
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC-terminal truncation of α-synuclein promotes amyloid fibril amplification at physiological pH†
Ingrid M.
van der Wateren
a,
Tuomas P. J.
Knowles
 ab,
Alexander K.
Buell
*c,
Christopher M.
Dobson
*a and
Céline
Galvagnion‡
*a
ab,
Alexander K.
Buell
*c,
Christopher M.
Dobson
*a and
Céline
Galvagnion‡
*a
aCentre for Misfolding Diseases, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: cmd44@cam.ac.uk; celine.galvagnion@dzne.de
bCavendish Laboratory, Department of Physics, University of Cambridge, J J Thomson Avenue, Cambridge, CB3 1HE, UK
cInstitute of Physical Biology, Heinrich Heine Universität, Universitätsstr. 1, 40225, Düsseldorf, Germany. E-mail: Alexander.Buell@uni-duesseldorf.de
First published on 24th May 2018
Abstract
Parkinson's disease is one of the major neurodegenerative disorders affecting the ageing populations of the modern world. One of the hallmarks of this disease is the deposition of aggregates, mainly of the small pre-synaptic protein α-synuclein (AS), in the brains of patients. Several very significantly modified forms of AS have been found in these deposits including those resulting from truncations of the protein at its C-terminus. Here, we report how two physiologically relevant C-terminal truncations of AS, AS(1-119) and AS(1-103), where either half or virtually all of the C-terminal domain, respectively, has been truncated, affect the mechanism of AS aggregation and the properties of the fibrils formed. In particular, we have found that the deletion of these C-terminal residues induces a shift of the pH region where autocatalytic secondary processes dominate the kinetics of AS aggregation towards higher pH values, from AS wild-type (pH 3.6–5.6) to AS(1-119) (pH 4.2–7.0) and AS(1-103) (pH 5.6–8.0). In addition, we found that both truncated variants formed protofibrils in the presence of lipid vesicles, but only those formed by AS(1-103) had the capacity to convert readily into mature fibrils. These results suggest that electrostatics play an important role in secondary nucleation, a key factor in aggregate proliferation, and in the conversion of AS fibrils from protofibrils to mature fibrils. In particular, our results demonstrate that sequence truncations of AS can shift the pH range where autocatalytic proliferation of fibrils is possible into the neutral, physiological regime, thus providing an explanation of the increased propensity of the C-truncated variants to aggregate in vivo.
1 Introduction
Parkinson's Disease (PD) is a neurodegenerative disorder characterised by the presence of intracellular inclusions, called Lewy Bodies (LBs) and Lewy neurites, mainly composed of the protein α-synuclein (AS).1–3 Although the physiological function of AS is not fully understood, the protein is primarily expressed at synaptic terminals in the central nervous system,4 and is proposed to play a role in synaptic function and plasticity.5–7 AS is estimated to represent ∼1% of the total protein content in soluble cytosolic brain fractions4 and has also been found in mitochondria and at mitochondria-associated ER membranes.8,9AS is largely disordered in solution but can adopt an α-helical conformation when bound to biological and synthetic membranes.10–13 The N-terminal region of AS consists of residues 1–60 and, together with the central segment (residues 61–95), contains seven imperfect repeats of 11 residues, which are characteristic of amphipathic helices.10,14 Residues 61–95 form the NAC (non-amyloid β component) region which is characterised by the presence of hydrophobic residues4 and is thought to form the core of AS amyloid fibrils.15,16 The C-terminal region (residues 96–140) contains 14 acidic residues, giving it an overall negative charge, and remains flexible and disordered when AS is bound to model membranes10–13,17–19 (Fig. 1).
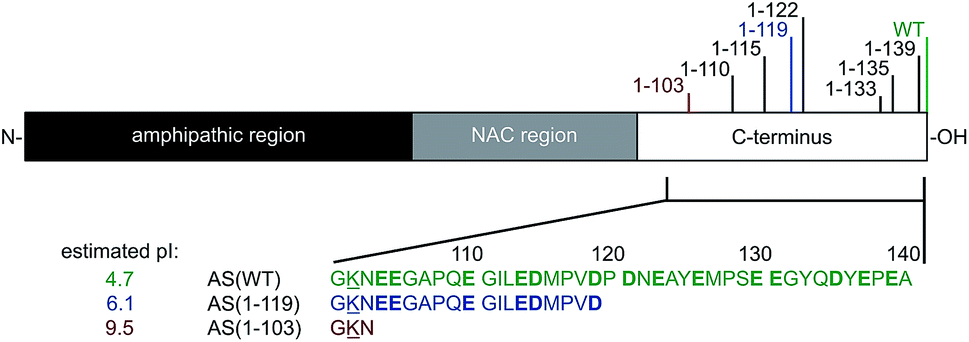 | ||
| Fig. 1 Schematic representation of AS showing the three main regions of the protein and the positions of the major C-terminal truncations found in vivo. The different variants of AS examined in this study, i.e. AS(WT), AS(1-119) and AS(1-103) are indicated in green, blue and red, respectively. Charged residues are underlined (positive) or shown in bold (negative) and the estimated pI values are indicated for each variant (http://www.protcalc.sourceforge.net). | ||
Monomeric AS is remarkably stable in solution at physiological concentration (50 μM)20 under quiescent conditions at neutral pH, and does not detectably convert into amyloid fibrils unless mechanical agitation and/or specific interfaces, e.g. air/water,21 detergent/water,22 lipid/water23–25 or polystyrene/water,26–28 are introduced. It is now well established that the first step in AS aggregation is the surface-induced nucleation at such interfaces, followed by the growth of the resultant species via monomer addition.24,25,28,29 Secondary processes can be observed if mechanical agitation is used24 or if the pH of the solution is decreased to 5.5 or below.28,29
C-terminal truncation of AS is one of the predominant modifications of AS associated with PD.30 C-terminal truncated (Ctt) variants represent ∼15% of all AS molecules found in LBs30 and are characterised by cleavage at positions in the vicinity of residue 12030–36 (see Fig. 1 and ESI, Table S1†). Moreover, N- and C-terminally truncated variants have also been found in the brains of healthy controls.37 Ctt variants of AS are thought to be generated from AS wild-type (WT) by processes that are independent of its aggregation both in brain tissues and in cultured cells31 as a result of incomplete degradation by the 20S proteasome,33,38 or by cleavage by calpain I,39 prolyl oligopeptidase40 and/or cathepsin D.41
The different forms of Ctt AS have been found to be more prone to aggregation in vitro33,42–45 and in vivo46 than AS(WT), showing a shorter lag phase, a higher level of fibril formation and different aggregate morphology; the length of the lag phase in kinetic experiments generally decreases as more of the C-terminal region is truncated (see Table S1†).
The disordered state of AS has been characterised as consisting of a broad distribution of conformers with a number of long-range interactions between the negatively charged C-terminal domain and the hydrophobic central region of the protein,47 and between the N- and C-terminal domains.48,49 It has been suggested that screening the negative charges at the C-terminal region of AS, by using small molecules,43 by lowering the pH,43 or by truncating parts of the C-terminus,42,44 can lead to an increased aggregation propensity of the protein. These findings suggest a protective role of the charges at the C-terminus against AS aggregation and that disruption of the long-range interactions via C-terminal truncations can facilitate aggregation.50
In this study, we have investigated the influence of C-terminal truncations on the different events involved in the process of AS aggregation.24,29 We have found that deletion of C-terminal residues significantly affects the properties of AS fibrils, including their ability to contribute to secondary processes. In particular, we have observed that truncation of segments of the C-terminus can induce a shift of the pH range at which secondary processes dominate the kinetics of AS aggregation, from acidic values for AS(WT) (3.6–5.6) to mildly acidic values for AS(1-119) (4.2–7.0) and to neutral values for AS(1-103) (5.6–8.0). Finally, we have found that C-terminal truncation of AS results in a change in the mechanism of lipid-induced aggregation as a result of the altered contributions of secondary processes to the overall mechanism of amyloid formation. The results suggest a critical role of the C-terminal acidic residues of AS both in secondary processes and in the conversion of AS protofibrils into mature fibrils. Moreover, the dramatic shift of the pH region where secondary processes contribute to the overall kinetics of amyloid formation caused by C-terminal truncations suggests an explanation for the increased propensity of the C-truncated variants to aggregate in vivo.
