
Amino acid-derived stimuli-responsive polymers and their applications
Kamal
Bauri
a,
Mridula
Nandi
b and
Priyadarsi
De
 *b
*b
aDepartment of Chemistry, Raghunathpur College, Raghunathpur – 723133, Purulia, West Bengal, India
bPolymer Research Centre and Centre for Advanced Functional Materials, Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur – 741246, Nadia, West Bengal, India. E-mail: p_de@iiserkol.ac.in
First published on 30th January 2018
Abstract
Natural biopolymers such as proteins and nucleic acids in living organisms possess an inherent ability to respond to local environmental stimuli, which motivated researchers to make biomolecule-derived non-biological macromolecules with a biomimetic structure having stimuli-responsive properties. This review mainly focuses on stimuli-responsive polymers having natural amino acid units either in the main-chain or in the side-chain, their self-assembled nanostructures and hydrogel networks. Recent advances in the design and synthesis of amino acid-derived polymers that are responsive to various physical, chemical, or biochemical stimuli such as temperature, light, pH, redox-, metal ions, gas, glucose, enzyme, proteins, DNA or a combination of these are illustrated. Their potential for use as stimuli-responsive “smart” nanomaterials in biomedical and biotechnological applications such as in controlled drug delivery, gene delivery, non-fouling materials, etc. is also highlighted. The primary aim of this review article is to motivate researchers towards the design and synthesis of novel stimuli-responsive biohybrid materials for making next generation smart materials.
 Kamal Bauri | Dr Kamal Bauri is an Assistant Professor in the Department of Chemistry at Raghunathpur College affiliated to Sidho-Kanho-Birsha University, West Bengal, India. He received his Ph.D. in 2016 from the Indian Institute of Science Education and Research Kolkata (IISER Kolkata), India, from Dr Priyadarsi De's group. He continued his research as a Research Associate in the same group until 2017. He has 18 peer-reviewed publications to his credit. His current research interests include stimuli-responsive polymers, and unconventional macromolecular luminogens for potential bioapplications. |
 Mridula Nandi | Mridula Nandi is currently a PhD student in Dr Priyadarsi De's research group at the Indian Institute of Science Education and Research Kolkata (IISER Kolkata), West Bengal, India. She received her BSc degree in 2013 in Chemistry from Calcutta University, West Bengal, India. In 2015, she obtained her MS degree from IISER Kolkata. Her research interests include synthesis of amino acid based macromolecular architectures and their solution properties. |
 Priyadarsi De | Dr Priyadarsi De is an Associate Professor in the Department of Chemical Sciences at the Indian Institute of Science Education and Research Kolkata (IISER Kolkata), India. He received his Ph.D. degree from the Indian Institute of Science, Bangalore, India. After his post-doctoral studies at UMASS Lowell (2002–2006) and Southern Methodist University (2007–2008), he worked in PhaseRx, Inc., Seattle, for fifteen months before joining IISER Kolkata in November 2009. His research group at IISER Kolkata mostly focuses on controlled synthesis of bio-inspired macromolecular architectures from naturally occurring amino acid and fatty acid based renewable resources for various applications. He is an Editorial Advisory Board Member of Macromolecules and ACS Macro Letters (American Chemical Society Publications, January 2017 to the present), and Polymer Chemistry (Royal Society of Chemistry, September 2015 to the present). |
1. Introduction
Proteins, which are essential for the structure and function of cells, tissues and organs, the transport of molecules, and the catalysis of biochemical reactions that are needed to sustain cellular processes, at molecular levels, are constituted by natural amino acids.1 The biological activity of proteins arises from their specific higher order conformations and also changes in conformation in response to micro-environmental cues such as changes in cellular pH, temperature, ionic strength, redox state, light, etc. Inspired by nature, researchers over the years have indulged in the design and construction of biomolecule-derived synthetic materials with structure and function similar to those of natural proteins.2 In this regard, polymeric materials with amino acids in the main-chain (polypeptides) or in the side-chain are very interesting biomimetic materials. Apart from being biodegradable and/or biocompatible materials, these biomimetic materials exhibit properties combined from both the amino acid sequences and the synthetic polymeric backbone. The application of certain stimuli brings in conformational, physical or chemical change of the polymeric material in response to them, and has been extensively utilized in biomedicine, bio- and nano-technology. One unique feature of the polypeptide polymers is their ability to adopt higher ordered secondary structures (α-helix, β-sheet, β-turn, etc.) driven by non-covalent forces like H-bonding, hydrophobic interactions, π–π stacking or electrostatic forces between the amino acid side-chains and covalent (disulfide) bonds.3 Each of these interactions is sensitive towards its local environment such as temperature, pH, ionic strength, solvent polarity and oxidising/reducing environment. Any change in the local environment is associated with structural reorganization, which in turn affects the biological activity.The past decade has witnessed rapid progress in the field of stimuli-responsive amino acid-based polymers because of their extensive applications in bio- and nano-technology. A variety of amino acid containing stimuli-responsive main-chain (polypeptide, polyesters, polyester urethane, etc.) and side-chain polymers have been exploited to date. Huang et al.4 and Li and co-workers5,6 have previously reviewed advances in stimuli-responsive polypeptides and highlighted their applications in tissue engineering, drug delivery and biodiagnostics. An amino acid is an excellent platform to construct a myriad of functionalized monomers for controlled living polymerizations. A number of new approaches have been reported for incorporation of amino acid moieties into the polymer chain.7,8
In this review article, the most recent advances in the synthesis of amino acid-derived various stimuli-responsive polymers and their use as stimuli-responsive “smart” nanomaterials for a broad range of applications such as in controlled drug delivery, gene delivery, tissue engineering, and regenerative medicine and as non-fouling materials are highlighted. The progress of various stimuli-responsive polypeptide hydrogels and side-chain amino acid-based hydrogels capable of sensing diverse biochemical signals, which can facilitate the environment-responsive drug release, modulate cell behavior, or disease diagnosis and treatment, is also discussed in this article. These stimuli can typically be classified into three different categories: physical stimuli such as temperature and light; chemical stimuli such as pH, redox (oxidation–reduction), ionic strength, metal ions, and gas; and biochemical stimuli such as glucose, enzyme, protein, DNA, etc. (Fig. 1).9
 | ||
| Fig. 1 Schematic illustration of amino acid-derived various stimuli-responsive polymers. | ||
2. Synthetic strategy for main-chain amino acid based polymers
Various synthetic strategies have been used so far for the synthesis of amino acid-based main-chain polymers. A few polymers such as polyamide or polypeptides, polyesters, and poly(depsipeptide) were prepared from amino acid-derived monomers. Polymers such as poly(ester amide), polyurethane and poly(disulfide amide) were also prepared by condensation reaction of amino acid-derivatives with some partner molecules (Fig. 2).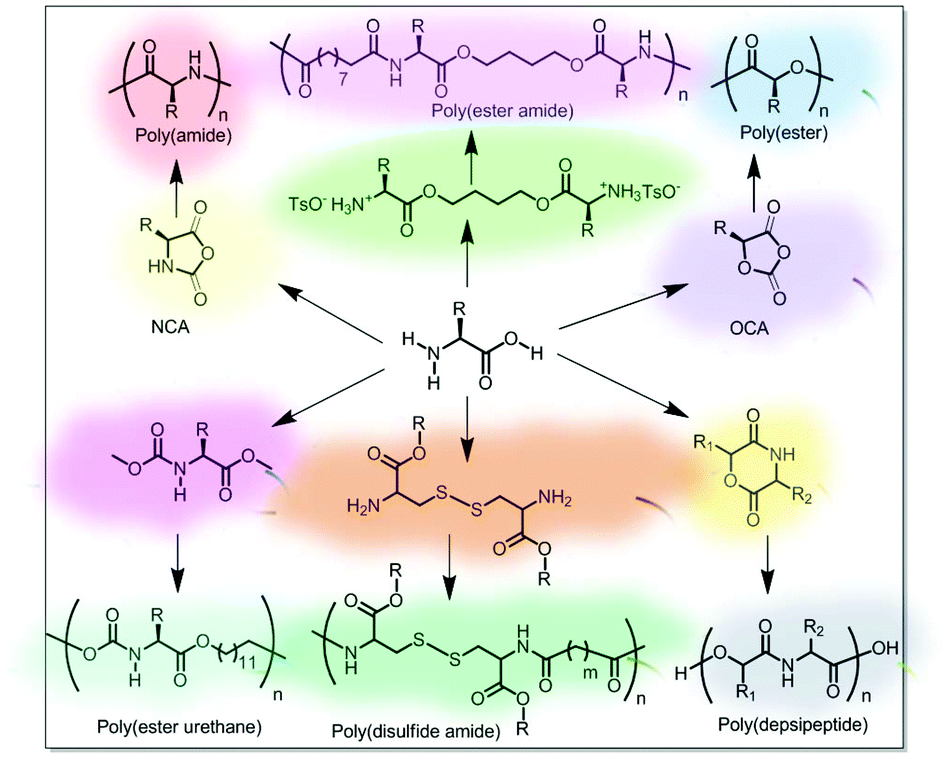 | ||
| Fig. 2 Various main-chain amino acid functional polymers. | ||
3. Main-chain amino acid-based stimuli-responsive polymers
Table 1 summarizes various stimuli-responsive synthetic polypeptides and their associated property changes upon the action of different stimuli. In this section we will highlight different classes of main-chain amino acid-based stimuli-responsive polymers.| Stimulus | Structure | Name/abbreviation | Response | Ref. |
|---|---|---|---|---|
| pH |

|
Poly(L-glutamic acid)-b-poly(L-lysine) (PGA-b-PLys) | pH-Tunable schizophrenic micellization | 14 |
|
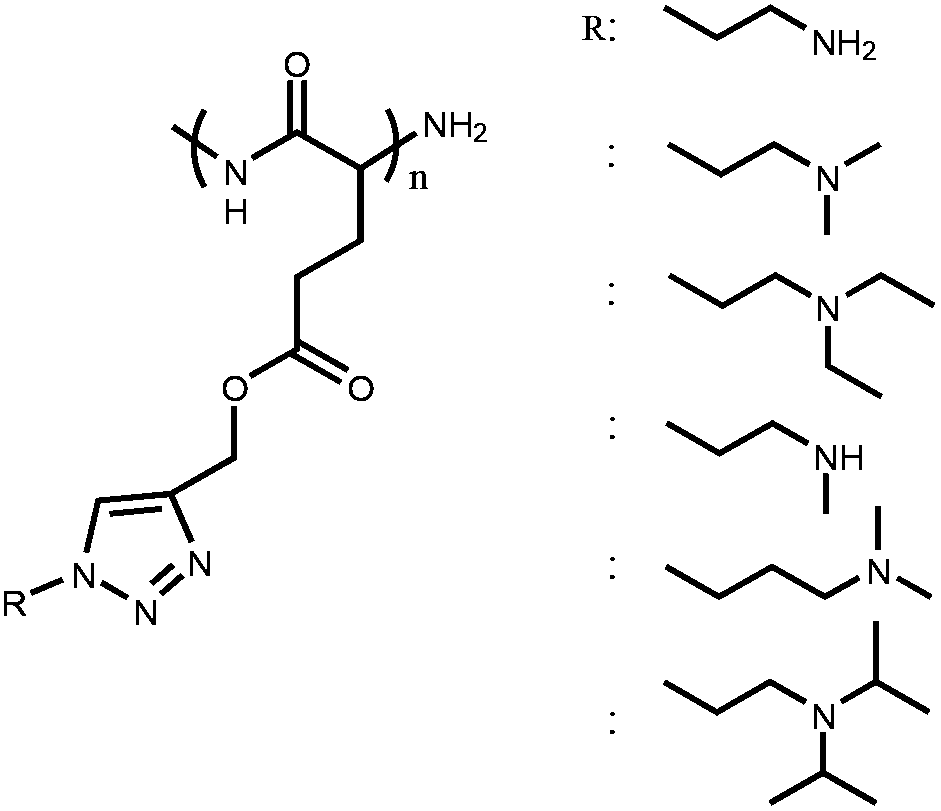
Primary, secondary and tertiary amines grafted on poly(γ-propargyl L-glutamate) (PPLG) homopolymers | pH-Reversible solubility transition behavior | 22 | |
| Thermo |
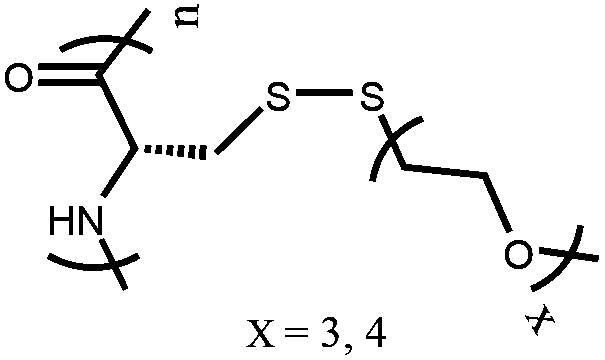
|
OEGylated poly (L-cysteine) [poly(L-EGx-SS-Cys)] | Irreversible LCST-type phase behavior | 33 |

|
OEGylated poly (L-glutamate) [poly(L-EGx-Glu)] | Reversible LCST-type phase behavior | 31 | |
|
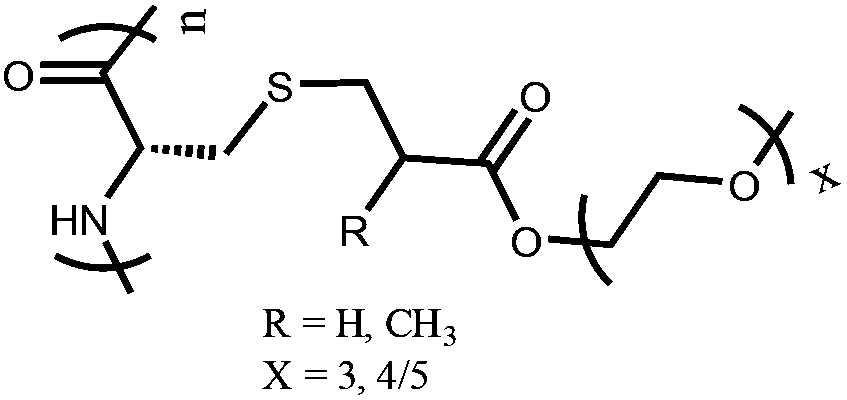
OEGylated poly(L-cysteine) (poly-EGxMA-C or poly-EGxA-C) | Reversible LCST-type phase behavior | 32 | |

|
PPLG-MSEA-Cl and PPLG-MSEA-BF4 | Reversible UCST-type phase behavior in methanol and ethanol | 44 | |
| pH/Thermo |

|
Diisopropylamine grafted on poly(γ-propargyl L-glutamate) (PPLG) homopolymers | pH-Tunable LCST behavior | 35 |
|
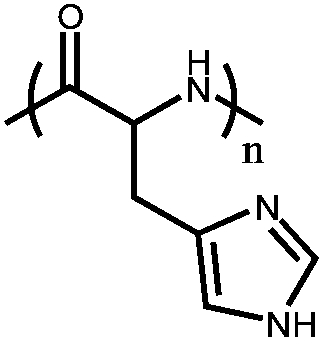
Poly(L-histidine) (PHis) | Conformational transition from random coil (low pH and temperature) to β-sheet (higher pH) to broken β-sheet (higher temperature) | 20 | |
| Redox |
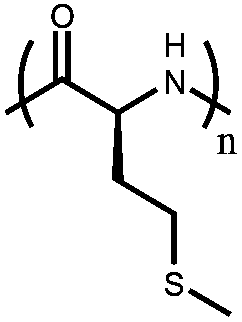
|
Poly(L-methionine) | α-Helix (thioether) to random coil (sulfoxide) to α-helix (sulfone) conformational transition | 56 |

|
Poly(α-gal-C) | α-Helix (thioether) to random coil (sulfone) conformational transition | 54 | |

|
OEGylated poly-L-cysteine [poly(LEGxMA-C)] | β-Sheet to random coil conformational transition; increased water solubility and LCST | 55 | |

|
Poly(α-D-galactose-L-homocysteine) | Reversible α-helix to random coil conformational switch upon reversible oxidation | 57 | |

|
Poly(S-OEGylated-L-homocysteine) | Reversible α-helix to random coil conformational switch upon reversible oxidation; increased water solubility and loss of LCST | 57 | |
| Photo |

|
Poly(S-(o-nitrobenzyl)-L-cysteine)-b-PEO (PNBC-b-PEO) | Photo-cleavage of o-nitrobenzyl (NB) groups | 67 |

|
Poly(γ-cinnamyl-L-glutamate) (PCLG) | Crosslinking due to photodimerization of cinnamyl groups | 65 | |
| Photo/Thermo |

|
P(OEGm-Azo) | Reversible photo-sensitivity and irreversible thermosensitivity | 73 |
| Glucose |

|
(mPEG-b-P(GA-co-GPBA)) | Hydrophilic phenylboronic acid–glucose complex formation at physiological pH | 78 |

|
PEG-Poly(Ser-Ketal) | Indirectly responsive to glucose; the ketal bond got cleaved in an acidic environment, producing a double hydrophilic block copolymer | 84 | |

|
mPEG-b-P(Ser-PBE) | Indirectly responsive to glucose; phenylboronic ester got degraded under H2O2-generated conditions, producing a double hydrophilic block copolymer | 85 | |

|
PEG-poly(Ser-S-NI) | Indirectly responsive to glucose; H2O2 converted thioether into sulfone and hypoxia bioreduced NI into 2-aminoimidazole with enhanced hydrophilicity | 86 | |
| Enzyme |

|
Poly(L-phosphotyrosine) | Macroscopic gel formation in the triblock copolymer with PEG in the presence of alkaline phosphatase | 88 |

|
Amphiphilic polypeptide | Bovine carbonic anhydrase II binds to the ligand, causing disassembly of the nanostructures | 87 | |
| Protein |

|
Mannose-6-phosphate glycopolypeptides (M6P-GPs) | Selectively target the lysosome because of mannose-6-phosphate-specific CI-MPR receptors present on MCF-7 cells | 100 |

|
Poly(α-manno-O-lys) | Precipitation upon binding with Con A | 93 | |
| Gas (CO2) |
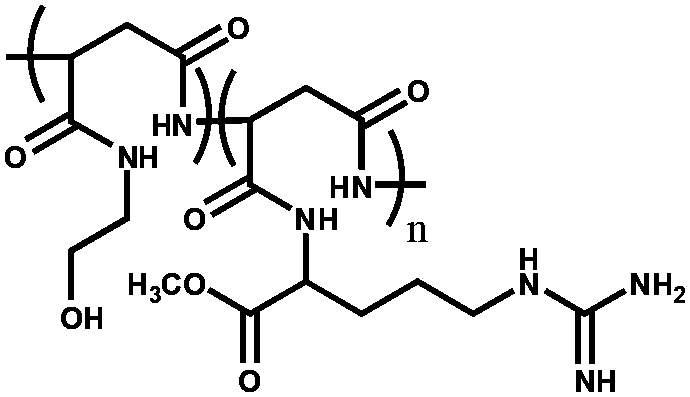
|
Poly(2-hydroxyethylaspartamide) derivative modified with L-arginine unit (PHEA-Larg) | Forms hydrogels, which showed CO2-reversible absorption behavior | 102 |
| Gas (SO2) |

|
(PPLG-PyBF4-r-OEG) | SO2-Triggered solubility transition | 104 |
3.1. pH-Responsive polypeptides
pH-Responsive polymers exhibit reversible changes in solubility or conformational properties as a function of the pH of the solution.10,11 These polymers generally contain ionizable moieties that are pendant to the polymer backbone, e.g., tertiary amino groups in poly(dimethyl amino ethyl methacrylate) (PDMAEMA) which is cationic at lower pH or carboxyl groups in poly(acrylic acid) (PAA) which is anionic at higher pH. Since amino acids possess both primary amino and carboxylic acid groups, it is expected that amino acid-generated polymers will display pH-responsive behavior, provided either of the functional groups remains free. Although polypeptides are amino acid-derived condensation polymers, where both primary amino and carboxyl acid functional groups are involved, pH-responsiveness can only result if the precursor amino acid itself is either an acidic or a basic amino acid such as glutamic acid or lysine. Most well studied pH-responsive polypeptides are poly(L-glutamic acid) (PGA) and poly(L-lysine) (PLys).The self-organization behavior of polybutadiene-b-poly(glutamic acid) (PB-b-PGA)12,13 and polybutadiene-b-poly(lysine) (PB-b-PLys)14 diblock copolymers were reported, where these two block copolymers self-assembled into a vesicle and a micelle, respectively. In both cases the pH-sensitivity of the secondary structure of polypeptide segments has been exploited to manipulate the size and shape of the supramolecular structures formed by self-assembly of these block copolymers in aqueous medium. At acidic pH, the poly(glutamic acid) segment is neutral and forms α-helical structure, but under basic conditions it transforms into charged random coiled conformation. Similarly, the PLys block undergoes a pH-reversible conformational transition from random coil to α-helix with increasing aqueous solution pH. Lecommandoux et al. have reported for the first time a novel pH-responsive schizophrenic vesicle from a zwitterionic diblock copolypeptides poly(L-glutamic acid)-b-poly(L-lysine) in pure water. At acidic pH, the poly(glutamic acid) segment is neutral and becomes insoluble. On the other hand the poly(L-lysine) block exists in a protonated form, forcing the polymers to self-assemble into some higher order structure, where PGA forms the core and PLys forms the shell in the aggregates. Under basic conditions, protonated –NH3+ moieties of the PLys block are transformed into neutral –NH2 groups, forming the core of the aggregates, and the PGA segment forms the shell (Fig. 3).15
 | ||
| Fig. 3 Schematic representation of pH-regulated schizophrenic vesicles from the self-assembly of the diblock copolypeptide PGA15-b-PLys15 in water. Reprinted with permission from ref. 15. Copyright (2005) American Chemical Society. | ||
Several other pH-responsive vesicles were formed from polypeptide-based block copolymers such as poly(L-lysine)-b-poly(γ-benzyl-L-glutamate)-b-poly(L-lysine),16 poly(L-lysine)-b-poly(L-phenylalanine),17 poly(L-lysine)-b-poly(L-glycine),18 and poly(L-glutamic acid)-b-poly(L-phenylalanine).19 A series of novel pH-triggered charge-reversal polypeptide nanoparticles were synthesized by random ring-opening copolymerization of γ-benzyl-L-glutamate N-carboxyanhydride (BLG-NCA) and 3-benzyloxycarbonyl-L-lysine N-carboxyanhydride (ZLys-NCA), followed by removal of the protecting group.20 These nanoparticles were used as intelligent drug delivery systems for cancer therapy. Solution pH, L-glutamic acid/L-lysine ratio and drug loading content have a great influence on the surface charge of the drug loaded nanoparticles. A protein-mimetic ampholytic triblock copolypeptide, poly(ethylene oxide)-block-poly(L-lysine)-block-poly(L-glutamate), was synthesized and its pH-regulated self-assembly behavior was investigated.21
Another interesting conformationally dynamic polypeptide is poly(L-histidine) (PHis). It was found that low molecular weight PHis is not only pH-responsive but also thermo-responsive. This homopolypeptide can adopt a random coil conformation at low pH and temperature, a β-sheet conformation at higher pH, and probably a broken β-sheet conformation at higher temperature.22 Yin and co-workers fabricated phospholipid liposomes mimetic polymersomes from a novel AB2-type miktoarm copolymer, mPEG2kDa-b-(PHis29kDa)2. The polymersome nano-structure was stable above pH 7.4, below which it transformed into a cylindrical micelle, a spherical micelle and finally to unimers as pH was decreased. The pH-induced structural transition of the polymeric nanostructure was attributed to the increased hydrophilicity of mPEG-b-(PHis)2 at lower pH.23
A new library of pH-responsive cationic polypeptides and block copolypeptides has been developed by Engler et al. Ring opening polymerization (ROP) of propargyl-functionalized NCA monomer followed by alkyne–azide click chemistry enables the formation of various synthetic polypeptides with primary, secondary and tertiary amine side-chain pendant moieties.24 They demonstrated that these new amine-functionalized polypeptides are strong candidates for drug and gene delivery.
3.2. Thermo-responsive polypeptides
In nature, several sequence-regulated peptides have been identified that exhibit stimuli responsive structural behavior. One such peptide motif that is found in the polymeric elastin-like polypeptide (ELP) of the mammalian elastin protein is the repeating pentameric sequence VPGXG (V = L-valine, P = L-proline, G = glycine). Here X can be any amino acid except proline. The ELP exhibits a sharp temperature-reversible hydrophilic–hydrophobic phase transition. The phase transition temperature, termed lower critical solution temperature (LCST), is tunable by changing the pH or ionic strength of the ELP solution. Below LCST, the ELP is soluble and exists in an extended chain conformation in water, while above LCST, the pentamer adopts an ordered β-spiral conformation that aggregates out of solution as precipitates. Generally, stimuli-sensitive peptide segments are ligated to some other proteins in order to make them more attractive for use in biotechnological applications. The most widely used of such peptides is ELP. For example, the temperature-responsive sharp solubility transition behavior of ELPs has been exploited to fabricate numerous systems for purification of biomolecules.25 ELPs have also been fused to appropriate peptides and proteins for remediation of toxic metals26 and targeted drug delivery.27,28 In addition, ELPs being genetically encoded biopolymers, their gene-level design makes them ideal candidates for tissue engineering scaffolds.Lecommandoux et al. have shown that chemoselective alkylation of methionine residues of recombinant ELP having repeating pentameric sequence of VPGXG (X = methionine) provides an easy way to modulate its LCST.29 Although ELP did exhibit LCST, no LCST was identified after methylation. A higher hydrophilicity of the resulting peptide chain due to the presence of cationic sulfonium moieties was believed to be the reason. Again, the benzyl derivative exhibited a higher LCST value, indicating the hydrophilic effect of positive charges partially counter balanced by the hydrophobic benzyl groups. Alkylated polypeptides adopt a less ordered conformation with increasing hydrophilicity of ELP (Fig. 4A and B).
 | ||
| Fig. 4 (A) Chemoselective alkylation of methionine residues of ELP and (B) their absorbance versus temperature plot. Reprinted with permission from ref. 29. Copyright (2015) American Chemical Society. (C) Selective oxidation of methionine residues of ELP for its precise LCST tuning. Reprinted with permission from ref. 31. Copyright (2017) American Chemical Society. | ||
Recently, in a detailed study they explored the dependence of the nature of the alkyl substituents and sulfonium counter-ions on the LCST of the recombinant ELP.30 Furthermore, they could tune the thermoresponsive properties of ELP by selective oxidation of methionine residues. The increased transition temperature of the oxidized polymer compared to the parent one is due to the increased hydrophilicity of the sulfoxide or sulfone groups compared to the thioether groups. A higher dipole moment of sulfone compared to sulfoxide groups can lead to sulfone–sulfone and sulfone–protein interactions. Consequently, a sulfone-derivative shows a decreased water solubility and a lower LCST value compared to the sulfoxide-analogue (Fig. 4C).31
In addition to natural ELPs and their modified-forms as thermo-responsive polypeptides, polymer chemists have developed various synthetic methods to prepare thermo-responsive polypeptides. Generally, oligo(ethylene glycol) (OEG)-based polymers show thermo-responsive behavior.32 Corresponding polypeptide polymers were prepared either through ROP of OEG-conjugated NCA monomers or by post polymerization modification of reactive polypeptide scaffolds. Chen et al. prepared three different OEGylated poly-L-glutamates poly(L-EGxGlu) with varying ethylene glycol (EG) chain length using the first synthetic approach. They found that poly-(L-EG1Glu) is insoluble in aqueous media and common organic solvents, while poly(L-EG2Glu) and poly(L-EG3Glu) displayed reversible LCST behaviors in water. Because of its longer EG chain length, poly(L-EG3Glu) shows a higher LCST compared to that of poly(L-EG2Glu).33 The same group reported another class of thermo-responsive polypeptides through ROP of OEGylated L-cysteine based NCA monomer, which was made via thiol–ene Michael addition between L-cysteine and monomethoxy OEG functionalized methacrylate (OEGxMA) and acrylate (OEGxA).34 Irreversible temperature-induced phase transition behavior was observed in the case of OEGylated disulfide bond-containing poly(L-cysteine)s. Such an irreversible thermal-responsive behavior was attributed to interchain disulfide bond formation upon heating.35
Using the second synthetic procedure, Cheng and co-workers made a series of temperature-sensitive polypeptides by ROP of γ-propargyl-L-glutamate NCA coupled with subsequent click chemistry between pendant alkyne groups and 1-(2-methoxyethoxy)-2-azidoethane (MEO2-N3) and 1-(2-(2-methoxyethoxy)ethoxy)-2-azidoethane (MEO3-N3). The graft polypeptide with MEO3 grafting units showed clearly a higher LCST value than that of the MEO2 side-chain owing to the greater hydrophilicity in the former case.36 The same synthetic strategy was used by Hammond's group to synthesize dual pH and thermo-responsive polypeptide. OEG and diisopropylamine side groups were incorporated into the polypeptide, where the OEG-functionalized system showed only temperature responsiveness and the diisopropylamine pendant system exhibited dual pH and thermo-responsiveness.37 Zhang et al. followed the same methodology to achieve thermo-responsive dendronized polyproline from a polypeptide precursor having azide pendant and alkyne-core OEG dendrons.38 Dynamic covalent chemistry between the primary amino groups of polylysine and aldehyde-terminated OEG-based dendrons enables the formation of another class of temperature-sensitive polypeptides.39 A new class of thermoresponsive dendronized polypeptides was accomplished through a highly efficient oxime ligation between oxyamino-substituted polylysines and aldehyde-cored OEG dendrons. In addition, phenylboronic acid moieties were introduced to afford dendronized copolypeptides with a thermally driven recognition ability of catechols.40 Recently, Meng and co-workers reported a facile and efficient approach to prepare a class of dual-responsive copoly(L-glutamate)s by random ring opening copolymerization of γ-propargyl-L-glutamate and γ-benzyl-L-glutamate, followed by post-modification steps. In this case, both OEG and glutamic acid residues were incorporated to offer thermal- and pH-responsive behavior simultaneously.41
Polymers bearing zwitterionic groups exhibit an upper critical solution temperature (UCST)-type phase behavior in water.42 Zwitterions appended polymers are insoluble in water at low temperature because of intra- and inter-chain electrostatic interactions between the adjacent zwitterionic moieties. Mostly, thermo-responsive polypeptides with UCST-type phase transition have been developed by incorporating charged species with associated counter-ions. For example, Tang et al. attached ionic liquid moieties (imidazolium43 or pyridinium44) to a functionalizable polypeptide chain by a post-polymerization modification technique followed by an ion exchange reaction to afford water-soluble UCST-type polypeptides. They compared the effect of polymer and salt concentrations on UCST of Y-shaped imidazolium pendant polypeptide with a traditional pendant one.45 Recently, they reported a new class of thermo and pH dual responsive polypeptides from the clickable precursor poly(γ-3-methylthiopropyl-L-glutamate) (PMTPLG). After alkylation of the PMTPLG, sulfonium moieties endow the resulting polypeptides with UCST-type thermo-responsiveness in alcoholic solvents. Their UCSTs are greatly influenced by the type of alkyl pendant (methyl, n-butyl and propargyl), which can also be modulated by adjusting the counter-anion via ion exchange reactions (Fig. 5A and B). Again a UCST-type doubly thermo and pH-responsive polypeptide was accomplished through the [2 + 3] cycloaddition reaction between an alkylated polypeptide having a propargyl pendant and 2-azidoethylamine followed by counter-anion exchange with tetrafluoroborate (BF4−).46
 | ||
| Fig. 5 (A) Synthetic route of PMTPLG and polypeptides bearing various alkyl methyl sulfonium moieties. (B) Synthetic route of polypeptides bearing methyl sulfonium linkages and ethyl ammonium pendants. Reproduced with permission from ref. 46. Copyright (2017) The Royal Society of Chemistry. | ||
3.3. Redox-responsive polypeptides
Because of the high redox potential gradient between the extra-cellular and intra-cellular environments, as well as between tumorous and normal tissues, redox (reduction–oxidation) stimulus has been recognized as a valuable strategy for rapid and effective drug release in tumor cells. The high reducing potential in cells is primarily due to the abundance of a tripeptide, glutathione (GSH). Recently, reactive oxygen species (ROS) have attracted much attention owing to their close connection with many diseases. That is why much effort has been devoted to the design and synthesis of redox-responsive polypeptides because of their potential applications in gene and drug delivery, especially in cancer therapy. The most extensively investigated redox-responsive polypeptide includes the incorporation of (a) a disulfide linkage into the main-chain and side-chain of the polypeptide or crosslinked matrices; (b) a thioether moiety into the side-chain of the macromolecules.Generally, two synthetic strategies have been developed for the synthesis of polypeptide containing disulfide bonds. This is achieved either by employing an amino group terminated disulfide functionalized mPEG macroinitiator or using poly(cystine) as a component. A series of novel reduction-responsive disulfide core-crosslinked nanogels were synthesized through one-step ring opening polymerization of L-phenylalanine NCA and L-cystine NCA with amino group terminated poly(ethylene glycol) monomethyl ether (mPEG-NH2) as a macroinitiator. It has been found that the nanogels (NGs) were biocompatible, and the cleavage of disulfide bonds triggered by intracellular GSH could accelerate the intracellular doxorubicin (DOX) release from DOX-loaded NGs, and thus enhance the in vitro cell proliferation inhibition.47
Shell-crosslinked micelles have been constructed using a triblock copolymer, poly(ethylene glycol)-b-poly(L-cysteine)-b-poly(L-phenylalanine), from disulfide-linked amino group terminated macroinitiator (mPEG-NH2) initiated successive ROP of L-cysteine and L-phenylalanine-derived NCAs. By the oxidation of the thiol groups of poly(L-cysteine) segment, a self-crosslinked core–shell–corona micellar aggregate was achieved in aqueous solution. This crosslinked shell was found to be helpful to reduce drug loss in the extracellular environment. Under intracellular conditions, disulfide bonds get cleaved, disrupting the shell, which is followed by an accelerated drug release from the micelle (Fig. 6).48
 | ||
| Fig. 6 Schematic illustration of preparation, drug loading and intracellular redox-responsive release of the anticancer drug DOX from PEG-poly(amino acid)s nanogels. Reproduced with permission from ref. 48. Copyright (2012) The Royal Society of Chemistry. | ||
By taking advantage of the disulfide cleavage reaction under a reductive environment, Kataoka and co-workers designed a novel block catiomer through the insertion of a biocleavable disulfide linkage between PEG and the polycation segment to act as a nonviral gene vector. The cationic segment, which acts as a buffering moiety inducing endosomal escape with minimal cytotoxicity, was achieved by reaction of benzyl aspartate with diethylenetriamine. These disulfide-linked cationic polyplex micelles showed 1–3 orders of magnitude higher gene transfection efficiency and a more rapid onset of gene expression than micelles without disulfide linkages. This is due to a much more effective endosomal escape triggered by the PEG detachment in the endosome.49 Similar results were reported by Cai et al. where transfection efficiency in HeLa cells increased about 3 to 6 fold when disulfide-linked polyplexes were used instead of disulfide-free delivery vectors.50 Similarly, various disulfide-linked block copolymers based on methoxy poly(ethylene glycol) (mPEG) and poly(amino acids) such as poly(phenylalanine)51 or poly(γ-benzyl L-glutamate)52 or poly(rac-leucine)53 or poly(3-benzyloxycarbonyl-L-lysine)54 were synthesized through ROP of amino acid-derived NCA monomers, using amino group terminated disulfide functionalized mPEG as a macroinitiator. It has been noted that fabricated nanocarriers from their self-assembly have vast potential in reduction-sensitive (GSH or dithiothreitol (DTT) as a reducing agent) targeted intracellular delivery of antitumor drugs to achieve enhanced efficacy in malignancy therapy. A unique design has been developed by Zhu and co-workers for efficient siRNA delivery and tumor therapy. The same macroinitiator was employed for the ROP of a mixture of a protected lysine and histidine-based NCA monomers. The disulfide bonds provide selective mPEG detachment under tumor relevant reduction conditions, while hydrophobic benzyl histidine enables remarkable endolysome escape and ensures siRNA stability.55
In addition to the reduction responsive disulfide bond, the thioether moiety can also be used to construct oxidation responsive polypeptides. Redox-triggered conformational switch of glycopolypeptide from α-helix to random coil has been encountered after the oxidation of the thioether moiety of L-cysteine to sulfone, without losing water solubility. In contrast, the analogous glycopolypeptide based on L-homocysteine did not display any conformational switching upon oxidation to sulfone.56 A similar kind of oxidation triggered secondary structure transition from β-sheet to random coil was encountered for OEGylated poly(L-cysteine) derivatives, poly(L-EGxMA-C)n, with x being 2, 3, 4/5 or 8/9. This oxidation-induced conformational switch was found to be accompanied by increased polarity of the side-chain of the polypeptide and eventually cloud point (CP) temperatures of the studied polymers. Moreover, self-assembled spherical micelles from the PEG45-b-poly(L-EG2MA-C)22 block copolymer could be disrupted upon oxidation of the thioether moiety.57 Such a class of oxidation-responsive micelles might be a promising platform for inflammation targeting drug delivery systems. Unlike the irreversible complete oxidation of the thioether motif to sulfone, partial oxidation of the thioether group to the sulfoxide can reversibly be reduced to the initial thioether motif in the presence of appropriate reducing agents or reductase enzymes. Deming's group reported the fabrication of copolypeptide vesicles from hydrophobic poly(L-methionine)65-b-poly(L-leucine0.5-stat-L-phenylalanine0.5)20 by partial oxidation of the oxidizable methionine motif to sulfoxide. Partial oxidation induces an α-helix to random coil conformational transition along with increased hydrophilicity. The DTT and methionine sulfoxide reductase (MSR) enzyme reducing agent couple can regenerate the hydrophobic thioether moiety, which promotes vesicle disruption and release of encapsulated cargoes. This class of MSR enzyme selective disrupted polypeptide vesicles might also provide a means for targeted cargo release in oxidatively stressed tissues. Again, an α-helix to random coil to α-helix conformational transition has been observed in poly(L-methionine) by stepwise oxidation of the methionine moiety to methionine sulfoxide to methionine sulfone.58 In a separate study, they showed a similar reversible secondary structural (α-helix ↔ random coil) switching capability of different poly(L-homocysteine) derivatives, poly(D-galactose-L-homocysteine) and poly(S-OEGylated-L-homocysteine) as poly(L-methionine), upon reversible oxidation of the alike thioether core structure of homocysteine to methionine. Tetraethylene glycol conjugated poly(L-homocysteine) exhibits temperature dependent phase transition which is lost after a partial oxidation to sulfoxide derivatives and remained water soluble over the wide range of temperatures tested.59 Recently, Fu et al. synthesized a new class of OEGylated thioether bond-containing poly(L-glutamate)s, PPLG-g-EGx (x = 2, 3, 4) through living/controlled ROP of clickable propargyl substituted NCA monomers followed by a subsequent thiol–yne photoaddition using methoxy oligo(ethylene glycol) thiol. The polypeptide, PPLG130-g-EG2, which was insoluble in water at room temperature, became water soluble and displayed reversible LCST behavior with a CP of 32 °C after 15% oxidation of the thioether moiety. The full extent of oxidation either to sulfoxide or to sulfone left the polymer water soluble at elevated temperatures up to 85 °C without exhibiting any phase transition behavior. Similarly, oxidation processes perturbed the thermo-responsive behavior of PPLG65-g-EG3 and PPLG65-g-EG4. Again, PPLG65-g-EG3 regains its reversible LCST behavior after reduction of the sulfoxide derivative with thioglycolic acid. But the CP is found to be somewhat higher than that of the parent PPLG130-g-EG3, indicating an incomplete reduction of the sulfoxide groups.60
Recently, Xu and co-workers developed a novel type of thermal and oxidation dual responsive hydrogel based on the methoxy poly(ethylene glycol)-b-poly(L-methionine) diblock copolymer mPEG-PMet.61 This block copolymer in aqueous solution exhibited a thermo-induced sol–gel phase transition depending on the polypeptide block length. As the poly(L-methionine) segment can respond to an H2O2-rich environment, oxidation-triggered gel erosion and release of the encapsulated dye molecule was observed in response to the H2O2 concentration. In addition, the hydrogel having antioxidative L-methionine residues showed good biocompatibility in vitro and in vivo and also possessed a unique cytoprotective ability against the damage of H2O2-induced oxidative stress. Moreover, the hydrogel could be made responsive to physiologically relevant ROS. This kind of smart hydrogels are expected to find applications in site-specific elimination of ROS, cell-based therapies with reduced cell damage, as well as drug carriers for diseases with ROS overproduction (Fig. 7).
 | ||
| Fig. 7 (A) The synthetic route of poly(ethylene glycol)-b-poly(L-methionine) (mPEG-PMet). (B) Oxidation of the mPEG-PMet diblock copolymer by treating with H2O2. (C) Photographs of the sol–gel transition with the increase of temperature and the gel disintegration by incubating with H2O2. Schematic illustration of ROS-responsive mPEG-PMet hydrogels for oxidation-triggered cargo release (D) and cytoprotective effects (E) under oxidative stress. Reprinted with permission from ref. 61. Copyright (2016) WILEY-VCH. | ||
L-3,4-Dihydroxyphenylalanine (DOPA) was used as an oxidizable entity to make oxidation-responsive copolypeptide vesicles. Poly(L-lysine-HBr)60-b-poly(L-3,4-dihydroxyphenylalanine)20 and poly(L-lysine-HBr)60-b-poly(L-3,4-dihydroxyphenylalanine0.25-random-L-leucine0.75)20 block copolymers were prepared by incorporation of oxidatively cross-linkable amino acid residues into hydrophobic α-helical segments of rod-coil diblock copolypeptides to give vesicles with dramatically improved membrane stability against freeze-drying, organic solvents, osmotic stress and complex media.62
3.4. Photoresponsive polypeptides
Among the various stimuli-responsive polymers, photoresponsive polymers have gained special attention in recent times since light stimulation can be spatiotemporally localized and controlled. Photo-responsive chemical moieties (e.g. coumarin, 2-nitrobenzyl, spiropyran, azide, 6 diazo, pyrene, cinnamyl and N-alkyldimethylmaleimide) can be positioned in the side-chain as pendant or onto the chain backbone in such a way that conformational changes can be triggered by exposure to UV, optical and near-infrared (NIR) electromagnetic radiations.63,64 Polypeptides being special classes of polymers with biodegradability and biocompatibility, different photo-active polypeptides have been used to construct photo-crosslinked polypeptide nanogels, a light-triggered drug delivery system, and photo-sensitive self-assembly. Photoresponsive polypeptide can be synthesized either through ROP of photochromic group-conjugated NCA monomers or attachment of light-active species to the side-chain by post-modification.Ohkawa and co-workers have reported a photochemical technique for the formation of photocrosslinked hydrogels from water soluble copolypeptides comprised of ornithine and coumarin-tethered ornithine-based monomers.65 Chen et al. have synthesized di- and triblock copolymers consisting of mPEG and the poly(L-glutamic acid-co-g-cinnamyl-L-glutamate) segment. Both these mPEG-b-P(LGA/CLG) copolypeptides self-assemble into a micelle with mPEG as shell and poly(L-glutamic acid-co-g-cinnamyl-L-glutamate) as core. Under 254 nm irradiation, the core of the micelles gets crosslinked due to photodimerization of the cinnamyloxy units, yielding nanogels (Fig. 8).66 These nanogels were used as a potent drug delivery system, where the block copolymer composition and the solution pH controlled the release behavior. Using a similar concept, Yan and co-workers were able to fabricate core-crosslinked micelles from the same diblock copolymer in aqueous medium. They showed that photocrosslinked micelles displayed a slow drug release in comparison with the non-crosslinked one.67
 | ||
| Fig. 8 Schematic illustration of micellization of mPEG-b-P(LGA/CLG) and the formation of the pH-responsive nanogel with UV-irradiation (A), photodimerization of the pendant cinnamyloxy groups (B), and synthesis procedure for mPEG-b-P(LGA/CLG) (C). Reproduced with permission from ref. 66. Copyright (2011) The Royal Society of Chemistry. | ||
In a different approach, various photo-cleavable moieties were attached to the polypeptide side-chain to develop photo-triggered drug delivery systems. For example, Kumar et al. fabricated fully biocompatible block copolymer micelles composed of PEO as a hydrophilic segment and a coumarin-conjugated polypeptide as a hydrophobic core. Taking advantage of the large two-photon absorption cross-section of the coumarin moiety, NIR-triggered disruption of the micelle was accomplished. They demonstrated that these photosensitive micelles could be used for NIR-induced release of an antibacterial drug, rifampicin, and an anticancer drug, paclitaxel, loaded into it.68 After that, Liu and Dong reported UV-triggered DOX release from photosensitive block copolymer micelles, where PEO chains act as shell and poly(S-(o-nitrobenzyl)-L-cysteine) (PNBC) formed the core. Cleavage of pendant ortho-nitrobenzyl groups upon UV exposure drives micellar disruption and a facile drug release.69 A L-lysine-based block copolypeptide hydrogel with UV light-triggered degradability has been reported using the photolabile ortho-nitrobenzyl protecting group.70
Polypeptides responsive to near-infrared light are more attractive than UV or visible light-responsive systems owing to a deeper penetration and a less harmful effect to tissues in the case of NIR. So, in order to circumvent the bad effect of UV light on cells, lanthanide-doped upconversion nanoparticles (UCNPs) were added as they can convert two or more low-energy photons (e.g., NIR light) into shorter wavelength emissions (e.g., UV and visible light). These NIR-responsive copolypeptide composite nanoparticles exhibited a fast and tunable NIR-triggered pulsatile or “on-off” drug release profile and an enhanced cytotoxicity, and they quickly penetrate into HeLa cells compared to free DOX.71
All the above photoresponsive polypeptides undergo light induced irreversible reactions or structural changes. Other photochromic molecules such as spiropyran (SP) and azobenzene moiety provided an opportunity to design and synthesize photoresponsive polypeptides with reversible structural change and hence phase transition in solution. The reversible light-responsive behavior of SP has been exploited by Mezzenga's group. They made spyropyran-decorated flower-like micelles in aqueous solution based on PEO and SP-bearing poly(L-glutamate) diblock copolymers. These self-assembled micelles could be disassembled upon exposure to UV-light and reassembled under visible light. This phenomenon is attributed to the light-induced isomerization of SP (closed form, hydrophobic motif) to merocyanine (open form, hydrophilic motif).72 Recently, a similar reversible photo-switchable behavior of SP has been utilized by fabricating UCNPs@ polymer nanocomposites through the self-assembly of the SP-functional amphiphilic polymers and encapsulation of the UCNPs in the core of the self-assemblies. These nanoparticles are not only NIR light-responsive but also pH-responsive. NIR radiation and acidic pH-triggered release of the hydrophobic molecule from the nanocomposites has been demonstrated. In addition, the cytotoxicity of the DOX loaded nanocomposites on U-87 MG cancer cells indicates that the loaded drugs can kill the cells effectively and the efficiency can be enhanced significantly upon NIR light irradiation.73 Again, azobenzene attached to the side-chain of a polypeptide may also respond to light, giving large photo-induced structural changes. On irradiation, the photochromic moiety azobenzene undergoes reversible stereochemical rearrangement from a trans to cis isomeric form, the direction being mainly dictated by the wavelength of the incident light. The azopolypeptides adopted α-helix conformation in trimethylphosphate. Although the helix content markedly depends on the azo content, it is not affected by the cis or trans forms of the side-chain azo moieties.74 However, due to the structural rigidity and strong π–π interactions, the resulting azopolypeptides often suffer from poor water solubility. That is why they are studied commonly in the form of random copolymers together with other water soluble polypeptides. Most frequently, the azo content in these polymers is reported to be below 50%; as a result, their photo-responsiveness usually produces a less prominent effect than polymers with higher azo contents. To examine the photo-switchable behavior of homo-azopolypeptides, Xiong and co-workers designed and synthesized OEG-installed azobenzene-bearing polypeptides of different OEG lengths, P(OEGm-Azo)n (m = 2, 4, or 6; n = feeding monomer/initiator ratios) with improved solubility in various solvents, especially water. At room temperature under ambient light, the polymer trans-P(OEG6-Azo)10 and trans-P(OEG6-Azo)50 adopted β-sheet and α-helical conformations, respectively, in trifluoroethanol. Upon UV-irradiation trans–cis isomerization takes place, which forces both the polypeptide chains cis-P(OEG6-Azo)10 and cis-P(OEG6-Azo)50 to adopt disordered conformations. Notably, this conformational transition was reversible upon heating the UV-treated cis-polymers at 70 °C. Additionally, P(OEG6-Azo)ns exhibited irreversible thermo-responsiveness in water.75 Interestingly, multi-stimuli-responsive polypeptides that could be used as nanocarriers triggered by the physiological cues in diseased sites or some clinic-related stimuli (e.g., light) might have high priority for potential clinical therapies. To this end, Dong's group designed and fabricated a novel multi-stimuli-responsive polypeptide-based vesicle (polypeptidosome), which could sense the combined effect of light and redox stimuli via two different paths (Fig. 9). The polypeptide segment of the designed block copolymer possess unique chemical and secondary structures, which contain (a) the pendant photo-sensitive o-nitrobenzyl (NB) groups, (b) the oxidizable thioether moieties, (c) the photocaged redox thiol groups (SH) on the parent poly(L-cysteine) (PLC) backbone, and (d) the tunable conformation (ordered β-sheet vs. disordered random coil), which enable the self-assembled polypeptidosome to show multiple responses. The anticancer drug DOX from the polypeptidosome could be released in a controlled or on–off manner by UV irradiation and the combination of UV irradiation and H2O2 oxidation produced a synergistic effect. Moreover, the combined effect of 3 min UV irradiation and H2O2 oxidation induced a large effect and a lower half-maximal inhibitory concentration (IC50) of 3.80 μg DOX equiv. per mL compared to 5.28 μg DOX equiv. per mL from H2O2 trigger alone.76
 | ||
| Fig. 9 Schematic depiction of the multi-responsive transformation of the polypeptidosome in an aqueous solution. Reprinted with permission from ref. 76. Copyright (2014) WILEY-VCH. | ||
Dong's group also prepared a series of comb-like graft copolypeptides utilizing photocleavage reaction on the side-chain groups of poly(S-(o-nitrobenzyl)-L-cysteine) (PNBC) followed by Michael-type thiol–ene addition reactions with three different hydrophilic acrylate moieties. The resulting comb-like polypeptide self-assembled into vesicles and/or micelles, of which the polypeptide vesicles exhibited sequential photo- and redox sensitivity under different stimulations. Notably, the DOX-loaded polypeptide nanoparticles presented photo- and reduction-sensitive drug release and triggered cytotoxicity.77
3.5. Glucose-responsive polypeptides
In recent times, there is growing impetus to the development of glucose-sensitive polymeric drug delivery systems, which can continuously and automatically release insulin in response to the elevated level of blood glucose of diabetic patients with minimal patient intervention.78,79 Among the many classes of polymer-based delivery systems, polypeptides are superior to others owing to their high biocompatibility and biodegradability. Two different strategies have been employed for the preparation of glucose-responsive polypeptides: (a) by incorporation of phenylboronic acid (PBA) moieties into the polypeptide side-chain, which is known to be an intelligent saccharide receptor due to its unique reversible six member cyclic boronic ester chelate complex forming ability with cis-diols and (b) by inclusion of pH-sensitive or oxidation-responsive units that can sense glucose indirectly; i.e. glucose oxidase (GOD) converts glucose into gluconic acid causing a decrease in local pH, which in turn affects pH-responsive units. By using the first strategy, Zhao et al. synthesized PBA functionalized block copolymers, monomethoxy poly(ethylene glycol)-b-poly(L-glutamic acid-co-N-3-L-glutamylamidophenylboronic acid) (mPEG-b-P(GA-co-GPBA)), by modifying mPEG-b-PGA with 3-aminophenylboronic acid (APBA). The as-prepared diblock copolymers self-assembled into micelles and could encapsulate insulin in phosphate buffer at physiological pH (pH 7.4). In vitro insulin release from the micelles could be finely regulated by glucose triggering, i.e. less insulin was released under a healthy blood glucose level (1.0 g L−1), while fast release occurred under a diabetic blood glucose level (above 2.0 g L−1).80 Thereafter, Liu and co-workers developed a complex polymeric micelle (CPM) with a novel core–shell–corona structure through the self-assembly of two types of diblock copolymers, poly(ethylene glycol)-b-poly(aspartic acid-co-aspartamidophenylboronic acid) (PEG-b-P(Asp-co-AspPBA)) and poly(N-isopropylacrylamide)-b-poly(aspartic acid-co-aspartamidophenylboronic acid) (PNIPAM-b-P(Asp-co-AspPBA)). The resultant CPM exhibited repeated on–off release of insulin regulated by the glucose level, and could also effectively protect the encapsulated insulin against protease degradation.81 They also synthesized core–shell–corona (CSC) complex micelles by adding a glucosamine (GA)-containing block copolymer, PEG-b-P(Asp-co-AspGA), into core–shell micelles with a PNIPAM core and a P(Asp-co-AspPBA) shell above the LCST of PNIPAM. The crosslinking reaction between PBA- and GA-containing blocks resulted in the formation core–shell–corona complex micelles, which were transformed into vesicles with a proposed structure of a PNIPAM inner cavity, a crosslinked P(Asp-co-AspPBA)/P(Asp-co-AspGA) shell, and a PEG outer corona. Fluorescein isothiocyanate (FITC)-insulin was encapsulated into the polymer vesicles and glucose-triggered drug release in both continuous and on–off manners was successfully examined.82 A similar kind of vesicles was fabricated based on the complexation between glucosamine (GA)-containing block copolymer poly(ethylene glycol)-block-poly(aspartic acid-co-aspart-glucosamine) (PEG45-b-P(Asp-co-AspGA)) and PEG114-b-P(Asp-co-AspPBA) via α-cyclodextrin/PEG inclusion complex as the sacrificial template. Vancomycin as a model drug was loaded into the polymer vesicles and sugar-triggered drug release was successfully performed.83 A polypeptide nanogel was prepared by crosslinking glycopolypeptides poly(ethylene glycol)-block-poly-γ-benzyl-L-glutamate-co-(γ-propargyl-L-glutamate-graft-glucose) (mPEG-b-P(BLG-co-(PLG-g-Glu))) using adipoylamidophenylboronic acid. The nanogel showed excellent glucose-sensitive insulin release through a competitive binding mechanism with the conjugated glucose.84 In another study, Kim et al. prepared a glycol chitosan (GC)/sodium alginate (SA)-poly(L-glutamate-co-N-3-L-glutamylphenylboronic acid) (PGGA) graft copolymer (GC/SA-PGGA) double-layered nanogel through an isotropic gelation method and electrostatic interaction between GC and SA-PGGA. In the presence of glucose, boronic acid groups of PGGA formed complexes with the glucose, which induced swelling of the bilayer nanogel, resulting in insulin release. In addition, insulin-loaded GC/SA-PGGA double layered nanogels could controllably release the encapsulated insulin at high glucose levels in vivo, and maintain low glucose levels for almost 3 h.85 Utilizing the second approach, Gu and co-workers have made significant contributions on glucose and multistimuli-responsive release of insulin for the treatment of diabetes. A biomimetic polymersome-based nanovesicle was fabricated from the self-assembly of the diblock copolymer consisting of PEG and Ketal-modified polyserine PEG-poly(Ser-Ketal). Cargoes, including recombinant human insulin, GOD and catalase, were faithfully entrapped in the core with negligible release through the closely packed bilayer membrane. Glucose being neutral and small in size, it was easy to transport it passively across the bilayer membrane of the polymersome. Once its local concentration increases, glucose diffuses across the membrane and interacts with GOx in the core, which leads to the catalytic conversion of glucose to gluconic acid. Moreover, catalase assists GOD's catalysis by breaking down the harmful byproduct H2O2 and providing oxygen for further promoting GOD's catalysis. Gluconic acid decreases the local pH value and hydrolyzes the pendant acid-labile ketal on the polyserine segment of PEG-poly(Ser-Ketal). Thus the resulting PEG-polyserine becomes water soluble, which causes nanovesicle disruption, leading to subsequent insulin release (Fig. 10A and B).86 The same group extended the study by incorporating phenylboronic ester (PBE)-conjugated polyserine as the H2O2-sensitive entity.87 Recently, a hypoxia and H2O2 dual-sensitive polypeptide-based nanocarrier was designed for glucose-sensitive drug delivery. The dual-responsive block copolymer, consisting of PEG and polyserine modified with 2-nitroimidazole (NI) via a thioether moiety (designated PEG-poly(Ser-S-NI)), self-assembled into bilayer vesicles with insulin and GOx encapsulation. H2O2 converted thioether into sulfone and hypoxia bioreduced NI into 2-aminoimidazole, enhancing the hydrophilicity of the polymer, which resulted in disruption of the vesicles and insulin release (Fig. 10C and D).88 | ||
| Fig. 10 (A) Schematic of the enzyme-based glucose-responsive nanovesicle. (B) The chemical structure of the pH-sensitive diblock copolymer PEG-poly(Ser-Ketal). Reprinted with permission from ref. 86. Copyright (2014) American Chemical Society [Direct link: <http://pubs.acs.org/doi/abs/10.1021/bm500364a>; further permissions related to the material excerpted should be directed to the ACS]. (C) Hypoxia and H2O2 dual-sensitive polymersome-based vesicles composed of PEG-poly(Ser-S-NI), (D) GOx converts glucose into gluconic acid and acidifies the aqueous core of polymersome nanovesicle, leading to hydrolysis of the polymeric bilayer shell and subsequent dissociation of vesicles. Reprinted with permission from ref. 88. Copyright (2017) American Chemical Society. | ||
3.6. Enzyme-responsive polypeptides
Enzyme-responsive polypeptides are particularly attractive since overexpression of enzymes has frequently been implicated in inflamed or tumor tissues, which allows preferential drug/gene delivery to the diseased sites by enzyme-triggered polypeptide-based assemblies. The common strategy for fabricating enzyme-responsive polypeptide-based assemblies is to covalently attach protein-specific ligand or enzymatic substrate moieties to amphiphilic copolymers. Thayumanavan's group explored protein-induced supramolecular disassembly of amphiphilic polypeptide nanoassemblies. An amphiphilic random copolypeptide is targeted from ROP of three different substituted glutamic acid based NCA monomers. The integration of biomolecules such as peptides, nucleic acids, and polysaccharides with synthetic polymers can further broaden the realm of enzyme-responsive polymers. Benzyl ester groups provide hydrophobicity, OEG moieties avoid nonspecific and electrostatic interactions with proteins and benzenesulfonamide ensures specific binding with a targeted protein, rendering an amphiphilic polypeptide. The resultant polypeptide could self-assemble into some nanostructures sequestering the hydrophobic guest molecule in aqueous media. They demonstrated that only bovine carbonic anhydrase II (bCA-II) can bind to the ligand, which causes disassembly of the nanostructures and release of the guest molecule (Fig. 11A and B).89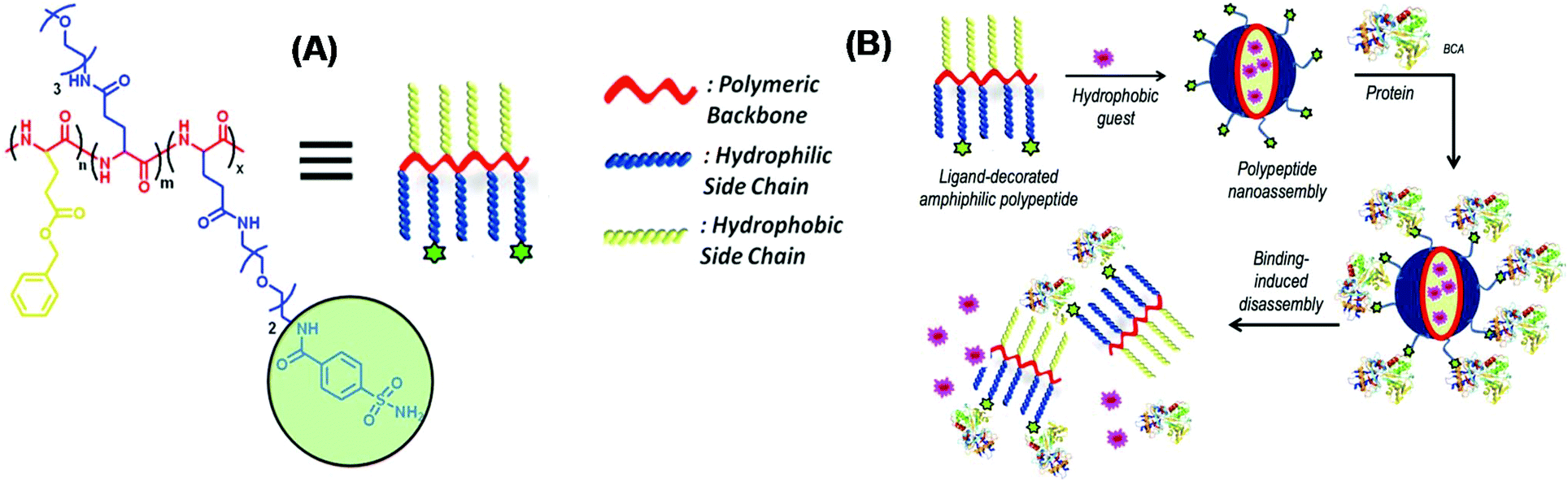 | ||
| Fig. 11 (A) Chemical structure of the enzyme-responsive amphiphilic polypeptide, (B) Schematic illustration of the protein binding induced disassembly of a polypeptide nanoassembly. Reprinted with permission from ref. 89. Copyright (2015) American Chemical Society. | ||
Sun et al. realized enzyme-induced hydrogelation of phosphatase-pendant polytyrosine, P(pOEt-Tyr). Bisamine-functionalized PEG (NH2-PEG-NH2) macroinitiator initiated ROP of O-diethylphospho L-tyrosine N-carboxyanhydride (pOEt-TyrNCA) enabled the synthesis of the triblock copolymer P(pOEt-Tyr)15-b-PEG-b-P(pOEt-Tyr)15. After deprotection the generated copolymer P(pTyr)15-b-PEG-b-P(pTyr)15 became water-soluble, which formed hydrogels (denoted TBP15-ALP hydrogels) in Tris-HCl buffer (pH ∼ 8.0) at 37 °C by protein tyrosine phosphatase such as alkaline phosphatase (ALP)-catalyzed dephosphorylation. The stiffness of the gel is reinforced by the addition of horseradish peroxidase and hydrogen peroxide that covalently crosslink phenol rings (Fig. 12).90
 | ||
| Fig. 12 (A) Synthesis of the triblock polymer TBP15, (B) photographs and cartoon illustration of TBP15 solution, TBP15-ALP (8 wt%), and TBP15-HRP (4 wt%) hydrogels. Reprinted with permission from ref. 90. Copyright (2015) American Chemical Society. | ||
In addition to the above covalent approach, there is also a supramolecular strategy for the preparation of enzyme-responsive polypeptide-based assemblies, which feature the non-covalent integration of enzymatic substrates into the assemblies. In this strategy, non-covalent interactions instead of covalent chemical bonds between the enzymatic substrates and the polymer building blocks enable the formation of superamhiphiles. This non-covalent approach, to some extent, saves the labor of organic synthesis and simplifies the introduction of complicated enzyme-responsive moieties, thus providing a promising alternative route to achieve enzyme-controlled self-assembly of polymers. Zhang and co-workers reported an enzyme-responsive polymeric superamphiphile by simply mixing a double-hydrophilic block copolymer, methoxy-poly(ethylene glycol)114-b-poly(L-lysine hydrochloride)200, and a natural multicharged enzyme-responsive molecule, adenosine 5′-triphosphate (ATP), in water. Negatively charged ATP molecules could noncovalently crosslink with the positively charged polylysine segments of the block copolymer, which introduces hydrophobic adenine groups resulting in the formation of self-assembled aggregates. Upon addition of calf intestinal alkaline phosphatase (CIAP), the multiple-charged ATP is hydrolyzed into single-charged phosphate and a neutral adenine group. Hence, the block copolymer–ATP complex dissociates, accompanied by aggregate disassembly and subsequent guest molecule release.91 Using the same diblock copolymer, they have reported pH and enzymatic double-stimuli-responsive multi-compartment supra-amphiphilic polymeric micelles. Pyridoxal phosphate (PLP), which is a natural water-soluble vitamin containing an aldehyde group and a phosphate group with negative charges, was used as an addendum to obtain the target supra-amphiphilic polymer. Faster disassembly was observed when two kinds of stimuli were exerted at the same time, which may be useful for cancer therapy.92 Börner and other groups have extensively applied various enzymes (phosphatase,93 tyrosinase94,95 or protease96) to regulate peptide-guided self-assembly, as well as to activate the formation of coatings.97
3.7. Protein-responsive polypeptides
We have discussed the enzyme-responsive polypeptide in a previous section. Although all enzymes are proteins but we made a separate section on protein-responsive polypeptides, since there are some proteins which do not possess enzymatic activity. One such protein with non-enzymatic activity is lectin. Lectins are carbohydrate binding plant proteins, which can interact with cell surface glycoproteins and are highly specific for sugar moieties. Their recognition motif on the cellular level has triggered research interest in glycopolypeptides as simplified synthetic counterparts of glycoproteins to study the lectin-responsive behavior, as well as their use as scaffolds for tissue engineering and as drug carriers. The most studied lectins in the investigation of glycopolypeptides are Concanavalin A (Con A) and Ricinuscommunis agglutinin (RCA). The lectin Con A binds with mannosyl and glucosyl groups; in contrast, RCA specially and selectively binds with galactosyl moieties. Just like the synthesis of other stimuli-responsive polypeptides, two different approaches have been employed for the synthesis of glycopolypeptides: (a) ROP of a glycosylated NCA monomer or (b) post-modification of the synthetic polypeptide precursors. Sugar moieties can be incorporated into the polypeptide chain either through a native O-linkage or a C-linkage. Okada's group was the first to prepare glycopolypeptides utilizing O-linked glyco-serine NCA in a controlled manner.98 Later on, Sen Gupta's group synthesized O-glycosylated lysine-NCA monomers, per-O-benzoylated-D-glyco-L-lysine99 and per-O-acetylated-D-glyco-L-lysine100 NCA from a stable glycosyl donor and a commercially available protected amino acid in very high yield. These sugar-based NCAs bearing protected mannose, galactose and lactose were easily polymerized to produce well defined, high molecular weight homoglycopolypeptides and diblock co-glycopolypeptides. Then, the polymers were deprotected to give water-soluble glycopolypeptides. They investigated the binding of the mannose-appended glycopolypeptide poly(α-manno-O-lys) with the lectin Con-A using precipitation and hemagglutination assays as well as by isothermal titration calorimetry. The binding affinity was found to be nearly the same between polypeptides having enantiomerically pure L-lysine and the corresponding racemic DL-lysine backbone. Wenz et al. synthesized a well-defined glycopolypeptide, O-glycosylated polylysine bearing per-acetylated sugars (glucose, mannose, galactose) via a thiourea linker. The synthesized galactosylated polypeptide showed special interaction with human T lymphocytes and internalized into the cytoplasm of T cells as assessed by flow cytometry and fluorescence microscopy.101 On the other hand, Deming and co-workers pioneered the synthesis of C-linked glycopeptides. Syntheses of the glycopeptides from protected β-D-glucose, β-D-galactose, or β-D-mannose-based lysine NCA monomers were achieved using (PMe3)4Co as a catalyst.102The post-polymerization modification approach provides a facile way to prepare glycopolypeptides, especially by utilizing highly efficient “click” chemistry, including copper-catalyzed azide–alkyne cycloaddition reaction,103 and thiol–ene and thiol–yne reactions.104 For example, a series of glycopolypeptides were prepared by combining the pendant alkyne groups of poly(γ-propargyl-L-glutamate) with azido sugars using Cu(I) catalyzed cycloaddition “click” reaction. In addition, glycopolypeptides of low dispersity and controlled length were also produced via anionic ROP of protected cyclic sugar-derived β-lactam monomers. Deprotected polymer possesses a regular secondary structure in aqueous solution and has an ability to selectively bind with natural carbohydrate receptors, Con A.105
The overexpression of the specific mannose binding receptor MRC2 (mannose receptor C-type 2) on MCF-7/MDA-MB-231 breast cancer cells has stimulated mannose-containing macromolecules for targeted drug delivery into the cancerous cells. Sen Gupta and co-workers prepared diverse morphologies such as micelles, nanorods and polymersomes from biocompatible miktoarm star copolymers comprising mannose-conjugated-glycopolypeptide and poly(ε-caprolactone). From Con A binding turbidity assay it was concluded that the mannose moieties present at the surface of the nanorods/polymersomes are accessible for interaction with cell-surface receptors. The hollow nanorods and polymersomes could sequester both hydrophobic as well as hydrophilic dyes. Rapid cellular endocytosis into MDA-MB-231 cells was observed within 2.0 h for mannosylated Rhodamine B octadecyl ester (RBOE) dye-encapsulated polymersomes and nanorods at 100 μg mL−1 concentration. The uptake efficiency of mannosylated polymersomes was compared with galactosylated polymersomes. Fluorescence intensity analysis showed that mannosylated polymersomes were preferentially taken up by MDA-MB-231 cells with respect to galactosylated polymersomes. This clearly indicates that ∼45% of the cellular uptake occurred through the overexpressed mannose-specific MRC2 receptor (Fig. 13).106 Recently, the same group designed and synthesized end-functionalized mannose-6-phosphate glycopolypeptides (M6P-GP) for lysosome targeting. The cellular uptakes of M6P-GP were conducted with cancerous (MDA-MB-231, MCF-7) as well as noncancerous (L929) cell lines. Fluorescence intensity quantification study revealed ∼3-fold higher uptake in MCF-7 cells compared to MDA-MB-231 and L929, which correlates well with the increased mannose-6-phosphate-specific CI-MPR receptors present on MCF-7 cells.107
 | ||
| Fig. 13 (a) General chemical structures of glycopolypeptide-based miktoarm star copolymers and nanostructures obtained by tuning the hydrophilic and hydrophobic block length. (b) Fluorescence microscopy images of MDA-MB-231 cells treated with RBOE-encapsulated mannosylated polymersomes, (c) treated with RBOE-encapsulated galactosylated polymersomes and (d) fluorescence intensity analysis after cellular uptake. Reprinted with permission from ref. 106. Copyright (2016) American Chemical Society. | ||
A systematic study of the effect with regard to the position of the glyco units in the polypeptide on the conformation and biological properties of synthetic glycopolypeptide has been done. To this end, Lavilla et al. prepared several glycopolypeptides with the same overall composition and number of galactose units, but having these distributed in different block sequences, by a block-sequence-controlled ROP approach followed by selective functionalization of preselected positions within the polypeptide chain. Circular dichroism measurements revealed some dependence of the secondary structure on the primary composition of the glycopolypeptides at physiological pH. While statistical, diblock, and tetrablock glycopolypeptides adopted a random coil conformation, the octablock glycopolypeptide was mostly α-helical. Although all galactopolypeptides were biologically active and have affinity to lectins, the extent of binding was shown to be dependent on the position of the galactose units and, thus, the primary glycopolypeptide structure. The octablock glycopolypeptide favored interaction with lectin RCA120 while the tetrablock polypeptide showed strongest binding activity to Galectin-3, suggesting glycocoding sensitivity of different lectins.108
3.8. Gas-responsive polypeptides
Among the various stimulus, gases are easy to operate in large volume systems; thus gas responsive polymers show promise for use in industrial applications. Tran et al. have developed CO2-responsive poly(amino acids) systems based on polyaspartamide: (a) novel CO2-responsive hydrogels and (b) amphiphilic polyaspartamide. The L-arginine-appended polyaspartamide was prepared by a facile aminolysis of poly(succinimide) with the semi-essential amino acid to install amidine or guanidine functionality, which showed reversible CO2-responsive characteristics. A hydrogel was made from poly(2-hydroxyethylaspartamide) derivative modified with the L-arginine unit (PHEA-Larg), and crosslinked by hexamethylene diisocyanate in the presence of dibutyltin dilaurate catalysts. The hydrogel not only possessed good gel strength but also showed reversible CO2 absorption characteristics. Again, an amphiphilic polyaspartamide derivative containing hydrophobic long alkyl moiety (octyl) and L-arginine unit was synthesized, which self-assembled into nanoparticles. The size and zeta potential of the nanoparticles could be reversibly changed by alternating bubbling of CO2 and N2 into the aqueous solution (Fig. 14).109 | ||
| Fig. 14 Synthesis of the CO2-responsive polypeptide. | ||
The same group expanded the study by incorporating histamine pendants as the CO2-responsive moiety in the hydrogel network.110 These hydrogels are not only CO2-sensitive, but also respond to changes in pH, and exhibit the ability to absorb metal ions such as Pb2+, Cu2+ and Ni2+ in water. Recently, Tang's laboratory showed SO2-responsive phase transition behavior of an α-helical random copolypeptide based on poly(L-glutamate) bearing pyridinium tetrafluoroborate (PyBF4) and OEG pendants. A multistep post-polymerization (including nucleophilic substitutions, copper mediated [2 + 3] alkyne–azide 1,3-dipolar cycloaddition, and ion-exchange reaction) on poly(γ-3-chloropropyl-L-glutamate) afforded the target amphiphilic random copolypeptide (PPLG-PyBF4-r-OEG), where the PyBF4 moiety offers hydrophobicity and OEG pendants hydrophilicity. The reported polymer showed SO2-reversible solution phase transition behavior because of the “crosslinking effect” of SO2. From the 1H NMR study it was observed that SO2 interacted with triazole groups and induced polymer aggregation and consequently solution phase transition. As a control experiment, they synthesized (a) PyBF4 and OEG grafted copoly(L-glutamate) devoid of the triazole moiety, and (b) triazole groups and OEG pendant poly(L-glutamate) without the PyBF4 moiety. No SO2-induced solution phase transition behaviour was observed in either case, indicating the indispensability of the PyBF4 pendants.111
3.9. Metal ion-responsive polypeptides
Like various biologically relevant external stimuli-triggered structural or conformational changes in polypeptides, metal ions also have been found to induce secondary structures of polypeptides. In metalloproteins, metal ions are usually coordinated by nitrogen, oxygen or sulfur centers belonging to amino acid residues of the protein. These donor groups are often provided by side-chains on the amino acid residues. Especially important are the imidazole substituents in histidine residues, thiolate substituents in cysteine residues, and carboxylate groups provided by aspartate. Recently, Bonduelle et al. found that synthetic poly(L-glutamic acid) polymers underwent reversible helix-to-coil transitions at pH = 7 under aqueous conditions upon coordination to Zn species.112 Kühnle et al. reported calcium ions (Ca2+) as bioinspired triggers to reversibly control the coil-to-helix transition in peptide–polymer conjugates.113,1143.10. Other responsive polypeptides
Zhang's group reported on the synthesis of cyclodextrin (CD)-responsive micelles based on poly(ethylene glycol)-polypeptide hybrid block copolymers as drug carriers. The four-armed block copolymer poly(ε-adamantane-L-lysine)2-b-poly(ethylene glycol)-b-poly-(ε-adamantane-L-lysine)2 has been synthesized from the tetraamino-modified PEG macroinitiator initiated ROP of adamantly appended NCA monomer. This as-made copolymer could self-assemble into a core–shell structure in aqueous solution and entrapped the hydrophobic anti-cancer drug DOX. These micelles underwent β-CD-triggered disassembly, resulting in the release of the encapsulated drug molecule from the micelles because of the host–guest interaction between the adamantane moieties and the β-CD.115 Recently, Nguyen et al. synthesized a new class of smart polypeptide polymers, called nucleopolypeptides, by post-polymerization coupling of the poly(γ-propargyl-L-glutamate) backbone with azide-terminated thymidine nucleobases. The as-prepared polypeptide having thymidine lateral chains exhibited a secondary structural transition from α-helix to β-sheet upon nucleic acid interaction and by its sequence variation.1163.11. Multiple stimuli-responsive polypeptides
Gao and Dong fabricated a novel reduction- and thermo-sensitive silica-crosslinked polypeptide hybrid micelle (CCM) for the triggered and intracellular release of the anticancer drug DOX. It was demonstrated that the DOX-loaded CCMs showed a greatly attenuated and reduction-triggered drug release profile compared to non-crosslinked counterparts and could quickly be internalized by HeLa cells.117 Unlike synthetic nanostructures with a single building block, supramolecular self-assemblies consisting of two or more building blocks are more advantageous as they could integrate more functions in a programmable manner. A dual pH and reduction-responsive fluorescent supramolecular drug delivery system has recently been reported by Cheng and co-workers.118 Fluorescent perylenediimide (PDI)-cored star cationic polylysine attached with camptothecin (CPT) through a reduction-responsive bond was integrated with a charge-reversal anionic copolymer to overcome the cationic cytotoxicity of polylysine generating P1@P2 (Fig. 15). In the tumor extracellular microenvironment, pH-induced disruption of P1@P2 self-assembly and intracellular GSH-triggered CPT release allowed the drug to show its cytotoxicity in the apoptosis of cancer cells (Fig. 15).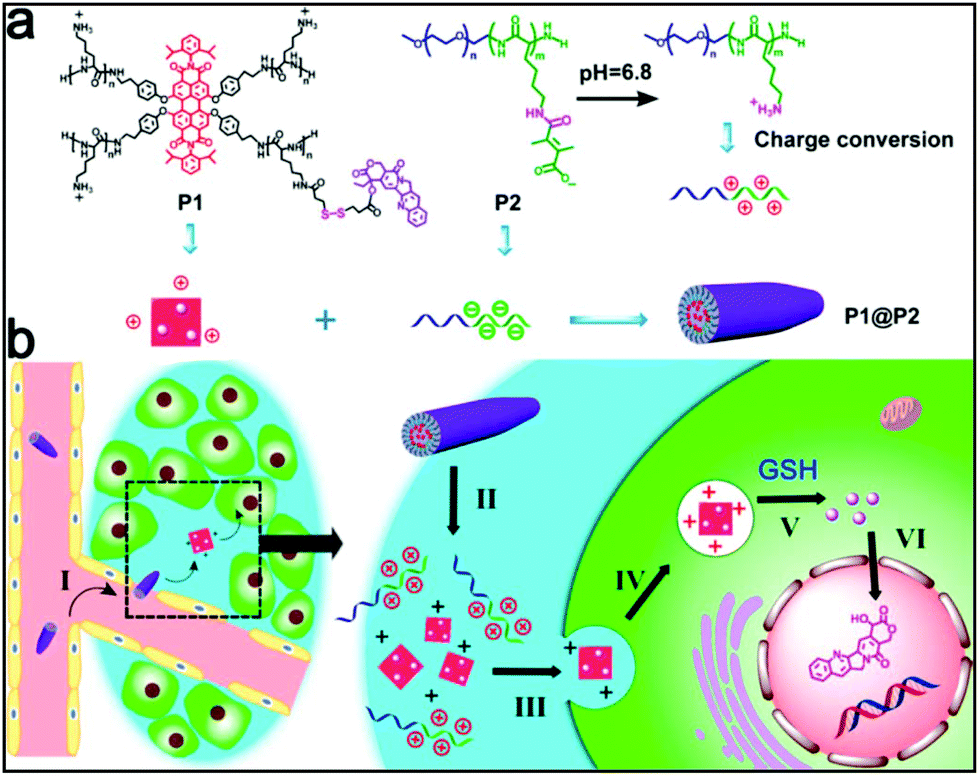 | ||
| Fig. 15 (a) Chemical structures of cationic star polymer (P1), charge-reversal anionic copolymer (P2) and their P1@P2 supramolecular complex, and (b) illustration of the CPT delivery pathway using the P1@P2 complex. Reprinted with permission from ref. 109. Copyright (2017) American Chemical Society. | ||
Very recently, homopolypeptide with pH and redox dual responsiveness has been developed by ROP of S-(2-(methoxycarbonyl)ethyl)-L-cysteine based NCA monomer and subsequent hydrolization. The doubly-responsive polymer has been utilized as a new “smart” single-walled carbon nanotube (SWCNT) dispersion agent, and the resultant SWCNT dispersed aqueous solution showed pH and oxidation dual responsiveness.119
4. Main-chain amino-acid-based polymers other than polypeptides
In the last decade, degradable non-peptide synthetic polymers with stimuli-responsive behavior derived from L-amino acid have attained significant importance due to their excellent biocompatibility and structural diversity. Although amino acid-based non-peptide polymers such as poly(ester-amide)s, poly(α-hydroxyurethane)s, poly(β-amino esters), poly(ester-urea), poly(ester-urea-urethane) and poly(acetal-urethane) were reported, only very few possess stimuli-responsive characteristics. Jayakannan's research group is active in fabricating amino acid-based various stimuli-responsive non-peptide polymeric nanocarriers for drug delivery applications by a melt condensation synthetic approach. A unique polymeric material based on L-cystine-derived polyester with reduction degradability, biocompatibility, and an ability to produce supramolecular helical structures in a single system has been reported.120 Natural L-aspartic acid-derived enzyme and pH dual responsive biodegradable polymer nanocarriers for multidrug delivery to cancer cells have been reported by the same group.121 Multiple anticancer drugs such as DOX, topotecan and curcumin were loaded into the polymer scaffold and their cytotoxicty was tested in both cervical (HeLa) and breast cancer (MCF-7) cell lines. The transformation of side-chain tert-butyloxycarbonyl (Boc) urethanes into cationic –NH3+ in the acidic endosomal environment disassembled the polymer nanoparticles (pH trigger-1). The biodegradation of the aliphatic polyester backbone by esterase enzyme ruptured the nanoassemblies and released the drugs in the cytoplasm (trigger-2). This single polymer scaffold showed unique dual pH and enzyme-responsiveness for delivering multiple anticancer drugs in cancer treatment (Fig. 16). Recently, Jayakannan's group developed an L-tyrosine-based enzyme-responsive polymer nanocarrier for the delivery of multiple anticancer drugs122 and an L-aspartic acid-based enzyme-responsive fluorescent amphiphilic polymer for color-tunable intracellular bioimaging in cancer cells.123 | ||
| Fig. 16 Aspartic acid-based pH-responsive and enzyme biodegradable polymer nanoassemblies for multiple-drug delivery at the intracellular compartments to cancer cells. Reprinted with permission from ref. 121. Copyright (2016) WILEY Periodicals, Inc. | ||
Lu and co-workers reported the synthesis of photosensitive polyesters from amino acids. Tyrosine-derived alkyne-functional O-carboxy anhydride monomer (OCA) was block copolymerized with mPEG and subsequently post-modified with an azide-containing azobenzene derivative to afford an amphiphilic diblock copolymer, which self-assembled to spherical micelles. The photoisomerization behavior of the azobenzene group has been exploited for reversible light-sensitive disruption and reconstruction of the micelles.124 The same group expanded the study by incorporating o-nitrobenzyl ester as the photo-responsive group.125 In addition, they studied the reversible self-assembly and disassembly behavior of block copolymer micelles in aqueous solution under alternative UV and visible light irradiation. Here spiropyran chromophore was introduced into the side chain on the poly(α-hydroxy acids) units on the same block copolymer skeleton.126
5. Side-chain amino acid-based stimuli-responsive polymers
Side-chain amino acid-based polymers have been synthesized by various controlled living polymerization techniques including atom transfer radical polymerization (ATRP), nitroxide-mediated polymerization (NMP), reversible addition–fragmentation chain transfer polymerization (RAFT), ROP method, ring opening metathesis polymerization (ROMP), living anionic polymerization, living cationic polymerization, acyclic diene metathesis (ADMET) polymerization and metal catalyzed metathesis or insertion polymerization. The monomers for side-chain amino acid pendant polymers are generally made by coupling the amino acids either through the –COOH functional group or –NH2 group to a polymerizable entity (Fig. 17). Considerable effort has been devoted to construct single or multi-stimuli responsive polymers with other functionalities such as chirality, bioactivity, etc. Table 2 summarizes side-chain amino acid-based stimuli-responsive polymers and their associated property changes upon the action of different stimuli. In the following section, we will discuss recent advances in the area of side-chain amino acid containing stimuli responsive polymers.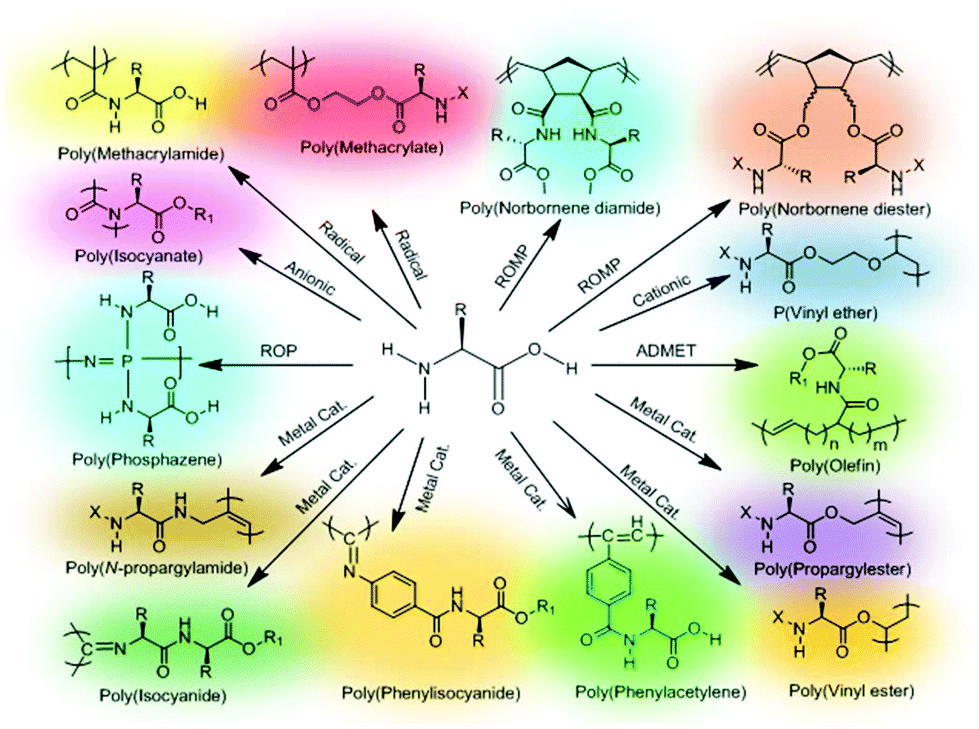 | ||
| Fig. 17 Different polymerization methods for the synthesis of various side-chain amino acid functional polymers. | ||
| Stimulus | Structure | Name/abbreviation | Response | Ref. |
|---|---|---|---|---|
| pH |
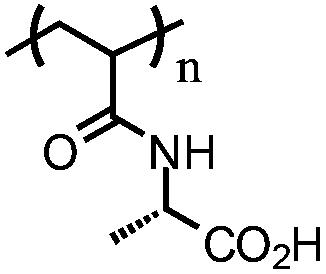
|
Poly(N-acryloylalanine) (PAAL) | Weak polyelectrolyte; soluble at basic pH | 137 |

|
Poly(N-acryloylvaline) (PAVAL) | Weak polyelectrolyte; soluble at basic pH | 138 | |
|
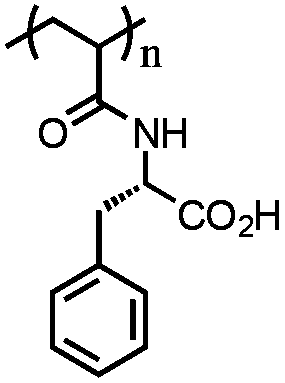
Poly(N-acryloylphenylalanine) (PAPHE) | Weak polyelectrolyte; soluble at basic pH | 141 | |

|
Poly(N-acryloyl-L-proline) [poly (A-Pro-OH)] | Weak polyelectrolyte; soluble at basic pH | 124 | |

|
Poly(+NH3-Gly-HEMA) | pH-Dependent swelling of cross-linked hydrogels | 167 | |

|
Poly(+NH3-Ala-HEMA) | pH-Reversible solubility transition; phase transition pH (pHtr) = 9.8 | 145 | |

|
Poly(+NH3-Val-HEMA) | pH-Dependent swelling of cross-linked hydrogel | 167 | |

|
Poly(+NH3-Phe-HEMA) | pH-Reversible solubility transition; pHtr = 5.4 | 145 | |
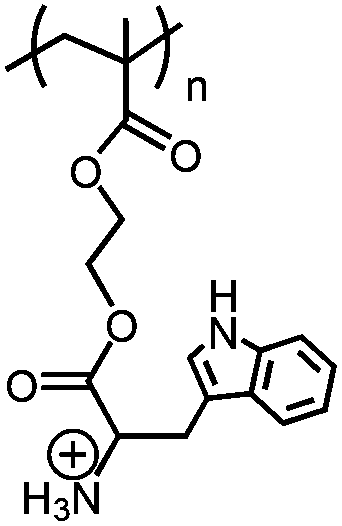
|
Poly(+NH3-Trp-HEMA) | pH-Reversible solubility transition; pHtr = 5.1 | 147 | |

|
PGluDMA | Zwitterionic polymer surface at neutral pH | 178 | |
| pH/CO2 |
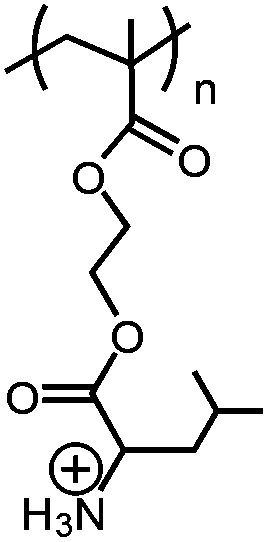
|
Poly(+NH3-Leu-HEMA) | pH-Reversible solubility transition; pHtr = 6.7 | 146 |
| pH/CO2 |
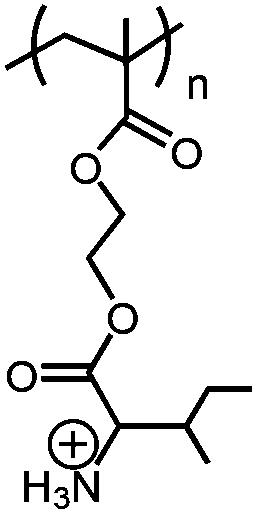
|
Poly(+NH3-Ile-HEMA) | pH-Reversible solubility transition; pHtr = 6.8 | 146 |
| Thermo |

|
Poly(N-acryloyl-L-proline methyl ester) [poly(A-Pro-OMe)] | Reversible LCST transition; LCST = 18 °C | 122 |

|
Poly-(N-acryloyl-4-trans-hydroxy-L-proline), [poly(A-Hyp-OH)] | Weak polyelectrolyte; soluble at basic pH; exhibits UCST upon block copolymerization with poly (A-Pro-OH) | 125 | |

|
Poly(N-acryloyl-L-hydroxy proline methyl ester) [poly(A-Hyp-OMe)] | Reversible LCST transition; LCST = 49.5 °C | 125 | |

|
OEG-lyated alanine-glutamic acid dipeptide-tethered polyisoyanide | Reversible LCST transition | 134 | |

|
Poly(NAAMe48-b-NAβAMem) | Dual LCST-phase transition; 1st LCST = 17 °C and 2nd LCST = 34–37 °C | 133 | |

|
PMETMASPS | Aqueous polymer solution showed UCST-transition in the presence of kosmotropic anions of the Hofmeister series | 136 | |
| pH/Thermo |

|
Poly(N-acryloyl-L-valine N′-methylamide) (PAVMA) | Reversible LCST transition; LCST = <9.0 °C | 130 |

|
Poly(MA-VPGVG) | pH-tunable LCST; LCST = 20 (pH = 1.5)-37 °C (pH = 3.2) | 120 | |

|
Val-Pro-Gly-Val-Gly and oligo(ethylene glycol) grafted polynorbornene | Comonomer-tunable LCST; LCST = 17–44 °C | 121 | |
|
PMAIPAC | pH-Tunable LCST; LCST = 29–60 °C | 126 | |

|
PMAIPGC | pH-Tunable LCST; LCST = 31–39 °C | 127 | |
| pH/Thermo |
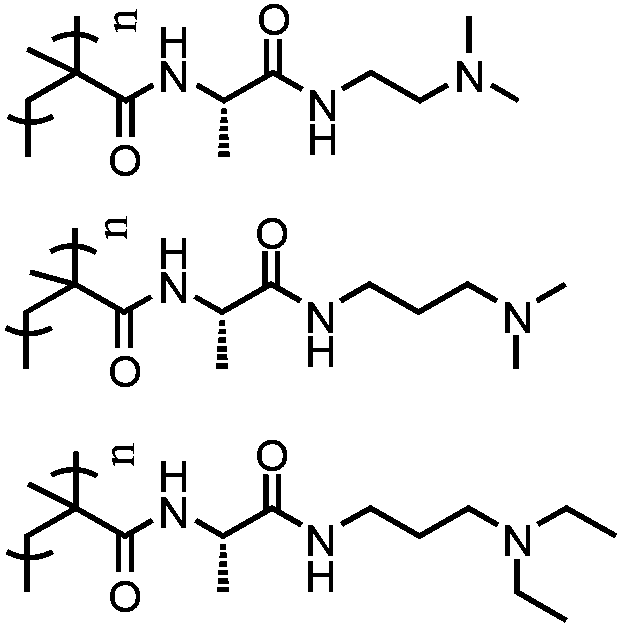
|
PMAME | pH-Tunable LCST with narrow pH window | 186 |
| PMAMP | ||||
| PMAEP | ||||
| pH/Thermo/CO2 |

|
PMAEE | pH-Tunable LCST, exhibited LCST at pH ≥ 9.0 | 186 |
| Light |

|
Glutamic acid- and azobenzene-containing poly(N-propargylamide) | UV-triggered trans–cis isomerization of azobenzene group with decreased helicity | 182 |
| Enzyme |

|
Specific peptide sequence grafted on the polynorbornene | Enzyme-regulated morphological switch of polymeric amphiphile aggregates | 184 |
5.1. Thermoresponsive polymers
Despite the numerous fascinating properties of ELPs as thermo-responsive polymer, their utilization seems to be limited because of their multistep preparation process, which is unsuitable for large scale production of these polymers. So this triggers mass production of thermoresponsive polymers with good biocompatibility. Several amino acids and short peptide-based monomers were used to make single or combined stimuli-responsive (such as pH and thermo) polymers. For example, van Hest et al. have synthesized elastin-based side-chain polymers (EBPs) from a methacrylate derivative of the pentapeptide valine-proline-glycine-valine-glycine (VPGVG) sequence via ATRP127,128 and RAFT.129 Similar to ELPs, these EBPs showed characteristic LCST behavior; the transition temperature decreased with increasing polymer concentration and molecular weight. Also, the transition temperature was observed to be strongly dependent on pH, due to the presence of the carboxylic acid from the C-terminal residue in the peptide side chains. Grubbs’ research group has shown that by ROMP random copolymerization of an elastin side-chain norbornene-monomer with a hydrophilic PEG based monomer, the LCST could be manipulated through the ratio of the co-monomers in the feed.130 But the LCST does not depend on the polymer molecular weight in contrast to Van Hest's findings.127,128 Endo and co-workers have made significant contributions in the development of amino acid-based thermoresponsive polymers. Controlled RAFT polymerization of an acrylamide bearing L-proline methyl ester group afforded the well-defined poly(N-acryloyl-L-proline methylester), poly(A-Pro-OMe), which exhibited thermosensitive phase separation at LCST = 15 °C in water. The transition temperature could be finely tuned in the range of 15–45 °C by copolymerizations with N,N-dimethylacrylamide at different comonomer feed ratios.131 Controlled synthesis of polyacrylamides containing proline and hydroxyproline moiety was achieved by RAFT polymerization of N-acryloyl-L-proline (A-Pro-OH) and N-acryloyl-4-trans-hydroxy-L-proline (A-Hyp-OH), respectively, without protecting group chemistry. Poly(A-Pro-OH) behaves as a weak polyelectrolyte, while poly(A-Hyp-OH) is water soluble. The methylation reaction of the carboxylic acid groups in poly(A-Pro-OH) and poly(A-Hyp-OH) resulted in thermoresposiveness of the polymers. Poly(A-Pro-OMe) exhibited phase separation at 18 °C, whereas poly(A-Hyp-OMe), which is more hydrophilic than the former one, had an LCST of 49.5 °C. The transition temperature of the random copolymer poly(A-Pro-OMe-co-A-Hyp-OMe) could be adjusted by the comonomer compositions.132Dual pH and thermoresponsive block copolymer comprising proline-containing segments were prepared through RAFT. Poly(A-Pro-OMe) acted as a thermoresponsive segment, whereas poly(A-Pro-OH) behaved as a weak anionic polyelectrolyte.133 Amino acid-based double thermoresponsive block copolymers, poly(A-Pro-OMe)-b-poly(A-Hyp-OH), exhibiting both LCST and UCST type transitions have been reported.134 Poly(A-Pro-OMe) was chosen as a thermoresponsive segment, whereas poly(A-Hyp-OH) could be regarded as a water-soluble polymer. A simple methylation of the carboxylic acid groups in poly(A-Pro-OMe)-b-poly(A-Hyp-OH) led to a unique kind of block copolymer system having two different LCSTs. A new type of dual thermo- and pH-responsive homopolymer was reported by Li and co-workers using RAFT polymerization of a methacrylamide-based monomer. In the methacrylamide-functional monomer the β-carboxyl group of L-aspartic acid was converted to an isopropyl amide group to mimic N-isopropylacrylamide for the purpose of thermoresponsive properties, whereas the α-carboxyl group was kept free to function as a pH-responsive entity.135 The analogous glutamic acid polymer had lower CP value and showed much narrower dual-responsive properties window than that of aspartic acid.136 They also made thermoresponsive meth(acrylamide)-based polymers from alanine derivatives.137 RAFT made poly(N-methacryloyl-L-alanine methyl ester) (MA-L-Ala-OMe) and poly(N-acryloyl-L-)alanine methyl ester (Ac-L-Ala-OMe) displayed thermal-responsive behavior in water. Replacing methyl ester with ethyl or isopropyl ester groups caused loss of thermal-responsive behavior. However, replacing the isopropyl ester with isopropyl amide groups can regain thermoresponsiveness such as in poly(MA-L-Ala-iPA). The CPs of these polymers were found to decrease with increasing polymer molecular weight, concentration, and NaCl concentrations. Utilizing this thermoresponsive property of L-alanine-based polymers, smart surfaces were fabricated by brush polymer synthesis from surface-initiated ATRP polymerization of the precursor monomers for temperature-induced cell capture and release.138 The poly(MA-L-Ala-OMe) brush modified substrate displayed a lower cell capture and release efficiency compared to the poly(MA-L-Ala-iPA) brushes, which is probably due to the different side chain functionality. Poly(N-acryloyl-L-valine N′-methylamide) (PAVMA) synthesized by Hu et al. exhibited temperature sensitive phase transition behavior with LCST = 9.0 °C.139 Higashi and co-workers unveiled that the LCST/UCST behaviors of amino acid-derived vinyl polymers depend on the nature of amino acid and the chemical modification of the pendant side-chain group.140 Alanine-based polyacrylamide bearing free –COOH groups displayed a UCST behavior in water below pH 2.0 due to thermoreversible hydrogen bonding of the pendant COOH groups, whereas the glycine/valine/phenylalanine-based polymers did not exhibit any phase separation. Nevertheless, simple methylation of COOH groups resulted in LCST in glycine-based polymers, while a unique kind of opposite phase transition behavior from UCST to LCST was observed in the case of alanine containing polymers.
Precisely tunable LCST from 18 to 73 °C was achieved by varying the methylated alanine to glycine-based comonomer ratio in the copolymer. In a recent study, they employed the same comonomers to make thermoresponsive polymer brushes via surface initiated ATRP for application in cell-sheet engineering.141 The same group reported a unique amino acid-derived block copolymer that showed thermosensitive dual phase transitions. The block copolymer is composed of poly(N-acryloyl-Ala-methylester) and poly(N-acryloyl-β-Ala-methylester) segments, both of which exhibited LCST-type transitions at 18 and 45 °C, respectively. Initially, upon heating the transparent block copolymer the aqueous solution becomes turbid, which turns almost transparent at 34 °C, but a further increase in temperature results in a turbid solution.142
Zhang's group reported the synthesis of a series of novel water-soluble polyisocyanides carrying OEG modified dipeptides as the pendant groups with characteristic thermoresponsive behavior. The dipeptide is made of alanine and glutamic acid. The alanine moiety is connected directly to the backbone to exhibit the strongest chiral induction, whereas the glutamic acid moiety acted as the linker between the alanine unit and the terminal OEG pendants. Polymers with ethoxyl-terminated triethylene glycol (TEG) units showed the lowest phase transition temperature, while the polymer with methoxyl-terminated TEG units displayed an intermediate CP value and that with methoxyl-terminated DEG units possessed the highest transition temperature (Fig. 18).143
 | ||
| Fig. 18 Chemical structures of polyisocyanides with different OEG pendant groups and their transmittance versus temperature plot to determine CP values in water. Reprinted with permission from ref. 143. Copyright (2013) American Chemical Society. | ||
Recently, Mandal's group have synthesized L-serine-based zwitterionic polymers, poly(L-serinyl acrylate)s (PSAs) with dual-stimuli-responsive UCST type phase transition properties via aqueous RAFT polymerization.144 The PSA exhibited an isoelectric point near pH 2.85 where it exists in its zwitterionic form. In the pH range of 2.3–3.5, the aqueous PSA solution appeared as a two-phase system, which became single phase upon heating, exhibiting distinct reversible UCST-type phase transition. The CP was found to increase with increasing polymer molecular weight, and decreased upon changing the solution pH, displaying the highest CP near the isoelectric point of PSA. In addition, the CP was shown to decrease with increasing ionic strength of the solution due to the antipolyelectrolyte effect. All these amino acid-derived zwitterionic polymers discussed above contain only nitrogen-based cations and carboxylate anions. The same group also designed a methionine-derived zwitterionic monomer, [Boc-L-methionine-(2-methacryloylethyl)]sulfoniopropanesulfonate (METMASPS), and subsequently polymerized it by the aqueous RAFT technique to afford the corresponding zwitterionic polymer (PMETMASPS) having sulfonium cations and sulfonate anions.145 Interestingly, the monomer exhibited both pH-responsive soluble–insoluble and UCST-type temperature induced insoluble–soluble phase transitions in water. Whereas the aqueous zwitterionic PMETMASPS solution exhibited a soluble–insoluble phase transition upon addition of different kosmotropic anions such as Ct3−, SO42−, H2PO4− and Ac− of the Hofmeister series without any pH-responsive behavior. But the turbid PMETMASPS solution in the presence of these anions showed a tunable UCST-type phase transition as a function of its molecular weight, the concentration of anions and the pH of the solution.
5.2. pH-Responsive polymers
Various core and shell crosslinked block copolymer micelles have been reported by several groups, involving either physically or chemically crosslinked micelles. McCormick's group made reversible shell “locked” nanoassemblies via interpolyelectrolyte complexation of the anionic poly(N-acryloyl-alanine) segments of triblock copolymer poly[(N,N-dimethylacrylamide)-b-(N-acryloyl-alanine)-b-(N-isopropylacrylamide)] micelles in the hydrated shell with the cationic homopolymer poly[(ar-vinylbenzyl)ammonium chloride] (PVBTAC) above the unimer to micelle phase transition temperature of the block copolymer in water. These interpolyelectrolyte complexed micelles remain intact upon cooling below the LCST, and were shown to be reversible in the presence of an added simple electrolyte (0.4 M NaCl in the present case).146 Likewise, interpolyelectrolyte core-crosslinked micelles were prepared from a pH and thermo-dual mode micelle and the same cationic polyelectrolyte PVBTAC at a certain pH and temperature. Here, the hydrophilic N,N-dimethylacrylamide (DMA) block acts as shell and doubly (temperature and pH) responsive statistical segments of N-isopropylacrylamide (NIPAM) and N-acryloyl-valine (AVAL) as the core of the micelle. This complexation can be reversed by addition of sufficient electrolyte.147 Aqueous RAFT method afforded a series of a pH/salt-responsive block copolymers comprising poly(sodium 2-acrylamido-2-methyl-1-propanesulfonate)-b-poly(N-acryloyl-L-alanine), which self-assemble to micelles through protonation of the alanine-based segment on lowering the solution pH. The micelles were crosslinked via interpolyelectrolyte complexation between the anionic shell and a cationic homopolymer, protonated poly(N-[3-(dimethylamino)-propyl]acrylamide) (PDMAPA) or poly(N,N-dimethylaminoethylmethacrylate) (PDMAEMA). On increasing the solution pH, the polycation crosslinker got deprotonated, resulting in the dissociation of the crosslinked micelles to their respective water-soluble unimer components.148 Using solutions of the same block copolymer micelles with anionic coronas and of the cationic homopolymer PDMAPA, layer-by-layer (LbL) films were assembled at low pH, maintaining the micelle structures. Pyrene-loaded LbL films were shown to have pH dependent release profiles.149O'Reilly's group reported the preparation of pH-responsive vesicles from a “schizophrenic” diblock copolymer, which was synthesized by RAFT polymerization of N-acryloyl-L-phenylalanine (A-Phe-OH) using the poly[2-(diethylamino)ethyl methacrylate] macro-chain transfer agent (CTA). The schizophrenic behavior of the vesicle with a switchable corona and membrane was achieved by simple direct dissolution or solvent switching in water at acidic and basic pH.150 Using the same block segment they showed that non-spherical morphologies could be attained by playing with the solution pH and highlighted the importance of end group for the resultant morphology.151 Recently, the development of non-cytotoxic and pH-sensitive nanostructured membranes was reported that consist of poly(N-methacryloyl glycine) and bacterial nanocellulose (BC). The as-prepared homogeneous and translucent nanocomposites exhibited good thermal, mechanical and viscoelastic properties, as well as a high water uptake capability. In vitro diclofenac (DCF) drug release from the nanocomposite was assessed at pH 2.1 and pH 7.4. DCF is essentially retained in the nanocomposites at pH 2.1 whereas at pH 7.4 the drug is efficiently released, revealing their potential for the controlled release of DCF in dermal as well as in oral drug delivery applications.152
Our group has made significant contributions on synthesizing various side-chain amino-acid/peptide-based pH responsive cationic chiral polymers, their macromolecular architectures, and crosslinked organo/hydro-gels and in using them in chiral recognition, drug delivery, and gene transfer applications.7,153 The polymers synthesized through N-terminus modification of an amino acid reported by Endo, O'Reilly and McCormick are all weak polyelectrolytes, which dissolve in water at basic pH. In 2012, we employed the RAFT polymerization technique to polymerize Boc-group protected L-alanine and L-phenylalanine-conjugated methacrylate-based monomers [(Boc-L-Ala-HEMA) and (Boc-L-Phe-HEMA)] to afford their corresponding polymers with controlled chain length, narrow dispersity and precise chain end functionality. Subsequent Boc-group expulsion from the synthesized polymers enabled the formation of water-soluble cationic polymers with primary ammonium groups (–NH3+) in the side-chains. These polymers showed pH-reversible soluble–insoluble phase transition behavior in water, which is attributed to the reversible switching of –NH3+ groups into –NH2 groups.154 As a part of our constant endeavor, we developed a library of pH-responsive synthetic polymeric architectures, pH/salt-responsive polymeric networks having natural amino acids or amino acid-derived biologically important peptide sequences by well controlled RAFT polymerization. Boc-L-leucine/isolucine155 and Boc-L/D-tryptophan156 appended polymethacrylates were afforded by the RAFT method. These homopolymers were employed for block copolymerization with methyl methacrylate. Boc-group deprotected leucine and isoleucine-based homopolymers exhibited the same transition pH value, which is because of the similar hydrophobicity imparted by isobutyl and sec-butyl groups of leucine and isoleucine, respectively. Similarly, L- and D-tryptophan containing polymers displayed the same pH transition value, which indicates that chirality has no influence on the phase transition behavior of these polymers. We also reported the synthesis of well-defined block copolymers composed of a polyisobutylene (PIB) segment and a Boc-protected alanine/leucine-based polymethacrylate block by a combination of living carbocationic and RAFT polymerizations.157 These double hydrophobic block copolymers self-assembled into core–shell type micellar structures in methanol. After Boc-group removal, the resultant amphiphilic block copolymer showed reversible pH-responsive behavior and formed stable spherical nanostructures. The transition pH values for the amphiphilic block copolymers were shown to be somewhat higher compared to that of the corresponding homopolymers due to the hydrophobic nature of the PIB block. In another article, we reported a promising vehicle for both drug delivery and gene transfer.158 Two amino acid-based amphiphilic block copolymers were synthesized from Boc-L-Ala-HEMA and Boc-L-Phe-HEMA monomers using monomethoxy poly(ethyleneglycol) (mPEG)-macro-CTA. These protected block copolymers self-assembled into nanostructures and encapsulated hydrophobic dye or drug molecules inside the core of the nanoaggregates. Again, the deprotected double hydrophilic block copolymers with cationic side-chain moieties showed effective binding capability with pDNA. This kind of smart carrier with excellent performance could be applied for an efficient dual drug and gene delivery system in biomedicine. Dual pH/thermo-responsive block copolymers comprising side-chain alanine/phenylalanine-tethered polymethacrylate and poly(di(ethylene glycol) methyl ether methacrylate) (PDEGMA) were synthesized by using pyrene-end capped fluorescent CTA.159 Tadpole-shaped organic/inorganic hybrid amphiphilic polymers were also synthesized by RAFT polymerization of leucine appended monomers with a polyhedral oligomeric silsesquioxane (POSS)-attached CTA. Morphological transformation from spherical nanoparticles to rod shaped nanoparticles was observed upon increasing the polymer chain length.160 Furthermore, Boc-L/D-leucine-derived methacrylate monomers (Boc-L-Leu-HEMA and Boc-D-Leu-HEMA) were copolymerized with 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) with varied comonomer compositions to obtain two different sets of chiral copolymer libraries, which on Boc deprotection showed dual pH and thermoresponsive characteristics. The LCST of these copolymers could easily be manipulated over a wide window between 26 and 74 °C by varying the pH of the aqueous solution and comonomer composition in the copolymers.161 A combination of self-condensing vinyl polymerization (SCVP) and RAFT polymerization has successfully enabled the formation of side-chain Boc-L-valine-HEA-based hyperbranched polymers. Star polymer architecture was constructed with a varied number of thermoresponsive (PMEO2MA/PPEGMA) arms and arm length using the as-made hyperbranched core. Boc-deprotected star polymers with pH-responsive core and thermoresponsive arms showed a self-assembled aggregation above the transition pH = 7.5 to a multi-micellar aggregation, which further fused together to form large aggregates above their LCST (Fig. 19).162
 | ||
| Fig. 19 Chemical structure and schematic representation of pH responsive hyperbranched polymer and their respective star polymers with hyperbranched core via successive RAFT process. Reproduced with permission from ref. 162. Copyright (2014) The Royal Society of Chemistry. | ||
Recently, we reported the synthesis of a new class of conventional fluorophore-free dual pH- and thermo-responsive fluorescent copolymers through sequence-controlled copolymerization of rationally designed monomers. In this case, leucine-based styrenic monomer having the primary ammonium group acted as a pH-responsive entity, whereas N-substituted maleimide monomer bearing a diethylene oxide side-chain is responsible for temperature sensitive behavior.163 We have employed this unconventional fluorophore-free water-soluble fluorescent copolymer for speedy, selective and sensitive detection of picric acid in 100% aqueous medium.164 In another study we explored how supramolecular host–guest interaction between randomly methylated β-cyclodextrin (RM β-CD) and the phenyl ring of side-chain phenylalanine-derived homopolymers amplified the fluorescence emission of the otherwise weakly fluorescent amino acid.165 Complexation-assisted broad tunability in cloud point temperature of the dual thermo- and pH-responsive copolymers made of side-chain pheylalanine and its dipeptide-based methacrylate monomers and NIPAM has been realized.
Various sequence-defined peptide side-chain containing polymers with smart pH-responsive behavior were synthesized by our group. Three different short peptide fragments from the central hydrophobic cluster (CHC) of the amyloid β-peptide Aβ1–42, namely Phe-Phe, Val-Phe and Ile-Phe based Boc-protected methacrylate monomers, were polymerized to make homo and block copolymers with mPEG as macro-CTA. Quantitative Boc-group removal gives double hydrophilic block copolymers. The self-assembly behaviors of the amphiphilic block copolymers and double hydrophilic block copolymers were investigated in detail.166 Likewise, another methacrylate monomer bearing a short peptide segment (Leu-Val-Phe) corresponding to the amyloid β-peptide Aβ1–42 was used to make homo and block copolymers. This tripeptide-based hydrophobic homopolymer forms a spherical morphology in methanol, whereas the deprotected one forms a toroid-like morphology in the same solvent. The Boc-protected amphiphilic block copolymer could encapsulate a hydrophobic dye or drug molecule, and the double hydrophilic block copolymer showed pH-responsiveness and strongly binds with pDNA.167 Furthermore, an interesting tetrapeptide segment (Leu-Val-Phe-Phe) from the central hydrophobic core of amyloid β-peptide, Aβ17–20, conjugated methacrylate monomer was polymerized by the RAFT technique. Boc-group deprotection results in a pH-responsive polymer, which was further modified to an amphiphilic graft copolymer-based nanocarrier after coupling with an aldehyde-terminated mPEG molecule under basic conditions through imine bond formation. This pH-sensitive dynamic covalent imine bond facilitates disruption of the nanostructure, leading to the release of the encapsulated dye molecule.168
Among stimulus-sensitive hydrogels, pH-responsible hydrogels derived from both natural and synthetic polymers have been investigated intensively because of holding considerable promise for controllable release of the drugs/gene at the target position.169 Many scientists have focused their research on stimuli-responsive amino acid-based polymer networks, because the presence of amino acid moieties may endow gels with new properties such as chirality, catalytic activity, sorption properties, sensitivity to pH and ionic strength, and the presence of specific ions. Karbarz and co-workers have made ampholytic macrogel170,171 and microgel172 networks by incorporating N-δ-acryloyl/methacryloyl ornithine/lysine with NIPAM as comonomer and N,N-methylenebisacrylamide as crosslinker. As the α-amino acid groups were unbound, the swelling and sorption behavior of the obtained macro- and microgels with respect to the amount of amino acid incorporated into the polymer network, temperature, concentration of ions and pH was investigated. Chirality and pH responsivity, two intriguing concepts, were combined in a single hydrogel by Deng et al. They made crosslinked polymeric hydrogel networks using N-acryloyl-L-alanine as chiral monomer with some comonomer and crosslinker. These hydrogels were found to be capable of chiral recognition and enantiodifferentiating release abilities toward chiral amino acids, and are expected to find practical applications as novel materials for chiral drug delivery.173,174 In 2014, our group synthesized side-chain Boc-protected L/D-alanine-based crosslinked polymeric organogels, which were transformed into hydrogels by subsequent Boc group removal under acidic conditions at room temperature. The resultant hydrogel exhibited superabsorbency in water with a pH-sensitive swelling behavior.175 We were the first to report the transformation of an organogel to a hydrogel by a simple deprotection strategy. We made a library of superabsorbent cationic polyelectrolyte hydrogels from several Boc-protected amino acids (glycine/alanine/valine/leucine/isoleucine/phenylalanine)-based monomers with different –R groups using a similar strategy to understand the effect of –R groups on the pH and ionic strength-induced swelling characteristics of those hydrogels.176 It was observed that the degree of swelling increases gradually as the bulkiness and hydrophobicity of the –R group of the pendant amino acid moiety increase. In addition, the swelling ratios of hydrogels decreased with increasing salt concentration and the value at a particular salt concentration decreased in the order NaCl > FeCl3 > AlCl3. A triple-responsive (pH, thermo and salt) hydrogel network has been constructed by RAFT copolymerization of Boc-protected valine-based monomer (Boc-L-Val-HEMA) with MEO2MA in the presence of a crosslinker followed by Boc-group expulsion. This polyelectrolyte hydrogel showed pH dependent thermoresponsive characteristics, which are suppressed at low pH (pH 4.0) but a drastic volume phase transition has been observed at pH 7.0.177
Casolaro and co-workers reported their research activity concerning the potential applications of multi-stimuli-responsive polyelectrolyte hydrogels containing α-amino acid (L-valine, L-phenylalanine and L-histidine) residues as carriers for therapeutic delivery; treatment of cancer,178,179 glaucoma,180 and mood disorders.181,182 Dynamic covalent chemistry has been employed to prepare tryptophan183 and leucine184 based polymeric gels with pH-reversible sol–gel transition behavior. Primary amine groups of amino acids residues crosslinked with the aldehyde or ketone functionality of a small organic/macro crosslinker to form a gel network having pH-sensitive imine bonds. A similar strategy was utilized to make a pH-responsive self-healing polymeric gel from a block copolymer comprising PIB and a side-chain leucine-based polymer and a dialdehyde-terminated telechelic PIB as a crosslinker.185 The L-hydroxyproline-based methacrylic polybetaines have been synthesized, where the native amino and carboxylic acid groups are preserved and have been shown to exhibit pH sensitivity with isoelectric point near 3.0. The polymer efficiently catalyzed aldol reactions under homogeneous conditions in N,N-dimethylformamide but not in water.186
Takahara and co-workers showed an amino acid-based pH-responsive zwitterionic polymer surface as a non-fouling material for proteins.187 A novel amphiphilic polymer (PGluDMA) containing glutamic acid grafted onto the end of a dodecyl polymer side chain, which contains the α-amine and the γ-carboxylic acid of the glutamic acid moiety. The polymer self-assembled into a multilayered structure in the thin film exposing the glutamic acid moieties to the polymer film/water interface. Surface charge was controllable by pH buffer solution at the water interface, resulting in a zwitterionic surface at neutral pH. Interestingly, the polymer film exhibited charge-selective protein adsorption as the synergistic interaction between the α-amine and the γ-carboxylic acid was weaker than in conventional amino acid-based zwitterionic systems (Fig. 20).
 | ||
| Fig. 20 (A) Chemical structure of PGluDMA. (B) pH-Responsiveness of the glutamic acid in the polymer side chains exposed to the polymer film/water interface. (C) Unknown protein adsorption behavior of the zwitterionic state on the polymer film at neutral pH. Reproduced with permission from ref. 187. Copyright (2015) The Royal Society of Chemistry. | ||
5.3. Redox-responsive polymers
A methionine-based poly(methacrylamide) was synthesized by radical polymerization. The thioether moiety was oxidized by H2O2 in chloroform (CHCl3) at room temperature to afford polymers with methionine sulfoxide and sulfone structures in the side chains.188 Recently, Karbarz and co-workers reported on the synthesis of a redox-responsive nanohydrogel for high drug loading. The nanogels were obtained by precipitation polymerization of NIPAM employing N,N-bis(acryloyl)cystine (BISS) as the crosslinker. Free carboxylic acid groups of cystine-derivatives provided pH sensitivity to the gels, enhanced their resistance to aggregation and allowed binding with the biomolecule having complementary functional groups. Nanogels with substantially increased BISS content possess a high loading capacity of anticancer drug versus dry gel and exhibited a much better stability and enhanced drug release under conditions that are similar to those inside cancer cells. The presence of a disulfide bond in the crosslinking agent allowed degradation of the nanogel by GSH at concentrations similar to that inside cancer cells.189 In 2013, the same group showed that introduction of a redox active group into the polymer network not only produces a redox-sensitive gel, but also it is possible to obtain an electrochemically responsive gel which is able to change in volume on changing the state of oxidation of the redox groups.190 NIPAM and BISS-based crosslinked hydrogels were prepared. Disulfide bridges present in the polymer network were used to modify the gels with ferrocene moieties. The ferrocene moieties attached to the polymer network were oxidized and reduced using Ce(IV) and ascorbic acid, respectively. It was observed that the percentage of electroactive groups and their oxidation state influenced the volume phase transition temperature (Fig. 21). | ||
| Fig. 21 Scheme of the synthesis and modification of gel and pictures of the corresponding gel samples. Reproduced with permission from ref. 190. Copyright (2013) The Royal Society of Chemistry. | ||
5.4. Photo-responsive polymers
Like photoresponsive polypeptides, a few light-sensitive side-chain amino acid based polymers have also been reported. Masuda's group reported the synthesis of a glutamic acid-derived photoresponsive polypropargylamide bearing azobenzene moiety as the side-chain pendant.191 The synthesized polymer adopted a helical conformation in tetrahydrofuran, CHCl3, dichloromethane and toluene. Solvent composition induced helix–helix transition is observed in CH2Cl2/CHCl3 and toluene/CHCl3 mixtures. The polymer also exhibited heat-induced helix–helix transition in CHCl3. The trans-azobenzene in the side chain when isomerized into the cis form upon UV irradiation underwent a decrease in helicity. The cis-azobenzene reisomerized into a trans form upon visible light irradiation, but the polymer did not recover the original helicity.5.5. Enzyme-responsive polymers
As discussed in an earlier section, enzymes could be a promising biological trigger in therapeutics, herein we highlighted one example where protease overexpression has been exploited for cancer-specific gene targeting by polymeric carriers. Hydrophilic polymers, grafted with cationic peptides, are used for the preparation of polyplex with pDNA. The grafted cationic peptides are substrates for the target signal transduction proteins (here protein kinase), which are activated specifically in disease cells and which decrease the peptide cationic charge, resulting in the release of pDNA. Conjugates were able to perform cellular transfection both in vitro and in vivo.192 An example of the enzymatic modulation of polymer morphologies has been reported by Gianneschi and his team. Micelles were prepared from two different amphiphilic polymer–peptide hybrid block copolymers containing substrates for four different cancer-associated enzymes: proteinkinase A (PKA), protein phosphatase-1 (PP1), and matrix metalloproteinases MMP-2 and MMP-9.193 These two amphiphilic peptide-brush copolymers differed only in the relative ordering of the peptide substrates. By incorporating the abovementioned enzyme substrates into the polar head groups of the copolymers, the micelle morphology and aggregation behavior of the materials were modified using the following mechanisms: (1) phosphorylation by PKA at serine residues, (2) dephosphorylation by PP1 at serine residues, (3) peptide cleavage by MMPs at Gly-Leu peptide bonds. Reversible switching of the micellar morphology through a phosphorylation–dephosphorylation cycle and peptide-sequence directed changes in morphology upon the actions of proteases were performed. In the case of both the hybrid micelles, phosphorylation by PKA and ATP produced larger amorphous aggregates, which regained their structure in the presence of phosphatase. Proteolytic cleavage at sites more proximal to the polymer backbone leads to more dramatic morphological changes. This is because of a larger change in the peptide shell structure (polymer–peptide amphiphile 2), whereas cleavage at sites away from the polymer backbone left the morphology unchanged (polymer–peptide amphiphile 2) (Fig. 22). | ||
| Fig. 22 Peptide–substrate polymeric amphiphiles assemble into spherical micelles. The peptide substrates within the micelle corona interact with enzymes to generate a variety of morphologies of polymeric amphiphile aggregates depending on the design of the peptide substrate and enzymes added. Reprinted with permission from ref. 193. Copyright (2011) American Chemical Society. | ||
5.6. CO2-Responsive polymers
Some of our synthesized side-chain amino acid-bearing pH-responsive polymers also exhibit CO2-responsive characteristics. The polymers having transition pH values in between 5.5–9.5 generally show CO2-responsiveness. Leucine and isoleucine-based polymethacrylates having primary amine pendant groups possess CO2-sensitive behavior, whereas side-chain phenylalanine and tryptophan appending polymers do not display any such properties since their transition pH values are 5.4 and 5.1, respectively. In addition, dual thermo and CO2-sensitive flip-flop micellization behavior has been observed in a block copolymer, made by chain extending the CO2-responsive leucine segment with a thermo-sensitive motif. This kind of novel CO2-responsive polymeric oscillating/phase changing micelle may have potential for use as an anticancer therapeutic agent (taking advantage of the increased production of CO2 in cancer cells).1945.7. Multi-stimuli-responsive polymers
Recently, Luo et al. used alanine as the building block to design multi-stimuli-responsive polymethacrylamide homopolymers. Four poly(N-methacryloyl-L-alanine) homopolymers containing different tertiary amine moieties, dimethylaminoethyl (PMAME), dimethylaminopropyl (PMAMP), diethylaminoethyl (PMAEE) and diethylaminopropyl (PMAEP), were synthesized by the RAFT method and their stimuli-responsive behaviors were compared (Fig. 23).195 All these polymers displayed pH-tunable LCST transitions in the basic pH range. The polymer PMAEE did not show any LCST transition at pH of 8.0 or lower, but phase transition was detected at pH of 9.0 or higher. A strong dependence of pH on LCST was observed, for example, LCST was found to be 66 °C at pH 9.0, which shifted to 44, 38, 32 and 27 °C at pH 9.5, 10.0, 11.0 and 13.0, respectively. But the other three polymethacrylamides, PMAME, PMAMP, and PMAEP, exhibited LCST-type thermo-responsive properties in water in rather narrower pH ranges compared with PMAEE. Moreover, the presence of tertiary amine moieties provides CO2-responsiveness to the PMAEE. At pH = 10.0 the CP of PMAEE85 was found to be 38 °C, and at 48 °C the solution was turbid. After bubbling CO2 into this solution, the sample turned clear in response to a decrease in pH from 10.0 to 5.9, and did not display any LCST. The solution became cloudy when it was purged with inert N2 gas at 48 °C. This periodical CO2/N2 bubbling process was performed several times, and the effect was reversible. | ||
| Fig. 23 Synthesis of tertiary amine-modified L-alanine-based methacrylamides and their corresponding polymethacrylamide homopolymers by RAFT polymerization. Reprinted with permission from ref. 195. Copyright (2016) Elsevier. | ||
Mori's group recently synthesized threonine-based chiral homopolymers having a multi-stimuli-responsive property, and showed how the amino acid structure is important in exhibiting stimuli-responsiveness.196 Methylated poly(N-acryloyl-L-threonine) showed pH-tunable LCST-type phase transition behavior in water. The LCST values were found to increase with increasing solution pH above pH 7.0. Whereas they increased with increasing pH value, since the number of hydrogen bonds between poly(A-Thr-OMe) chains decreases in water at pH < 4. Although urea addition could influence the phase transition value, no such phenomenon was observed in the case of electrolyte addition. Moreover, the analogous serine-based polymer remained water soluble and did not exhibit a similar response. This indicates that a hydrophilic/hydrophobic balance is the key to dictate temperature-induced phase transition behavior.
6. Conclusion and outlook
Nature has its own ways of creating and maintaining a balance amongst the cellular processes to sustain life and this has inspired researchers to understand the basis and mechanism of various life processes. The contemporary work on the development of ‘stimuli-responsive’ smart materials to understand the structure–property relationship of naturally-occurring proteins as well as their possible application in bio-medicine, bio- and nano-technology has brought us a step closer to the direction of realizing how the complex processes take place inside our body. By meticulously programming amino acids and/or amino acid sequences into synthetic polymeric backbones or side-chains, one can prepare synthetic amino-acid derived polymers. These polymers have good biocompatibility and/or biodegradability and bio-mimicking properties such as the ability to form higher order structures and their sensitivity towards micro-environmental stimuli such as changes in cellular pH, temperature, ionic strength, redox state, and light.197,198 Additionally, functionalities such as charged moieties that can recognize small biomolecules and certain stimuli can also be conveniently incorporated into the backbone as well as side chains. Yet there is a long way to go in producing synthetic biomolecules-derived materials on a large scale for practical applications, partly because, to obtain protein-mimicking properties in the stimuli-responsive amino acid-derived materials, precise control over the higher order structure is required. With the current synthetic procedures adopted for obtaining these materials, sequence control and monodispersity of the synthetic biomaterials become difficult to achieve. Also high costs, extensive and tedious synthetic procedures for preparation, and cautious handling of moisture sensitive monomers such as NCA which requires strictly anhydrous polymerization conditions for generating synthetic polypeptides with high purity are analytical barriers.199,200 However, there is still room for more development in the field of synthetic stimuli-responsive biomolecule-derived materials with a better understanding and by adopting economical pathways for application in therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to the Department of Science and Technology (DST), International Division, New Delhi, India (Project DST/INT/BLR/P-14/2016) for financial support under the Indo-Belarus joint research programme.Notes and references
- D. L. Nelson, A. L. Lehninger and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, New York, 2008 Search PubMed.
- C. Deng, J. Wu, R. Cheng, F. Meng, H.-A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330–364 CrossRef CAS.
- R. Jones, Nat. Nanotechnol., 2008, 3, 699–700 CrossRef CAS PubMed.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC.
- S. Zhang and Z. Li, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 546–555 CrossRef CAS.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- K. Bauri, S. G. Roy and P. De, Macromol. Chem. Phys., 2016, 217, 365–379 CrossRef CAS.
- Z. Song, Z. Han, S. Lv, C. Chen, L. Chen, L. Yin and J. Cheng, Chem. Soc. Rev., 2017, 46, 6570–6599 RSC.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- K. Dan and S. Ghosh, Macromol. Rapid Commun., 2012, 33, 127–132 CrossRef CAS PubMed.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- F. Chécot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef.
- H. Kukula, H. Schlaad, M. Antonietti and S. Förster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef CAS PubMed.
- J. Babin, J. Rodriguez-Hernandez, S. Lecommandoux, H.-A. Klok and M.-F. Achard, Faraday Discuss., 2005, 128, 179–192 RSC.
- J. Rodríguez-Hernández and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026–2027 CrossRef PubMed.
- H. Iatrou, H. Frielinghaus, S. Hanski, N. Ferderigos, J. Ruokolainen, O. Ikkala, D. Richter, J. Mays and N. Hadjichristidis, Biomacromolecules, 2007, 8, 2173–2181 CrossRef CAS PubMed.
- J. Sun, Y. B. Huang, Q. Shi, X. S. Chen and X. B. Jing, Langmuir, 2009, 25, 13726–13729 CrossRef CAS PubMed.
- J. Gaspard, J. A. Silas, D. F. Shantz and J.-S. Jan, Supramol. Chem., 2010, 22, 178–185 CrossRef CAS.
- M. S. Kim, K. Dayananda, E. K. Choi, H. J. Park, J. S. Kim and D. S. Lee, Polymer, 2009, 50, 2252–2257 CrossRef CAS.
- Y. Huang, Z. Tang, X. Zhang, H. Yu, H. Sun, X. Pang and X. Chen, Biomacromolecules, 2013, 14, 2023–2032 CrossRef CAS PubMed.
- J. Sun, P. Černoch, A. Völkel, Y. Wei, J. Ruokolainen and H. Schlaad, Macromolecules, 2016, 49, 5494–5501 CrossRef CAS.
- D. Mavrogiorgis, P. Bilalis, A. Karatzas, D. Skoulas, G. Fotinogiannopoulou and H. Iatrou, Polym. Chem., 2014, 5, 6256–6278 RSC.
- H. Yin, H. C. Kang, K. M. Huh and Y. H. Bae, J. Mater. Chem., 2012, 22, 19168–19178 RSC.
- A. C. Engler, D. K. Bonner, H. G. Buss, E. Y. Cheung and P. T. Hammond, Soft Matter, 2011, 7, 5627–5637 RSC.
- M. R. Banki, L. Feng and D. W. Wood, Nat. Methods, 2005, 2, 659–661 CrossRef CAS PubMed.
- J. Kostal, A. Mulchandani, K. E. Gropp and W. Chen, Environ. Sci. Technol., 2003, 37, 4457–4462 CrossRef CAS PubMed.
- D. Y. Furgeson, M. R. Dreher and A. Chilkoti, J. Controlled Release, 2006, 110, 362–369 CrossRef CAS PubMed.
- M. F. Shamji, J. Chen, A. H. Friedman, W. J. Richardson, A. Chilkoti and L. A. Setton, J. Controlled Release, 2008, 129, 179–186 CrossRef CAS PubMed.
- J. R. Kramer, R. Petitdemange, L. Bataille, K. Bathany, A. L. Wirotius, B. Garbay, T. J. Deming, E. Garanger and S. Lecommandoux, ACS Macro Lett., 2015, 4, 1283–1286 CrossRef CAS.
- R. Petitdemange, E. Garanger, L. Bataille, K. Bathany, B. Garbay, T. J. Deming and S. Lecommandoux, Bioconjugate Chem., 2017, 28, 1403–1412 CrossRef CAS PubMed.
- R. Petitdemange, E. Garanger, L. Bataille, W. Dieryck, K. Bathany, B. Garbay, T. J. Deming and S. Lecommandoux, Biomacromolecules, 2017, 18, 544–550 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- C. Chen, Z. Wang and Z. Li, Biomacromolecules, 2011, 12, 2859–2863 CrossRef CAS PubMed.
- X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2013, 46, 3753–3760 CrossRef CAS.
- Y. Ma, X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2014, 47, 4684–4689 CrossRef CAS.
- Y. Cheng, C. He, C. Xiao, J. Ding, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2627–2634 RSC.
- C. M. Chopko, E. L. Lowden, A. C. Engler, L. G. Griffith and P. T. Hammond, ACS Macro Lett., 2012, 1, 727–731 CrossRef CAS PubMed.
- X. Zhang, W. Li, X. Zhao and A. Zhang, Macromol. Rapid Commun., 2013, 34, 1701–1707 CrossRef CAS PubMed.
- J. Yan, K. Liu, X. Zhang, W. Li and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 33–41 CrossRef CAS.
- J. Yan, K. Liu, W. Li, H. Shi and A. Zhang, Macromolecules, 2016, 49, 510–517 CrossRef CAS.
- F. Meng, X. Fu, Y. Ni, J. Sun and Z. Li, Polymer, 2017, 118, 173–179 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4190 CrossRef CAS PubMed.
- Y. Deng, Y. Xu, X. Wang, Q. Yuan, Y. Ling and H. Tang, Macromol. Rapid Commun., 2015, 36, 453–458 CrossRef CAS PubMed.
- Y. Wu, X. Wang, Y. Ling and H. Tang, RSC Adv., 2015, 5, 40772–40778 RSC.
- C. Ge, S. Liu, C. Liang, Y. Ling and H. Tang, Polym. Chem., 2016, 7, 5978–5987 RSC.
- C. Ge, L. Zhao, Y. Ling and H. Tang, Polym. Chem., 2017, 8, 1895–1905 RSC.
- J. Ding, F. Shi, C. Xiao, L. Lin, L. Chen, C. He, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2857–2864 RSC.
- K. Wang, G. F. Luo, Y. Liu, C. Li, S. X. Cheng, R. X. Zhuo and X. Z. Zhang, Polym. Chem., 2012, 3, 1084–1090 RSC.
- S. Takae, K. Miyata, M. Oba, T. Ishii, N. Nishiyama, K. Itaka, Y. Yamasaki, H. Koyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 6001–6009 CrossRef CAS PubMed.
- X. J. Cai, H. Q. Dong, W. J. Xia, H. Y. Wen, X. Q. Li, J. H. Yu, Y. Y. Li and D. L. Shi, J. Mater. Chem., 2011, 21, 14639–14645 RSC.
- T. Waku, M. Matsumoto, M. Matsusaki and M. Akashi, Chem. Commun., 2010, 46, 7025–7027 RSC.
- T. Thambi, H. Y. Yoon, K. Kim, I. C. Kwon, C. K. Yoo and J. H. Park, Bioconjugate Chem., 2011, 22, 1924–1931 CrossRef CAS PubMed.
- T. B. Ren, W. J. Xia, H. Q. Dong and Y. Y. Li, Polymer, 2011, 52, 3580–3586 CrossRef CAS.
- J. X. Ding, J. J. Chen, D. Li, C. S. Xiao, J. C. Zhang, C. L. He, X. L. Zhuang and X. S. Chen, J. Mater. Chem. B, 2013, 1, 69–81 RSC.
- H. Zhu, C. Dong, H. Dong, T. Ren, X. Wen, J. Su and Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10393–10407 CAS.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- X. Fu, Y. Ma, Y. Shen, W. Fu and Z. Li, Biomacromolecules, 2014, 15, 1055–1061 CrossRef CAS PubMed.
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS PubMed.
- X. Fu, Y. Ma, J. Sun and Z. Li, RSC Adv., 2016, 6, 70243–70250 RSC.
- Q. Xu, C. He, K. Ren, C. Xiao and X. Chen, Adv. Healthcare Mater., 2016, 5, 1979–1990 CrossRef CAS PubMed.
- E. P. Holowka and T. J. Deming, Macromol. Biosci., 2010, 10, 496–502 CAS.
- Q. Yan, D. Han and Y. Zhao, Polym. Chem., 2013, 4, 5026–5037 RSC.
- D. Roy and B. S. Sumerlin, Macromol. Rapid Commun., 2014, 35, 174–179 CrossRef CAS PubMed.
- K. Ohkawa, K. Shoumura, M. Yamada, A. Nishida, H. Shirai and H. Yamamoto, Macromol. Biosci., 2001, 1, 149–156 CrossRef CAS.
- J. X. Ding, X. L. Zhuang, C. S. Xiao, Y. L. Cheng, L. Zhao, C. L. He, Z. H. Tang and X. Chen, J. Mater. Chem., 2011, 21, 11383–11391 RSC.
- L. Yan, L. Yang, H. He, X. Hu, Z. Xie, Y. Huang and X. Jing, Polym. Chem., 2012, 3, 1300–1307 RSC.
- S. Kumar, J. F. Allard, D. Morris, Y. L. Dory, M. Lepage and Y. Zhao, J. Mater. Chem., 2012, 22, 7252–7257 RSC.
- G. Liu and C.-M. Dong, Biomacromolecules, 2012, 13, 1573–1583 CrossRef CAS PubMed.
- G. E. Negri and T. J. Deming, ACS Macro Lett., 2016, 5, 1253–1256 CrossRef CAS.
- G. Liu, N. Liu, L. Zhou, Y. Su and C.-M. Dong, Polym. Chem., 2015, 6, 4030–4039 RSC.
- V. K. Kotharangannagari, A. Sanchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2011, 44, 4569–4573 CrossRef CAS.
- S. Chen, Y. Gao, Z. Cao, B. Wu, L. Wang, H. Wang, Z. Dang and G. Wang, Macromolecules, 2016, 49, 7490–7496 CrossRef CAS.
- O. Pieroni, J. L. Houben, A. Fissi, P. Costantino and F. Ciardelli, J. Am. Chem. Soc., 1980, 102, 5913–5915 CrossRef CAS.
- W. Xiong, X. Fu, Y. Wan, Y. Sun, Z. Li and H. Lu, Polym. Chem., 2016, 7, 6375–6382 RSC.
- G. Liu, L. Zhou, Y. Guan, Y. Su and C.-M. Dong, Macromol. Rapid Commun., 2014, 35, 1673–1678 CrossRef CAS PubMed.
- X. Wu, L. Zhou, Y. Su and C.-M. Dong, Polym. Chem., 2015, 6, 6857–6869 RSC.
- L. Zhao, C. Xiao, L. Wang, G. Gai and J. Ding, Chem. Commun., 2016, 52, 7633–7652 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- L. Zhao, J. X. Ding, C. S. Xiao, P. He, Z. H. Tang, X. Pang, X. L. Zhuang and X. S. Chen, J. Mater. Chem., 2012, 22, 12319–12328 RSC.
- G. Liu, R. J. Ma, J. Ren, Z. Li, H. X. Zhang, Z. K. Zhang, Y. L. An and L. Shi, Soft Matter, 2013, 9, 1636–1644 RSC.
- H. Yang, R. Ma, J. Yue, C. Li, Y. Liu, Y. An and L. Shi, Polym. Chem., 2015, 6, 3837–3846 RSC.
- H. Yang, C. Zhang, C. Li, Y. Liu, Y. An, R. Ma and L. Shi, Biomacromolecules, 2015, 16, 1372–1381 CrossRef CAS PubMed.
- L. Zhao, C. Xiao, J. Ding, X. Zhuang, G. Gai, L. Wang and X. Chen, Polym. Chem., 2015, 6, 3807–3815 RSC.
- D. Lee, K. Choe, Y. Jeong, J. Yoo, S. M. Lee, J. H. Park, P. Kim and Y. C. Kim, RSC Adv., 2015, 5, 14482–14491 RSC.
- W. Tai, R. Mo, J. Di, V. Subramanian, X. Gu, J. B. Buse and Z. Gu, Biomacromolecules, 2014, 15, 3495–3502 CrossRef CAS PubMed.
- X. Hu, J. Yu, C. Qian, Y. Lu, A. R. Kahkoska, Z. Xie, X. Jing, J. B. Buse and Z. Gu, ACS Nano, 2017, 11, 613–620 CrossRef CAS PubMed.
- J. Yu, C. Qian, Y. Zhang, Z. Cui, Y. Zhu, Q. Shen, F. S. Ligler, J. B. Buse and Z. Gu, Nano Lett., 2017, 17, 733–739 CrossRef CAS PubMed.
- M. R. Molla, P. Prasad and S. Thayumanavan, J. Am. Chem. Soc., 2015, 137, 7286–7289 CrossRef CAS PubMed.
- Y. Sun, Y. Hou, X. Zhou, J. Yuan, J. Wang and H. Lu, ACS Macro Lett., 2015, 4, 1000–1003 CrossRef CAS.
- C. Wang, Q. Chen, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 8612–8615 CrossRef CAS PubMed.
- C. Wang, Y. Kang, K. Liu, Z. Li, Z. Wang and X. Zhang, Polym. Chem., 2012, 3, 3056–3059 RSC.
- H. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2009, 48, 6431–6434 CrossRef PubMed.
- P. Wilke and H. G. Börner, ACS Macro Lett., 2012, 1, 871–875 CrossRef CAS.
- P. Wilke, N. Helfricht, A. Mark, G. Papastavrou, D. Faivre and H. G. Börner, J. Am. Chem. Soc., 2014, 136, 12667–12674 CrossRef CAS PubMed.
- M. Meissler, A. Taden and H. G. Börner, ACS Macro Lett., 2016, 5, 583–587 CrossRef CAS.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- K. Aoi, K. Tsutsumiuchi and M. Okada, Macromolecules, 1994, 27, 875–877 CrossRef CAS.
- D. Pati, A. Y. Shaikh, S. Hotha and S. Sen Gupta, Polym. Chem., 2011, 2, 805–811 RSC.
- D. Pati, A. Y. Shaikh, S. Das, P. K. Nareddy, M. J. Swamy, S. Hotha and S. Sen Gupta, Biomacromolecules, 2012, 13, 1287–1295 CrossRef CAS PubMed.
- T. Stohr, A.-R. Blaudszun, U. Steinfeld and G. Wenz, Polym. Chem., 2011, 2, 2239–2248 RSC.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS PubMed.
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS PubMed.
- K.-S. Krannig and H. Schlaad, J. Am. Chem. Soc., 2012, 134, 18542–18545 CrossRef CAS PubMed.
- E. L. Dane and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 16255–16264 CrossRef CAS PubMed.
- D. Pati, S. Das, N. G. Patil, N. Parekh, D. H. Anjum, V. Dhaware, A. V. Ambade and S. Sen Gupta, Biomacromolecules, 2016, 17, 466–475 CrossRef CAS PubMed.
- S. Das, N. Parekh, B. Mondal and S. Sen Gupta, ACS Macro Lett., 2016, 5, 809–813 CrossRef CAS.
- C. Lavilla, G. Yilmaz, V. Uzunova, R. Napier, C. R. Becer and A. Heise, Biomacromolecules, 2017, 18, 1928–1936 CrossRef CAS PubMed.
- B. N. Tran, Q. T. Bui, Y. S. Jeon, H. S. Park and J.-H. Kim, Polym. Bull., 2015, 72, 2605–2620 CrossRef CAS.
- B. N. Tran, J. Y. Kim, Y.-C. Kim, Y. J. Kim and J.-H. Kim, J. Appl. Polym. Sci., 2016, 133, 43305 Search PubMed.
- M. Zhu, Y. Wu, C. Ge, Y. Ling and H. Tang, Macromolecules, 2016, 49, 3542–3549 CrossRef CAS.
- C. Bonduelle, F. Makni, L. Severac, E. Piedra-Arroni, C.-L. Serpentini, S. Lecommandoux and G. Pratviel, RSC Adv., 2016, 6, 84694–84697 RSC.
- R. I. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2011, 50, 4499–4502 CrossRef PubMed.
- R. I. Kühnle, D. Gebauer and H. G. Börner, Soft Matter, 2011, 7, 9616–9619 RSC.
- K. Wang, Y. Liu, C. Li, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, ACS Macro Lett., 2013, 2, 201–205 CrossRef CAS.
- M. Nguyen, J.-L. Stigliani, G. Pratviel and C. Bonduelle, Chem. Commun., 2017, 53, 7501–7504 RSC.
- Y. Gao and C.-M. Dong, Polym. Chem., 2017, 8, 1223–1232 RSC.
- W. Cheng, H. Cheng, S. Wan, X. Zhang and M. Yin, Chem. Mater., 2017, 29, 4218–4226 CrossRef CAS.
- J. Xiao, J. Tan, R. Jiang, X. He, Y. Xu, Y. Ling, S. Luan and H. Tang, Polym. Chem., 2017, 8, 7025–7032 RSC.
- S. Anantharaj and M. Jayakannan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2864–2875 CrossRef CAS.
- S. Saxena and M. Jayakanna, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3279–3293 CrossRef CAS.
- R. Aluri and M. Jayakannan, Biomacromolecules, 2017, 18, 189–200 CrossRef CAS PubMed.
- S. Saxena and M. Jayakanna, Biomacromolecules, 2017, 18, 2594–2609 CrossRef CAS PubMed.
- Y. Li, Y. Niu, D. Hu, Y. Song, J. He, X. Liu, X. Xia, Y. Lu and W. Xu, Macromol. Chem. Phys., 2015, 216, 77–84 CrossRef CAS.
- X. Liu, J. He, Y. Niu, Y. Li, D. Hu, X. Xia, Y. Lu and W. Xu, Polym. Adv. Technol., 2015, 26, 449–456 CrossRef CAS.
- Y. Niu, Y. Li, Y. Lu and W. Xu, RSC Adv., 2014, 4, 58432–58439 RSC.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- L. Ayres, K. Koch, P. H. H. M. Adams and J. C. M. van Hest, Macromolecules, 2005, 38, 1699–1704 CrossRef CAS.
- F. Fernández-Trillo, A. Dureault, J. P. M. Bayley, J. C. M. van Hest, J. C. Thies, T. Michon, R. Weberskirch and N. R. Cameron, Macromolecules, 2007, 40, 6094–6099 CrossRef.
- R. M. Conrad and R. H. Grubbs, Angew. Chem., Int. Ed., 2009, 48, 8328–8330 CrossRef CAS PubMed.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872–4874 RSC.
- H. Mori, I. Kato, M. Matsuyama and T. Endo, Macromolecules, 2008, 41, 5604–5615 CrossRef CAS.
- H. Mori, I. Kato and T. Endo, Macromolecules, 2009, 42, 4985–4992 CrossRef CAS.
- H. Mori, I. Kato, S. Saito and T. Endo, Macromolecules, 2010, 43, 1289–1298 CrossRef CAS.
- C. Luo, Y. Liu and Z. Li, Macromolecules, 2010, 43, 8101–8108 CrossRef CAS.
- C. Luo, B. Zhao and Z. Li, Polymer, 2012, 53, 1725–1732 CrossRef CAS.
- D. Yu, C. Luo, W. Fu and Z. Li, Polym. Chem., 2014, 5, 4561–4568 RSC.
- Y. Shen, G. Li, Y. Ma, D. Yu, J. Sun and Z. Li, Soft Matter, 2015, 11, 7502–7506 RSC.
- Z. Liu, J. Hu, J. Sun, G. He, Y. Li and G. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3573–3586 CrossRef CAS.
- N. Higashi, R. Sonoda and T. Koga, RSC Adv., 2015, 5, 67652–67657 RSC.
- N. Higashi, A. Hirata, S. Nishimura and T. Koga, Colloids Surf., B, 2017, 159, 39–46 CrossRef CAS PubMed.
- N. Higashi, D. Sekine and T. Koga, J. Colloid Interface Sci., 2017, 500, 341–348 CrossRef CAS PubMed.
- G. Hu, W. Li, Y. Hu, A. Xu, J. Yan, L. Liu, X. Zhang, K. Liu and A. Zhang, Macromolecules, 2013, 46, 1124–1132 CrossRef CAS.
- T. Maji, S. Banerjee, Y. Biswas and T. K. Mandal, Macromolecules, 2015, 48, 4957–4966 CrossRef CAS.
- T. Maji, S. Banerjee, A. Bose and T. K. Mandal, Polym. Chem., 2017, 8, 3164–3176 RSC.
- B. S. Lokitz, A. J. Convertine, R. G. Ezell, A. Heidenreich, Y. Li and C. L. McCormick, Macromolecules, 2006, 39, 8594–8602 CrossRef CAS.
- B. S. Lokitz, A. W. York, J. E. Stempka, N. D. Treat, Y. Li, W. L. Jarrett and C. L. McCormick, Macromolecules, 2007, 40, 6473–6480 CrossRef CAS.
- M. G. Kellum, A. E. Smith, S. K. York and C. L. McCormick, Macromolecules, 2010, 43, 7033–7040 CrossRef CAS.
- M. G. Kellum, C. A. Harris, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1104–1111 CrossRef CAS.
- J. Du and R. K. O'Reilly, Macromol. Chem. Phys., 2010, 211, 1530–1537 CrossRef CAS.
- J. Du, H. Willcock, N. S. Ieong and R. K. O'Reilly, Aust. J. Chem., 2011, 64, 1041–1046 CrossRef CAS.
- L. Saïdi, C. Vilela, H. Oliveira, A. J. D. Silvestre and C. S. R. Freire, Carbohydr. Polym., 2017, 169, 357–365 CrossRef PubMed.
- S. G. Roy and P. De, J. Appl. Polym. Sci., 2014, 131, 41084 CrossRef.
- S. Kumar, S. G. Roy and P. De, Polym. Chem., 2012, 3, 1239–1248 RSC.
- K. Bauri, S. G. Roy, S. Pant and P. De, Langmuir, 2013, 29, 2764–2774 CrossRef CAS PubMed.
- S. G. Roy, R. Acharya, U. Chatterji and P. De, Polym. Chem., 2013, 4, 1141–1152 RSC.
- K. Bauri, P. De, P. N. Shah, R. Li and R. Faust, Macromolecules, 2013, 46, 5861–5870 CrossRef CAS.
- S. Kumar, R. Acharya, U. Chatterji and P. De, Langmuir, 2013, 29, 15375–15385 CrossRef CAS PubMed.
- S. Kumar and P. De, Polymer, 2014, 55, 824–832 CrossRef CAS.
- U. Haldar, M. Nandi, B. Ruidas and P. De, Eur. Polym. J., 2015, 67, 274–283 CrossRef CAS.
- K. Bauri, S. Pant, S. G. Roy and P. De, Polym. Chem., 2013, 4, 4052–4060 RSC.
- S. G. Roy and P. De, Polym. Chem., 2014, 5, 6365–6378 RSC.
- B. Saha, K. Bauri, A. Bag, P. K. Ghorai and P. De, Polym. Chem., 2016, 7, 6895–6900 RSC.
- K. Bauri, B. Saha, J. Mahanti and P. De, Polym. Chem., 2017, 8, 7180–7187 RSC.
- M. Nandi, B. Maiti, K. Srikanth and P. De, Langmuir, 2017, 33, 10588–10597 CrossRef CAS PubMed.
- S. Kumar, R. Acharya, U. Chatterji and P. De, J. Mater. Chem. B, 2013, 1, 946–957 RSC.
- S. Kumar, R. Acharya, U. Chatterji and P. De, Polym. Chem., 2014, 5, 6039–6050 RSC.
- S. Kumar, V. Bheemireddy and P. De, Macromol. Biosci., 2015, 15, 1447–1456 CrossRef CAS PubMed.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, Polymers, 2017, 9, 137 CrossRef.
- M. Karbarz, K. Pyrzynska, J. Romanski, J. Jurczak and Z. Stojek, Polymer, 2010, 51, 2959–2964 CrossRef CAS.
- J. Romanski, M. Karbarz, K. Pyrzynska, J. Jurczak and Z. Stojek, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 542–550 CrossRef CAS.
- M. Mackiewicz, J. Romanski and M. Karbarz, RSC Adv., 2014, 4, 48905–48911 RSC.
- P. Xie, X. Liu, R. Cheng, Y. Wu and J. Deng, Ind. Eng. Chem. Res., 2014, 53, 8069–8078 CrossRef CAS.
- R. Cheng, J. Liu, P. Xie, Y. Wu and J. Deng, Polymer, 2015, 68, 246–252 CrossRef.
- S. G. Roy, U. Haldar and P. De, ACS Appl. Mater. Interfaces, 2014, 6, 4233–4241 CAS.
- S. G. Roy and P. De, Polymer, 2014, 55, 5425–5434 CrossRef CAS.
- S. G. Roy, A. Kumar and P. De, Polymer, 2016, 85, 1–9 CrossRef CAS.
- M. Casolaro, R. Cini, B. D. Bello, M. Ferrali and E. Maellaro, Biomacromolecules, 2009, 10, 944–949 CrossRef CAS PubMed.
- M. Casolaro, I. Casolaro, S. Bottari, B. D. Bello, E. Maellaro and K. D. Demadis, Eur. J. Pharm. Biopharm., 2014, 88, 424–433 CrossRef CAS PubMed.
- M. Casolaro, I. Casolaro and S. Lamponi, Eur. J. Pharm. Biopharm., 2012, 80, 553–561 CrossRef CAS PubMed.
- M. Casolaro and I. Casolaro, Gels, 2016, 2, 24 CrossRef.
- M. Casolaro and I. Casolaro, J. Biomed. Eng. Med. Device, 2016, 1, 111 Search PubMed.
- S. G. Roy, K. Bauri, S. Pal and P. De, Polym. Chem., 2014, 5, 3624–3633 RSC.
- B. Maiti, B. Ruidas and P. De, React. Funct. Polym., 2015, 93, 148–155 CrossRef CAS.
- U. Haldar, K. Bauri, R. Li, R. Faust and P. De, ACS Appl. Mater. Interfaces, 2015, 7, 8779–8788 CAS.
- E. G. Doyaguez, F. Parra, G. Corrales, A. F. -Mayoralas and A. Gallardo, Polymer, 2009, 50, 4438–4446 CrossRef CAS.
- S. Fujii, M. Kido, M. Sato, Y. Higaki, T. Hirai, N. Ohta, K. Kojio and A. Takahara, Polym. Chem., 2015, 6, 7053–7059 RSC.
- F. Sanda, F. Ogawa and T. Endo, Polymer, 1998, 39, 5543–5547 CrossRef CAS.
- M. Mackiewicz, J. Romanski, E. Drozd, B. G. -Bzura, P. Fiedor, Z. Stojek and M. Karbarz, Int. J. Pharm., 2017, 523, 336–342 CrossRef CAS PubMed.
- K. Kaniewska, J. Románski and M. Karbarz, RSC Adv., 2013, 3, 23816–23823 RSC.
- H. Zhao, F. Sanda and T. Masuda, Polymer, 2006, 47, 2596–2602 CrossRef CAS.
- J.-H. Kang, D. Asai, J.-H. Kim, T. Mori, R. Toita, T. Tomiyama, Y. Asami, J. Oishi, Y. T. Sato, T. Niidome, B. Jun, H. Nakashima and Y. Katayama, J. Am. Chem. Soc., 2008, 130, 14906–14907 CrossRef CAS PubMed.
- T.-H. Ku, M.-P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS PubMed.
- K. Bauri, A. Pan, U. Haldar, A. Narayanan and P. De, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2794–2803 CrossRef CAS.
- C. Luo, W. Fu, Z. Li and B. Zhao, Polymer, 2016, 101, 319–327 CrossRef CAS.
- K. Shoji, M. Nakayama, T. Koseki, K. Nakabayashi and H. Mori, Polymer, 2016, 97, 20–30 CrossRef CAS.
- K. E. Gebhardt, S. Ahn, G. Venkatachalam and D. A. Savin, Langmuir, 2007, 23, 2851–2856 CrossRef CAS PubMed.
- K. E. Gebhardt, S. Ahn, G. Venkatachalam and D. A. Savin, J. Colloid Interface Sci., 2008, 317, 70–76 CrossRef CAS PubMed.
- S. S. Naik and D. A. Savin, Macromolecules, 2009, 42, 7114–7121 CrossRef CAS.
- S. S. Naik, J. G. Ray and D. A. Savin, Langmuir, 2011, 27, 7231–7240 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |