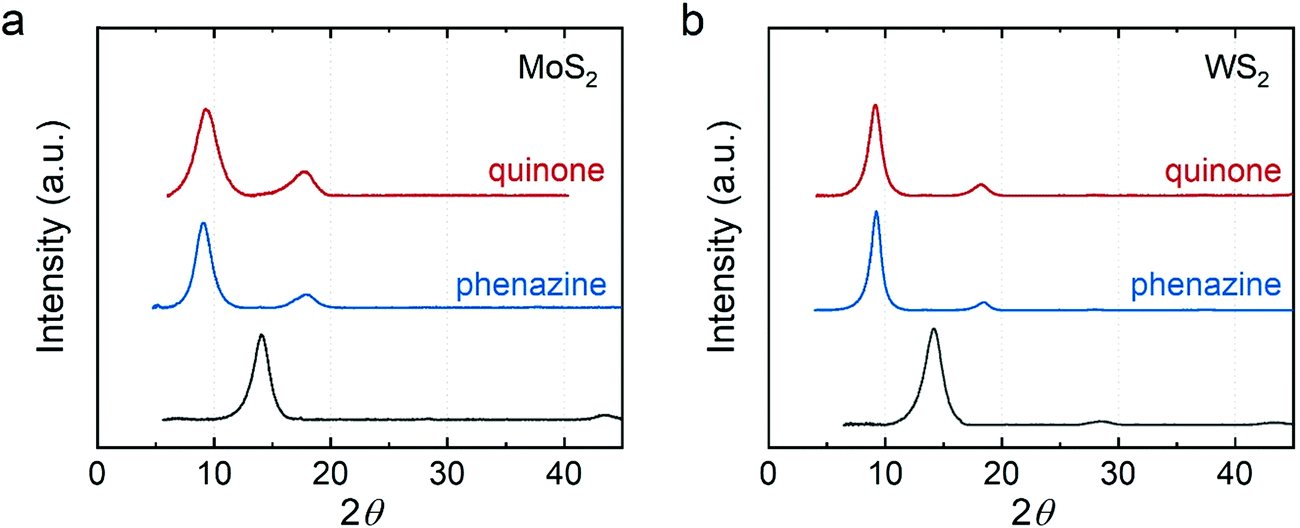 | ||
| Fig. 4 XRD of electron-proton transfer mediator intercalated (a) MoS2 and (b) WS2. Red: p-benzoquinone-intercalated; blue: phenazine-intercalated; dark gray: restacked. | ||
We quantified the percentage of phenazine in the intercalation compounds using CHN combustion elemental analysis (Table 3). We found that the ratio of phenazine in the intercalation compounds is around 7 to 8%. Interestingly, the mass percentage of carbon is a few percent higher than the calculated value from the proposed formula, similar to what we observed in the alkylamine intercalated MoS2. We hypothesized that toluene molecules may adsorb and/or intercalate into the MoS2 and WS2. To confirm whether toluene can intercalate into MoS2 and WS2, we removed the phenazine and only added toluene in the process of vacuum filtration. The XRD of toluene treated MoS2 and WS2 (Fig. S5, ESI†) shows no shift in the diffraction peak position indicating no intercalation of toluene. While co-intercalation of toluene and phenazine could occur, we expect that the lattice expansion from co-intercalation would be larger. After including toluene (presumed to be adsorbed to the material surface) in the proposed formula, the ratio of phenazine remains ∼7 to 8% (Table S2, ESI†).
| Sample | Mass percentage | Approximate formula | ||
|---|---|---|---|---|
| C | H | N | ||
| Phenazine–MoS2 | 9.154 | 0.76 | 1.326 | (C12H8N2)0.083MoS2 |
| (6.834) | (0.382) | (1.328) | ||
| Phenazine–WS2 | 5.467 | 0.654 | 0.745 | (C12H8N2)0.069WS2 |
| (3.819) | (0.214) | (0.742) | ||
Due to the residual hydrocarbons, we cannot quantify the ratio of p-benzoquinone in the intercalation compounds. We estimate its ratio by assuming a constant ratio of residual toluene (i.e., 0.05) in all intercalation compounds. Our estimation shows that the ratio of the p-benzoquinone is roughly 10% (Table S3, ESI†). While the structure of the intercalation compounds requires further characterization, our XRD and elemental analysis results confirm the intercalation of phenazine and p-benzoquinone in the MoS2 and WS2.
Overall, our approach is applicable to guest species whether they are electron-donating (i.e., reducing agents or Lewis bases). Similarly, Morrison et al. reported the intercalation of guest species that are not electron-donating using a biphasic method.27 They observed the formation of intercalation compounds at the interface of two immiscible solvents and they attributed the driving force for this process to the hydrophobic nature of the basal plane of MoS2. Unlike their approach, the intercalation in our method occurs on a wet restacked TMDC film. Ries et al. have shown that water molecules remain in between MoS2 layers in the restacked film while it is still wet.33 These water molecules can potentially weaken the van der Waals interactions between host layers. Additionally, the expanded interlayer spacing may reduce the energy barrier for the diffusion of the guest species.9 Here, we propose the mechanism of intercalation as follows: the guest species (and solvent molecules) replace water molecules in the van der Waals gap of the host materials due to the hydrophobic surface of TMDCs and the pressure gradient generated by the vacuum filtration. After the drying process, the volatile solvent molecules escape from the intercalation compounds due to weaker van der Waals interactions with the TMDC layers whereas the guest species remain in between the TMDC layers.
Conclusions
We presented a versatile approach for directly intercalating guest species into restacked thin films of MoS2 and WS2. A key feature of our intercalated materials is the easily transferrable thin-film morphology. We synthesized known ferrocene and n-alkylamine intercalated MoS2 and WS2 compounds via vacuum filtration. Moreover, we intercalated two electron–proton transfer mediators (i.e., phenazine and benzoquinone) into MoS2 and WS2 for the first time. This method should be broadly applicable to intercalating other exfoliated layered materials (e.g., graphene oxide and MXenes) with a broad range of guest species, which will accelerate the study of intercalation compounds in catalysis, sensing, energy storage, and beyond.Experimental methods
Materials
Molybdenum(IV) sulfide powder (<2 μm, 99%), tungsten(IV) sulfide powder (2 μm, 99%), n-butyllithium solution (1.6 M in hexane), ferrocene (98%), 1,1′-dimethylferrocene (95%), bis(pentamethylcyclopentadienyl)iron(II) (97%), and phenazine (98%) were purchased from Sigma-Aldrich and used as received without further purification. p-Benzoquinone (99.5%) was purchased from Sigma-Aldrich and purified via sublimation. Octylamine (99%), dodecylamine (98%), and hexadecylamine (98%) were purchased from Sigma-Aldrich and heated at 100 °C for 10 min under nitrogen prior to use. Pentane was purchased anhydrous and stored over 3 Å molecular sieves prior to use. Hexane (98.5%, Sigma Aldrich), toluene (99.8%, Fisher Chemical), and methanol (99.9%, Fisher Chemical) were used as received without further purification.Exfoliation of MoS2 and WS2
The exfoliation was performed by using the lithium intercalation method that has been reported previously.34 Briefly, 320 mg of MoS2 was charged into an oven-dried 50 mL Schlenk flask. After degassing and cycling with nitrogen three times, 5 mL of 1.6 M n-butyllithium solution in hexane was added. The flask was at room temperature and stirring at 300 rpm for 48 h under nitrogen. After 48 h, the flask was transferred into a nitrogen glovebox. Pentane was added to wash the materials at least 3 times. The LixMoS2 was stored under nitrogen. Similarly, 497 mg of WS2 was added into an oven-dried three-neck round bottom flask. 5 mL of 1.6 M n-butyllithium solution in hexane was added. The solution was refluxed at 70 °C with magnetic stirring at 300 rpm for 48 h under nitrogen. After 48 h, the flask was transferred into a nitrogen glovebox. Pentane was added to wash the materials at least 3 times. The LixWS2 was stored under nitrogen.∼67.5 mg of LixMoS2 (∼100 mg of LixWS2) was transferred into a centrifuge tube in a nitrogen glovebox. Deionized water (> 18 MΩ cm) that had been degassed for at least 15 min was added into the tube immediately after the tube was transferred out of the glovebox. The tube was sonicated for 1 h and the temperature was kept below 30 °C. The mixture was centrifuged at 2500 rpm to remove the non-exfoliated TMDC sheets and 11000 rpm for 15 minutes three times to remove the lithium salt byproducts. The exfoliated MoS2 and WS2 suspensions were used immediately after preparation.
Synthesis of metallocene intercalated MoS2 and WS2
15 mL of exfoliated MoS2/WS2 suspension was restacked on a nitrocellulose membrane (0.025 μm, MF-Millipore™) by vacuum filtration. After the suspension was filtered, 3 mL of a saturated hexane solution containing the metallocene was added into the filtration funnel while the membrane was still wet. The vacuum filtration proceeded for 1 hour. The material was washed with hexane 4 to 5 times to remove residual metallocene.Synthesis of n-alkylamine intercalated MoS2 and WS2
15 mL of exfoliated MoS2/WS2 suspension was restacked on a nylon membrane (0.2 μm, MF-Millipore™) by vacuum filtration. After the suspension was filtered, 0.2 mmol of n-alkylamine in 6 mL of methanol was added into the filtration funnel while the membrane was still wet. The filtration was carried out under static vacuum. Before the solution in the flask completely dried out, the material was washed with methanol 4 to 5 times to remove residual alkylamine. The filtration time was not controlled but typically this process takes less than 30 min.Synthesis of phenazine and p-benzoquinone intercalated MoS2 and WS2
15 mL of exfoliated MoS2/WS2 suspension was restacked on a nitrocellulose membrane (0.025 μm, MF-Millipore™) by vacuum filtration. After the suspension was filtered, 3 mL of saturated phenazine toluene solution (1.5 mmol of p-benzoquinone in 3 mL of toluene) was added into the filtration funnel while the membrane was still wet. The vacuum filtration proceeded for 1 hour. The material was washed with toluene 4 to 5 times to remove residual guest species.Sample characterization
X-ray diffraction data were obtained from a Bruker D8 Discover instrument with the IμS 2-D XRD system. Raman spectra were recorded on a Renishaw Raman Confocal and a 514 nm laser was used. Scanning electron microscopy images were obtained from an Apreo variable-pressure SEM. A PerkinElmer Optima 8300 inductively coupled plasma-optical emission spectrophotometer was used for elemental analysis. Solutions of MoS2 samples were prepared by dissolving the materials in aqua regia whereas WS2 intercalated samples were digested using a mixture of HF, HNO3, and H2O (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1). Microanalysis was conducted by the CENTC Elemental Analysis Facility at the University of Rochester. Microanalysis samples were weighed with a PerkinElmer Model AD6000 Autobalance and their compositions were determined with a PerkinElmer 2400 Series II Analyzer.
:1:1). Microanalysis was conducted by the CENTC Elemental Analysis Facility at the University of Rochester. Microanalysis samples were weighed with a PerkinElmer Model AD6000 Autobalance and their compositions were determined with a PerkinElmer 2400 Series II Analyzer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. D.-Y. K. acknowledges postdoctoral funding support from the University of Washington. Part of this work was conducted at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington, which is supported in part by funds from the National Science Foundation (awards NNCI-2025489, NNCI-1542101), the Molecular Engineering & Sciences Institute, and the Clean Energy Institute. Analytical data were obtained from the CENTC Elemental Analysis Facility at the University of Rochester, funded by NSF CHE-0650456. We thank Max Friedfeld for UV-Vis spectroscopy measurements and Scott Braswell for SEM characterization.References
- F. R. Gamble, F. J. DiSalvo, R. A. Klemm and T. H. Geballe, Science, 1970, 168, 568–570 CrossRef CAS PubMed.
- F. R. Gamble, J. H. Osiecki, M. Cais, R. Plsharody, F. J. DiSalvo and T. H. Geballe, Science, 1971, 174, 493–497 CrossRef CAS PubMed.
- C. Wan, X. Gu, F. Dang, T. Itoh, Y. Wang, H. Sasaki, M. Kondo, K. Koga, K. Yabuki, G. J. Snyder, R. Yang and K. Koumoto, Nat. Mater., 2015, 14, 622–627 CrossRef CAS.
- Q. Zhang, L. Mei, X. Cao, Y. Tang and Z. Zeng, J. Mater. Chem. A, 2020, 8, 15417–15444 RSC.
- Y. Jung, Y. Zhou and J. J. Cha, Inorg. Chem. Front., 2016, 3, 452–463 RSC.
- N. Feng, R. Meng, L. Zu, Y. Feng, C. Peng, J. Huang, G. Liu, B. Chen and J. Yang, Nat. Commun., 2019, 10, 1–11 CrossRef CAS.
- M. Mattinen, M. Leskelä and M. Ritala, Adv. Mater. Interfaces, 2021, 8, 2001677 CrossRef CAS.
- D. Saha and P. Kruse, J. Electrochem. Soc., 2020, 167, 126517 CrossRef CAS.
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. Robles Hernandez, L. C. Grabow and Y. Yao, Nano Lett., 2015, 15, 2194–2202 CrossRef CAS.
- J. T. Ye, Y. J. Zhang, R. Akashi, M. S. Bahramy, R. Arita and Y. Iwasa, Science, 2012, 338, 1193–1196 CrossRef CAS PubMed.
- I. H. Kwak, H. G. Abbas, I. S. Kwon, Y. C. Park, J. Seo, M. K. Cho, J. P. Ahn, H. W. Seo, J. Park and H. S. Kang, J. Mater. Chem. A, 2019, 7, 8101–8106 RSC.
- N. H. Attanayake, A. C. Thenuwara, A. Patra, Y. V. Aulin, T. M. Tran, H. Chakraborty, E. Borguet, M. L. Klein, J. P. Perdew and D. R. Strongin, ACS Energy Lett., 2018, 3, 7–13 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
- R. R. Chianelli, J. C. Scanlon, M. S. Whittingham and F. R. Gamble, Inorg. Chem., 1975, 14, 1691–1696 CrossRef CAS.
- F. R. Gamble, J. H. Osiecki and F. J. DiSalvo, J. Chem. Phys., 1971, 55, 3525–3530 CrossRef CAS.
- G. Zhu, J. Liu, Q. Zheng, R. Zhang, D. Li, D. Banerjee and D. G. Cahill, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Zheng, H. Zhang, S. Dong, Y. Liu, C. Tai Nai, H. Suk Shin, H. Young Jeong, B. Liu and K. Ping Loh, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., 2011, 123, 11289–11293 CrossRef.
- C. Wang, Q. He, U. Halim, Y. Liu, E. Zhu, Z. Lin, H. Xiao, X. Duan, Z. Feng, R. Cheng, N. O. Weiss, G. Ye, Y. C. Huang, H. Wu, H. C. Cheng, I. Shakir, L. Liao, X. Chen, W. A. Goddard, Y. Huang and X. Duan, Nature, 2018, 555, 231–236 CrossRef CAS PubMed.
- Z. Lin, Y. Liu, U. Halim, M. Ding, Y. Liu, Y. Wang, C. Jia, P. Chen, X. Duan, C. Wang, F. Song, M. Li, C. Wan, Y. Huang and X. Duan, Nature, 2018, 562, 254–258 CrossRef CAS.
- M. Laipan, L. Xiang, J. Yu, B. R. Martin, R. Zhu, J. Zhu, H. He, A. Clearfield and L. Sun, Prog. Mater. Sci., 2020, 109, 100631 CrossRef CAS.
- K. E. Dungey, M. D. Curtis and J. E. Penner-Hahn, Chem. Mater., 1998, 10, 2152–2161 CrossRef CAS.
- A. A. Jeffery, C. Nethravathi and M. Rajamathi, J. Phys. Chem. C, 2014, 118, 1386–1396 CrossRef.
- J. P. Lemmon and M. M. Lerner, Chem. Mater., 1994, 6, 207–210 CrossRef CAS.
- I. Saada and R. Bissessur, J. Mater. Sci., 2012, 47, 5861–5866 CrossRef CAS.
- R. Bissessur, J. Heising, W. Hirpo and M. Kanatzidis, Chem. Mater., 1996, 8, 318–320 CrossRef CAS.
- W. M. R. Divigalpitiya, R. F. Frindt and S. R. Morrison, Science, 1989, 246, 369–371 CrossRef CAS.
- H. Tagaya, T. Hashimoto, M. Karasu, T. Izumi and K. Chiba, Chem. Lett., 1991, 2113–2116 CrossRef CAS.
- L. Kosidowski and A. V. Powell, Chem. Commun., 1998, 2201–2202 RSC.
- A. A. Jeffery, C. Nethravathi and M. Rajamathi, RSC Adv., 2015, 5, 51176–51182 RSC.
- S. Jeong, D. Yoo, M. Ahn, P. Miro, T. Heine and J. Cheon, Nat. Commun., 2015, 6, 1–7 CrossRef.
- A. Klechikov, J. Yu, D. Thomas, T. Sharifi and A. V. Talyzin, Nanoscale, 2015, 7, 15374–15384 RSC.
- L. Ries, E. Petit, T. Michel, C. C. Diogo, C. Gervais, C. Salameh, M. Bechelany, S. Balme, P. Miele, N. Onofrio and D. Voiry, Nat. Mater., 2019, 18, 1112–1117 CrossRef CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS.
- I. Janica, D. Iglesias, S. Ippolito, A. Ciesielski and P. Samorì, Chem. Commun., 2020, 56, 15573–15576 RSC.
- N. Wakabayashi, H. G. Smith and R. M. Nicklow, Phys. Rev. B: Solid State, 1975, 12, 659–663 CrossRef CAS.
- W. J. Schutte, J. L. De Boer and F. Jellinek, J. Solid State Chem., 1987, 70, 207–209 CrossRef CAS.
- M. B. Dines, Science, 1975, 188, 1210–1211 CrossRef CAS PubMed.
- C. Morrison, H. Sun, Y. Yao, R. A. Loomis and W. E. Buhro, Chem. Mater., 2020, 32, 1760–1768 CrossRef CAS.
- M. S. Whittingham and A. J. Jacobson, Intercalation chemistry, Academic Press, 1982 Search PubMed.
- A. Paul, R. Borrelli, H. Bouyanfif, S. Gottis and F. Sauvage, ACS Omega, 2019, 4, 14780–14789 CrossRef CAS PubMed.
- P. Gonzalez Rodriguez, H. Yuan, K. J. H. Van Den Nieuwenhuizen, W. Lette, D. J. Schipper and J. E. Ten Elshof, ACS Appl. Mater. Interfaces, 2016, 8, 28926–28934 CrossRef CAS.
- N. Chen, L. Ni, J. Zhou, G. Zhu, Y. Zhang, S. Chen, F. Gao, C. Lu, H. Ji, J. Chen, X. Wang, X. Guo, L. Peng, W. Ding and W. Hou, Sustainable Energy Fuels, 2018, 2, 2788–2798 RSC.
- A. Espina, C. Trobajo, S. A. Khainakov, J. R. Garcia and A. I. Bortun, J. Chem. Soc., Dalton Trans., 2001, 499–501 Search PubMed.
- R. Schöllhorn and A. Weiss, J. Less-Common Met., 1974, 36, 229–236 CrossRef.
- R. Schöllhorn, E. Sick and A. Weiss, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1973, 28, 168–171 CrossRef.
- F. Hof, R. A. Schäfer, C. Weiss, F. Hauke and A. Hirsch, Chem. – Eur. J., 2014, 20, 16644–16651 CrossRef CAS PubMed.
- X. S. Chu, A. Yousaf, D. O. Li, A. A. Tang, A. Debnath, D. Ma, A. A. Green, E. J. G. Santos and Q. H. Wang, Chem. Mater., 2018, 30, 2112–2128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Raman and UV-Vis spectra, additional XRD data, tabulated lattice constants, and tabulated compositional data. See DOI: 10.1039/d1mh01193f |
| This journal is © The Royal Society of Chemistry 2022 |