2 Experimental
Materials
1,2-Dimyristoyl-sn-glycero-3-phospho-L-serine (sodium salt; DMPS) was purchased from Avanti Polar Lipids (Alabama, USA). Sodium phosphate monobasic (NaH2PO4, BioPerformance Certified, >99.0%), sodium phosphate dibasic (Na2HPO4, ReagentPlus, >99.0%) and sodium azide (NaN3, ReagentPlus, >99.5%) were purchased from Sigma Aldrich (Poole, UK). Thioflavin T UltraPure Grade (ThT, >95%) was purchased from Eurogentec (Southampton, UK).Protein preparation
AS(WT), AS(1-119) and AS(1-103) were expressed and purified using essentially the same protocol as that reported previously,24,29 with very minor alterations (see ESI for more details†).Vesicle preparation
Small unilamellar vesicles (SUV) were prepared from DMPS using sonication as described previously.24Fibril formation at pH 3–9
Solutions containing ca. 260 μM of protein were incubated in phosphate buffer (20 mM H3PO4, 0.01% NaN3) at pH values ranging from 3 to 9, adjusted using NaOH (Breckland Scientific, Stafford, UK). The solutions were stirred at 45 °C for 48 h, then sonicated for 1 min (at 30% “on time”, 10% power) using a Bandelin Sonopuls probe sonicator (HD2070) (Berlin, Germany) to homogenise the solution. At this stage, aliquots were taken for the preparation of samples for AFM and TEM (see details below). The rest of the solution was ultracentrifuged for 1 h at 90 krpm and 4 °C (Beckman Coulter Optima™ TLX, High Wycombe, UK) and the supernatant was used to determine the concentration of soluble protein (using an extinction coefficient of 5600 M−1 cm−1 for AS(WT) and 1400 M−1 cm−1 for AS(1-119) and AS(1-103) at UV 278 nm (determined using the Scripps protein calculator at http://www.protcalc.sourceforge.net)).Preparation of fibrillar seeds at pH 6.5
Solutions of 300 μM monomeric protein (AS(WT), AS(1-119) and AS(1-103)) were incubated in phosphate buffer (20 mM NaH2PO4/Na2HPO4, 0.01% NaN3, pH 6.5) (unless stated otherwise) at 45 °C with maximum stirring for 50 h with sonication at 24, 48 and 50 h for 10 s (at 30% “on time”, 10% power) (Bandelin Sonopuls HD2070). We also explored different sonication times and quantified the concentrations of soluble protein at the end of the 50 h incubation period (see ESI for more details†).The resulting fibrils were used in experiments to measure elongation rates and these aliquots were diluted in phosphate buffer at pH 6.5, sonicated for 1 min (at 30% “on time”, 10% power) and added immediately to solutions of monomeric protein.
Atomic force microscopy (AFM) measurements
Samples containing fibrils were diluted to give a fibril concentration of 2 μM (monomer equivalent) in 20 mM phosphate buffer at the same pH as that of the sample. Diluted samples (30–50 μl) were deposited on cleaved mica (G 250-1, Agar Scientific, Stansted, UK), which was fixed onto glass microscope slides (Thermo Scientific, Paisley, UK) and left to dry. Once dry, the slides were washed twice using 100–200 μl of double-distilled water (Elga, High Wycombe, UK) and allowed to dry before storage at room temperature (RT).AFM images were acquired using a NanoWizard II AFM (JPK Instruments, Berlin, Germany) equipped with HQ:NSC36/No Al 65–130 kHz 0.6–2 N m−1 chips from μmasch (Innovative Solutions Bulgaria Ltd., Sofia, Bulgaria) in intermittent-contact mode in air and processed using Gwyddion (https://www.gwyddion.net)51 and ImageJ.52
Transmission electron microscopy (TEM) measurements
Samples containing fibrils were diluted to give a fibril concentration of 5–10 μM (monomer equivalents) in 20 mM phosphate buffer at the same pH as that of the sample. Samples (5 μl) were deposited on grids and left for 1 min before being blotted using filter paper. The grids were washed once with double-distilled water, then incubated with 2% w/v uranyl acetate (Agar Scientific, Stansted, UK) for 45 s and finally washed again, twice, with double-distilled water. The grids were allowed to dry at RT, and imaged at the Cambridge Advanced Imaging Centre using a Tecnai G2 80–200 kV. Images were saved as 8-bit TIF files and processed using ImageJ.52The details of the grids used are as follows: C169/050 carbon support film; thin clear films of carbon 400 mesh 3 mm copper grid (TAAB Laboratories Equipment Ltd, Aldermaston, UK); C400Cu carbon film, 400 mesh copper (EM Resolutions Ltd, Sheffield, UK), C169/N050 carbon support film; thin clean films of carbon, 400 mesh, 3 mm nickel grid (TAAB Laboratories Equipment Ltd).
Time-resolved aggregation kinetics
The changes in the ThT fluorescence intensity during the different aggregation experiments described in this study were measured using multi-well plate-readers (POLARstar Omega, FLUOstar Omega, FLUOstar OPTIMA, CLARIOstar) from BMG Labtech (Ortenberg, Germany) using the bottom reading mode. Two types of plates were used in this work:(i) Corning® 96-well half area black with clear flat bottom made of polystyrene treated with NBS™ (#3881, Corning Ltd, Corning, USA), referred to as “PEG-ylated plate” in the text, for the elongation and lipid-induced aggregation experiments (Fig. 2, 6, S12 and S13†), and
(ii) Corning® 96-well black with clear flat bottom made of polystyrene with high-binding surface (Corning #3601), referred to as “polystyrene plate” in the text, for the non-seeded aggregation experiments performed at different pH values (Fig. 5, S8, S9 and S10†).
Samples were incubated in the presence of 50 μM ThT (#88306, Anaspec Inc., Fremont, USA), unless stated otherwise, as triplicates or duplicates in the desired buffer under quiescent conditions. The wavelengths used were 440 nm (excitation)/480 nm (emission). Note: the stock solutions of ThT were prepared by dissolving ThT powder in phosphate buffer at the desired pH at about 1 mM final concentration (w/v) and passed through a 0.22 μm syringe filter (SLGP033RS, Millipore, Darmstadt, Germany). The concentrations of samples generated by dilution in buffer were determined by absorbance spectroscopy using extinction coefficient of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for absorbance at 412 nm (Abs412,ThT)53 correcting for buffer background, using a CLARIOstar multi-well plate-reader.
000 M−1 cm−1 for absorbance at 412 nm (Abs412,ThT)53 correcting for buffer background, using a CLARIOstar multi-well plate-reader.
Determination of insoluble protein concentration at the end of kinetic experiments
The concentrations of insoluble protein at the end of the aggregation reactions were determined using absorbance measurements such that [insoluble protein] = [total protein] − [soluble protein], where [total protein] is the concentration of AS used at the beginning of the reaction and [soluble protein] is the concentration of protein soluble in the reaction mixture at the plateau phase. The reaction mixtures were collected and ultracentrifuged (Beckman Coulter Optima® TLX) for 30 min to 1 h at 90 krpm at RT. The supernatant was either stored at −20 °C or directly transferred to a UV-transparent plate (Corning #3679) for absorbance measurements (CLARIOstar). The concentrations of soluble protein were calculated from the absorbance at 278 nm using the extinction coefficients given above, after correcting for the buffer background signal. In the case of samples containing ThT, the absorbance of the supernatant measured at 278 nm (Abs278,sup) is the sum of the absorbance from ThT (Abs278,ThT) and that from the protein (Abs278,protein): Abs278,sup = Abs278,ThT + Abs278,protein. Abs278,ThT was determined using the Abs412,ThT and the following equation: Abs278,ThT = 0.147 × Abs412,ThT (see standard curve in Fig. S1†).Determination of ThT quantum yield
The ThT quantum yield was determined from the ratio of the ThT fluorescence intensity to the concentration of insoluble protein at the plateau phase.Circular dichroism (CD) experiments
Solutions of 20 μM protein were incubated in the presence of DMPS vesicles in phosphate buffer at pH 6.5 and 30 °C. CD spectra were measured using a ChiraScan (Applied Photophysics, Leatherhead, UK) collecting five repeats using a step size of 0.2 nm and a bandwidth of 0.5 nm. The CD spectra acquired for solutions containing 20 μM protein + 8 mM DMPS were analyzed using DichroWeb/CD Pro to extract the content of α-helix, β-sheet and random coil structure (input units: millidegrees/theta (machine units), K2D analysis program without a reference set).54–56 The signals measured at 222 nm were acquired using the same equipment, collecting 50 repeats and bandwidth 0.5 nm. The binding curves were fitted to a Langmuir isotherm as described previously.24Differential scanning calorimetry (DSC) experiments
SUV made from DMPS at a concentration of 1 mM were incubated in the absence or presence of 100 μM protein in Eppendorf tubes and degassed for several minutes at RT before use in a MicroCal VP-DSC (Malvern, UK). DSC thermograms were acquired from 10–60 °C at a scan rate of 90 °C h−1 for buffer and 45 °C h−1 for the samples under investigation.Microscopy experiments in glass microcapillaries
Fibrils (100 μM, monomer equivalents) were first sonicated for 1 min at 30% “on time” and 10% power and then diluted to a final concentration of 5 μM in 20 mM phosphate buffer, pH 6.5, 50 μM ThT, in the absence or presence of 50 μM monomeric protein. The reaction mixtures were transferred through capillary action into capillaries (square boro tubing # 8250-050 with 0.50 mm internal diameter and 0.10 mm wall (CM Scientific, Silsden, UK)). Wax (Hawksley, Lancing, UK) was used to seal off the capillary ends and the capillaries were fixed to standard glass microscopy slides (Thermo Scientific) using two-component epoxy resin (Araldite, Basel, Switzerland). The slides were fixed to a heating plate set to 37.5 °C in order to perform the experiments at or near the desired temperature of 37 °C. A Zeiss inverted microscope (Axio Observer A1, Zeiss, Cambridge, UK) using multidimensional acquisition was used to observe fluorescence intensity over time using a filter cube 49001 ET-CFP (excitation 426–446 nm, dichroic 455 nm, emission 460–500 nm). Images were taken every 20 s.3 Results and discussion
C-terminal truncation of AS affects the properties of amyloid fibrils and decreases their seeding efficiency
Amyloid fibril formation occurs through a nucleated polymerisation mechanism and consists of distinct individual microscopic steps.57,58 The elongation of fibrils is by far the fastest process in most cases and therefore it is responsible for the bulk of the formation of fibril mass.58 It is also a straightforward process to study experimentally in isolation, as pre-formed fibrils can be added to soluble protein in a well-defined manner (“seeding”) and the resulting fibril elongation can be measured with a variety of experimental techniques, such as ThT fluorescence or biosensing techniques.59,60 When the concentration of added seeds is high enough, all microscopic processes other than fibril elongation can be neglected. We have previously performed such strongly seeded experiments for AS(WT), which has allowed us to determine the average molecular rate constant of fibril elongation,29 as well as its degree of heterogeneity.61In order to compare fibril elongation rates of Ctt AS species to that of AS(WT), we performed seeded experiments using AS(WT), AS(1-119) and AS(1-103) fibrils and monomers. Pre-formed amyloid fibrils were incubated in solutions of monomeric protein molecules of the same variant (seeding) or one of the other variants (cross-seeding) (see Experimental section for details), and changes in ThT fluorescence were used to monitor fibril formation over time (Fig. 2). We found, as previously observed,29 that when AS(WT) monomers were incubated in the presence of AS(WT) seeds, the ThT fluorescence intensity increased rapidly, with a monotonically decreasing rate due to the depletion of the soluble protein as the aggregation progresses (Fig. 2a). The concentration of insoluble protein present at the plateau phase of the reaction was found to correspond directly to the initial concentration of monomeric protein (Fig. 2b). This observation confirmed the fact that the majority of the monomeric protein molecules had been incorporated into fibrils29 and provided a reference for comparison of equivalent experiments with the truncated variants.
 | ||
Fig. 2 Seeded aggregation of AS(WT), AS(1-119) and AS(1-103). (a) Change in the ThT fluorescence when seeds of AS(WT) (left), AS(1-119) (middle) or AS(1-103) (right) at a concentration of 5 μM (monomer equivalent) were incubated in the presence of increasing concentrations of monomeric AS(WT) (top), AS(1-119) (middle), AS(1-103) (bottom) at pH 6.5, 37 °C and under quiescent conditions. (b) Change in the concentration of insoluble proteins measured at the plateau phase of the aggregation reaction for different initial concentrations of monomers. (c) Change in the elongation rate when seeds of AS(WT) (5 μM in monomer equivalent) were incubated with increasing concentrations of AS(WT) (green), AS(1-119) (blue), AS(1-103) (red) monomers. The dotted lines show the fits of the data using the following equation, which describes the saturation behaviour of the elongation rate:62 . The linear fits to the low concentration data (solid lines) were used to obtain a robust measure of the elongation rate constant (see ESI for more details†). . The linear fits to the low concentration data (solid lines) were used to obtain a robust measure of the elongation rate constant (see ESI for more details†). | ||
In experiments in which the Ctt AS monomers were added to AS(WT) seeds (Fig. 2a, left column), the concentration of insoluble protein at the end of the reaction was found to correspond to the initial concentration of monomeric protein only for [Ctt AS] ≤ 50 μM (Fig. 2b). This result suggests that Ctt variants can elongate AS(WT) seeds but that the elongation reaction only reaches completion for lower concentrations of Ctt AS under these conditions. We evaluated the rate by which the three AS variants elongate AS(WT) seeds at the different initial monomer concentrations (Fig. 2c) by analysing the slopes of the aggregation curves at early time points. In this way, we obtained the product, k+P0, of the elongation rate constant, k+, and the number concentration of seeds, P0, for each variant. By estimating P0 from an analysis of the length distribution of the seeds at t0 (ref. 29) (Fig. S2 and S3, see ESI for more details†), we found that the elongation rate constant decreases as the degree of C-terminal truncation increases, from 3616 M−1 s−1 (AS(WT)) to 2779 M−1 s−1 (AS(1-119)) and 1963 M−1 s−1 (AS(1-103)). This finding suggests that the WT seeds do not provide as good a template for the truncated sequences as for the WT protein. Alternatively, C-terminal truncation of the monomers could perturb the interaction of monomers with fibril ends, as the successful attachment trajectory of a monomer to a growing fibril is likely to involve contacts outside the regions that form the β-sheet core. Thus, although the C-terminal region is not being incorporated into the β-sheet core of the fibrils, it plays a role in defining absolute fibril elongation rates.
We observed different behaviour when Ctt AS fibrils were used to seed the aggregation reaction (Fig. 2a middle and right columns). When Ctt AS seeds were incubated in solution in the absence of monomer, we observed an increase of up to a factor of two in the ThT-fluorescence intensity, suggesting either that the fibril samples initially contained unreacted or dissociated monomeric protein that was able to add onto the fibril ends more efficiently after the sonication step immediately prior to the experiment (see Experimental section for details), or that the binding of ThT to the fibrils made from the Ctt variant is slow compared to that to the WT fibrils. Additionally, when monomeric protein was added to solutions containing the Ctt AS seeds, we found that the kinetics of fibril elongation were slower for fibrils containing the truncated variants. This finding was confirmed by the observation that the concentration of insoluble species formed when samples of the monomeric proteins (AS(WT), AS(1-119) and AS(1-103)) were incubated in the presence of Ctt AS seeds was much smaller than that formed under the same conditions using AS(WT) seeds (Fig. 2b).
Images of the seed fibrils prepared from the different AS variants, obtained using TEM and AFM, showed the presence of higher order assemblies for the Ctt AS (Fig. S2†), suggesting that the accessibility of monomers to the ends of these seeds might be reduced by fibril association. Such phenomena prevent an accurate estimation of the elongation rate constant, as the number of accessible growing fibril ends is not likely to remain constant during the initial parts of the seeded aggregation curves. Due to this phenomenon, we only analysed quantitatively the data obtained by using AS(WT) seeds. Taken together, these results suggest that Ctt AS monomers elongate AS(WT) seeds at similar rates to those of AS(WT) monomers, but that Ctt AS fibrils are less able than AS(WT) fibrils to seed the aggregation of AS because of their higher propensity to cluster together.
Finally, we observed that the ThT fluorescence yield, defined as the ThT fluorescence intensity at the plateau phase per mole of insoluble protein, decreased with increasing values of concentrations of insoluble protein when AS(WT) fibrils were used to seed the aggregation reaction (Fig. S4a†). Furthermore, the ThT fluorescence yield of fibrils formed from elongation of AS(1-103) seeds was found to be smaller for all concentrations of insoluble protein than that of those formed from elongation of AS(1-119) seeds (Fig. S4b†). The fact that we observed different ThT fluorescence yields for the same concentrations of insoluble protein indicates that the binding of ThT to the fibrils is altered by the truncation.
C-terminal truncation of AS affects the pH-dependence of higher order assembly of amyloid fibrils
In order to investigate if the observed change in seeding efficiency and ThT fluorescence yield associated with C-terminal truncations could be attributable to an alteration in the overall morphology of the fibrils formed, we monitored the seeding of AS aggregation in glass micro-capillaries under quiescent conditions at 37 °C. This experimental procedure allows the observation of the higher order assembly behaviour of the aggregate species formed during the reaction29 (Fig. 3 and S5†). When AS(WT) seeds were incubated in the absence of monomeric protein, the ThT fluorescence was found to be essentially uniform throughout the capillary (relative standard deviation of ∼10%) and to remain approximately constant over time (Fig. 3). When AS(WT) seeds were incubated in the presence of AS(WT) monomers, we observed an increase of a factor of 1.4 in the mean ThT fluorescence within the first 4 h (Fig. 3); the relatively modest increase in ThT fluorescence in this experiment can be explained by the time delay between the addition of the seed fibrils to the monomeric protein and the start of the measurements. If the ThT fluorescence is compared with that of the sample containing only seeds, the relative increase in intensity as a result of the addition of monomer is 2.6 (Fig. 3).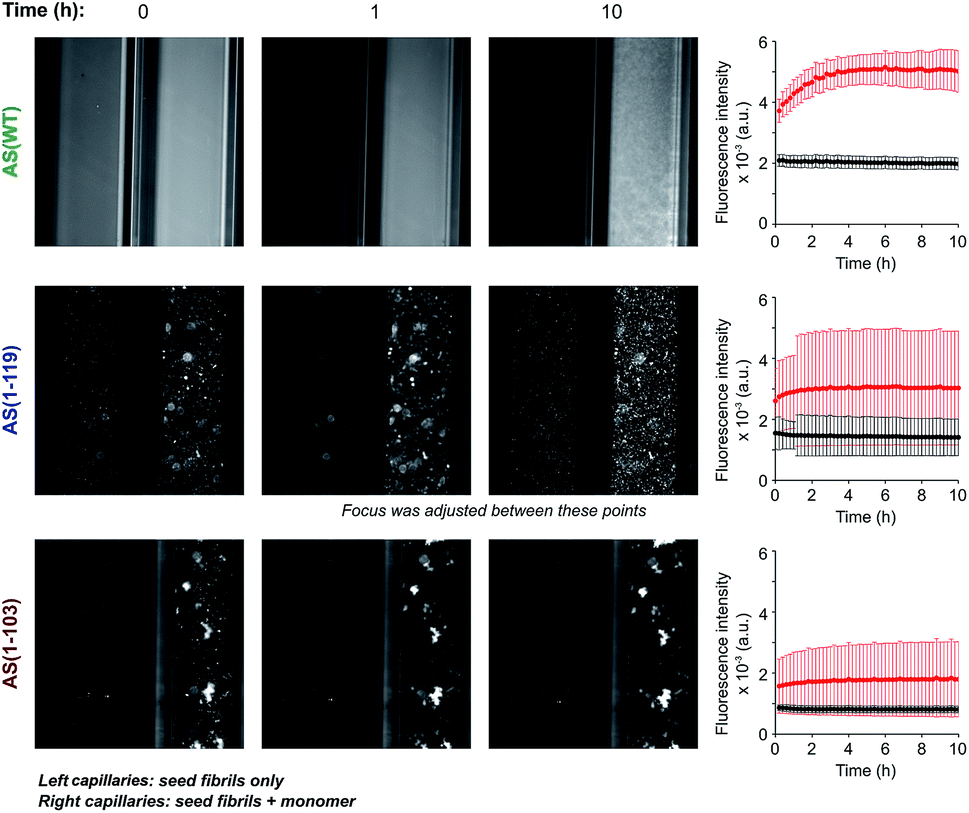 | ||
| Fig. 3 Aggregation of AS(WT), AS(1-119) and AS(1-103) in micro-capillaries. Fluorescence images of capillaries containing seed fibrils (5 μM in monomer equivalent) of AS(WT) (top), AS(1-119) (middle) or AS(1-103) (bottom) in the absence (left capillaries) or presence (right capillaries) of 50 μM monomeric protein. The solution conditions used were 20 mM sodium phosphate pH 6.5, 50 μM ThT, 37 °C. For each AS variant, the change in the average fluorescence intensity with time in the left (black) and right (red) capillaries are shown on the right side of the images. Scale: the internal diameter of the capillaries was 0.5 mm. | ||
The solution containing only Ctt AS seeds displayed a slightly (AS(1-103), relative standard deviation 14%) or much greater (AS(1-119), relative standard deviation 45%) degree of heterogeneity in the distribution of fluorescence intensity along the capillaries (Fig. 3 and S5†) relative to that observed for AS(WT). Addition of monomeric protein resulted in a similar average increase in fluorescence intensity over time (2.6 fold for AS(1-119) and 2 fold for AS(1-103)) (see Fig. S5† for images with increased brightness/contrast) but led to a more heterogeneous mixture (relative standard deviation of 68% for AS(1-103) and 61% for AS(1-119)) (Fig. 3 and S5†). The capillary experiments therefore suggest that the fibrils formed from the Ctt variants of AS have a stronger tendency than those of AS(WT) to form higher order assemblies.
In order to examine this conclusion further, the overall morphology and distribution of the fibrils formed by AS(WT), AS(1-119) and AS(1-103) was studied at higher resolution using TEM and AFM techniques over a range of pH values (Fig. 4 and S6†). Overall, we observed the maximal tendency of fibril clustering to shift towards higher pH values from AS(WT) to AS(1-119) and AS(1-103). This trend can be rationalised in terms of the increase in the value of the isoelectric point (4.7 for AS(WT), 6.1 for AS(1-119) and 9.5 for AS(1-103) monomeric proteins) with increasing truncation of the largely acidic C-terminal domain. The isoelectric point of the fibrillar state can be expected to be similar, albeit not identical, to that of the monomeric state of AS; differences in the isoelectric point can be expected as a result of the more compact structure of the monomer inside the fibril compared to the disordered soluble state. Bringing the fibrils closer to their isoelectric point decreases their net charge and destabilises the colloidal suspension of individually dispersed fibrils, leading to more pronounced higher order assembly process.29
 | ||
| Fig. 4 Transmission electron microscopy images of the insoluble species formed by AS(WT), AS(1-119) and AS(1-103) at pH values ranging from 3 to 9. Scale bar: 1 μm. | ||
It is, however, likely that electrostatic effects are not the only determinants of the higher order assembly of AS fibrils. The presence of a disordered C-terminal region could also contribute to repulsion between fibrils as a result of entropic forces, such as those observed in colloidal suspensions stabilised through grafted polymers;63 indeed, it has been pointed out previously that the disordered regions of proteins in amyloid fibrils can behave like polymer brushes.64,65 Overall, therefore, these observations confirm that C-terminal truncation of AS enhances the formation of higher order assemblies of AS fibrils at pH 6.5. This process is likely to reduce the accessibility of the fibril ends for interaction with monomeric protein, and of the fibril surfaces for binding to ThT molecules, thus providing a rationale for (i) the inability of Ctt AS seeds to initiate efficient aggregation (Fig. 2a) and (ii) the decrease in the ThT fluorescence yield in the presence of Ctt AS fibrils (Fig. S4†). Furthermore, the smaller degree of conversion of soluble protein molecules into amyloid fibrils during the time scale of our experiments can be explained through a significant reduction in the kinetics of fibril elongation due to the higher order assembly of the fibrils. This effect has the potential to slow down the aggregation process to such a large degree that it can appear already to have reached equilibrium during the timescale of the experimental measurements.
Solution pH differentially affects the ability of Ctt AS fibrils to proliferate through autocatalytic secondary nucleation
We next investigated the effect of C-terminal truncations on the ability of AS fibrils to promote autocatalytic secondary nucleation. We have described in previous studies that the formation of new aggregates of AS(WT) by secondary nucleation at the surface of existing fibrils is very strongly dependent on the pH value of the solution.28,29 The secondary nucleation rate of AS(WT) changes by several orders of magnitude between pH 6.0 and pH 5.5, an effect attributed to a change in the protonation state of the acidic residues in the C-terminal region of AS.29 In the light of this conclusion, C-terminal truncations of AS could very significantly influence the rate of autocatalytic secondary nucleation. We therefore studied the aggregation behaviour of AS(WT), AS(1-119) and AS(1-103) at pH values ranging from 2 to 8 (Fig. 5 and S8†).In our previous studies of AS(WT), we probed secondary nucleation by performing aggregation experiments in the presence of very low concentrations of seed fibrils.29,66,67 Such experiments with the Ctt variants of AS are much more difficult to perform in a quantitative manner as a result of the tendency for higher order assembly of the Ctt AS fibrils described above, and so we used a different strategy. We incubated monomeric protein in uncoated polystyrene plates (also called “binding plates”, see Experimental section for details), which were found to be able to initiate the aggregation of AS(WT).26,28 Under these conditions, amyloid fibril formation by AS is nucleated at the polymer surface. However, under quiescent conditions, where fibril fragmentation as a pathway of autocatalytic fibril amplification57 is not significant,29 aggregation can only be detected under conditions where secondary nucleation is able to amplify the small number of nuclei that form through heterogeneous primary nucleation. This concept is illustrated by the data for AS(WT) (Fig. 5a, S8a and b†), where we observed a sigmoidal increase in ThT fluorescence at pH values ranging from 3.6 to 5.6. The upper bound of this range coincides with that of the conditions under which secondary nucleation is detectable in weakly seeded reactions.29 We therefore used the observation of aggregation in polystyrene plates under quiescent conditions as indicative of a significant contribution of secondary nucleation to the overall self-assembly process.
 | ||
| Fig. 5 Aggregation of AS(WT), AS(1-119), AS(1-103) in polystyrene plates at different pH values. (a–c) Change in the ThT fluorescence when 10 μM monomeric AS(WT) (a), AS(1-119) (b) or AS(1-103) (c) were incubated in polystyrene plates (using 20 μM ThT) at pH values ranging from 2 to 8 and 37 °C. (d) Variation in thalf of the reaction of amyloid formation as a function of pH for AS(WT) (green), AS(1-119) (blue) and AS(1-103) (red). (e) pH range where secondary nucleation was found to contribute to the aggregation process. The estimated net charges of the different peptides at the boundaries of the pH range are indicated. | ||
On this assumption, we studied the pH dependence of the aggregation of the Ctt AS species under these conditions and we found detectable aggregation between pH 4.2 and 7.0 for AS(1-119) and between pH 5.6 to 8.0 for AS(1-103) (Fig. 5b, c and S8c, d†). Outside these pH values, the formation of ThT-active species was not measurable (Fig. 5b, c and S8c, d†). Within the pH range where aggregation was observed, we found that the half time of the reaction, i.e. the time required for half of the soluble protein to be converted into aggregates, decreased significantly as the solution pH approached the pI values (Fig. 5d). Moreover, the lag time of the reaction, i.e. the time at which a threshold fraction (we here used 0.5%) of the ThT fluorescence at the plateau phase has been reached, was found to decrease with increasing concentrations of monomeric protein (Fig. S9†).
These results suggest that the pH range where secondary nucleation contributes to amyloid formation by AS shifts to higher values as the number of residues truncated from the C-terminal region of the protein is increased (Fig. 5e). The finding that C-terminal truncation has such significant effects on the pH dependence of the secondary nucleation process of AS provides strong support for the hypothesis that this effect is principally determined by changes in ionisation of the C-terminal region.29 Interestingly, C-terminal truncation does not abolish the strong pH-dependence of secondary nucleation, but rather significantly shifts the pH window within which it can be observed.
In order to confirm that the efficient proliferation of aggregates within the distinct pH-regions observed here is indeed due to the almost complete lack of electrostatic repulsion, we performed aggregation experiments in the same setup at varying salt concentrations, for each AS variant at the pH where aggregation was found to be most efficient (pH 5 for AS(WT), pH 6 for AS(1-119), pH 7 for AS(1-103), see Fig. S10†). We found a very weak dependence of the lag times of the aggregation reaction on the ionic strength of the solution, therefore confirming that the elimination of the majority of the electrostatic repulsion is a necessary requirement for proliferation through secondary nucleation.
C-terminal truncation affects the mechanism of lipid-induced aggregation of AS
In order to probe directly whether or not the de novo formation of amyloid fibrils is also affected by C-terminal truncations of AS, we performed aggregation experiments in the presence of SUV made from DMPS at pH 6.5 under quiescent conditions in PEG-ylated plates (also called “non-binding plates”), according to previously established protocols.24 We have shown that for AS(WT) under these conditions, no secondary processes occur at appreciable rates, while at the same time the rate of primary nucleation is increased by several orders of magnitude compared to that in the absence of lipids.24 Therefore, this system is well-suited to study the primary nucleation reaction associated with amyloid formation by AS.We first characterised the binding of AS(1-119) and AS(1-103) to the vesicles. Since AS adopts an α-helical structure upon membrane binding, we used CD to study the interaction between the Ctt variants and DMPS. The CD spectra of AS(1-119) and AS(1-103) in the presence of an excess of lipid vesicles are characterised by two minima, at 208 and 222 nm, as previously reported for AS(WT),24 and are nearly superimposable on the spectrum of AS(WT) (Fig. S11a†). This observation suggests that the number of residues involved in the formation of the amphipathic α-helix is similar68 for the three AS variants, a conclusion supported by an estimation using CDPro of the percentage of residues in α-helical structure: 80, 97 and 100% for AS(WT), AS(1-119), AS(1-103), respectively.
The binding curves of the AS variants to DMPS vesicles are all well described by a single step binding model24 (Fig. S11b†); we found that, as with AS(WT), both AS(1-119) and AS(1-103) bind to DMPS vesicles with sub-micromolar affinity and that the stoichiometry (the number of lipid molecules per bound protein molecule) was 23 compared to 28 for AS(WT) (Fig. S11b†). This observation can be rationalised by the fact that the degree of steric and electrostatic repulsion among monomers on the surface of vesicles is likely to decrease as the length of the highly negatively charged C-terminus decreases.
We then studied the effects of the C-terminal truncations on the lipid-induced aggregation of AS by incubating each variant either at increasing concentrations of monomeric protein with a fixed concentration of DMPS (Fig. 6a–c) or at a fixed concentration of monomeric protein and increasing concentrations of DMPS (Fig. 6d–f). As observed for AS(WT),24 both AS(1-119) (Fig. 6d) and AS(1-103) (Fig. 6e) alone were not found to form detectable quantities of fibrils in the absence of DMPS vesicles under these experimental conditions, but the lipid vesicles were able to initiate the aggregation of both Ctt variants. The aggregation curves of AS(1-119) were characterised by the absence of a lag phase and by a sharp increase in fluorescence intensity, the rate of which was dependent on the initial concentration of protein and that of DMPS (Fig. 6a and d). At the plateau phase, the concentration of insoluble AS(1-119) increased with the initial concentration of monomeric protein without reaching more than 20% of its value, suggesting that a large fraction of monomeric protein remains in solution at this stage (Fig. 6c). Increasing the initial concentration of DMPS also led to an increase in the concentration of insoluble AS(1-119) at the plateau phase, and the majority of monomers were found to convert to amyloid fibrils for concentration of DMPS of 500 μM (Fig. 6f). This aggregation behaviour is similar to that reported for AS(WT)24 and can be rationalised through the cessation of both elongation and primary nucleation, as well as a negligible contribution of secondary processes that would be able to create new aggregation competent fibril ends.24
 | ||
| Fig. 6 Lipid-induced aggregation of AS(1-103) and AS(1-119). (a and b) Change in the ThT fluorescence when increasing concentrations of AS(1-119) (a) or AS(1-103) (b) were incubated in the presence of 100 μM DMPS at 30 °C and pH 6.5 under quiescent conditions (black line: 100 μM protein without lipid, grey line: 100 μM DMPS). (d and e) Changes in the ThT fluorescence intensity when 100 μM AS(1-119) (d) or AS(1-103) (e) was incubated in the presence of increasing concentrations of DMPS at 30 °C and pH 6.5 under quiescent conditions. (c and f) Concentration of insoluble protein measured at the plateau phase for different initial concentrations of protein (mean, standard deviation, n = 3 (filled symbols), n = 1 (open symbols)) (c) or DMPS (mean, standard deviation, n = 3 (except for AS(1-103) 0 μM DMPS: n = 2)) (f). The determination of the protein concentration was performed using samples with (empty symbols) or without (filled symbols) ThT in solution. Note: the DMPS membrane is in the fluid phase for all these measurements (see Fig. S11c†). (g) Schematic representation of the mechanism of lipid-induced aggregation of AS(1-119) and AS(1-103) based on the results described in this study. | ||
Since AS(1-119) appears to aggregate through the same mechanism as that of AS(WT), we analysed the early time points of the lipid-induced aggregation data using the one-step nucleation model, which describes well the lipid-induced aggregation of AS(WT)24 (Fig. S12†). We found that AS(1-119) aggregates an order of magnitude more rapidly than AS(WT) under these conditions (knk+ = 3.01 ± 0.04 × 10−4 (AS(WT)) and 2.10 ± 0.03 × 10−3 (AS(1-119)) mol−1.2 s−2). The fit to a single step model for the AS(1-119) kinetic data is less good than that of AS(WT) (Fig. S12†). It could be improved by including a conversion step, representing an extension of the single step model that we have derived previously.24 However, in order to be able to compare the rate constants directly, and to avoid overfitting, we decided to use the simpler model in both cases.
In the case of AS(1-103), we observed a different aggregation behaviour from that of AS(WT) and AS(1-119). First, the aggregation curves of AS(1-103) are characterised by a short lag phase followed by multiple increases and plateaus (Fig. 6b and e). Then, the concentrations of insoluble species at the final plateau phase were found to be proportional to the initial concentration of monomeric AS(1-103) (Fig. 6c) but independent of the concentration of DMPS (Fig. 6f). These results suggest that all the soluble protein has been consumed at the plateau phase of the aggregation reaction of AS(1-103).
In order to obtain additional insights into the origin of the differences in the lipid-induced aggregation behaviour of AS(WT) and AS(1-119) on the one hand and AS(1-103) on the other, we characterised the morphology of the fibrils formed at different time points during the aggregation process using TEM (Fig. S13†). At the plateau phase of AS(1-119), the majority of fibrils were found to be thin and curly, clearly similar to those observed for AS(WT).24 We have previously suggested that these structures are protofibrils that are kinetically trapped in an elongation incompetent state; we also observed that they can be converted into mature fibrils if mechanical forces, e.g. generated by sonication, are applied,24 releasing new elongation competent fibril ends. Interestingly, such protofibrils were also detected at early times during the lipid-induced aggregation of AS(1-103) but they were no longer visible at the final plateau phase, where the dominant species were observed to be straight and with the thickness characteristic of mature fibrils (Fig. S13†).
The data shown in Fig. 5 indicate that at pH 6.5 in the absence of vesicles, secondary processes contribute significantly to the overall aggregation reaction of AS(1-103) and AS(1-119), a result observed for AS(WT) at pH values below 5.5.29 Our results therefore suggest that the lipid-induced protofibrils formed by AS(1-103) are able to act as suitable templates for secondary nucleation. The finding that the lipid-induced protofibrils of AS(1-119) do not show secondary processes at pH 6.5 but that those produced by the polymer surface-induced process reveal the existence of such events, indicates that the surface properties of the fibrils nucleated in the presence of lipids or at the surface of plates have significant differences. This conclusion is corroborated by the very different fluorescence intensity associated with ThT binding that these two types of fibrils display (Fig. S14†).
4 Conclusions
We have investigated in detail the effects that C-terminal truncations have on the overall mechanism of amyloid formation by AS. To that end, we have applied a strategy that enables us to study individual processes that constitute the overall aggregation mechanism, such as fibril elongation, higher order assembly and autocatalytic proliferation, and the lipid-induced aggregation.Overall our results show that the most significant effect of the C-terminal truncations of the AS sequence is the dramatic change in the pH range within which autocatalytic secondary nucleation plays a role in the aggregation process. While the WT sequence displays secondary nucleation only at mildly acidic pH values, which are indeed encountered within a range of organelles, such as lysosomes and endosomes, the secondary nucleation pH range of the Ctt AS variants extends into the neutral region, therefore giving aggregates formed from these sequences the potential to proliferate within the cytosol (Table S3†). This insight is significant in the light of the finding that considerable fractions of the aggregated forms of AS in vivo display C-terminal truncation and therefore this post-translational modification may play a crucial role in the overall potential of AS to form aggregates that are able to proliferate exponentially and spread throughout the diseased brain.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Beata Blaszczyk, Sam Ness, Ewa Klimont and Swapan Preet for producing and purifying AS(WT) used during this study. The authors would also like to thank Dr Aviad Levin for assistance with the microcapillary measurements, and the Cambridge Advanced Imaging Centre for their assistance with TEM imaging. The authors thank Magdalene College, Cambridge (AKB), the Leverhulme Trust (AKB), the European Commission for a Marie Skłodowska-Curie Actions – Individual Fellowship (CG) and the Cambridge Centre for Misfolding Diseases (IMV, TPJK, CMD and CG) for support of this work.Notes and references
- K. Wakabayashi, K. Tanji, S. Odagiri, Y. Miki, F. Mori and H. Takahashi, Mol. Neurobiol., 2013, 1–14 Search PubMed.
- M. G. Spillantini, M. L. Schmidt, V. M. Lee, J. Q. Trojanowski, R. Jakes and M. Goedert, Nature, 1997, 388, 839–840 CrossRef PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef PubMed.
- A. Iwai, E. Masliah, M. Yoshimoto, N. Ge, L. Flanagan, H. A. Rohan de Silva, A. Kittel and T. Saitoh, Neuron, 1995, 14, 467–475 CrossRef PubMed.
- S. Bellani, V. L. Sousa, G. Ronzitti, F. Valtorta, J. Meldolesi and E. Chieregatti, Commun. Integr. Biol., 2010, 3, 106–109 CrossRef PubMed.
- D. D. Murphy, S. M. Rueter, J. Q. Trojanowski and V. M. Lee, J. Neurosci., 2000, 20, 3214–3220 CrossRef PubMed.
- J. Burré, J. Parkinson's Dis., 2015, 5, 699–713 CrossRef PubMed.
- K. Nakamura, V. M. Nemani, E. K. Wallender, K. Kaehlcke, M. Ott and R. H. Edwards, J. Neurosci., 2008, 28, 12305–12317 CrossRef PubMed.
- C. Guardia-Laguarta, E. Area-Gomez, E. A. Schon and S. Przedborski, Mov. Disord., 2015, 30, 1026–1033 CrossRef PubMed.
- W. S. Davidson, A. Jonas, D. F. Clayton and J. M. George, J. Biol. Chem., 1998, 273, 9443–9449 CrossRef PubMed.
- C. R. Bodner, C. M. Dobson and A. Bax, J. Mol. Biol., 2009, 390, 775–790 CrossRef PubMed.
- S. Chandra, X. Chen, J. Rizo, R. Jahn and T. C. Südhof, J. Biol. Chem., 2003, 278, 15313–15318 CrossRef PubMed.
- G. Fusco, A. De Simone, T. Gopinath, V. Vostrikov, M. Vendruscolo, C. M. Dobson and G. Veglia, Nat. Commun., 2014, 5, 3827 CrossRef PubMed.
- J. M. George, H. Jin, W. S. Woods and D. F. Clayton, Neuron, 1995, 15, 361–372 CrossRef PubMed.
- G. Comellas, L. R. Lemkau, A. J. Nieuwkoop, K. D. Kloepper, D. T. Ladror, R. Ebisu, W. S. Woods, A. S. Lipton, J. M. George and C. M. Rienstra, J. Mol. Biol., 2011, 411, 881–895 CrossRef PubMed.
- J. A. Rodriguez, M. I. Ivanova, M. R. Sawaya, D. Cascio, F. E. Reyes, D. Shi, S. Sangwan, E. L. Guenther, L. M. Johnson, M. Zhang, L. Jiang, M. A. Arbing, B. L. Nannenga, J. Hattne, J. Whitelegge, A. S. Brewster, M. Messerschmidt, S. Boutet, N. K. Sauter, T. Gonen and D. S. Eisenberg, Nature, 2015, 525, 486–490 CrossRef PubMed.
- D. Eliezer, E. Kutluay, R. Bussell and G. Browne, J. Mol. Biol., 2001, 307, 1061–1073 CrossRef PubMed.
- T. S. Ulmer, A. Bax, N. B. Cole and R. L. Nussbaum, J. Biol. Chem., 2005, 280, 9595–9603 CrossRef PubMed.
- T. Bartels, L. S. Ahlstrom, A. Leftin, F. Kamp, C. Haass and M. F. Brown, Biophys. J., 2010, 99, 2116–2124 CrossRef PubMed.
- B. G. Wilhelm, S. Mandad, S. Truckenbrodt, K. Kröhnert, C. Schäfer, B. Rammner, S. J. Koo, G. A. Classen, M. Krauss, V. Haucke, H. Urlaub and S. O. Rizzoli, Science, 2014, 344, 1023–1028 CrossRef PubMed.
- S. Campioni, G. Carret, S. Jordens, L. Nicoud, R. Mezzenga and R. Riek, J. Am. Chem. Soc., 2014, 136, 2866–2875 CrossRef PubMed.
- L. Giehm, C. L. P. Oliveira, G. Christiansen, J. S. Pedersen and D. E. Otzen, J. Mol. Biol., 2010, 401, 115–133 CrossRef PubMed.
- C. Galvagnion, J. W. P. Brown, M. M. Ouberai, P. Flagmeier, M. Vendruscolo, A. K. Buell, E. Sparr and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7065–7070 CrossRef PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. T. Michaels, M. Vendruscolo, T. P. J. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef PubMed.
- M. Grey, C. J. Dunning, R. Gaspar, C. Grey, P. Brundin, E. Sparr and S. Linse, J. Biol. Chem., 2015, 290, 2969–2982 CrossRef PubMed.
- M. Grey, S. Linse, H. Nilsson, P. Brundin and E. Sparr, J. Parkinson's Dis., 2011, 1, 359–371 Search PubMed.
- R. Vácha, S. Linse and M. Lund, J. Am. Chem. Soc., 2014, 136, 11776–11782 CrossRef PubMed.
- R. Gaspar, G. Meisl, A. K. Buell, L. Young, C. F. Kaminski, T. P. J. Knowles, E. Sparr and S. Linse, Q. Rev. Biophys., 2017, 50 DOI:10.1017/s0033583516000172.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. J. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7671–7676 CrossRef PubMed.
- M. Baba, S. Nakajo, P. H. Tu, T. Tomita, K. Nakaya, V. M. Lee, J. Q. Trojanowski and T. Iwatsubo, Am. J. Pathol., 1998, 152, 879–884 Search PubMed.
- W. Li, N. West, E. Colla, O. Pletnikova, J. C. Troncoso, L. Marsh, T. M. Dawson, P. Jakala, T. Hartmann, D. L. Price and M. K. Lee, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2162–2167 CrossRef PubMed.
- B. C. Campbell, C. a. McLean, J. G. Culvenor, W. P. Gai, P. C. Blumbergs, P. Jäkälä, K. Beyreuther, C. L. Masters and Q. X. Li, J. Neurochem., 2001, 76, 87–96 CrossRef PubMed.
- C.-W. Liu, B. I. Giasson, K. a. Lewis, V. M. Lee, G. N. Demartino and P. J. Thomas, J. Biol. Chem., 2005, 280, 22670–22678 CrossRef PubMed.
- J. P. Anderson, D. E. Walker, J. M. Goldstein, R. De Laat, K. Banducci, R. J. Caccavello, R. Barbour, J. Huang, K. Kling, M. Lee, L. Diep, P. S. Keim, X. Shen, T. Chataway, M. G. Schlossmacher, P. Seubert, D. Schenk, S. Sinha, W. P. Gai and T. J. Chilcote, J. Biol. Chem., 2006, 281, 29739–29752 CrossRef PubMed.
- K. A. Lewis, Y. Su, O. Jou, C. Ritchie, C. Foong, L. S. Hynan, C. L. White, P. J. Thomas and K. J. Hatanpaa, Am. J. Pathol., 2010, 177, 3037–3050 CrossRef PubMed.
- A. Ohrfelt, H. Zetterberg, K. Andersson, R. Persson, D. Secic, G. Brinkmalm, A. Wallin, E. Mulugeta, P. T. Francis, E. Vanmechelen, D. Aarsland, C. Ballard, K. Blennow and A. Westman-Brinkmalm, Neurochem. Res., 2011, 36, 2029–2042 CrossRef PubMed.
- G. Muntané, I. Ferrer and M. Martinez-Vicente, Neuroscience, 2012, 200, 106–119 CrossRef PubMed.
- C.-W. Liu, Science, 2003, 299, 408–411 CrossRef PubMed.
- A. J. Mishizen-Eberz, R. P. Guttmann, B. I. Giasson, G. A. Day, R. Hodara, H. Ischiropoulos, V. M. Y. Lee, J. Q. Trojanowski and D. R. Lynch, J. Neurochem., 2003, 86, 836–847 CrossRef PubMed.
- I. Brandt, M. Gerard, K. Sergeant, B. Devreese, V. Baekelandt, K. Augustyns, S. Scharpe, Y. Engelborghs and A.-M. Lambeir, Peptides, 2008, 29, 1472–1478 CrossRef PubMed.
- D. Sevlever, P. Jiang and S.-H. C. Yen, Biochemistry, 2008, 47, 9678–9687 CrossRef PubMed.
- I. V. J. Murray, B. I. Giasson, S. M. Quinn, V. Koppaka, P. H. Axelsen, H. Ischiropoulos, J. Q. Trojanowski and V. M.-Y. Lee, Biochemistry, 2003, 42, 8530–8540 CrossRef PubMed.
- W. Hoyer, D. Cherny, V. Subramaniam and T. M. Jovin, Biochemistry, 2004, 43, 16233–16242 CrossRef PubMed.
- K. Levitan, D. Chereau, S. I. A. Cohen, T. P. J. Knowles, C. M. Dobson, A. L. Fink, J. P. Anderson, J. M. Goldstein and G. L. Millhauser, J. Mol. Biol., 2011, 411, 329–333 CrossRef PubMed.
- W. Wang, L. T. T. Nguyen, C. Burlak, F. Chegini, F. Guo, T. Chataway, S. Ju, O. S. Fisher, D. W. Miller, D. Datta, F. Wu, C.-X. Wu, A. Landeru, J. A. Wells, M. R. Cookson, M. B. Boxer, C. J. Thomas, W. P. Gai, D. Ringe, G. A. Petsko and Q. Q. Hoang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9587–9592 CrossRef PubMed.
- A. Ulusoy, F. Febbraro, P. H. Jensen, D. Kirik and M. Romero-Ramos, Eur. J. Neurosci., 2010, 32, 409–422 CrossRef PubMed.
- M. M. Dedmon, K. Lindorff-Larsen, J. Christodoulou, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2005, 127, 476–477 CrossRef PubMed.
- P. Bernadó, C. W. Bertoncini, C. Griesinger, M. Zweckstetter and M. Blackledge, J. Am. Chem. Soc., 2005, 127, 17968–17969 CrossRef PubMed.
- C. W. Bertoncini, Y.-S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef PubMed.
- C. M. Ritchie and P. J. Thomas, Health, 2012, 04, 1167–1177 CrossRef.
- D. Nečas and P. Klapetek, Cent. Eur. J. Phys., 2012, 10, 181–188 Search PubMed.
- J. Schindelin, C. T. Rueden, M. C. Hiner and K. W. Eliceiri, Mol. Reprod. Dev., 2015, 82, 518–529 CrossRef PubMed.
- G. V. De Ferrari, W. D. Mallender, N. C. Inestrosa and T. L. Rosenberry, J. Biol. Chem., 2001, 276, 23282–23287 CrossRef PubMed.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 1993, 209, 32–44 CrossRef PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef PubMed.
- S. I. A. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- T. P. J. Knowles, W. Shu, G. L. Devlin, S. Meehan, S. Auer, C. M. Dobson and M. E. Welland, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10016–10021 CrossRef PubMed.
- A. K. Buell, C. M. Dobson and M. E. Welland, Methods Mol. Biol., 2012, 849, 101–119 Search PubMed.
- D. Pinotsi, A. K. Buell, C. Galvagnion, C. M. Dobson, G. S. Kaminski Schierle and C. F. Kaminski, Nano Lett., 2014, 14, 339–345 CrossRef PubMed.
- A. K. Buell, J. R. Blundell, C. M. Dobson, M. E. Welland, E. M. Terentjev and T. P. J. Knowles, Phys. Rev. Lett., 2010, 104, 228101 CrossRef PubMed.
- T. A. Witten and P. A. Pincus, Macromolecules, 1986, 19, 2509–2513 CrossRef.
- S. Wegmann, I. D. Medalsy, E. Mandelkow and D. J. Muller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E313–E321 CrossRef PubMed.
- K. K. M. Sweers, K. O. van der Werf, M. L. Bennink and V. Subramaniam, ACS Nano, 2012, 6, 5952–5960 CrossRef PubMed.
- P. Flagmeier, G. Meisl, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson, A. K. Buell and C. Galvagnion, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10328–10333 CrossRef PubMed.
- J. W. P. Brown, A. K. Buell, T. C. T. Michaels, G. Meisl, J. Carozza, P. Flagmeier, M. Vendruscolo, T. P. J. Knowles, C. M. Dobson and C. Galvagnion, Sci. Rep., 2016, 6, 36010 CrossRef PubMed.
- G. Holzwarth and P. Doty, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary methods, supplementary results, supplementary tables, supplementary figures. See DOI: 10.1039/c8sc01109e |
| ‡ Present address: German Center for Neurodegenerative Diseases (DZNE), Sigmund-Freud-Str. 27, 53127, Bonn, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |