
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective benzylic Csp3–H bond activations mediated by a phosphorus–nitrogen PN3P-nickel complex†
Changguang
Yao
 ab,
Tonghuan
Zhang
bc,
Théo P.
Gonçalves
b and
Kuo-Wei
Huang
*b
ab,
Tonghuan
Zhang
bc,
Théo P.
Gonçalves
b and
Kuo-Wei
Huang
*b
aSchool of Resource, Environmental and Chemical Engineering, Nanchang University, Nanchang 330031, Jiangxi, China
bKAUST Catalysis Center and Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia. E-mail: hkw@pincer.systems
cLab of Computational Chemistry and Drug Design, State Key Laboratory of Chemical Oncogenomics, Peking University, Shenzhen Graduate School, Shenzhen 518055, China
First published on 30th December 2021
Abstract
In contrast to the typical Csp2–H activation, a PN3P-Nickel complex chemoselectively cleaved the benzylic Csp3–H bond of toluene in the presence of KHMDS, presumably via an in situ generated potassium benzyl intermediate. Under similar conditions, CO underwent deoxygenation to afford the corresponding nickel cyano complex, and ethylbenzene was dehydrogenated to give styrene and a nickel hydride compound. 2,6-Xylyl isocyanide was transformed into an unprecedented indolyl complex, likely by trapping the activated benzyl species with an isocyanide moiety.
C–H bond activation has emerged as one of the most revolutionary trends in organic chemistry because it provides straightforward, atom-economically synthetic routes and permits previously unachievable synthetic disconnections. This approach has been extensively used to directly install a wide range of new carbon–carbon and carbon–heteroatom bonds.1–4 Nonetheless, the selective and predictable transformation of specific C–H bonds into various complicated compounds remains challenging since most organic compounds contain more than one kind of C–H bond.3,5–8 As a unique hydrocarbon substrate, toluene possesses one benzylic and three different aromatic C–H bonds, oxidation of the former of which serves a critical role in yielding industrially important chemicals such as benzyl alcohols, benzaldehydes and benzoic acids.9–13 A series of new reactions have been developed in recent years through benzylic C–H bond functionalization of toluene and its derivatives. Matsuzaka and co-workers reported catalytic dehydrative condensation of benzylic C–H bonds of toluene and xylene with aromatic aldehydes to afford stilbenes.14 The Guan group discovered potassium-mediated benzylic C–H bond addition of diarylmethanes or alkylpyridines to styrenes, as well as lithium-promoted benzylic aroylation of toluenes.15,16 Walsh et al. reported the synthesis of diarylmethanes and diarylethylamine via benzylic arylation/amination of toluene derivatives by using the cation-π interactions.17–19 The catalytic addition reaction of toluenes with imines and carbonyls was also demonstrated by Kobayashi20 and Kondo,21 respectively. Within these developments, however, very few well-defined intermediates were isolated from benzylic Csp3–H bond activation (Fig. 1). For example, benzyl complexes could be obtained via the radical exchange mediated by a Fe–Sn bimetallic complex.22 The cyclometalated rhodium complex through Csp3–H activation of 8-methylquinoline was achieved by Zhou et al.23 Very recently, Sergeev and co-workers realized selective and radical-free activation of benzylic C–H bonds in methylarenes, to afford several iridium-benzyl complexes with one to five methyl groups in the aromatic ring.24
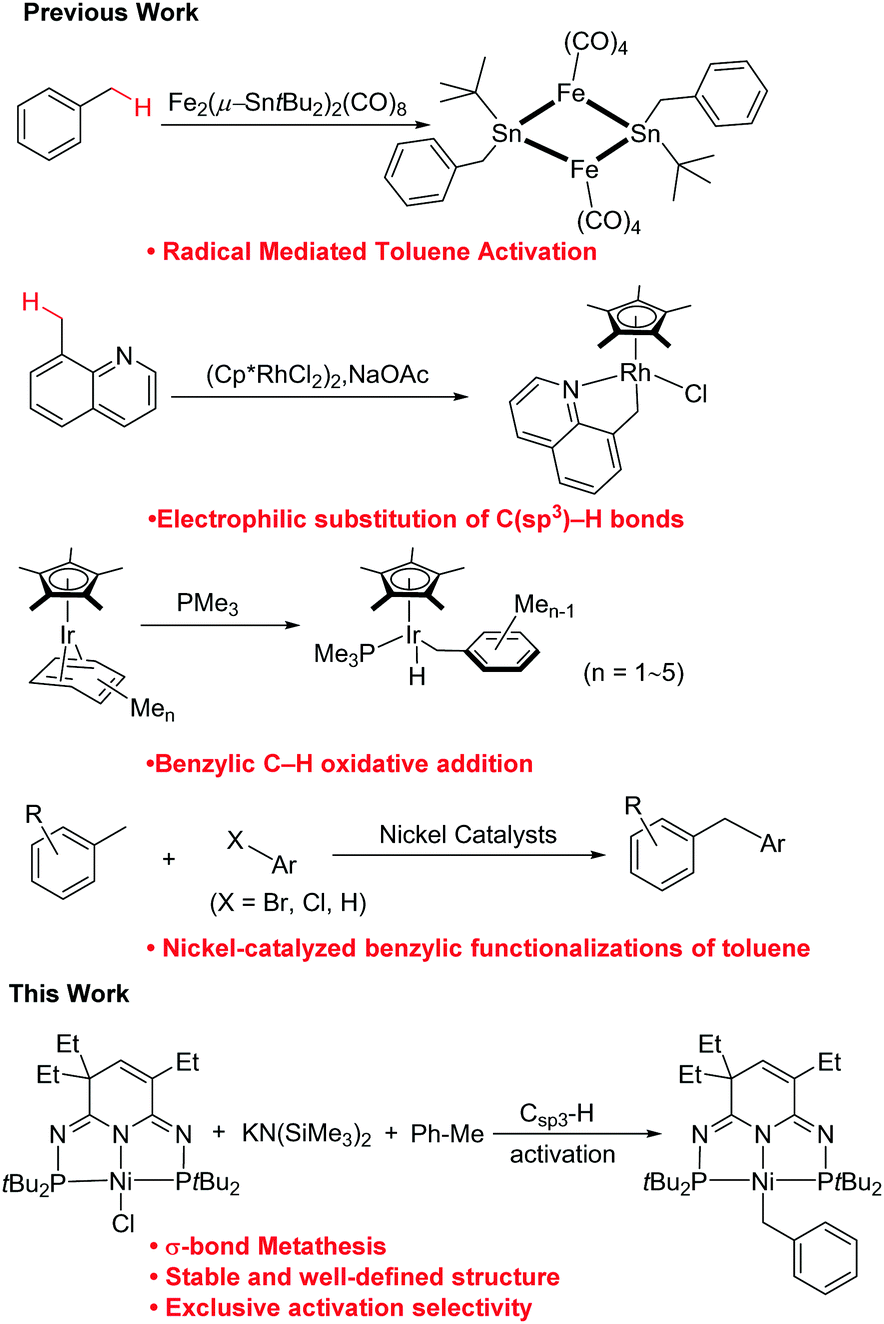 | ||
| Fig. 1 Well-defined metal-benzyl complex and nickel-catalyzed benzylic C–H functionalization of toluene derivatives. | ||
We have worked on the design and preparation of transition metal compounds bearing a novel class of pincer-type PN3(P)-ligands.25,26 These compounds have shown rich reactivities and catalytic activities through metal–ligand cooperation. Compared to Milstein's CH2 spacers in pyridine-based pincer complexes, our seemingly small changed NH analogs have enabled the observation of distinct catalytic reactivities and various thermodynamic and kinetic properties.27–29 Very recently, further extensions of the PN3P system to a new series of second-generation PN3P pincer complexes have also been achieved in our group through a ligand post-synthetic modification strategy.30–34 This new class of compounds have shown unprecedented thermal stability, and thus the corresponding Ni azide, hydroxide and triflate complexes were synthesized and studied.32–34 Herein, we further demonstrate the benzylic C–H bond activations of toluene, ethylbenzene and 2,6-dimethylphenyl isocyanide for the synthesis of nickel benzyl, hydride and indolyl complexes by utilizing the PN3P-nickel complex with the combination of KN(SiMe3)2. Different from previous reports on nickel-catalyzed benzylic C–H functionalization as shown in Fig. 1, which involved a radical intermediate, light irradiation or cation-π interaction, with no nickel benzyl intermediate isolated, the present work affords a well-defined Ni-benzyl compound via an overall σ-bond metathesis reaction under mild conditions.19,35,36
Both NiII{N(SiMe3)2}2 and (L)NiIIN(SiMe3)2 have been described as unstable and reactive species.37,38 Therefore, we attempted to use our thermostable 2nd-generation of pincer ligand to preprare the nickel silylamido complex (PN3P)NiN(SiMe3)2 (2) through a salt metathesis of nickel chloride 1 and KN(SiMe3)2 (KHMDS) (Scheme 1). Unfortunately, the target silylamido compound was not successfully obtained even at an elevated temperature (80 °C). Interestingly, when the reaction was carried out under a CO atmosphere, a new compound formed slowly as proven by the new signals at δ 124.02 and 123.64 ppm in the 31P NMR spectrum. By monitoring the 1H NMR spectrum, a new singlet could be found at δ 0.10 ppm, consistent with the formation of Me3Si–O–SiMe3. Full characterizations clarified the identity of this new compound as nickel cyanate 3 (Scheme 1), exactly identical to that of our previous report from the reaction of nickel carbodiimides and tBuNC.32 While complex 3 might be a result of the formation of 2 as the intermediate followed by subsequent deoxygenation of CO to produce the –CN moiety as reported by Sellmann,37 the reaction of KHMDS with CO to afford KCN and O(SiMe3)2 cannot be ruled out as recently reported by the Stephan group during our study.39
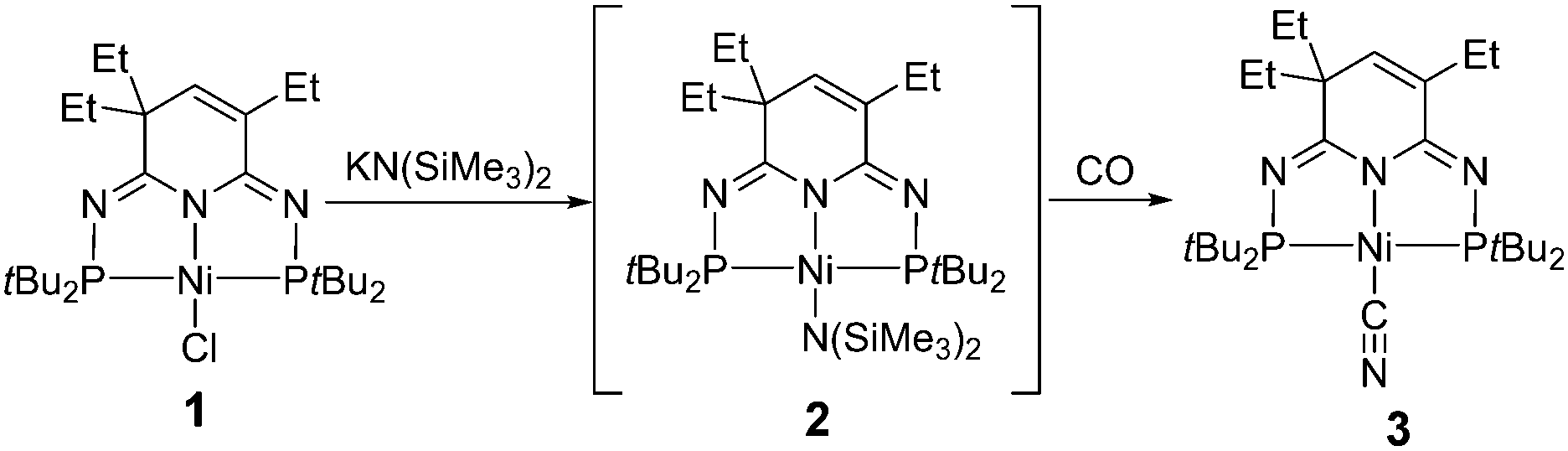 | ||
| Scheme 1 Reaction of 1/KN(SiMe3)2 and CO. | ||
To further probe the reactivity of complex 1/KHMDS, we thus continued to explore more in situ reactions towards different substrates. When the toluene solution of 1/KHMDS was heated to 80 °C, the reaction proceeded smoothly as corroborated by the appearance of a new doublet at δ 100.27 and 99.66 ppm in the 31P NMR spectrum. By removing all the volatiles in vacuo, an apparent triplet at δ 2.57 ppm (J = 8.8 Hz) was observed in 1H NMR, likely attributed to the nickel benzylic protons. In agreement with the proton assignment, the 13C NMR spctrum also displayed a well-resolved upfield triplet at −2.94 ppm with a coupling constant of 19.5 Hz corresponding to the directly coordinated benzylic carbon (Scheme 2). Crystallographic analysis of compound 4 indicates that it consists of the ligated Ni(II) center with a PN3P ligand and a benzylic moiety (Fig. 2). The benzylic C atom is trans to the central N atom of the ring with a N(1)–Ni(1)–C(28) angle of 175.39(8)°. The Ni(1)–C(28) bond lenth of 1.967(2) Å is within the range of Ni–C bonds reported in the literature.30,40
 | ||
| Scheme 2 Reactions of 1/KN(SiMe3)2 with toluene and ethylbenzene. | ||
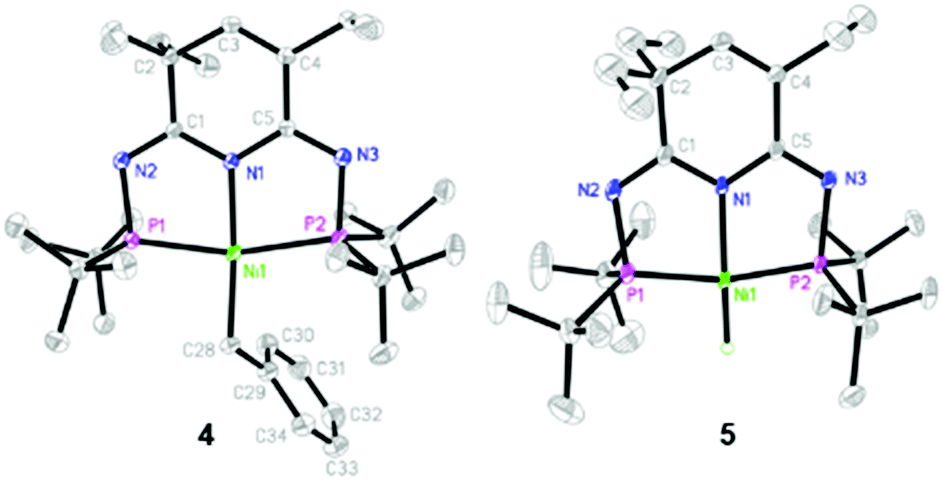 | ||
| Fig. 2 Molecular structures of complexes 4 and 5 with ellipsoids set at 50% probability. Hydrogen atoms except the Ni–H moiety are omitted for clarity. Selected bond lengths [Å] and angles [°]: For 4: Ni(1)–N(1) 1.9383(16), Ni(1)–P(1) 2.1841(5), Ni(1)–P(2) 2.2353(5), Ni(1)–C(28) 1.967(2), C(28)–C(29) 1.511(3); N(1)–Ni(1)–C(28) 175.39(8), Ni(1)–C(28)–C(29) 118.65(16), C(28)–Ni(1)–P(1) 94.40(6), C(28)–Ni(1)–P(2) 101.30(6), P(1)–Ni(1)–P(2) 163.87(2). For 5: Ni(1)–N(1) 1.904(3), Ni(1)–P(1) 1.9256(10), Ni(1)–P(2) 1.9317(10), Ni(1)–H(1) 1.41(6); N(1)–Ni(1)–H(1) 178(2), H(1)–Ni(1)–P(1) 98(2), H(1)–Ni(1)–P(2) 96(2), P(1)–Ni(1)–P(2) 166.47(5). | ||
As the involvement of 2 could not be confirmed experimentally, we have conducted density functional theory calculations to evaluate the process of toluene activation (Scheme 3). The activation energy barrier was found to be unprecedentedly high (ΔG‡ = 66.2 kcal mol−1 from 2 to TS1), suggesting that such a reaction pathway is less plausible under the current reaction conditions (see ESI†). In this regard, Guan and co-workers reported a series of weak base-catalyzed benzylic C–H bond additions of alkylarenes via “kinetic deprotonative functionalization”.15,16,41,42 While no KCH2Ar species were observed by 1H NMR spectrum, the kinetic isotope effect (KIE) suggested that the cleavage of the benzylic C–H bond of alkylarene is the rate-determining step.41 The KCH2Ph intermediate may play a similar role in promoting the production of complex 4 in our reaction.
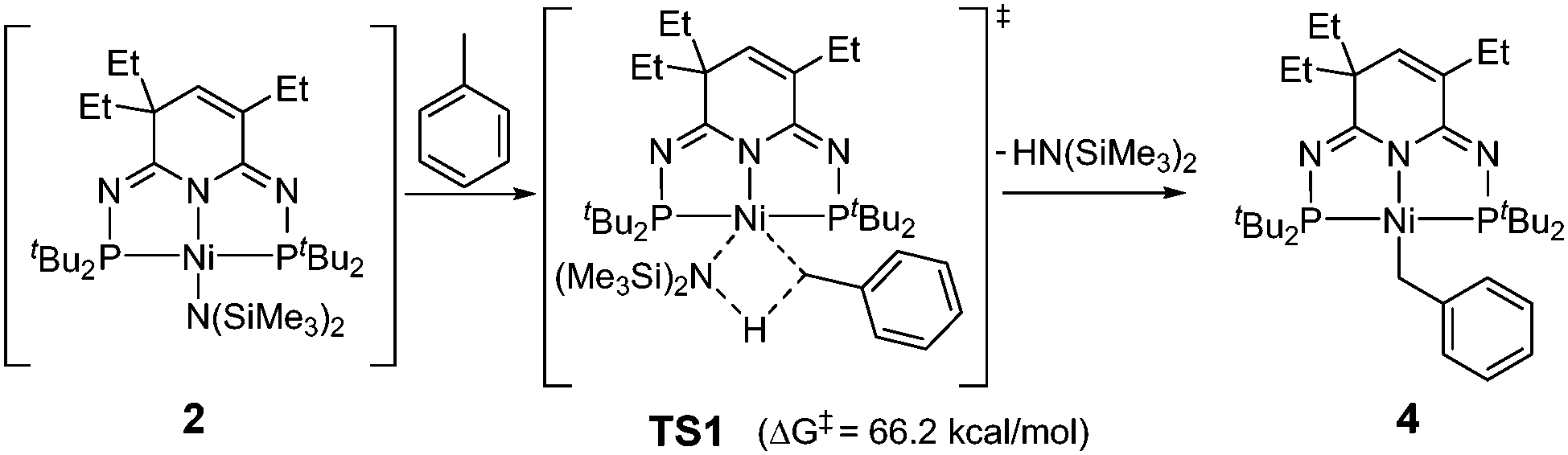 | ||
| Scheme 3 Calculated toluene activation pathway by complex 2. | ||
With the observed overall Csp3–H bond activation, we further examined relative benzylic activation reactions. By switching the substrate to ethylbenzene, nickel hydride 5 was formed concomitant with styrene as proven by the characteristic upfield triplet Ni–H signals at δ −15.88 ppm and three sets of olefinic signals at δ 6.58, 5.60 and 5.07 ppm in the 1H NMR spectrum. The formation of these two compounds involves a benzylic C–H bond activation of ethylbenzene and a subsequent β–H elimination of the resultant (1-phenylethyl)nickel species. To support the role of the (1-phenylethyl)nickel species, ethylbenzene was replaced by tert-butylbenzene and no reaction occurred in the latter case under the same conditions. This result also indicated that the C–H bonds in the methyl group of ethylbenzene could not be activated by 1/KHMDS. The molecular structure of complex 5 was further confirmed by X-ray diffraction analysis (Fig. 2).
We next extended the substrate scope to a functional toluene derivative. Adding 2,6-dimethylphenyl isocyanide to the C6D6 solution of 1/KHMDS and stirring at 80 °C for 48 h led to the formation of a new activation product 7 as supported by the set of peaks at δ 108.09 and 107.58 ppm in the 31P NMR spectrum. Only one of the xylyl methyl groups was found at δ 2.53 ppm accompanied by a broad resonance at δ 7.87 ppm and an olefinic quartet at δ 6.67 ppm in the 1H NMR spectrum. In addition, aromatic hydrogens of the isocyanide ligand appeared as an asymmetric ABC pattern. The NMR data were in absolute agreement with the report by Jones,43 suggesting the formation of a bound 2-substituted 7-methylindole nickel complex 6 (Scheme 4). The molecular structure was unambiguously confirmed by X-ray diffraction analysis (Fig. 3). Although the utilization of o-alkylphenylisocyanides is a well-known pathway to synthesizing indoles via transition metal compounds,44 to the best of our knowledge, complex 6 is the first structurally confirmed example of a metal indole compound and, therefore, provides direct intermediate evidence of indole synthesis from isonitrile derivatives.
 | ||
| Scheme 4 Reaction of 1/KHMDS with 2,6-dimethylphenyl isocyanide. | ||
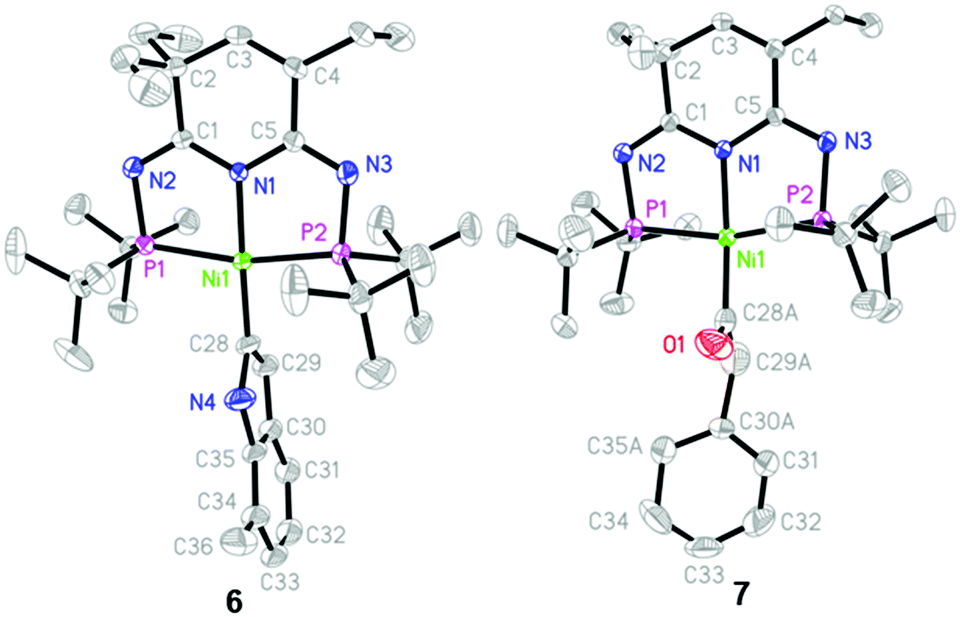 | ||
| Fig. 3 Molecular structures of complexes 6 and 7 with ellipsoids set at 50% probability. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: For 6: Ni(1)–N(1) 1.9152(18), Ni(1)–P(1) 2.1930(7), Ni(1)–P(2) 2.2068(7), Ni(1)–C(28) 1.889(2); N(1)–Ni(1)–C(28) 176.52(12), C(28)–Ni(1)–P(1) 97.65(8), C(28)–Ni(1)–P(2) 96.99(8), P(1)–Ni(1)–P(2) 165.15(3). For 7: Ni(1)–N(1) 1.9425(18), Ni(1)–P(1) 2.2090(7), Ni(1)–P(2) 2.2123(7), Ni(1)–C(28A) 1.877(6); N(1)–Ni(1)–C(28A) 167.1(3), C(28A)–Ni(1)–P(1) 98.71(17), C(28A)–Ni(1)–P(2) 96.21(18), P(1)–Ni(1)–P(2) 164.79(3). | ||
To generate a toluene derivative by directly installing a new C–C bond via benzylic C–H functionalization, we introduced CO into the resultant C6D6 solution of complex 4 and heated the solution to 60 °C for 3 days. The benzyl complex 4 was transformed into phenylacetyl nickel complex 7, which was fully characterized by NMR spectroscopy, elemental analysis, HRMS and X-ray diffraction (Scheme 5). To the best of our knowledge, this is the first example of a synthesis of 2-phenylacetyl compound directly by benzylic C–H functionalization of toluene. In sharp contrast to other acyl metal species, 7 was found to be stable in the presence of water.
 | ||
| Scheme 5 Synthesis of 7 by carbonylation of 4. | ||
In summary, we have demonstrated a benzylic C–H bond activation of toluene via the PN3PNiCl/KHMDS binary system. The activation of ethylbenzene results in the formation of nickel hydride 5 along with styrene likely by β–H elimination of the (1-phenylethyl)nickel species. Furthermore, a rare intermediate 2-coordinated 7-methylindole nickel complex (6) for the transition metal complex-mediated indole synthesis was also obtained by the activation of 2,6-dimethylphenyl isocyanide. Direct carbonylation of complex 4 afforded a phenylacetyl nickel complex (7). Further extension of the scope of application and the mechanistic investigation of nickel-mediated Csp3–H activation are ongoing in our laboratories.
This work was supported by the National Natural Science Foundation of China (22061027), Nanchang University and the King Abdullah University of Science and Technology (KAUST). We thank two anonymous reviewers for providing helpful comments to improve our mechanistic discussion significantly.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- S. Rousseaux, B. Liégault and K. Fagnou, Chapter 1: C–H functionaliszation: A New Strategy for the Synthesis of Biologically Active Natural Products, 2012 Search PubMed.
- J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed.
- O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC.
- T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- K. S. Chan, P. F. Chiu and K. S. Choi, Organometallics, 2007, 26, 1117–1119 CrossRef CAS.
- C. W. Cheung and K. S. Chan, Organometallics, 2008, 27, 3043–3055 CrossRef CAS.
- K. S. Choi, P. F. Chiu, C. S. Chan and K. S. Chan, J. Chin. Chem. Soc., 2013, 60, 779–793 CrossRef CAS.
- W. W. Kaeding, R. O. Lindblom, R. G. Temple and H. I. Mahon, I&EC Process Des. Dev., 1965, 4, 97–101 Search PubMed.
- G. Gunduz and O. Akpolat, Ind. Eng. Chem. Res., 1990, 29, 45–48 CrossRef CAS.
- S. Takemoto, E. Shibata, M. Nakajima, Y. Yumoto, M. Shimamoto and H. Matsuzaka, J. Am. Chem. Soc., 2016, 138, 14836–14839 CrossRef CAS PubMed.
- Y.-F. Liu, D.-D. Zhai, X.-Y. Zhang and B.-T. Guan, Angew. Chem., Int. Ed., 2018, 57, 8245–8249 CrossRef CAS PubMed.
- C.-C. Bao, Y.-L. Luo, H.-Z. Du and B.-T. Guan, Sci. China: Chem., 2021, 64, 1349–1354 CrossRef CAS.
- S.-C. Sha, S. Tcyrulnikov, M. Li, B. Hu, Y. Fu, M. C. Kozlowski and P. J. Walsh, J. Am. Chem. Soc., 2018, 140, 12415–12423 CrossRef CAS PubMed.
- Z. Wang, Z. Zheng, X. Xu, J. Mao and P. J. Walsh, Nat. Commun., 2018, 9, 3365 CrossRef PubMed.
- H. Jiang, S.-C. Sha, S. A. Jeong, B. C. Manor and P. J. Walsh, Org. Lett., 2019, 21, 1735–1739 CrossRef CAS PubMed.
- Y. Yamashita, H. Suzuki, I. Sato, T. Hirata and S. Kobayashi, Angew. Chem., Int. Ed., 2018, 57, 6896–6900 CrossRef CAS PubMed.
- M. Shigeno, K. Nakaji, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2019, 21, 2588–2592 CrossRef CAS PubMed.
- L. Zhu, V. Yempally, D. Isrow, P. J. Pellechia and B. Captain, J. Cluster Sci., 2012, 23, 627–648 CrossRef CAS.
- W. Hou, Y. Yang, Y. Wu, H. Feng, Y. Li and B. Zhou, Chem. Commun., 2016, 52, 9672–9675 RSC.
- A. P. Y. Chan, M. Jakoobi, C. Wang, Y. Tian, N. Halcovitch, R. Boulatov and A. G. Sergeev, Chem. Commun., 2021, 57, 7894–7897 RSC.
- H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS.
- T. P. Gonçalves, I. Dutta and K.-W. Huang, Chem. Commun., 2021, 57, 3070–3082 RSC.
- D. Milstein, Top. Catal., 2010, 53, 915–923 CrossRef CAS.
- C. Gunanathan and D. Milstein, Science, 2013, 341, 249–259 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- X. F. Wang, L. F. Yao, Y. P. Pan and K.-W. Huang, J. Organomet. Chem., 2017, 845, 25–29 CrossRef CAS.
- M.-H. Huang, J. Hu and K.-W. Huang, J. Chin. Chem. Soc., 2018, 65, 60–64 CrossRef CAS.
- C. Yao, X. Wang and K.-W. Huang, Chem. Commun., 2018, 54, 3940–3943 RSC.
- C. Yao, P. Chakraborty, E. Aresu, H. Li, C. Guan, C. Zhou, L.-C. Liang and K.-W. Huang, Dalton Trans., 2018, 47, 15997–16360 RSC.
- C. Yao, T. Zhang, C. Zhou and K.-W. Huang, Dalton Trans., 2019, 48, 12817–12821 RSC.
- V. Soni, S. M. Khake and B. Punji, ACS Catal., 2017, 7, 4202–4208 CrossRef CAS.
- Z.-Y. Xu, Y.-Y. Jiang, H.-Z. Yu and Y. Fu, Chem. – Asian J., 2015, 10, 2479–2483 CrossRef CAS PubMed.
- D. Sellmann, F. Geipel and F. W. Heinemann, Chem. – Eur. J., 2000, 6, 4279–4284 CrossRef CAS.
- M. Faust, A. M. Bryan, A. Mansikkamaki, P. Vasko, M. M. Olmstead, H. M. Tuononen, F. Grandjean, G. J. Long and P. P. Power, Angew. Chem., Int. Ed., 2015, 54, 12914–12917 CrossRef CAS PubMed.
- M. Xu, B. Kooij, T. Wang, J. H. Lin, Z.-W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2021, 60, 16965–16969 CrossRef CAS PubMed.
- Z. L. Lu, S. Abbina, J. R. Sabin, V. N. Nemykin and G. D. Du, Inorg. Chem., 2013, 52, 1454–1465 CrossRef CAS PubMed.
- Y. Yamashita, H. Suzuki, I. Sato, T. Hirata and S. Kobayashi, Angew. Chem., Int. Ed., 2018, 57, 6896–6900 CrossRef CAS PubMed.
- B. Guan and Z. Shi, Sci. Sin.: Chim., 2021, 51, 201–212 Search PubMed.
- W. D. Jones and W. P. Kosar, J. Am. Chem. Soc., 1986, 108, 5640–5641 CrossRef CAS.
- J. Campo, M. Garcia-Valverde, S. Marcaccini, M. J. Rojo and T. Torroba, Org. Biomol. Chem., 2006, 4, 757–765 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the preparation and characterization of compounds 4–7; additional NMR spectra. CCDC 1891459, 1891460, 1891465 and 1891466. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06507f |
| This journal is © The Royal Society of Chemistry 2022 |